
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deuteration of Pd-activated C(sp2)–H bonds in the solid state†
Alen
Bjelopetrović‡
 a,
Dajana
Barišić‡
ab,
Marina
Juribašić Kulcsár
*a,
Ivan
Halasz
a,
Manda
Ćurić
a and
Stipe
Lukin
*a
a,
Dajana
Barišić‡
ab,
Marina
Juribašić Kulcsár
*a,
Ivan
Halasz
a,
Manda
Ćurić
a and
Stipe
Lukin
*a
aDivision of Physical Chemistry, Ruđer Bošković Institute, Bijenička 54, HR-10000, Zagreb, Croatia. E-mail: marina.juribasic@irb.hr; stipe.lukin@irb.hr
bSelvita Ltd. Zagreb, Prilaz Baruna Filipovića 29, HR-10000 Zagreb, Croatia
First published on 31st August 2023
Abstract
We report mechanochemically-induced deuteration of Pd-activated aromatic C(sp2)–H bonds at ambient temperature under solvent-free conditions. Deuterium was sourced from cysteine-d4 to obtain mono- or dideuterated products from various aromatic palladacycles. Next to good deuteration yields, based on time-resolved in situ Raman monitoring and DFT calculations, we present a detailed view of the reaction course in the solid state. The obtained knowledge could lead to the broader application of this methodology for the deuteration of organics.
Introduction
Development of methods for direct and selective conversion of the carbon–hydrogen (C–H) bonds to the carbon–deuterium (C–D) bonds continues to be a challenge for the chemical and life sciences, as D-labeled compounds are widely used in mass spectrometry and chromatography, as well as in mechanistic and metabolic studies.1 Moreover, such compounds can alter the ADME (absorption, distribution, metabolism, and excretion) properties of the drug.2 The pinnacle of this is the recent approval of the first deuterated drug, deutetrabenazine, for treating chorea associated with Huntington's disease.2,3Over the past 15 years, metal-catalyzed functionalization of the C–H bond has emerged as a superior approach that significantly simplifies synthesis and allows a direct hydrogen isotope exchange (HIE) in the substrate.4a In contrast to heterogeneously catalyzed HIE, the homogeneous metal catalysis of HIE installs D atoms at specific positions relative to the directing group.1a,b,4a The most frequently used metal-based catalysts for HIE of C–H bonds are Ir, Rh, Pd, and Ru compounds, whereas common deuterium sources are D-rich organic molecules, heavy water, or deuterium gas.4
Since most of the current homogeneous HIE reactions proceed in solutions and require high temperature, we were prompted to explore mechanochemistry as an approach to HIE. Indeed, mechanochemistry5–9 is emerging as a sustainable and efficient synthetic approach that aims to avoid harsh reaction conditions while maintaining molecular and atomic mobility.8 Since the reactions are carried out in the solid state, problems such as solubility or costly workup can sometimes be avoided.
In addition, mechanochemically-induced processes allow reactions at ambient temperature, often in shorter reaction time than analogous processes in solution.6 Moreover, a recent discovery of the C–H bond activation and functionalization under mechanochemical conditions6 enabled the preparation of stable metalated products.7 We found it particularly interesting to use mechanochemically-prepared palladated products with the Pd-activated C–H bond(s) as precursors for the deuteration reaction. This way two steps performed in a ball mill would achieve HIE. Without using solvents and additional gaseous reactants, the deuteron source (D-source) should ideally be a molecule capable of breaking the C–Pd bond, and thereby transferring a deuteron to the carbon atom.
Here we report the first mechanochemical deuteration of the Pd-activated C(sp2)–H bonds in various precursors (Scheme 1) and show that mechanochemical milling is a viable and efficient approach for HIE reactions. Deuteration using a deuterated L-cysteine (Cys4D, Scheme 1) as the D-source was performed in the solid state at ambient temperature without using solvents and additional gaseous reactants. This work shows that cysteine can break the C–Pd bond and transfer a deuteron to the carbon atom. Using a simple strategy, various deuterated organic molecules were easily isolated in good to excellent yields.
