DOI:
10.1039/D3QI00861D
(Research Article)
Inorg. Chem. Front., 2023,
10, 5337-5346
Highly stable drone-shaped lanthanide clusters: structure, assembly mechanism, and crystalline–amorphous transitions†
Received
9th May 2023
, Accepted 12th July 2023
First published on 13th July 2023
Abstract
Great progress has been made in the design and synthesis of high-nuclear lanthanide clusters with impressive structural connections, high stability, and special topologies, but still lack reliable regularity. Herein, a drone-shaped lanthanide cluster (1) was obtained by mixing stoichiometric ratios of 2-pyridinecarbohydrazide, 2,3,4-trihydroxybenzaldehyde, and a mixed-metal dysprosium salt (Dy(NO3)3·6H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DyCl3·6H2O) under solvothermal conditions. Notably, a four-coordinated bridging chloride ion (μ4-Cl−) was found in the structure of cluster 1. The eight ligands were distributed on the periphery of the cluster core and tightly wrapped around the cluster core. The freely rotatable pyridine ring at the end of the ligand can effectively prevent the attack of solvent molecules on the cluster core and promote the stability of cluster 1. The self-assembly process of cluster 1 was tracked by time-dependent high-resolution electrospray-ionization mass spectrometry, and the trend of various intermediate molecular ion peaks over time was identified. We speculated that the possible self-assembly mechanism of cluster 1 was L + Dy → DyL → Dy2L2 → Dy3L3 → Dy4L4 → 2Dy4L4 → Dy8L8 → Dy9L8. This study finds that the self-assembly mechanism of drone-shaped lanthanide clusters can guide the construction of high-nuclear lanthanide clusters with special shapes. Interestingly, cluster 1 can undergo obvious color transitions in air and under UV light conditions. IR and PXRD results indicated that the above color change was caused by a transition from the crystalline to amorphous state and did not trigger the change in functional groups. This work explores the crystalline-to-amorphous transition of high-nuclear lanthanide clusters. Magnetic studies revealed that cluster 1 exhibited distinct SMM behavior under zero-field conditions. We provided a vivid example of the synthesis of high-nuclear lanthanide clusters with special shapes and provided insights into building multifunctional lanthanide clusters.
:DyCl3·6H2O) under solvothermal conditions. Notably, a four-coordinated bridging chloride ion (μ4-Cl−) was found in the structure of cluster 1. The eight ligands were distributed on the periphery of the cluster core and tightly wrapped around the cluster core. The freely rotatable pyridine ring at the end of the ligand can effectively prevent the attack of solvent molecules on the cluster core and promote the stability of cluster 1. The self-assembly process of cluster 1 was tracked by time-dependent high-resolution electrospray-ionization mass spectrometry, and the trend of various intermediate molecular ion peaks over time was identified. We speculated that the possible self-assembly mechanism of cluster 1 was L + Dy → DyL → Dy2L2 → Dy3L3 → Dy4L4 → 2Dy4L4 → Dy8L8 → Dy9L8. This study finds that the self-assembly mechanism of drone-shaped lanthanide clusters can guide the construction of high-nuclear lanthanide clusters with special shapes. Interestingly, cluster 1 can undergo obvious color transitions in air and under UV light conditions. IR and PXRD results indicated that the above color change was caused by a transition from the crystalline to amorphous state and did not trigger the change in functional groups. This work explores the crystalline-to-amorphous transition of high-nuclear lanthanide clusters. Magnetic studies revealed that cluster 1 exhibited distinct SMM behavior under zero-field conditions. We provided a vivid example of the synthesis of high-nuclear lanthanide clusters with special shapes and provided insights into building multifunctional lanthanide clusters.
Introduction
The design and synthesis of lanthanide clusters with specific structural connections, impressive topologies, and rich properties are favored by chemists.1–5 Many high-nuclear lanthanide clusters with elegant structures have emerged.6–10 For example, in 2017, Zheng and co-workers reported a giant wheel-like structure Gd140 with a diameter of 6.0 nm and exhibiting 10-fold symmetry.6 In 2014, Zheng's group finally obtained Gd104 with a spherical structure by inducing the hydrolysis of Ln(ClO4)3 in acetate.7 In 2018, Bu and his colleagues achieved the synthesis of a hamburger-shaped lanthanide cluster Dy76, which is very rare among high-nuclear lanthanide clusters, by using a mixed anion template; it can be regarded as an assembly of two Dy48 clusters.8 In 2015, Zhao et al. formed eight CO32− ions through in situ ligand decomposition and finally obtained the cluster Ln60 with a cage structure.11 The above-mentioned high-nuclear lanthanide clusters have certain structural aesthetics, clear chemical composition, and rich chemical and physical properties, indicating application prospects in the fields of molecular magnetism, solid-state lighting, and biological diagnosis treatment.12–17 Although great progress has been made in constructing high-nuclear lanthanide clusters with impressive structures and rich properties, examples that deeply dissect the self-assembly mechanism of high-nuclear lanthanide clusters are still scarce.18,19 Given that high-valent lanthanide metal ions with large coordination radii often have difficulty in forming stable M–L coordination bonds with ligands, they usually show poor stability.20–22 Usually, high-nuclear lanthanide clusters form in a “one-pot” closed system with a certain temperature and pressure.23,24 Therefore, capturing and identifying extremely short-lived and complex intermediate fragments in the formation process are extremely challenging.
The formation process of high-nuclear lanthanide clusters is very complex and their intermediates cannot be easily captured and identified. Nevertheless, a series of progress have been made.25–28 For example, in 2022, Zheng and co-workers exemplified the assembly of a 60-metal complex of Er(III) with histidine and demonstrated the rational assembly of polynuclear lanthanide hydroxide clusters synthesized under the guidance of the combination of I− and CO32− as mixed templates.25 In 2023, Tong et al. used N-methylbenzimidazole-2-methanol to react with Ln(III) under solvothermal conditions to obtain a spherical high-nuclear lanthanide cluster Ln32, and the formation process of Ho32 was monitored by high-resolution electrospray-ionization mass spectrometry (HRESI-MS) analysis.26 Our research group has also made some progress in exploring the formation of high-nuclear lanthanide clusters.29–34 For example, in 2019, we obtained Dy10 nanoclusters with multiple relaxation behaviors by using Schiff base organic ligands having multidentate chelation coordination sites. After exploring their formation process, a step-by-step assembly mechanism was proposed by HRESI-MS.29 We then constructed a cage-shaped cluster Dy14 by connecting disc-shaped lanthanide clusters with strong hydrogen bonds and proposed its assembly mechanism.30 In 2020, our research group successfully synthesized double-caged Dy60 clusters under solvothermal conditions by using bisacylhydrazone ligands with multidentate chelate coordination. Their formation process was monitored by HRESI-MS analysis.31 Moreover, we proposed for the first time the out-to-in growth mechanism and the annular/linear growth mechanism of the self-assembly process of high-nuclear lanthanide clusters.24,34 Overall, research on the formation process and assembly mechanism of high-nuclear lanthanide clusters is only beginning. Given that the intermediate fragments in the formation process are captured and screened, speculating on the formation process and assembly mechanism is important to explore the chemical reaction nature and elementary reactions of high-nuclear lanthanide clusters.35,36 Therefore, analyzing the formation process of high-nuclear lanthanide clusters in detail and speculating on their assembly mechanism have great significance.
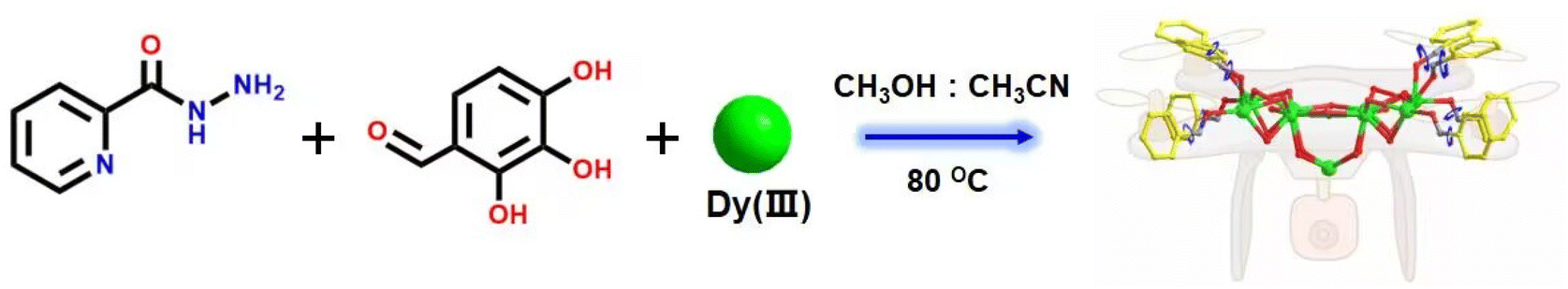
In the present study, we reacted 2-pyridinecarbohydrazide, 2,3,4-trihydroxybenzaldehyde and a mixed-metal dysprosium salt to obtain the drone-shaped lanthanide cluster 1, whose molecular formula was [Dy9(L)8(μ4-Cl)(CH3O)4(CH3OH)4(H2O)4]·Cl·3CH3OH. The rare μ4-Cl− in lanthanide clusters was found in the structure of cluster 1. Furthermore, the eight ligands in cluster 1 have freely rotatable pyridine rings at their ends, and the ligands around them can be regarded as the wings of drones. Thus, cluster 1 can be regarded as a drone-shaped lanthanide cluster, and this bare and freely rotatable rotor structure had potential application in the construction of dynamic lanthanide luminescent materials and smart molecular devices. Notably, the eight coordinated ligands were all located on the periphery of the cluster core and tightly wrapped around the cluster core and the exposed rotor at the end of the ligands to ensure the stability of cluster 1. HRESI-MS further revealed highly stable Dy9 cluster cores. The changes in species during the formation of Dy9 clusters in the solution state were further tracked by time-dependent HRESI-MS, and the possible assembly mechanism was proposed as follows: L + Dy → DyL → Dy2L2 → Dy3L3 → Dy4L4 → 2Dy4L4 → Dy8L8 → Dy9L8. Magnetic studies revealed that cluster 1 had a clear frequency dependence under zero-field conditions, indicating its single-molecule magnet (SMM) behavior, with an effective energy barrier and magnetic relaxation time of Ueff = 52.4 K and τ0 = 2.24 × 10−10 s, respectively. To the best of our knowledge, this study is the first to obtain drone-shaped nanoclusters with a rotor structure and high stability. This work provided a vivid example of the design and synthesis of high-nuclear lanthanide clusters with high stability and special shapes, and it is expected to open new avenues for the construction of high-nuclear lanthanide clusters with rotor structures.
