
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu-based nanoparticles supported on carbon nanotubes for electrocatalytic hydrogen evolution: structural and electronic effects†
Nuria
Romero‡
 *ab,
Dídac A.
Fenoll‡
a,
Laia
Gil
a,
Sergi
Campos
a,
Jordi
Creus
ab,
Gerard
Martí
a,
Javier
Heras-Domingo
a,
Vincent
Collière
b,
Camilo A.
Mesa
c,
Sixto
Giménez
c,
Laia
Francàs
a,
Luis
Rodríguez-Santiago
a,
Xavier
Solans-Monfort
*a,
Mariona
Sodupe
a,
Roger
Bofill
a,
Karine
Philippot
*b,
Jordi
García-Antón
a and
Xavier
Sala
*a
*ab,
Dídac A.
Fenoll‡
a,
Laia
Gil
a,
Sergi
Campos
a,
Jordi
Creus
ab,
Gerard
Martí
a,
Javier
Heras-Domingo
a,
Vincent
Collière
b,
Camilo A.
Mesa
c,
Sixto
Giménez
c,
Laia
Francàs
a,
Luis
Rodríguez-Santiago
a,
Xavier
Solans-Monfort
*a,
Mariona
Sodupe
a,
Roger
Bofill
a,
Karine
Philippot
*b,
Jordi
García-Antón
a and
Xavier
Sala
*a
aDepartament de Química, Universitat Autònoma de Barcelona, Bellaterra, Cerdanyola del Vallès, 08193, Catalonia, Spain. E-mail: xavier.sala@uab.cat; xavier.solans@uab.cat
bCNRS, LCC (Laboratoire de Chimie de Coordination), UPR8241, Université de Toulouse, UPS, INPT, F-31077 Toulouse cedex 4, France. E-mail: nuria.romero@lcc-toulouse.fr; karine.philippot@lcc-toulouse.fr
cInstitute of Advanced Materials (INAM), Universitat Jaume I, Avenida de Vicent Sos Baynat, s/n, 12006 Castelló de la Plana, Castellón, Spain
First published on 21st June 2023
Abstract
The performance of Ru-based nanoparticles (NPs) in the hydrogen evolution reaction (HER) relies on both their structural properties and the oxidation state of the metal. Herein, the versatility of the organometallic approach for the synthesis of metal-based nanostructures is combined with thermal oxidation treatments to prepare carbon-nanotube (CNT)-supported Ru-containing nanomaterials for their use as electrocatalysts after dropcasting onto a glassy carbon rotating disk electrode. This strategy allowed access to a series of hybrid nanomaterials of different Ru/RuO2 compositions and different structural order. Linear sweep voltammetry experiments show that the relative disposition of the Ru/RuO2 phases, their interconversion under reductive turnover conditions and the degree of structural order affect the HER electrocatalytic performance of different materials. The electrode containing NPs consisting of a RuO2 core and metallic Ru at the surface outperforms that containing either pure RuO2 NPs or Ru NPs. Impedance spectroscopy studies and DFT calculations suggest that this catalytic activity enhancement arises from improved charge transport properties and from the structure of the exposed metallic Ru shell, which is partially oxidized and highly amorphous.