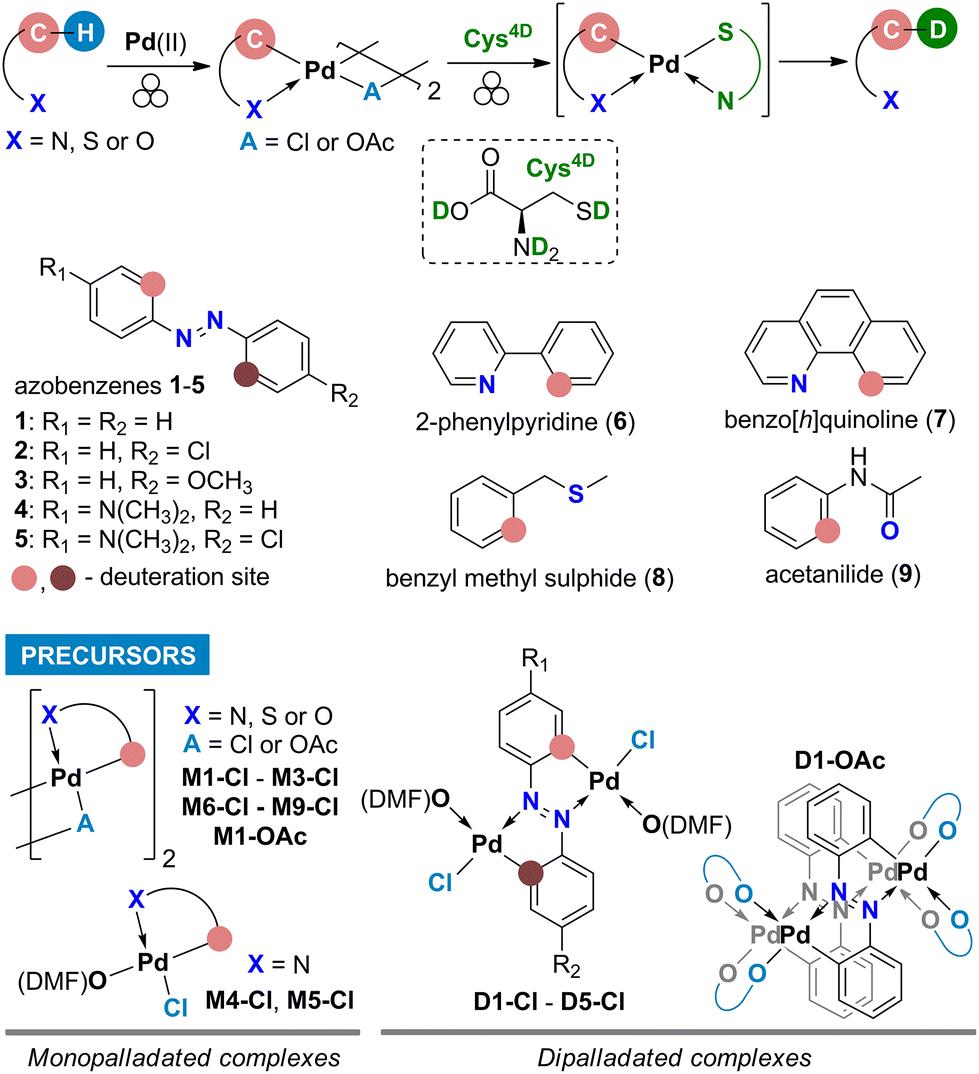 | ||
| Scheme 1 General reaction route for two-step mechanochemical HIE in aromatic substrates 1–9 along with the structures of their palladated precursors. | ||
Results and discussion
The deuterated L-cysteine (Cys4D) was chosen as a D-source for the palladated precursors because it is a solid at room temperature and a readily available biologically relevant amino acid that coordinates strongly to the Pd centers.10 Common inorganic D-sources (D2O, ND4Cl) and alcohols (ethanol-d) were tested and gave no deuteration whereas other deuterated solid thiols, aliphatic (1-adamantanethiol-d) as well as aromatic (4-chlorothiophenol-d), were less efficient regarding both the yield and the deuteration percentage (data in Table S1†).For the test reactions of palladacycles with Cys4D, we chose mono- and dicyclopalladated azobenzenes7b,11 as precursors (M1-Cl and D1-Cl, Scheme 2). These complexes exhibit strong characteristic Raman bands7b allowing in situ Raman monitoring of their reactions which is essential for a deeper understanding of the ball-milling reaction. Namely, collected Raman data provides an insight into the reaction course as well as the nature and reactivity of the intermediates.9
 | ||
| Scheme 2 Reaction conditions using three characteristic azobenzene complexes. | ||
The monopalladated precursor M1-Cl was reacted with Cys4D first. The reaction proceeded nicely if an excess of Cys4D was used. Azobenzene 1D was isolated in a 77% yield after 15 hours of milling M1-Cl with 4 equivalents (equiv.) of Cys4D per palladium center (Pd). 1H NMR data confirmed deuterium incorporation at the ortho-position to the azo group in 61% (Chart 1). To our delight, a doubly deuterated product 12D (84%) was obtained in 83% yield by reacting D1-Cl with 4 equiv. of Cys4D per Pd.
 | ||
| Chart 1 Products, reaction time and isolated yields of the mechanochemical deuteration of mono- and dipalladated chloride precursors (marked bellow each product in italic). Percentage of deuteration (average for all magnetically equivalent nuclei) is marked at the site of the D incorporation. aData for the acetate precursors M1-OAc and D1-OAc. bCombined yield for two isomeric monodeuterated products. | ||
The amount of the added Cys4D was screened on M1-Cl and D1-Cl using up to 20 equiv. of Cys4D per Pd (Table S1†). The optimal results were obtained for 4 equiv. of Cys4D per Pd. Thus, this ratio was further used. We note that reaction time was adjusted to maximize the yield of the isolated product.