Results and discussion
Structural analysis of cluster 1
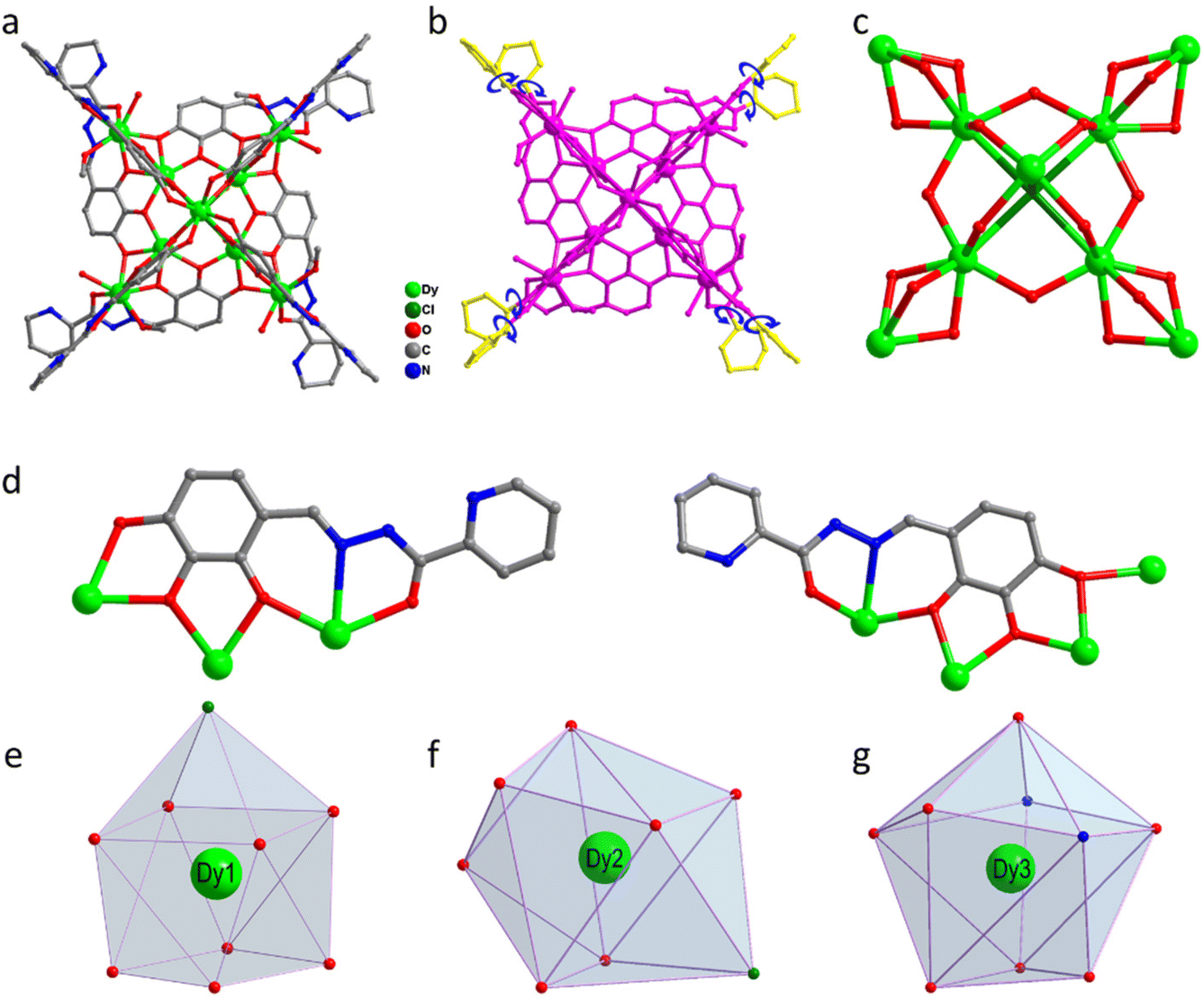
About 0.25 mmol of mixed-metal dysprosium salt (Dy(NO3)3·6H2O:DyCl3·6H2O = 1:1), 0.2 mmol of 2-pyridinecarbohydrazide, 0.2 mmol of 2,3,4-trihydroxybenzaldehyde, and 60 μL of triethylamine were accurately weighed and dissolved in a mixed solvent of MeOH:MeCN = 1:1. The mixture was reacted under solvothermal conditions at 80 °C for 48 h to obtain brown-red blocky crystals (1) (Scheme 1). The single-crystal X-ray structure diffraction results showed that cluster 1 crystallized in the tetragonal P4/n space group (Table S1†), which contained nine Dy(III) ions and eight deprotonated (L)3− ligands, one μ4-Cl−, four CH3O− ions, four terminally coordinated H2O molecules, and four CH3OH molecules (molecular formula is: [Dy9(L)8(CH3O)4(μ4-Cl)Cl(CH3OH)4(H2O)4]Cl·3CH3OH) (Fig. 1a). It is well known that most of the widely reported Ln9 complexes are hourglass-shaped, in which the hourglass-shaped nine-nuclear cluster core can be regarded as formed by two square pyramidal units sharing vertices.37,38 However, the eight Dy(III) ions in the structure of cluster 1 were distributed on the equatorial plane and tightly connected through the oxygen atoms on the ligand and μ4-Cl− bridges. Conversely, the Dy(III) ions above the equatorial plane were connected to the eight Dy(III) ions through the oxygen atoms on the ligand to form a {Dy/O/Cl} cluster nucleus (Fig. 1c). Notably, μ4-Cl− bridges were realized in the structure of cluster 1, which is very rare in lanthanide clusters (Fig. S1†).39,40 Additionally, the terminal pyridine rings of the eight ligands in the structure of cluster 1 can freely rotate as rotors. Thus, cluster 1 can be regarded as a drone-shaped lanthanide cluster, where each side can be regarded as the wings of the drone, and the exposed rotor can be regarded as the rotor of the drone (Fig. 1b). Notably, the eight peripheral ligands were all located at the periphery of the cluster core, tightly wrapping the cluster core from the attack of foreign solvent molecules. The exposed rotor structure at the end of the ligands also protects the cluster core. The above two factors together ensured the stability of cluster 1. The eight ligands adopted two different coordination modes to chelate Dy(III) ions: μ3-η1:η2:η2:η1:η1 and μ4-η2:η2:η2:η1:η1 (Fig. 1d). The nine-metal-centered Dy(III) ions in cluster 1 had three different coordination environments. The metal center Dy1 was in the O8Cl coordination environment provided by the (L)3− ligand and a Cl− ion (Fig. 1e). SHAPE calculations showed that its coordination configuration was a capped sq. antiprism with a C4v symmetry environment (Table S3†). The metal center Dy2 was in the O7Cl coordination environment provided by the (L)3− ligands, μ4-Cl− ions, and CH3O− ions (Fig. 1f). SHAPE calculations showed that the coordination configuration was a Johnson biaugmented trigonal prism (J50) with a C2v symmetric environment (Table S3†). The metal center Dy3 was in the O7N2 coordination environment provided by the (L)3− ligands, CH3O− ions, and terminally coordinated H2O molecules and CH3OH molecules (Fig. 1g). SHAPE calculations revealed that its coordination configuration was a tricapped trigonal prism (J51) with a D3h symmetric environment (Table S3†). Structural analysis demonstrated that all Dy–O/N bond lengths were within the normal range (Table S2†).