1. Introduction
The imperative need for supplanting fossil fuels with clean and renewable energy sources to mitigate climate change enforces the development of new renewable energy technologies.1–3 The production of hydrogen (H2) by the water splitting (WS) process that can be triggered either by green electricity or direct sunlight conversion is a promising strategy.4 In this endeavor, the design of cheap, active, and stable electrocatalysts to accelerate the involved processes, namely, the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER), is essential for achieving a worthwhile complete catalytic WS process.Metal-based nanoparticles (NPs), with high surface to volume ratios and good stability, are known to be very efficient and selective (electro)catalytic materials for many transformations,5 among which HER stands as a highly attractive example.6,7 In acidic media, Pt remains as the state-of-the-art metal. However, Ru-based nanomaterials have recently emerged as potential alternatives to Pt,8 with improved stability along the overall pH range and competitive kinetics (appropriate Gibbs free energy of the M–H bond)9 when properly designed.10 However, compared to Pt NPs, Ru NPs show larger cohesive energy,11 making them prone to agglomeration under turnover conditions. Thus, the dispersion and immobilization of NPs onto a conductive support is a key strategy to achieve an electrocatalyst with optimal long-term performance, not only preventing the aggregation of metal sites but also improving the electronic communication between the catalyst and the electrode.6,7,10
Among conductive supports, carbon-based nanomaterials are commonly used in electrocatalytic applications due to their robustness, chemical inertness, and availability on a large scale in a wide range of well-defined shapes and sizes.12 Of particular interest are carbon-nanotubes (CNTs), which show a highly exposed surface area, metal-like conductivity, good chemical resistance and stability, as well as appropriate hydrophilicity,13 which are suitable properties for electrocatalytic applications in aqueous media.14 In this context, CNTs have been shown to be effective at stabilizing Ru NPs on hybrid electrodes for the HER.15–22 Globally, the effect of (i) functionalizing the CNT surface by oxidative methods,17,19 (ii) doping the CNT support with one or more elements (i.e. N, B, Co/N),20,22 and (iii) combining Ru with another element to modulate the NP electronic properties (i.e. RuP2 or B-doped Ru), on the overall electrocatalytic performance, has been reported.18,21 It is worth mentioning that even though the as-synthesized mixtures of Ru and RuO2 phases have been identified as the best-performing Ru–CNT electrocatalysts,16,17 their interconversion under reductive turnover conditions and their respective (or synergistic) role in the ultimate HER performance have not been studied in detail. Also, the effect of the structural properties (crystalline/amorphous) of the CNT-supported Ru/RuO2 electrocatalysts on the HER activity has not yet been discussed. To study the influence of these parameters, we combined the organometallic approach for the synthesis of nanostructures,23,24 an efficient method to obtain small NPs with a narrow size-distribution and controllable surface properties, with thermal oxidation treatments of CNTs in order to obtain a series of CNT-supported Ru-based NPs of varied Ru/RuO2 compositions and different structural orders, and test their electrocatalytic activity in the HER. The monitoring of the evolution of these nanomaterials under reductive turnover conditions, combined with the DFT modelling of their structures and computed H-adsorption energies, has allowed us to establish a correlation between the characteristics of the CNT-supported Ru-based NPs and their electrocatalytic properties in the HER.
2. Experimental
2.1. Synthetic procedures
All operations for the synthesis of Ru/CNT nanomaterials were carried out using standard Schlenk tubes, Fisher Porter bottle techniques or in a glove box (MBraun) under an argon atmosphere. Solvents (THF and hexane) were purified before use by distillation under a N2 atmosphere with a drying agent (sodium + benzophenone or CaH2, respectively) and degassed according to a freeze–pump–thaw process. The ruthenium precursor, [Ru(COD)(COT)] (COD = 1,5-cyclooctadiene; COT = 1,3,5-cyclooctatriene) was purchased from Nanomeps, Toulouse. Ru(IV) oxide 99.9% purity was purchased from Merck. Hydrogen gas with a purity of 5.0 was purchased from Abelló Linde, S.A. CNTs were purchased from Nanostructured & Amorphous Materials, Inc. (Texas, USA) with 99.9% purity and an OD of 50–80 nm and a length of 10–20 μm. High purity deionized water was obtained by passing distilled water through a nanopore Milli-Q water purification system.2.2. Characterization techniques
A Unisense H2-NP Clark type electrode was used to measure the produced hydrogen in the gas phase during the chronopotentiometry experiments by placing it together with the WE (in this case a FTO electrode) and RE (in this case Ag/AgCl) in the same compartment. Faradaic efficiencies were calculated for each system. The sensor was calibrated by adding different known volumes of 99% pure hydrogen at the end of the experiment.
Linear sweep voltammetry (LSV). For LSV, a 20 mL vial was used as an electrochemical cell. For HER experiments, the system was scanned from Ei = 0.400 V to Ef = −0.360 V vs. RHE at a scan rate of 10 mV s−1 unless otherwise stated.
Chronopotentiometry (CP). Controlled current intensity experiments were performed at japp = −10 mA cm−2 for the time indicated in each case.