After the successful reaction with M1-Cl, we applied the same methodology to unsymmetrically substituted monopalladated azobenzenes. Milling of the dimeric monopalladated complexes of 4-chloroazobenzene (M2-Cl) or 4-methoxyazobenzene (M3-Cl) with Cys4D led to a mixture of two monodeuterated products (2D or 3D, Chart 1). The formation of mixtures is attributed to the presence of two isomeric forms in the precursors M2-Cl and M3-Cl (Fig. S2–S4†). In contrast, monopalladated aminoazobenzenes are monomeric precursors obtained as a single isomer in which the aminophenyl ring is palladated. Therefore, products 4D and 5D were deuterated specifically in the ortho position of the aminosubstituted phenyl ring by 56 and 52%, respectively (Chart 1). These experiments proved the crucial role of the substituents on the palladated substrate for determining the deuteration site, as it depends on the site of palladation.
Control reactions of the native azobenzene 1 (1 equiv.) with Cys4D (8 equiv.) without PdCl2 or with PdCl2 added stoichiometrically (1 equiv.) or catalytically (5 mol%) gave no deuteration product in 15 h. Also, reactions of the precursor D1-Cl with glycine or L-alanine instead of Cys4D produced dipalladated complexes in which, according to the 1H NMR spectra of the reaction mixture (Fig. S52†), one molecule of the amino acid is coordinated to each Pd.
Adding an excess of D2O (8 equiv. per Pd) to the reaction of D1-Cl and Cys4D did not affect the yield or the deuteration outcome. However, adding an excess of base (8 equiv. per Pd), sodium acetate (NaOAc), or 4-(N,N-dimethylamino)pyridine (DMAP) strongly hindered the reaction ending after 15 hours of milling in traces of the isolated 12D (Table S1†).
Deuterations of monomeric complexes M4-Cl, M5-Cl, and D1-Cl–D5-Cl are liquid-assisted grinding (LAG) reactions since these palladacycles contain one (M4-Cl and M5-Cl) or two (D1-Cl–D5-Cl) molecules of DMF that are released when Cys4D binds to Pd. To test the DMF effect, we used the LAG reaction of the dimeric M1-Cl (that does not contain DMF). 1 equiv. of DMF per Pd was added to mimic the reactions of M4-Cl and M5-Cl. The isolated yield of 1D was lowered to 40%, and the deuteration percentage was 60% (compared to 61% without DMF) (Table S1†). This result can be ascribed partly to the stickier reaction mixture due to the presence of DMF and to the basic properties of DMF.
To extend our concept to other aromatics, we used chloride palladacycles of benzo[h]quinoline (6), 2-phenylpyridine (7), benzyl methyl sulfide (8), and acetanilide (9) containing N, S, or O donor atoms as precursors (Scheme 1). As summarized in Chart 1, our approach proved efficient allowing 61–71% single-site deuteration of these commonly used aromatics.
To explore the possibility of multiple deuteration in the same molecule, we used dipalladated azobenzenes as precursors because these compounds contain two Pd-activated C–H bonds. We were pleased to see that ortho-deuteration of both phenyl rings occurred, and we could isolate targets 12D–52D in 65–84% yield (Chart 1). NMR data confirmed that D was incorporated at both sites of palladation.
Since the acetate palladated complexes are also frequently used, we attempted the deuteration using M1-OAc and D1-OAc as precursors under the same experimental conditions as for the chloride precursors. These reactions resulted in 34% and 36% yields of 1D and 12D, respectively (Chart 1). The product 1D obtained from the acetate precursor M1-OAc was only 34% ortho-deuterated. In contrast, the deuteration of product 12D obtained from D1-OAc was comparable to that of the product obtained from the reaction with the chloride precursor D1-Cl.
At the end of the reaction, the palladium metal is retrieved from the reaction mixture as a highly insoluble and amorphous complex with the empirical formula Pd(Cys3D)2 (see ESI† for details). Recovery of up to 85% of the elemental palladium was achieved by heating the solid Pd(Cys3D)2 above 800 °C.
Raman monitoring
To shed some light on the reaction course, we used Raman monitoring and density-functional theory (DFT) calculations.The chloride monopalladated azobenzene (M1-Cl) was studied first. M1-Cl is a dimer with the most prominent bands in Raman spectra at 1389 and 1209 cm−1 belonging dominantly to ν(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and ν(C–N) vibrations, respectively (Fig. 1a). These bands shift to 1380 and to 1200 cm−1 within 22 min of milling with Cys4D, as the first intermediate M1-1 is formed by ligand exchange in M1-Cl where cysteine becomes bound to Pd via its amino and the deprotonated thiol groups (Scheme 3). 1H NMR spectrum of the reaction mixture recorded ex situ after 75 min of milling supports its presence (Fig. S63 and S64†). According to the DFT data, isomer M1-1 is more stable than M1-2, suggesting that M1-1 is formed in the reaction (Scheme 3).