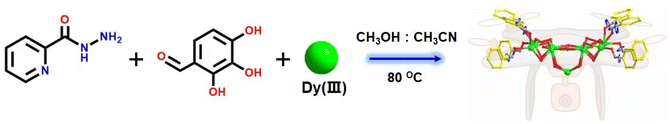 |
| | Scheme 1 Schematic of the synthesis of drone-shaped cluster 1. | |
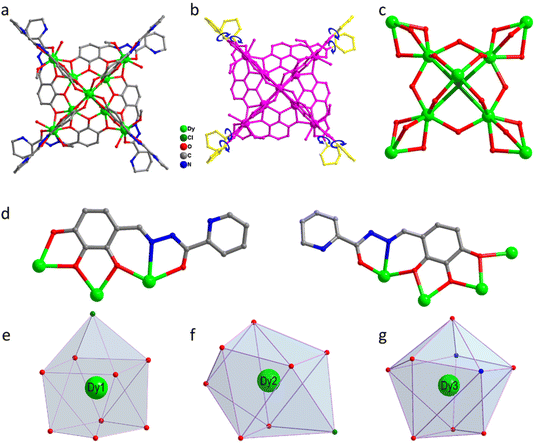 |
| | Fig. 1 Structure of drone-shaped cluster 1 (a), schematic of the rotor structure (b), cluster core connection (c), ligand coordination mode (d), and metal center coordination configuration (e–g). | |
Thermogravimetric analysis of cluster 1 was performed under a flowing N2 atmosphere, and the temperature was slowly increased from 35 °C to 1000 °C at a rate of 5 °C min−1 (Fig. S2†). Cluster 1 had three weightlessness processes. First, when the temperature gradually increased from 35 °C to 180 °C, the weight loss rate was 8.6%, which corresponds to the loss of four terminally coordinated water molecules, seven methanol molecules, and a free chloride ion (theoretical value is 8.1%). Then, upon gradually increasing the temperature from 180 °C to 327 °C, the weight loss rate was 4.7%, which corresponds to the loss of four terminally coordinated CH3O− ions and a chloride ion (theoretical value is 4.1%). Finally, when the temperature exceeded 345 °C, the framework of cluster 1 began to collapse. Additionally, the experimental and theoretical values of PXRD for cluster 1 agreed well, indicating that it was a pure phase (Fig. 4). Scanning electron micrographs showed that the fresh sample of cluster 1 and the sample after 720 min were bulk crystals with a clean surface (Fig. S3†).
Assembly mechanism analysis of cluster 1
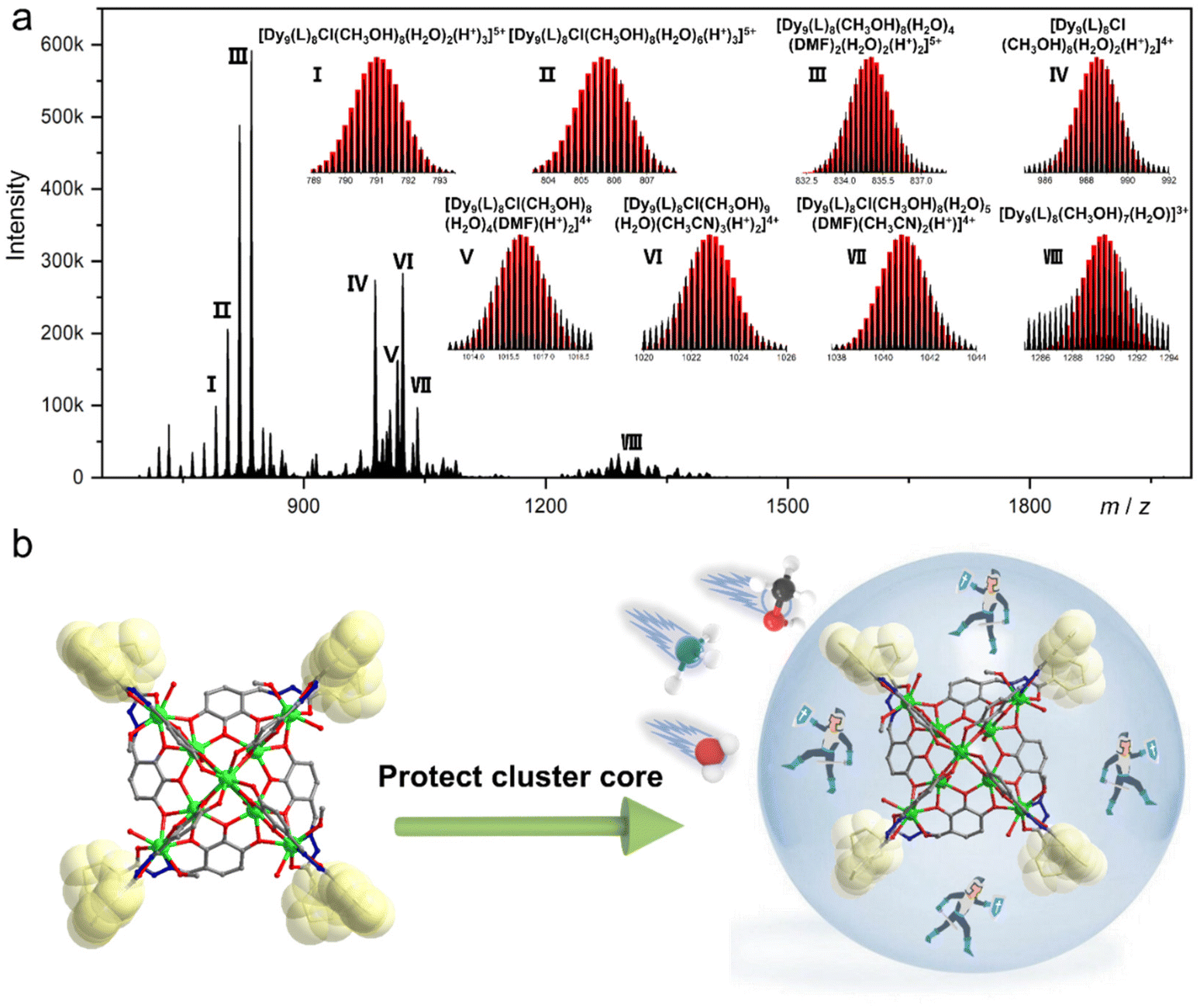
Over the past two decades, HRESI-MS has been extensively used to identify fragments in solution and has developed into an important technique for tracking fragment transitions and reaction mechanisms in solution.27,31 HRESI-MS can also be used to evaluate the stability, fragmentation mechanism, and degree of ionization of clusters in solution.29,30 Accordingly, on the premise of analyzing the structural characteristics of cluster 1 in detail, we used HRESI-MS to verify the integrity and stability of cluster 1 in solution. A small amount of pure crystals of cluster 1 was dissolved in chromatographic N,N-dimethylformamide (DMF), and HRESI-MS testing was performed after extensive dilution using chromatographic methanol. Data were collected within the range of m/z = 400–4000 (Fig. 2a). The results showed that under the HRESI-MS ion source voltage, the main framework of cluster 1 could be detected, indicating that it had better stability in solution. We observed a series of fragments containing the main framework of Dy9L8. For example, at the position of m/z = 835.20, a framework fragment with the highest intensity appeared, and its molecular formula was obtained by fitting [Dy9(L)8(CH3OH)8(H2O)4(DMF)2(H2O)2(H+)2]5+ (calc. 835.24); the position at m/z = 790.99 was [Dy9(L)8Cl(CH3OH)8(H2O)2(H+)3]5+ (calc. 790.99); the position at m/z = 805.61 was [Dy9(L)8Cl(CH3OH)8(H2O)6(H+)3]5+ (calc. 805.61); the position at m/z = 988.49 was [Dy9(L)8Cl(CH3OH)8(H2O)2(H+)2]4+ (calc. 988.49); the position at m/z = 1015.99 was [Dy9(L)8Cl(CH3OH)8(H2O)4(DMF)(H+)2]4+ (calc. 1016.01); the position at m/z = 1022.77 was [Dy9(L)8Cl(CH3OH)9(H2O)(CH3CN)3(H+)2]4+ (calc. 1022.76); the position at m/z = 1040.76 was [Dy9(L)8Cl(CH3OH)8(H2O)5(DMF)(CH3CN)2(H+)]4+ (calc. 1040.78,); and the position at m/z = 1289.95 was [Dy9(L)8(CH3OH)7(H2O)]3+ (calc. 1289.99) (Fig. 2a). Combined with the structural analysis of cluster 1, its stability can be attributed to the fact that the eight ligands were located at the periphery of the cluster core and tightly wrapped around the cluster core and the eight “rotors” around the cluster core. Both protected the cluster core from the attack of foreign solvent molecules and jointly promoted the stability of the cluster core. The combination of the HRESI-MS technique and crystallography demonstrated the excellent solution stability of cluster 1 (Fig. 2b).
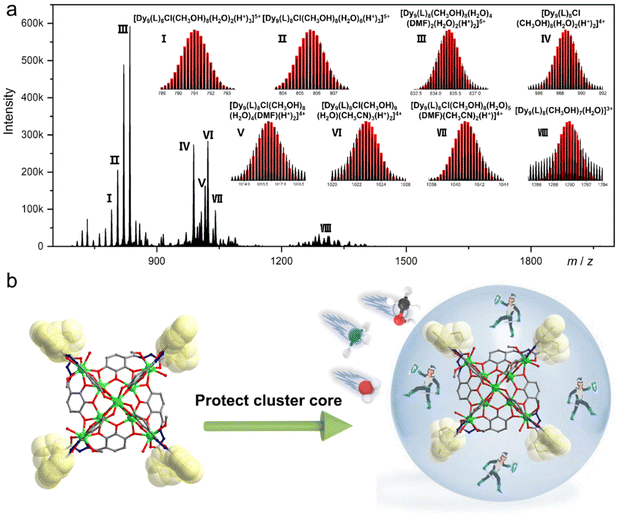 |
| | Fig. 2 HRESI-MS spectrum of cluster 1 in the positive-ion mode in DMF (a) and schematic of the molecular rotor structure protecting the cluster core (b). | |
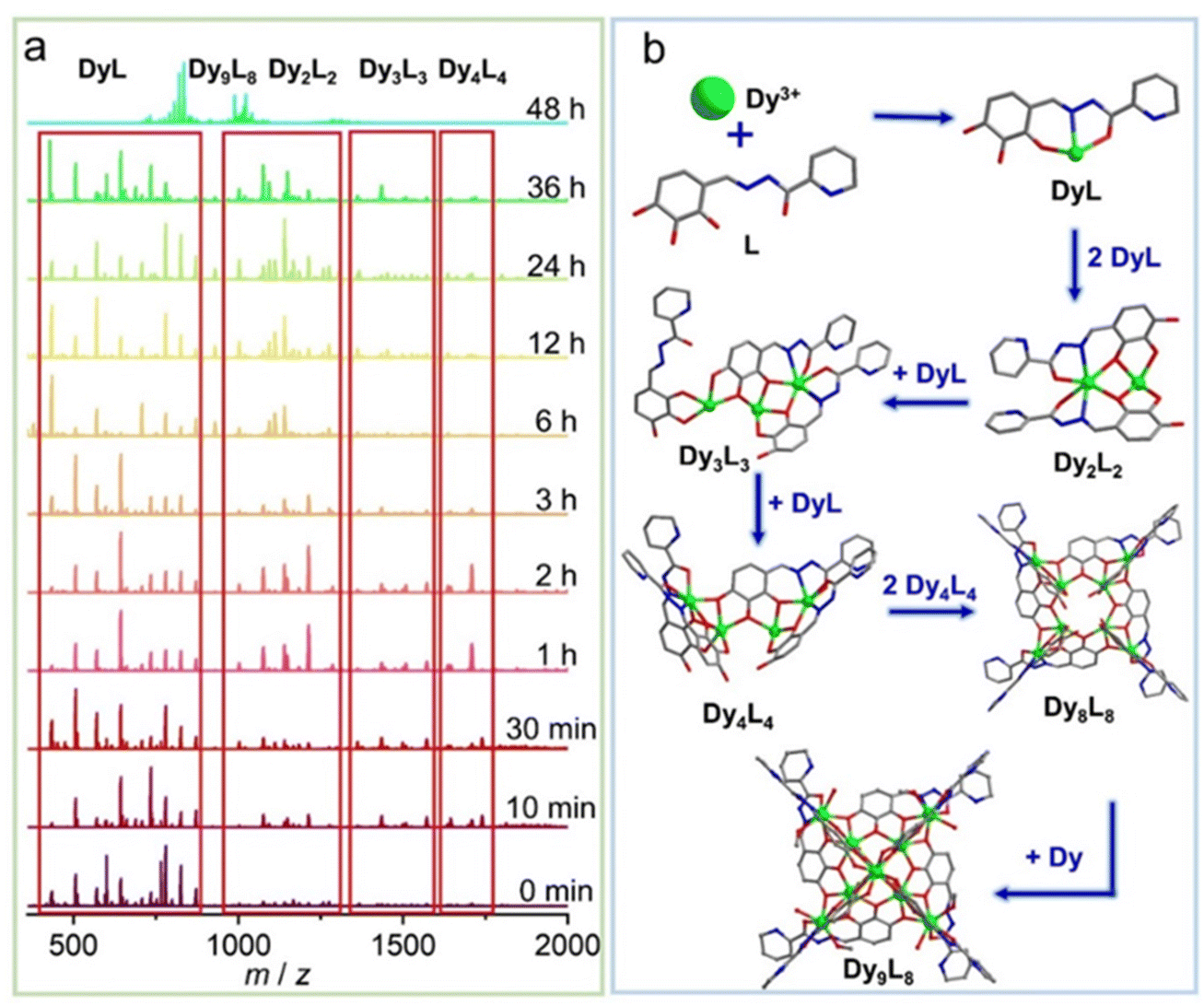
To further explore the formation mechanism of cluster 1, we tracked the species changes during the formation of cluster 1 in solution by using time-dependent HRESI-MS, collecting data in the positive- and negative-ion modes, and analyzing them in the positive-ion mode (Fig. 3). HRESI-MS data suggested that cluster 1 formed through a step-by-step assembly. First, the reaction system was stirred at room temperature for 5 min (0 min). Then, 2,3,4-trihydroxybenzaldehyde was reacted with 2-pyridinecarbohydrazide in situ to generate the H3L ligand, which immediately reacted with metal Dy(III) ions to generate multiple [Dy(L)/(HL)] fragments. The ligand fragment with the highest intensity appeared in the position m/z = 780.25, which can be assigned to [Dy(L)(DMF)3(H2O)7(H+)]+ (Fig. 3a and S4†). The other similar fragments appeared at positions m/z = 434.01, 507.06, 571.04, 644.14, 735.15, 826.22, and 872.19, respectively. By fitting, the molecular formulas of the above fragments were [Dy(L)] (calc. 433.98; exp. 434.01), [Dy(HL)(H2O)4]+ (calc. 507.03; exp. 507.06), [Dy(L)(DMF)(CH3OH)(H+)]+ (calc. 571.09; exp. 571.04), [Dy(L)(DMF)2(CH3OH)2(H+)]+ (calc. 644.14; exp. 644.14), [Dy(L)(DMF)5(H2O)4(H+)]+ (calc. 735.18; exp. 735.15), [Dy(L)(CH3OH)2(DMF)4(H2O)2(H+)]+ (calc. 826.27; exp. 826.22), and [Dy(L)(DMF)5(H2O)4(H+)]+ (calc. 872.26; exp. 872.19) (Fig. 3a, S4 and Table S4†). The experimental results showed that after the generation of the Schiff base ligand, the organic ligand deprotonated under the control of triethylamine, and then combined with the metal center Dy(III) ion to form [Dy(L)/(HL)]+ fragments. With a prolonged reaction time, the intensity of [Dy(L)/(HL)]+ fragments gradually decreased, and the [Dy(L)/(HL)]+ fragments disappeared after heating for 48 h. The maximum intensity was reached at 36 h, indicating that [Dy(L)/(HL)]+ fragments were gradually consumed under thermodynamic action, and [Dy2(L)2/(HL)(H2L)2]+ fragments were obtained by continuous chelation of metal Dy(III) ions and the L ligand. Moreover, [Dy(L)/(HL)]+ fragments were transformed into a small amount of [Dy2(L)2/(HL)(H2L)2]+ fragments with a prolonged heating time. It began to appear at 5 min, formed in large quantities at 1 h, reached the maximum intensity at 24 h, and weakened until it disappeared after 36 h. The experimental results showed that m/z = 1076.07–1213.10, and the molecular ion peaks belonged to [Dy2(L)n/(HL)n(H2L)n]+ fragments. By fitting, their molecular formulas were [Dy2(L)2(DMF)(CH3CN)2(H2O)3(H+)]+ (calc. 1076.11; exp. 1076.07), [Dy2(L)(HL)(H2O)8(CH3CN)2]+ (calc. 1093.10; exp. 1093.06), [Dy2(L)2(HL)2(H2L)]+ (calc. 1140.01; exp. 1139.96), [Dy2(L)2(DMF)2(CH3CN)2(H2O)3(H+)]+ (calc. 1149.16; exp. 1149.13), and [Dy2(HL)2(H2L)2(DMF)]+ (calc. 1213.10; exp. 1213.09) (Table S4†). Furthermore, a small amount of [Dy2(L)n/(HL)n(H2L)n]+ fragments chelated with metal Dy(III) ions and L to form [Dy3(L)n/(HL)n/(H2L)n]+ fragments. It began to appear at 5 min, and its molecular ion peak appeared at m/z = 1367.00–1740.25. By fitting, their molecular formulas were [Dy3(L)3(CH3OH)(H2O)2(H+)]+ (calc. 1367.00; exp. 1367.05), [Dy3(L)2(HL)(CH3OH)2(H2O)8]+ (calc. 1508.09; exp. 1508.03), [Dy3(L)2(HL)(H2O)8(CH3OH)4]+ (calc. 1571.14; exp. 1571.14), [Dy3(L)3(DMF)3(CH3CN)2(H2O)2(H+)]+ (calc. 1636.08; exp. 1636.03), [Dy3(L)2(H2L)2(CH3OH)2(H2O)4]+ (calc. 1709.14; exp. 1709.12), and [Dy3(L)2(H2L)2(CH3OH)3(H2O)4]+ (calc. 1741.14; exp. 1741.14) (Table S4†). With further prolonged heating time, the [Dy4(L)4]+ fragment appeared at 10 min, which was formed by a [Dy3(L)n/(HL)n/(H2L)n]+ fragment chelating a [Dy(L)/(HL)]+ fragment. The molecular ion peaks of [Dy4(L)4]+ appeared at m/z = 1771.95 and 1844.99, and the molecular formulas were [Dy4(L)4(CH3CN)(H+)]+ (calc. 1771.95; exp. 1771.93) and [Dy4(L)4(CH3CN)(H2O)4(H+)]+ (calc. 1844.99; exp. 1844.96) (Table S4†). With a prolonged reaction time, the intensity of [Dy4(L)4]+ gradually increased, and the intensity reached the maximum at 2 h. The above fragments gradually disappeared when the heating time reached 48 h. This finding indicated that two [Dy4(L)4]+ fragments formed [Dy8(L)8]2+ fragments through template assembly. Finally, with the reaction time reaching 48 h, at m/z = 791.01–1289.99, the molecular ion peaks belonged to [Dy9(L)8], which was the main frame of the structure. It was formed by the [Dy8(L)8]2+ fragment chelating a metal Dy(III) ion. By fitting, their molecular fragment peaks were [Dy9(L)8Cl(CH3OH)8(H2O)2(H+)3]5+ (calc. 790.99; exp. 790.99), [Dy9(L)8Cl(CH3OH)8(H2O)6(H+)3]5+ (calc. 805.61; exp. 805.61), [Dy9(L)8(CH3OH)8(H2O)4(DMF)2(H2O)2(H+)2]5+ (calc. 835.24; exp. 835.20), [Dy9(L)8Cl(CH3OH)8(H2O)2(H+)2]4+ (calc. 988.49, exp. 988.49), [Dy9(L)8Cl(CH3OH)8(H2O)4(DMF)(H+)2]4+ (calc. 1016.01; exp. 1015.99), [Dy9(L)8Cl(CH3OH)9(H2O)(CH3CN)3(H+)2]4+ (calc. 1022.76; exp. 1022.77), [Dy9(L)8Cl(CH3OH)8(H2O)5(DMF)(CH3CN)2(H+)]4+ (calc. 1040.78; exp. 1040.77), and [Dy9(L)8(CH3OH)7(H2O)]3+ (calc. 1289.99; exp. 1289.95) (Fig. 2a and Table S4†). In addition, the changes of the above fragments at different temperatures were tracked by HRESI-MS (Fig. S5†). The experimental results showed that with the change of time, the [Dy9L8] fragment gradually strengthened, and reached the strongest at 80 °C, and the remaining fragments increased first and then decreased with the increase of temperature, and were the lowest at 80 °C (Fig. S5†). Therefore, by analyzing the changing trend of the cation mode, the formation process of cluster 1 proceeded through a stepwise assembly, and its possible assembly mechanism was L + Dy → DyL → Dy2L2 → Dy3L3 → Dy4L4 → 2Dy4L4 → Dy8L8 → Dy9L8 (Fig. 3b).