N) and ν(C–N) vibrations, respectively (Fig. 1a). These bands shift to 1380 and to 1200 cm−1 within 22 min of milling with Cys4D, as the first intermediate M1-1 is formed by ligand exchange in M1-Cl where cysteine becomes bound to Pd via its amino and the deprotonated thiol groups (Scheme 3). 1H NMR spectrum of the reaction mixture recorded ex situ after 75 min of milling supports its presence (Fig. S63 and S64†). According to the DFT data, isomer M1-1 is more stable than M1-2, suggesting that M1-1 is formed in the reaction (Scheme 3).
 | ||
| Fig. 1 (a) Selected Raman spectra and (b) 2D plot of the time-resolved Raman monitoring of the reaction of M1-Cl (0.14 mmol) with Cys4D (1.12 mmol). | ||
 | ||
| Scheme 3 Ligand exchange followed by deuteration of M1-Cl. M1-2 is not formed. | ||
As the reaction progressed, Raman data indicated the formation of the deuterated azobenzene 1D (Fig. 1b). Namely, bands at 1145 and 1180 cm−1 belonging to ν(C–N) vibrations and the bands at 1436, 1470 and 1488 cm−1 of ν(NN) vibrations appeared. The final in situ Raman spectrum agrees with the ex situ data for 1D (Fig. S84†).
Raman monitoring of the reaction of D1-Cl with Cys4D (Fig. 2b) revealed the presence of several intermediates preceding the formation of 12D (Chart 1). The broad band observed in the first Raman spectrum of the reaction is at 1309 cm−1 and is attributed to the ν(NN) bond of the parent compound D1-Cl12a and/or the first intermediate D1-1 (Scheme 4). According to the DFT calculations, the Raman spectra of the starting D1-Cl and its planar derivative with two coordinated cysteines D1-1 are similar, as are the spectra of M1-Cl and M1-1.
 | ||
| Fig. 2 (a) Selected Raman spectra and (b) 2D plot of the time-resolved Raman monitoring of the reaction of D1-Cl (0.14 mmol) with Cys4D (1.12 mmol). | ||
 | ||
| Scheme 4 Proposed reaction route from D1-Cl to 12D. | ||
At ca. 30 min of milling, isomerization of D1-1 and formation of the bridged intermediate are apparent from the emergence of a strong band at 1375 cm−1, consistent with structural changes induced by the breaking of one Pd–Nazo bond in D1-1 and positioning of both Pd atoms on the same side of the azobenzene ligand in the bridged intermediates (Scheme 4).12a According to DFT calculations, two structurally similar bridged complexes B1-1 and B1-2, that differ in the coordination mode of the non-bridging cysteine ligand, are most likely formed. Intensity loss and shift of the Raman band to 1389 cm−1 is observed after 2 hours and is related to the first deuteration and formation of M1D-2. M1D-2 enters the second deuteration which leads to the product 12Dortho-deuterated 84% (Chart 1). We note that due to the structure of the most stable isomers of D1-1 and B1-1, formation of M1D-2 is predicted even though it is less stable than M1D-1. Isomerization of M1D-2 to M1D-1 would include breaking of the Pd–N bond and is not expected.
Insight into the dynamics of the formation of intermediates and 12D, obtained by in situ Raman monitoring, shows that the second deuteration is the slowest step in the deuteration of D1-Cl (Fig. 2b). According to our previous work,11,12 Raman spectra of the monopalladated complexes M1-Cl, M1D-1, M1D-2 and of the bridged species B1-1 and B1-2 are remarkably similar. However, the higher observed consumption rate of the bridged dipalladated complex than of the monopalladated complex allowed us to distinguish their ν(NN) bands at 1378 and 1389 cm−1, respectively, and thus to characterize each deuteration step.