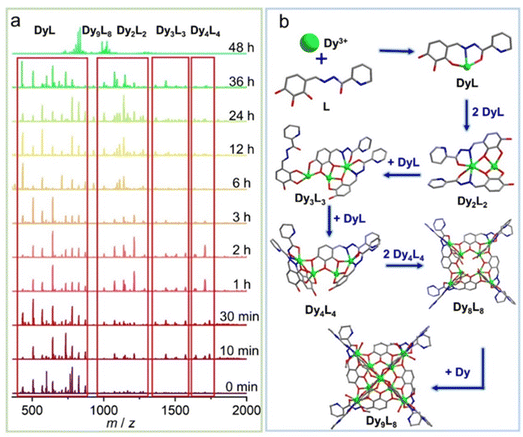 |
| | Fig. 3 Time-correlated HRESI-MS spectrum of cluster 1 (a) and schematic of the possible assembly mechanism of cluster 1 (b). | |
Transition from the crystalline to amorphous state of cluster 1
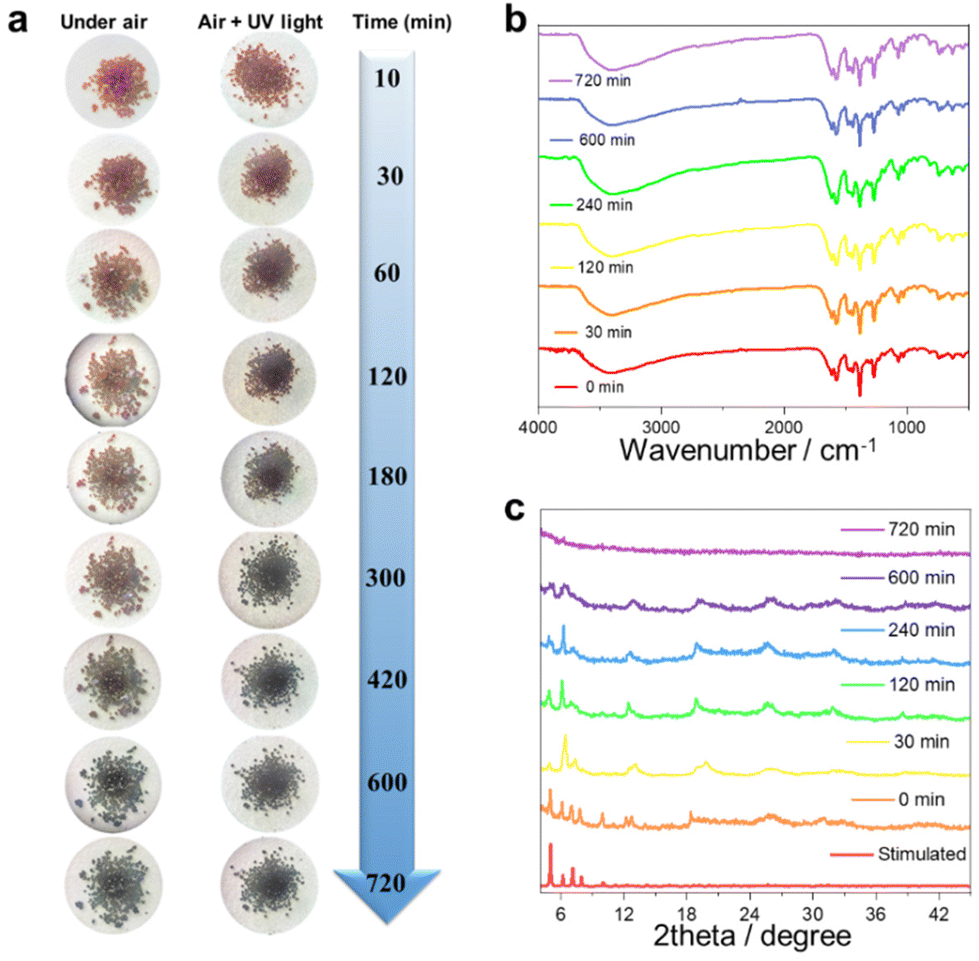
Single-crystal to single-crystal conversion is extensively reported.41–43 However, achieving a crystalline-to-amorphous transition in lanthanide clusters is very rare. The potential discoloration behavior of cluster 1 may be due to air exposure. We investigated the color-changing behavior of cluster 1 before and after exposure to UV light in air at different time scales. Specifically, we tracked and photographed the color change of cluster 1 at 10, 30, 60, 120, 180, 300, 420, 600, and 720 min (Fig. 4a). The results showed that cluster 1 maintained a bright brown-red color from 10 min to 60 min under the atmospheric conditions. With a gradually prolonged time, cluster 1 completely changed from reddish brown to dark black after 700 min. We also tried to irradiate the freshly collected pure sample of cluster 1 under 365 nm UV light for 4 min and then removed the UV light and placed it in air. Surprisingly, we found that the discoloration speed of cluster 1 was accelerated after being irradiated with UV light. Cluster 1 gradually changed from reddish-brown to black within 30 min and completely blackened after 420 min. To further explore the discoloration mechanism of cluster 1, we selected cluster 1 at 0, 30, 120, 240, 600, and 700 min for FTIR (Fig. 4b), PXRD (Fig. 4c), and solid-state UV tests (Fig. S6†). 0 to 720 minutes all show similar infrared absorption, and their absorption peaks are mainly located at 3400, 1660, 1430, 1380, 1266, and 1100 cm−1, respectively. The broad absorption peak around 3400 cm−1 can be attributed to the stretching vibration of ν(HO–H) in the H2O molecule. The peak at around 1660 cm−1 can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration of the imine group (–CN–). The strong peak at around 1430 cm−1 can be attributed to the stretching vibrations of the aromatic ring (CN and CC). The strong peak at around 1380 cm−1 can be attributed to the stretching vibration of –CH3. The weaker absorption peak around 1100 cm−1 can be attributed to the stretching vibration between the alcoholic hydroxyl groups (C–O) in the ligand. Altogether, these FTIR results indicate that the functional groups within the structure were not destroyed during the transition of cluster 1 from the crystalline to amorphous state (Fig. 4b). The PXRD experimental results further showed that the PXRD spectra from 0 min to 240 min were highly similar to the simulated spectra, indicating that cluster 1 remained stable until 240 min. The diffraction peaks at all angles disappeared completely with the passage of time up to 700 min, indicating that cluster 1 may have transformed from crystalline to amorphous (Fig. 4c). We further explored the solid-state UV-Vis spectra of cluster 1 at different time intervals. The experimental results showed that cluster 1 had absorption peaks at 800, 908, 1101, and 1291 nm within the range of 0–8 h. With a gradually prolonged time, the intensity of the absorption peaks at 800 and 908 nm gradually weakened until they disappeared after 10 h. Beyond 10 h, the absorption peaks of cluster 1 disappeared completely (Fig. S6†). In summary, we speculated that cluster 1 may have undergone physical changes that induced an obvious change in its absorption, and the crystal color changed from bright brown-red to dark black. The transformation process of lanthanide clusters from crystalline to amorphous states is indicated by PXRD, IR spectroscopy, and visual visualization.
N stretching vibration of the imine group (–CN–). The strong peak at around 1430 cm−1 can be attributed to the stretching vibrations of the aromatic ring (CN and CC). The strong peak at around 1380 cm−1 can be attributed to the stretching vibration of –CH3. The weaker absorption peak around 1100 cm−1 can be attributed to the stretching vibration between the alcoholic hydroxyl groups (C–O) in the ligand. Altogether, these FTIR results indicate that the functional groups within the structure were not destroyed during the transition of cluster 1 from the crystalline to amorphous state (Fig. 4b). The PXRD experimental results further showed that the PXRD spectra from 0 min to 240 min were highly similar to the simulated spectra, indicating that cluster 1 remained stable until 240 min. The diffraction peaks at all angles disappeared completely with the passage of time up to 700 min, indicating that cluster 1 may have transformed from crystalline to amorphous (Fig. 4c). We further explored the solid-state UV-Vis spectra of cluster 1 at different time intervals. The experimental results showed that cluster 1 had absorption peaks at 800, 908, 1101, and 1291 nm within the range of 0–8 h. With a gradually prolonged time, the intensity of the absorption peaks at 800 and 908 nm gradually weakened until they disappeared after 10 h. Beyond 10 h, the absorption peaks of cluster 1 disappeared completely (Fig. S6†). In summary, we speculated that cluster 1 may have undergone physical changes that induced an obvious change in its absorption, and the crystal color changed from bright brown-red to dark black. The transformation process of lanthanide clusters from crystalline to amorphous states is indicated by PXRD, IR spectroscopy, and visual visualization.
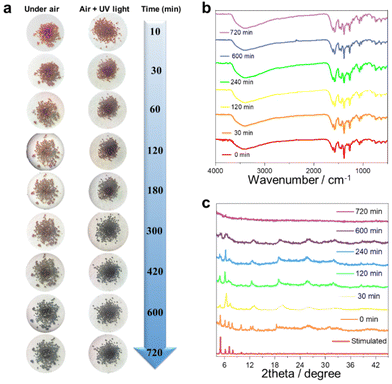 |
| | Fig. 4 Color-change map of cluster 1 at different time points under the conditions of air and UV light exposure (a). FTIR spectra and PXRD patterns of cluster 1 at different time points in air (b and c). | |
Magnetic analysis of cluster 1
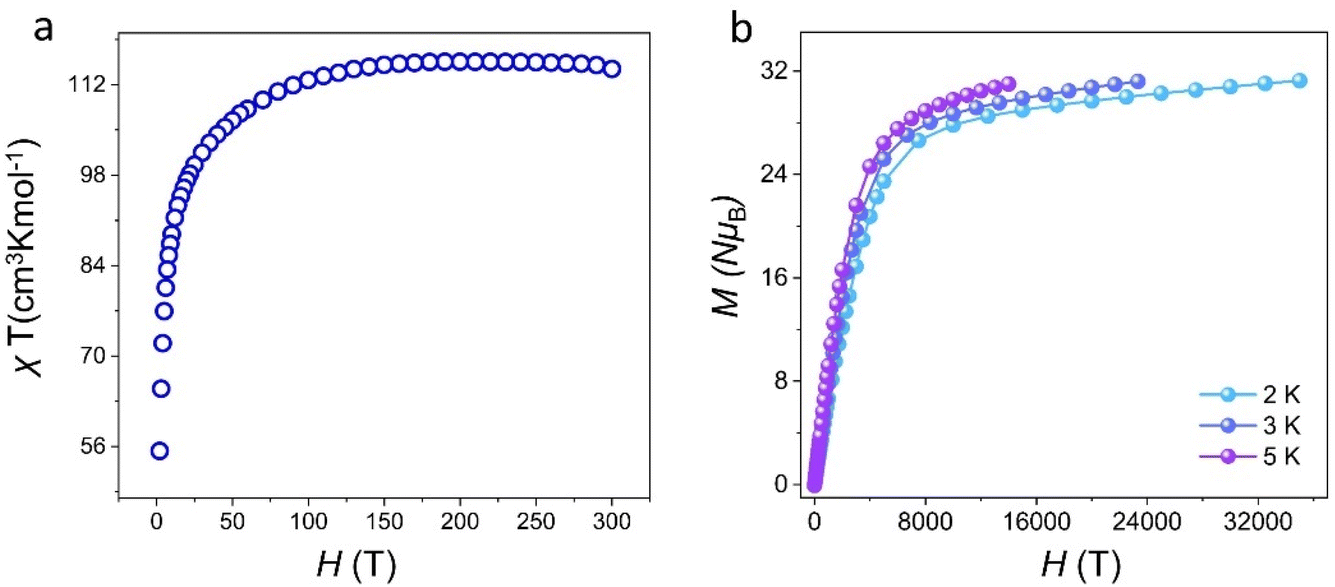
Lanthanide metal ions with high spin, especially Dy(III) ions with large intrinsic magnetic anisotropy, are ideal 4f transition-metal ions for the construction of SMMs.12 Dysprosium-based SMMs usually exhibit excellent energy barriers and relaxation times.44–46 Thus, we further investigated the magnetic behavior of cluster 1. The temperature-varying molar susceptibility of the pure phase of cluster 1 was tested under an external DC magnetic field of 1000 Oe at 2–300 K (Fig. 5a). At 300 K, the χmT value of cluster 1 was 115.79 cm3 K mol−1 (6H15/2, S = 5/2, g = 4/3, J = 15/2, L = 5), which was less than that of nine Dy(III) ions (theoretical value was 127.53 cm3 K mol−1). With a gradually decreased temperature to about 100 K, the χmT of cluster 1 slightly decreased. When the temperature reached 2 K, χmT decreased rapidly to 55.86 cm3 K mol−1. These findings showed that this temperature-decreasing χmT value may be due to the decrease in the Stark sublevel of the Dy(III) excited state and the induced crystal-field effect.33 Furthermore, the field-dependent magnetization of cluster 1 was tested at different temperatures (2–5 K) and applied fields of 0–7 T, and the M vs. H/T curves were plotted (Fig. 5b). Their measured magnetization at 2 K temperature increased rapidly in the low-field region and gradually flattened in the high-field region. Cluster 1 reached a maximum value of 28.97Nβ when a static field of 15 kOe was reached. The M vs. H/T curves of cluster 1 at different temperatures did not overlap with each other. The test results revealed that Dy(III) ions may have low excited states and/or magnetic anisotropy. As shown in Fig. S7,† the hysteresis loop of cluster 1 was not obvious at 2 K, which may be attributed to the existence of the crystal-field effect and the strong quantum-tunneling effect.
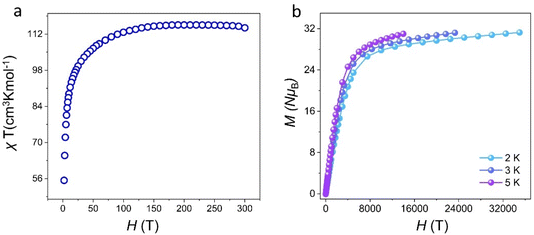 |
| | Fig. 5 Temperature dependence of χmT for cluster 1 (a) and M vs. H/T plots of cluster 1 (b). | |
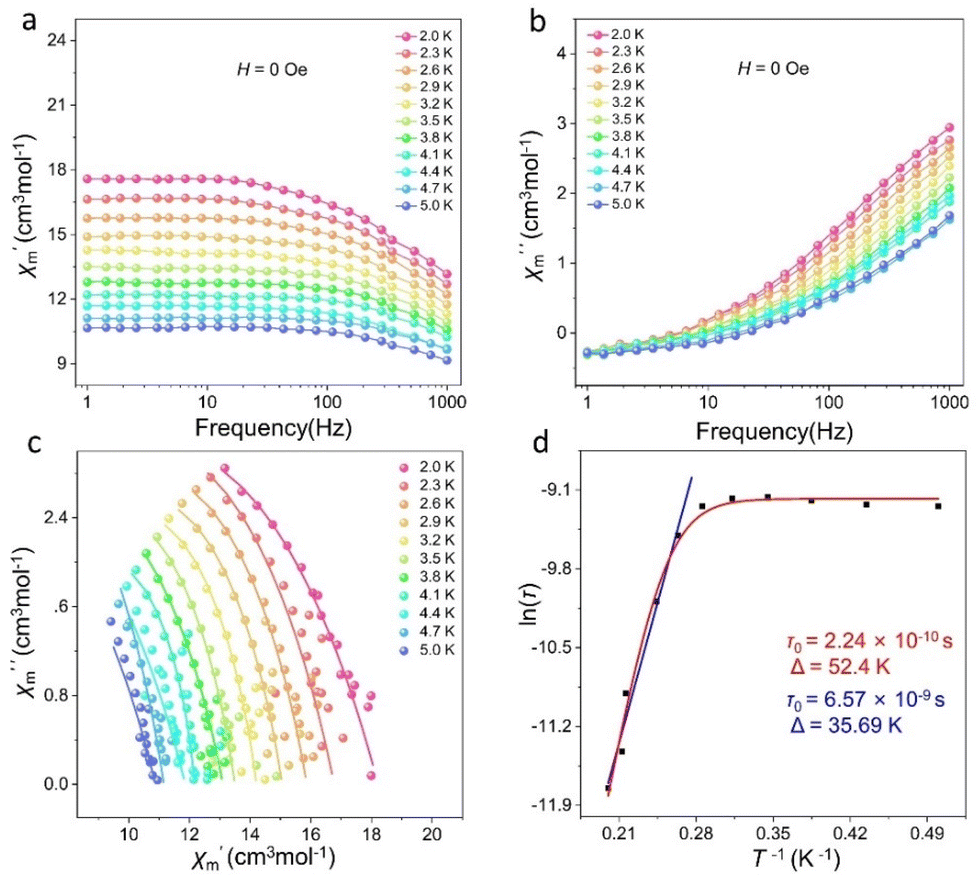
We further tested the alternating current (ac) susceptibility of cluster 1 to explore its dynamic magnetic behavior. As shown in Fig. 6 and S8,† under zero DC field, the out-of-phase (χ′′) signals of cluster 1 all exhibited frequency- and temperature-dependent behaviors. The temperature-dependent out-of-phase AC susceptibility signal of cluster 1 was not obvious, but it can still have frequency-dependent behavior under zero field. This may be due to the strong quantum-tunneling effect. As shown in Fig. 6c, the Cole–Cole plots of cluster 1 all presented a relatively semicircular shape, and the fitted values were consistent with the generalized Debye model. The anisotropic energy barrier and magnetic relaxation time of cluster 1 were analyzed based on the thermal activation process according to the Arrhenius law (τ = τ0exp(Ueff/kBT)).47 The optimum results demonstrated that the effective energy barrier and magnetic relaxation time of cluster 1 were Ueff = 35.69 K and τ0 = 6.57 × 10−9 s, respectively, at zero DC field (Fig. 6d). The relaxation mechanism of Dy(III)-based SMMs is known to be usually complex because of Orbach processes (τ0−1 = exp(−Ueff/kBT)), Raman processes (CTn), direct relaxation processes (AHmT), or quantum tunneling processes (QTM).29 Therefore, when we fit cluster 1 at all temperatures based on the above considerations, we obtained effective energy barriers and relaxation times of Ueff = 52.4 K and τ0 = 2.24 × 10−10 s, respectively (Fig. 6d).