DFT calculations
In situ and ex situ monitoring suggested a complex reaction route to the deuterated products but could not determine which of the available D-sources would most likely engage in the reaction. Moreover, Raman data gave limited information about the involved reaction intermediates for D1-Cl. We thus modeled the deuteration of mono- and dipalladated azobenzene precursors.After elaborate analysis of the possible options for the deuteration of the monopalladated azobenzenes M1-1 (Fig. 3) and M1-2 (Table S3†), Cys4D·DCl was identified as the preferred D-source for the deuteration of the chloride precursors, whereas Cys4D is the D-source for the acetate precursors and at the beginning of the reaction. Calculations show that both ND3+ and COOD groups can deuterate the palladated ligand. The preference for one over the other depends on the intra- and intermolecular interactions near the deuteration site and is mainly on the side of the COOD as a donor group.
 | ||
| Fig. 3 D-transfer to M1-1 from various D-sources. B3LYP-D3/6-311+G**/SDD(Pd)/gas phase free energies reported relative to M1-1 (in kcal mol−1). | ||
The calculated free-energy profile for the deuteration of M1-1 by various D-sources (Fig. 3) agrees with the experimental findings. Deuteration by Cys4D·DCl is a less energy-demanding than deuteration using Cys4D as a D-source. The lowest-lying transition states for deuteration of M1-1 are at 9.8 kcal mol−1 for Cys4D·DCl and at 17.5 kcal mol−1 for Cys4D, which agrees with the reaction of M1-Cl being faster than the reaction using M1-OAc as the precursor.
In the S-bridged dipalladated cysteine complexes, the five- and six-membered palladated rings have to be deuterated to obtain 12D, Scheme 5. Two paths can be envisaged depending on which ring is broken first, and both were considered. Both paths end in the doubly ortho-palladated 12D.
 | ||
| Scheme 5 Paths leading from the dipalladated complex to the dideuterated azobenzene. | ||
In path I, the phenyl group involved in the six-membered palladated ring of the S-bridged dipalladated complex is deuterated first, Scheme 6. Following this path, B1-1 transforms to the monodeuterated intermediate M1D-2 with the conserved five-membered cyclopalladated ring via transition state TS3-1. After the elimination of one palladium center, M1D-2 undergoes the second deuteration that breaks the five-membered ring to give 12D after Pd elimination.
 | ||
| Scheme 6 Deuteration of B1-1 by Cys4D·DCl to 12D following path I and path II. Experimentally characterized compounds are highlighted in dashed rectangles. | ||
Path II starts with the deuteration of the five-membered cyclopalladated ring in the S-bridged dipalladated complex, Scheme 6. Monopalladated S-bridged intermediate with one empty coordination place (I4-1) is formed. Cys4D or chloride can fill this place and form complexes I5-1 or I5-2, respectively. In addition, I5-2 can be deprotonated in presence of Cys4D and form ion pair I5-3. This process is spontaneous for the tested geometries both in the gas phase and using the PCM model for DMF. All three species (I5-1–I5-3) can enter the second D-transfer and after Pd elimination form 12D.
The first step of the path I is less energy-demanding than the first step of the path II for B1-1 (Fig. 4) and B1-2 (Fig. S138†). Deuteration of the planar D1-1 is the most energetically-demanding process that would occur via transition state TS1 located ca. 10 kcal mol−1 higher in energy than transition states TS3-1 and TS3-2 in the path I and is thus not expected to occur. Therefore, the path I is preferred.
 | ||
| Fig. 4 Free-energy profile for deuteration of B1-1 by Cys4D·DCl to 12D. B3LYP-D3/6-311+G**/SDD(Pd)/gas phase data (in kcal mol−1) reported relative to B1-1. Palladium is eliminated as PdCl(Cys3D)(Cys4D). | ||
To account for the possible influence of the reaction medium, we have used the B3LYP-D3/6-311+G**/SDD(Pd) level of theory with PCM modeling of propanoic acid (ε = 3.44) and DMF (ε = 37.22) as solvents. We note that the examined reaction mixtures might likely have a lower dielectric constant than DMF and thus, PCM-DMF data should be regarded as a guide for how a polar medium can influence the reaction.
Data obtained by PCM calculations for deuteration of M1-1 show that the difference in the free energies of the transition states for the D-transfer by Cys4D·DCl and Cys4D got smaller as the medium polarity increased (Table S2 and Fig. S136†). This difference is 7.7 kcal mol−1 in the gas phase (free energies of the transition states are 9.8 kcal mol−1 for Cys4D·DCl and 17.5 kcal mol−1 for Cys4D, Fig. 3), 4.8 kcal mol−1 in the propanoic acid (14.2 for Cys4D·DCl and 19.0 kcal mol−1 for Cys4D), and 0.3 kcal mol−1 in DMF (18.1 kcal mol−1 for Cys4D·DCl and 18.4 kcal mol−1 for Cys4D). Results suggest that Cys4D, along with the still preferred Cys4D·DCl, might also act as a D-source for deuteration in DMF.
To further test the presented results in the gas phase, we have performed PCM-DMF calculations for the deuteration of D1-Cl (Fig. S138†). Free energies of the reactants, transition states and products are lifted relative to B1-1. General trend that was observed in the gas phase is preserved in DMF. Data agree with the facile reaction that still shows a small preference to the path I leading from D1-Cl to 12D.