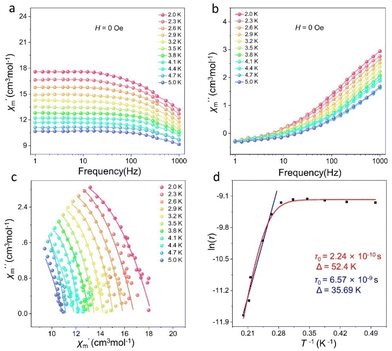 |
| | Fig. 6
χ′ and χ′′ vs. frequency plots of cluster 1 (a and b), Cole–Cole plots of cluster 1 (c), and energy barrier fits of cluster 1 (d). | |
Conclusions
We successfully synthesized a drone-shaped lanthanide cluster by reacting 2-pyridinecarbohydrazide, 2,3,4-trihydroxybenzaldehyde, and a mixed-metal dysprosium salt under mixed solvent conditions. Structural analysis showed that the overall structure of cluster 1 can be regarded as a “drone”, in which the eight ligands can be regarded as the wings of the drone, and the pyridine ring at the end of the ligand can be regarded as the rotor of the drone. The rare μ4-Cl− appeared in the structure. The eight ligands distributed on the periphery of the cluster core and the pyridine ring at the end of the ligand can effectively block the attack of foreign solvent molecules and jointly guaranteed the stability of cluster 1. Additionally, the formation process of cluster 1 was monitored by time-dependent HRESI-MS, and the possible formation mechanism of cluster 1 was proposed as L + Dy → DyL → Dy2L2 → Dy3L3 → Dy4L4 → 2Dy4L4 → Dy8L8 → Dy9L8. The reddish-brown crystals of cluster 1 gradually turned dark black with time. The above color change was caused by the gradual transformation of the crystals of cluster 1 into an amorphous state. To our knowledge, this study is one of the rare examples of monitoring the transition from the crystalline to amorphous state of lanthanide clusters. Magnetic studies further revealed that cluster 1 had frequency- and temperature-dependent correlation behavior. This work can serve as a reference for the design and synthesis of high-nuclear lanthanide clusters with high stability and special shape and provided a new way to construct multifunctional lanthanide clusters.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22271068, 22061005, and 22075058) and Innovation Project of Guangxi Graduate Education (YCBZ2023059).
References
- X. Z. Li, C. B. Tian and Q. F. Sun, Coordination-Directed Self-Assembly of Functional Polynuclear Lanthanide Supramolecular Architectures, Chem. Rev., 2022, 122, 6374–6458 CrossRef CAS PubMed.
- H. Schnöckel, Structures and Properties of Metalloid Al and Ga Clusters Open Our Eyes to the Diversity and Complexity of Fundamental Chemical and Physical Processes during Formation and Dissolution of Metals, Chem. Rev., 2010, 110, 4125–4163 CrossRef PubMed.
- X. M. Luo, Y. K. Li, X. Y. Dong and S. Q. Zang, Platonic and Archimedean solids in discrete metal-containing clusters, Chem. Soc. Rev., 2023, 52, 383–444 RSC.
- Y. Z. Zheng, G. J. Zhou, Z. Zheng and R. E. P. Winpenny, Molecule-based magnetic coolers, Chem. Soc. Rev., 2014, 43, 1462–1475 RSC.
- L. Qin, Y. Z. Yu, P. Q. Liao, W. Xue, Z. Zheng, X. M. Chen and Y. Z. Zheng, A “Molecular Water Pipe”: A Giant Tubular Cluster {Dy72} Exhibits Fast Proton Transport and Slow Magnetic Relaxation, Adv. Mater., 2016, 28, 10772–10779 CrossRef CAS PubMed.
- X. Y. Zheng, Y. H. Jiang, G. L. Zhuang, D. P. Liu, H. G. Liao, X. J. Kong, L. S. Long and L. S. Zheng, A Gigantic Molecular Wheel of {Gd140}: A New Member of the Molecular Wheel Family, J. Am. Chem. Soc., 2017, 139, 18178–18181 CrossRef CAS PubMed.
- J. B. Peng, X. J. Kong, Q. C. Zhang, M. Orendáč, J. Prokleška, Y. P. Ren, L. S. Long, Z. Zheng and L. S. Zheng, Beauty, Symmetry, and Magnetocaloric Effect-Four-Shell Keplerates with 104 Lanthanide Atoms, J. Am. Chem. Soc., 2014, 136, 17938–17941 CrossRef CAS PubMed.
- X. Y. Li, H. F. Su, Q. W. Li, R. Feng, H. Y. Bai, H. Y. Chen, J. Xu and X. H. Bu, A Giant Dy76 Cluster: A Fused Bi-Nanopillar Structural Model for Lanthanide Clusters, Angew. Chem., Int. Ed., 2019, 58, 10184–10188 CrossRef CAS PubMed.
- L. Qin, G. J. Zhou, Y. Z. Yu, H. Nojiri, C. Schröder, R. E. P. Winpenny and Y. Z. Zheng, Topological Self-Assembly of Highly Symmetric Lanthanide Clusters: A Magnetic Study of Exchange-Coupling “Fingerprints” in Giant Gadolinium(III) Cages, J. Am. Chem. Soc., 2017, 139, 16405–16411 CrossRef CAS PubMed.
- W. P. Chen, P. Q. Liao, P. B. Jin, L. Zhang, B. K. Ling, S. C. Wang, Y. T. Chan, X. M. Chen and Y. Z. Zheng, The Gigantic {Ni36Gd102} Hexagon: A Sulfate-Templated “Star-of-David” for Photocatalytic CO2 Reduction and Magnetic Cooling, J. Am. Chem. Soc., 2020, 142, 4663–4670 CrossRef CAS PubMed.
- J. Dong, P. Cui, P. F. Shi, P. Cheng and B. Zhao, Ultrastrong Alkali-Resisting Lanthanide-Zeolites Assembled by [Ln60] Nanocages, J. Am. Chem. Soc., 2015, 137, 15988–15991 CrossRef CAS PubMed.
- Z. Zhu, M. Guo, X. L. Li and J. Tang, Molecular magnetism of lanthanide: Advances and perspectives, Coord. Chem. Rev., 2019, 378, 350–364 CrossRef CAS.
- J. L. Liu, Y. C. Chen, F. S. Guo and M. L. Tong, Recent advances in the design of magnetic molecules for use as cryogenic magnetic coolants, Coord. Chem. Rev., 2014, 281, 26–49 CrossRef CAS.
- D. N. Woodru, R. E. P. Winpenny and R. A. Lay, Lanthanide Single-Molecule Magnets, Chem. Rev., 2013, 113, 5110–5148 CrossRef PubMed.
- J. H. Jia, Q. W. Li, Y. C. Chen, J. L. Liu and M. L. Tong, Luminescent single-molecule magnets based on lanthanides: Design strategies, recent advances and magneto-luminescent studies, Coord. Chem. Rev., 2019, 378, 365–381 CrossRef CAS.
- A. D'Aléo, F. Pointillart, L. Ouahab, C. Andraud and O. Maury, Charge transfer excited states sensitization
of lanthanide emitting from the visible to the near-infra-red, Coord. Chem. Rev., 2012, 256, 1604–1620 CrossRef.
- F. Habib and M. Murugesu, Lessons learned from dinuclear lanthanide nano-magnets, Chem. Soc. Rev., 2013, 42, 3278–3288 RSC.
- X. Y. Zheng, J. Xie, X. J. Kong, L. S. Long and L. S. Zheng, Recent advances in the assembly of high-nuclearity lanthanide clusters, Coord. Chem. Rev., 2019, 378, 222–236 CrossRef CAS.
- L. G. Christie, S. Asche, J. S. Mathieson, L. Vilà-Nadal and L. Cronin, Investigating the Formation of Giant {Pd72}Prop and {Pd84}Gly Macrocycles Using NMR, HPLC, and Mass Spectrometry, J. Am. Chem. Soc., 2018, 140, 9379–9382 CrossRef CAS PubMed.