In the gas phase PdCl(Cys3D)(Cys4D) is by 5.5 kcal mol−1 more stable than the monomeric form of Pd(Cys3D)2 (Chart S1†). However, Pd(Cys3D)2 is preferred by 1.0 and 6.1 kcal mol−1 in propanoic acid and DMF, respectively. Thus, PdCl(Cys3D)(Cys4D) could form during reaction and change to the isolated solid Pd(Cys3D)2 during work-up.
Experimental
Experimental details, spectral data for new compounds, in situ Raman experiments and additional computational data and details can be found in the ESI.†Ball-milling reactions
Experiments were performed using an IST500 mixer mill operating at 30 Hz with a built-in fan. Deuteration of precursors was carried out sequentially under analogous conditions at an ambient temperature of 22 ± 2 °C. These reactions were performed in transparent 14 mL poly(methyl methacrylate (PMMA) jars that allowed in situ Raman monitoring. One milling ball made of zirconium(IV)oxide (ZrO2, 12 mm, 4.5 g) was used. Reactions were performed using approximately 225 mg of the reaction mixture.In situ Raman monitoring without interrupting the milling process was performed using a portable Raman system with PD-LD (now Necsel) BlueBox laser source with an excitation wavelength of 785 nm, equipped with a B&W-Tek BAC102 fiber-optic Raman probe and coupled to OceanOptics Maya2000Pro spectrometer. The most intense band of the PMMA vessel at 2955 cm−1, corresponding to the ν(C–H) bond, was used as a scaling reference to subtract its contribution from the experimental Raman spectra. The asymmetric least squares (AsLS) method13a was used to correct the spectra baseline. To extract the spectra of observed species, multivariate curve resolution with alternative least squares method (MCR-ALS)13b in MATLAB was used. The literature data14 and quantum chemical calculations were used to assign experimental spectra.
Deuteration of precursors
0.14 mmol of the dimeric monopalladated (M1-Cl–M3-Cl, M6-Cl–M9-Cl, M1-OAc) or the monomeric dipalladated complexes (D1-Cl–D5-Cl, D1-OAc) was milled with 1.12 mmol of Cys4D (4 equiv. per Pd). 0.28 mmol of the precursor and 1.12 mmol of Cys4D (4 equiv. per Pd) were used in reactions of the monomeric monopalladated complexes (M4-Cl and M5-Cl). Completeness of the reactions was confirmed by in situ Raman monitoring and ex situ1H NMR spectra of the reaction mixtures. No change in the reaction mixture was observed after milling was stopped. Reaction duration, isolated yields, and deuteration site and the appropriate percentage are given in Chart 1.Deuteration of thiols
3.0 g of native L-Cys was dissolved in 16 mL of D2O. The solution was refluxed for 1 h, evaporated under reduced pressure, and cooled to ambient temperature. This procedure was repeated twice each time using a new portion of D2O. The deuteration was confirmed by IR and Raman spectroscopies (Fig. S83, S91 and S92†).1.0 g of native 1-adamantanethiol or 4-chlorothiophenol was dissolved in 15 mL ethanol-d (EtOD). The solution was refluxed for 1 h, evaporated under reduced pressure, and cooled to ambient temperature. This procedure was repeated twice each time using a new portion of EtOD. The deuteration was confirmed by IR and 1H NMR spectroscopies (Fig. S17, S18, S93 and S94†).
DFT calculations
Calculations were carried out in Gaussian1615 using a functional with Grimme's empirical correction (B3LYP-D3).16 Pd atoms were modeled by the Stuttgart–Dresden (SDD) pseudopotential and the accompanying SDD basis set.17 Standard 6-311+G** basis set was used for C, H/D, N, O, S and Cl atoms. Other functionals (ωB97x-D,18 B3LYP), basis sets (6-31G*/SDD(Pd) and def2tzvp) and polarizable continuum modeling19 (DMF and propanoic acid) were also tested (see Table S2†). Full geometry optimizations were accompanied by vibrational frequency calculations that identified calculated stationary points as minima (reactants and products) or first-order saddle points (transition states). Nature of the transition states was confirmed by intrinsic reaction coordinate (IRC) searches20 followed by full geometry optimizations. Reported energies are free energies given at 298.15 K and 1 atm. No additional corrections were applied. Data for deuteration of the mono- and dipalladated azobenzene are reported relative to M1-1 and B1-1, respectively.Conclusions
We have developed a novel approach for the deuteration of C(sp2)–H bonds by reacting palladacycles with Cys4D in a ball mill at ambient temperature and without solvents. The advantage of this simple approach is that the reactions are carried out in the solid state, thus avoiding solubility problems and undesirable solvent effects. The incorporation of one or two deuterons at predictable sites of structurally diverse aromatics makes this method practical and offers a fresh approach to an old demand. The presented work thus paves the way for similar concepts for solid-state hydrogen exchange reactions that could be extended to various transition metals and substrates with sp2 and sp3 C–H bonds, which will be the focus of our future work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Croatian Science Foundation (grants IP-2019-04-9951 and IP-2020-02-1419). Computations were done on the Isabella cluster at SRCE, Zagreb, Croatia. We thank Dr Darko Babić for his valuable assistance and discussion, Dr Tatjana Šumanovac and Dr Senada Muratović for experimental help, and Dr Sunčica Roca and Ms NIkolina Višić for a part of the NMR measurements.References
- (a) J. Atzrodt and V. Derdau, Pd- and Pt-catalyzed H/D exchange methods and their application for internal MS standard preparation from a Sanofi-Aventis perspective, J. Labelled Compd. Radiopharm., 2010, 53, 674–685 CrossRef CAS; (b) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, The Renaissance of H/D Exchange, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed; (c) J. Yang, Deuterium: Discovery and Applications in Organic Chemistry, Elsevier, Amsterdam, 2016 Search PubMed; (d) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Deuterium- and Tritium-Labelled Compounds: Applications in the Life Sciences, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed; (e) G. C. Lloyd-Jones and M. P. Muñoz, Isotopic labelling in the study of organic and organometallic mechanism and structure: an account, J. Labelled Compd. Radiopharm., 2007, 50, 1072–1087 CrossRef CAS; (f) S. A. Blum, K. L. Tan and R. G. Bergman, Application of Physical Organic Methods to the Investigation of Organometallic Reaction Mechanisms, J. Org. Chem., 2003, 68, 4127–4137 CrossRef CAS PubMed; (g) M. Gómez-Gallego and M. A. Sierra, Kinetic Isotope Effects in the Study of Organometallic Reaction Mechanisms, Chem. Rev., 2011, 111, 4857–4963 CrossRef PubMed.
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, Applications of Deuterium in Medicinal Chemistry, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed.
- C. Schmidt, First deuterated drug approved, Nat. Biotechnol., 2017, 35, 493–494 CrossRef CAS PubMed.
- (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, C–H Functionalisation for Hydrogen Isotope Exchange, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed; (b) W. J. Kerr, D. M. Lindsay, P. K. Owens, M. Reid, T. Tuttle and S. Campos, Site-Selective Deuteration of N-Heterocycles via Iridium-Catalyzed Hydrogen Isotope Exchange, ACS Catal., 2017, 7, 7182–7186 CrossRef CAS; (c) S. Chen, G. Song and X. Li, Chelation-assisted rhodium hydride-catalyzed regioselective H/D exchange in arenes, Tetrahedron Lett., 2008, 49, 6929–6932 CrossRef CAS; (d) H. Xu, M. Liu, L.-J. Li, Y.-F. Cao, J.-Q. Yu and H.-X. Dai, Palladium-Catalyzed Remote meta-C–H Bond Deuteration of Arenes Using a Pyridine Template, Org. Lett., 2019, 21, 4887–4891 CrossRef CAS PubMed; (e) L. Piola, J. A. Fernandez-Salas, S. Manzini and S. P. Nolan, Regioselective ruthenium catalysed H–D exchange using D2O as the deuterium source, Org. Biomol. Chem., 2014, 12, 8683–8688 RSC.
- (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: opportunities for new and cleaner synthesis, Chem. Soc. Rev., 2012, 41, 413–417 RSC; (b) T. Friščić, Supramolecular concepts and new techniques in mechanochemistry: cocrystals, cages, rotaxanes, open metal-organic frameworks, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC; (c) G.-W. Wang, Mechanochemical organic synthesis, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC; (d) D. Braga, L. Maini and F. Grepioni, Mechanochemical preparation of co-crystals, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC; (e) N. R. Rightmire and T. P. Hanusa, Advances in organometallic synthesis with mechanochemical methods, Dalton Trans., 2016, 45, 2352–2362 RSC.
- (a) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, Metal-Mediated and Metal-Catalyzed Reactions under Mechanochemical Conditions, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS; (b) J. G. Hernández and C. Bolm, Altering Product Selectivity by Mechanochemistry, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed; (c) J. G. Hernandez, C–H Bond Functionalization by Mechanochemistry, Chem. – Eur. J., 2017, 23, 17157–17165 CrossRef CAS PubMed; (d) S. Zhao, Y. Li, C. Liu and Y. Zhao, Recent advances in mechanochemical C–H functionalization reactions, Tetrahedron Lett., 2018, 59, 317–324 CrossRef CAS.