- D. Dalessio, A. N. Sobolev, B. W. Skelton, R. O. Fuller, R. C. Woodward, N. A. Lengkeek, B. H. Fraser, M. Massi and M. I. Ogden, Lanthanoid “Bottlebrush” Clusters: Remarkably Elongated Metal−Oxo Core Structures with Controllable Lengths, J. Am. Chem. Soc., 2014, 136, 15122–15125 CrossRef CAS PubMed.
- L. Zhang, L. Wan, N. Chang, J. Liu, C. Duan, Q. Zhou, X. Li and X. Wang, Removal of phosphate from water by activated carbon fiber loaded with lanthanum oxide, J. Hazard. Mater., 2011, 190, 848–855 CrossRef CAS PubMed.
- X. Y. Li, Y. Jing, J. Zheng, H. Ding, Q. Li, M. H. Yu and X. H. Bu, Two Luminescent High-Nuclearity Lanthanide Clusters Ln48 (Ln=Eu and Tb) with a Nanopillar Structure, Cryst. Growth Des., 2020, 20, 5294–5301 CrossRef CAS.
- J. M. Peng, H. L. Wang, Z. H. Zhu, J. Bai, F. P. Liang and H. H. Zou, Series of the Largest Dish-Shaped Dysprosium Nanoclusters Formed by In Situ Reactions, Inorg. Chem., 2022, 61, 6094–6100 CrossRef CAS PubMed.
- H. L. Wang, T. Liu, Z. H. Zhu, J. M. Peng, H. H. Zou and F. P. Liang, A series of dysprosium clusters assembled by a substitution effect-driven out-to-in growth mechanism, Inorg. Chem. Front., 2021, 8, 2136–2143 RSC.
- W. Huang, W. Chen, Q. Bai, Z. Zhang, M. Feng and Z. Zheng, Anion-guided Stepwise Assembly of High-nuclearity Lanthanide Hydroxide Clusters, Angew. Chem., Int. Ed., 2022, 61, e202205385 CAS.
- H. L. Wang, D. L. Liu, J. H. Jia, J. L. Liu, Z. Y. Ruan, W. Deng, S. P. Yang, S. G. Wu and M. L. Tong, High-Stability Spherical Lanthanide Nanoclusters for Magnetic Resonance Imaging, Natl. Sci. Rev., 2023, 10, nwad036 CrossRef PubMed.
- Y. L. Li, H. L. Wang, Z. H. Zhu, F. P. Liang and H. H. Zou, Recent advances in the structural design and regulation of lanthanide clusters: Formation and self-assembly mechanisms, Coord. Chem. Rev., 2023, 493, 215322 CrossRef CAS.
- H. Zheng, M. H. Du, S. C. Lin, Z. C. Tang, X. J. Kong, L. S. Long and L. S. Zheng, Assembly of a Wheel-Like Eu24Ti8 Cluster under the Guidance of High-Resolution Electrospray Ionization Mass Spectrometry, Angew. Chem., Int. Ed., 2018, 57, 10976–10979 CrossRef CAS PubMed.
- H. L. Wang, X. F. Ma, J. M. Peng, Z. H. Zhu, B. Li, H. H. Zou and F. P. Liang, Tracking the Stepwise Formation of the Dysprosium Cluster (Dy10) with Multiple Relaxation Behavior, Inorg. Chem., 2019, 58, 9169–9174 CrossRef CAS PubMed.
- Z. H. Zhu, J. M. Peng, H. L. Wang, H. H. Zou and F. P. Liang, Assembly Mechanism and Heavy Metal Ion Sensing of Cage-Shaped Lanthanide Nanoclusters, Cell Rep. Phys. Sci., 2020, 1, 100165 CrossRef CAS.
- Z. R. Luo, H. L. Wang, Z. H. Zhu, T. Liu, X. F. Ma, H. F. Wang, H. H. Zou and F. P. Liang, Assembly of Dy60 and Dy30 cage-shaped Nanoclusters, Commun. Chem., 2020, 3, 1–9 CrossRef PubMed.
- Y. L. Li, H. L. Wang, Z. H. Zhu, J. Li, H. H. Zou and F. P. Liang, A Series of High-Nuclear Gadolinium Cluster Aggregates with a Magnetocaloric Effect Constructed through Two-Component Manipulation, Inorg. Chem., 2021, 60, 16794–16802 CrossRef CAS PubMed.
- H. L. Wang, Y. L. Li, Z. H. Zhu, X. L. Lu, F. P. Liang and H. H. Zou, Anion-Manipulated Hydrolysis Process Assembles of Giant High-Nucleation Lanthanide-Oxo Cluster, Inorg. Chem., 2022, 61, 20169–20176 CrossRef CAS PubMed.
- B. F. Long, S. Yu, Z. H. Zhu, Y. L. Li, F. P. Liang and H. H. Zou, Coordination site manipulation of the annular growth mechanism to assemble chiral lanthanide clusters with different shapes and magnetic properties, Inorg. Chem. Front., 2022, 9, 5950–5959 RSC.
- F. S. Guo, Y. C. Chen, L. L. Mao, W. Q. Lin, J. D. Leng, R. Tarasenko, M. Orendáč, J. Prokleška, V. Sechovský and M. L. Tong, Anion-Templated Assembly and Magnetocaloric Properties of a Nanoscale {Gd38} Cage versus a {Gd48} Barrel, Chem. – Eur. J., 2013, 19, 14876–14885 CrossRef CAS PubMed.
- Z. Zheng, Ligand-controlled self-assembly of polynuclear lanthanide–oxo/hydroxo complexes: from synthetic serendipity to rational supramolecular design, Chem. Commun., 2001, 1, 2521–2529 RSC.
- Z. Li, D. Wang, Z. Zhou, G. Zhao, Q. Li, Y. Bi and Z. Zheng, Thiacalix[4]arene-Sandwiched Sandglass-like Ln9 Clusters (Ln=Tb and Eu): Insights into the Selective Luminescence Quenching Properties by p-Nitrobenzene Derivatives, Inorg. Chem., 2022, 61, 20814–20823 CrossRef CAS PubMed.
- H. F. Wang, J. X. Tang, H. H. Zou and F. P. Liang, Sandglass-Shaped Ln9III Cluster Containing Two Quadrangular Pyramids (Ln5O5) Shared by Vertices: Structure, Magnetic and Photoresponsive Metal Ion Sensing, J. Cluster Sci., 2020, 31, 1155–1161 CrossRef CAS.
- B. D. Chandler, J. O. Yu, D. T. Cramb and G. K. H. Shimizu, Series of Lanthanide-Alkali Metal-Organic Frameworks Exhibiting Luminescence and Permanent Microporosity, Chem. Mater., 2007, 19, 4467–4473 CrossRef CAS.
- A. Stein, G. A. Ozin, P. M. Macdonald, G. D. Stucky and R. Jelinek, Silver, Sodium Halosodalites: Class A Sodalites, J. Am. Chem. Soc., 1992, 114, 5171–5186 CrossRef CAS.
- Q. Liu, Y. Wu, M. Feng, W. Chen, Z. Zheng and M. Feng, Rare Silver−Histidine Cluster Complex and Its Single-Crystal-to-Single-Crystal Phase-Transition Behavior, ACS Omega, 2022, 7, 8141–8149 CrossRef CAS PubMed.
- R. Z. Lange, G. Hofer, T. Weber and A. Dieter Schlüter, A Two-Dimensional Polymer Synthesized through Topochemical [2+2]-Cycloaddition on the Multigram Scale, J. Am. Chem. Soc., 2017, 139, 2053–2059 CrossRef CAS PubMed.
- J. Kim, J. Park, D. Kim, M. Di Serio and O. S. Jung, Stepwise coordination isomerism of 2D networks: adsorption of diiodomethane into crystals and recognition in SCSC mode, Inorg. Chem. Front., 2021, 8, 3292–3300 RSC.
- Z. Zhu, C. Zhao, T. Feng, X. Liu, X. Ying, X. L. Li, Y. Q. Zhang and J. Tang, Air-Stable Chiral Single-Molecule Magnets with Record Anisotropy Barrier Exceeding 1800 K, J. Am. Chem. Soc., 2021, 143, 10077–10082 CrossRef CAS PubMed.
- J. Liu, Y. C. Chen, J. L. Liu, V. Vieru, L. Ungur, J. H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X. M. Chen and M. L. Tong, A Stable Pentagonal Bipyramidal Dy(III) Single-Ion Magnet with a Record Magnetization
Reversal Barrier over 1000 K, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- P. Zhang, Q. C. Luo, Z. Zhu, W. He, N. Song, J. Lv, X. Wang, Q. G. Zhai, Y. Z. Zheng and J. Tang, Radical-Bridged Heterometallic Single-Molecule Magnets Incorporating Four Lanthanoceniums, Angew. Chem., Int. Ed., 2022, 62, e202218540 CrossRef PubMed.
- Z. H. Zhu, X. F. Ma, H. L. Wang, H. H. Zou, K. Q. Mo, Y. Q. Zhang, Q. Z. Yang, B. Li and F. P. Liang, A triangular Dy3 single-molecule toroic with high inversion energy barrier: magnetic properties and multiple-step assembly mechanism, Inorg. Chem. Front., 2018, 5, 3155–3162 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2260488. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00861d |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Fu-Pei
Liang
and
Hua-Hong
Zou
*,
Fu-Pei
Liang
and
Hua-Hong
Zou