- (a) M. Juribašić, K. Užarević, D. Gracin and M. Ćurić, Mechanochemical C–H bond activation: rapid and regioselective double cyclopalladation monitored by in situ Raman spectroscopy, Chem. Commun., 2014, 50, 10287–10290 RSC; (b) A. Bjelopetrović, S. Lukin, I. Halasz, K. Užarević, I. Đilović, D. Barišić, A. Budimir, M. Juribašić Kulcsár and M. Ćurić, Mechanism of Mechanochemical C–H Bond Activation in an Azobenzene Substrate by PdII Catalysts, Chem. – Eur. J., 2018, 24, 10672–10682 CrossRef PubMed; (c) D. Barišić, I. Halasz, A. Bjelopetrović, D. Babić and M. Ćurić, Mechanistic Study of the Mechanochemical PdII-Catalyzed Bromination of Aromatic C–H Bonds by Experimental and Computational Methods, Organometallics, 2022, 41, 1284–1294 CrossRef; (d) D. Barišić, M. Pajić, I. Halasz, D. Babić and M. Ćurić, Mechanochemical halogenation of unsymmetrically substituted azobenzenes, Beilstein J. Org. Chem., 2022, 18, 680–687 CrossRef PubMed.
- S. Lukin, M. Tireli, T. Stolar, D. Barišić, M. V. Blanco, M. Di Michiel, K. Užarević and I. Halasz, Isotope Labeling Reveals Fast Atomic and Molecular Exchange in Mechanochemical Milling Reactions, J. Am. Chem. Soc., 2019, 141, 1212–1216 CrossRef CAS PubMed.
- (a) S. Lukin, L. Germann, T. Friščić and I. Halasz, Toward Mechanistic Understanding of Mechanochemical Reactions Using Real-Time In Situ Monitoring, Acc. Chem. Res., 2022, 55, 1262–1277 CrossRef CAS PubMed; (b) S. Lukin, K. Užarević and I. Halasz, Raman spectroscopy for real-time and in situ monitoring of mechanochemical milling reactions, Nat. Protoc., 2021, 16, 3492–3521 CrossRef CAS PubMed.
- (a) N. Yoshinari, N. Kuwamura, T. Kojima and T. Konno, Development of coordination chemistry with thiol-containing amino acids, Coord. Chem. Rev., 2023, 474, 214857 CrossRef CAS; (b) V. K. Jain and L. Jain, The chemistry of tri- and high-nuclearity palladium(II) and platinum(II) complexes, Coord. Chem. Rev., 2010, 254, 2848–2903 CrossRef CAS.
- (a) M. Juribašić, A. Budimir, S. Kazazić and M. Ćurić, Dicyclopalladated Complexes of Asymmetrically Substituted Azobenzenes: Synthesis, Kinetics and Mechanisms, Inorg. Chem., 2013, 52, 12749–12757 CrossRef PubMed; (b) A. Bjelopetrović, M. Robić, D. Babić, M. Juribašić Kulcsár and M. Ćurić, Facile Mechanochemical Anion Substitution in Cyclopalladated Azobenzenes, Organometallics, 2019, 38, 4479–4484 CrossRef; (c) A. Bjelopetrović, D. Barišić, Z. Duvnjak, I. Džajić, M. Juribašić Kulcsár, I. Halasz, M. Martinez, A. Budimir, D. Babić and M. Ćurić, A Detailed Kinetico-Mechanistic Investigation on the Palladium C–H Bond Activation in Azobenzenes and Their Monopalladated Derivatives, Inorg. Chem., 2020, 59, 17123–17133 CrossRef PubMed.
- (a) M. Juribašić Kulcsár, I. Halasz, A. Budimir, K. Užarević, S. Lukin, A. Monas, F. Emmerling, J. Plavec and M. Ćurić, Reversible Gas-Solid Ammonia N–H Bond Activation Mediated by an Organopalladium Complex, Inorg. Chem., 2017, 56, 5342–5351 CrossRef PubMed; (b) M. Juribašić, I. Halasz, D. Babić, D. Cinčić, J. Plavec and M. Ćurić, Aging and Ball-Milling as Low-Energy and Environmentally Friendly Methods for the Synthesis of Pd(II) Photosensitizers, Organometallics, 2014, 33, 1227–1234 CrossRef.
- (a) P. H. C. Eilers and H. F. M. Boelens, Baseline Correction with Asymmetric Least Squares Smoothing, Leiden University Medical Center Report, 2015 Search PubMed; (b) J. Jaumot, A. de Juan and R. Tauler, MCR-ALS GUI 2.0: New features and applications, Chemom. Intell. Lab. Syst., 2015, 140, 1–12 CrossRef CAS.
- (a) N. Biswas and S. Umapathy, Structures, Vibrational Frequencies, and Normal Modes of Substituted Azo Dyes: Infrared, Raman, and Density Functional Calculations, J. Phys. Chem. A, 2000, 104, 2734–2745 CrossRef CAS; (b) G. Socrates, Infrared and Raman Characteristic Group Frequencies, J. Wiley & Sons, Chichester, 3rd edn, 2001 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef.
- K. Fukui, The path of chemical reactions - the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi00932g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2023 |