2.3. Computational details
H adsorption energies reported along the text are mean values that were computed as proposed by Nørskov and co-workers39 and summarized in eqn (1):
| Eads = [EnH-surf − (Esurf + nEH2/2)]/n, | (1) |
| Eads = [E(N+1)H-surf − (ENH-surf + EH2/2)], | (2) |
3. Results and discussion
3.1. Synthesis of CNT-supported Ru-based nanomaterials
Ru(0) NPs were synthesized on the surface of CNTs following the organometallic method,24,40 which consists of decomposition under mild conditions (3 bar H2, R.T. 2 h of vigorous stirring) of [Ru(COD)(COT)] (COD = 1,5-cyclooctadiene; COT = 1,3,5-cyclooctatriene) in the presence of multi-walled CNTs in THF (step 1 in reaction (1), Fig. 1, detailed synthetic conditions can be found in the Experimental section). The obtained nanomaterials were then exposed to ambient air (step 2 in reaction (1), Fig. 1) during the purification process (washing with THF and hexane, centrifugation), which induced surface oxidation, yielding core–shell Ru@RuO2/CNT nanomaterials. Ru@RuO2/CNT was then subjected to oxidative treatment under air at 300 °C for 10 (RuO2-10′/CNT) (reaction (2), Fig. 1) or 120 minutes (RuO2-120′/CNT) (reaction (3), Fig. 1) to access fully oxidized RuO2 NPs of different sizes and morphologies (see below).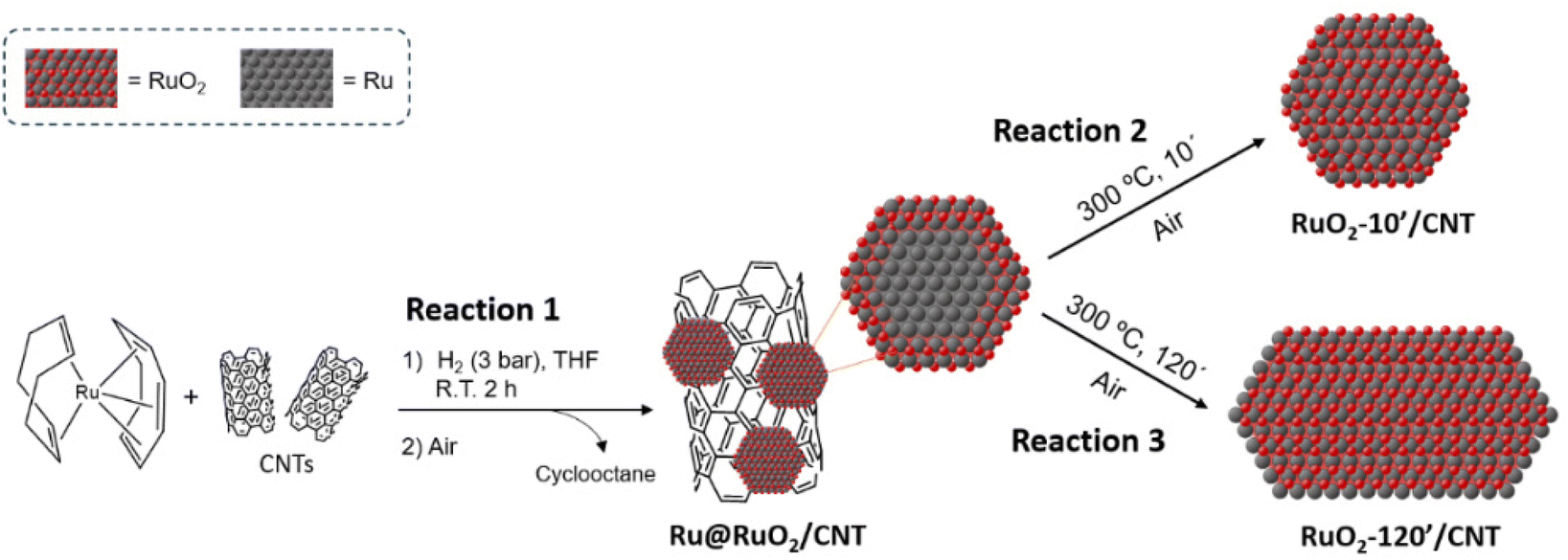 | ||
| Fig. 1 Scheme of the synthesis of CNT-supported Ru-based nanomaterials. | ||
3.2. Structural, morphological and compositional characterization of CNT-supported Ru-based nanomaterials
The obtained CNT-supported Ru-based nanomaterials were characterized by TEM, HRTEM-EDX and XPS techniques (Fig. 2 and Fig. S1–S5†) to evaluate the dispersion, shape, size distribution and composition of Ru NPs.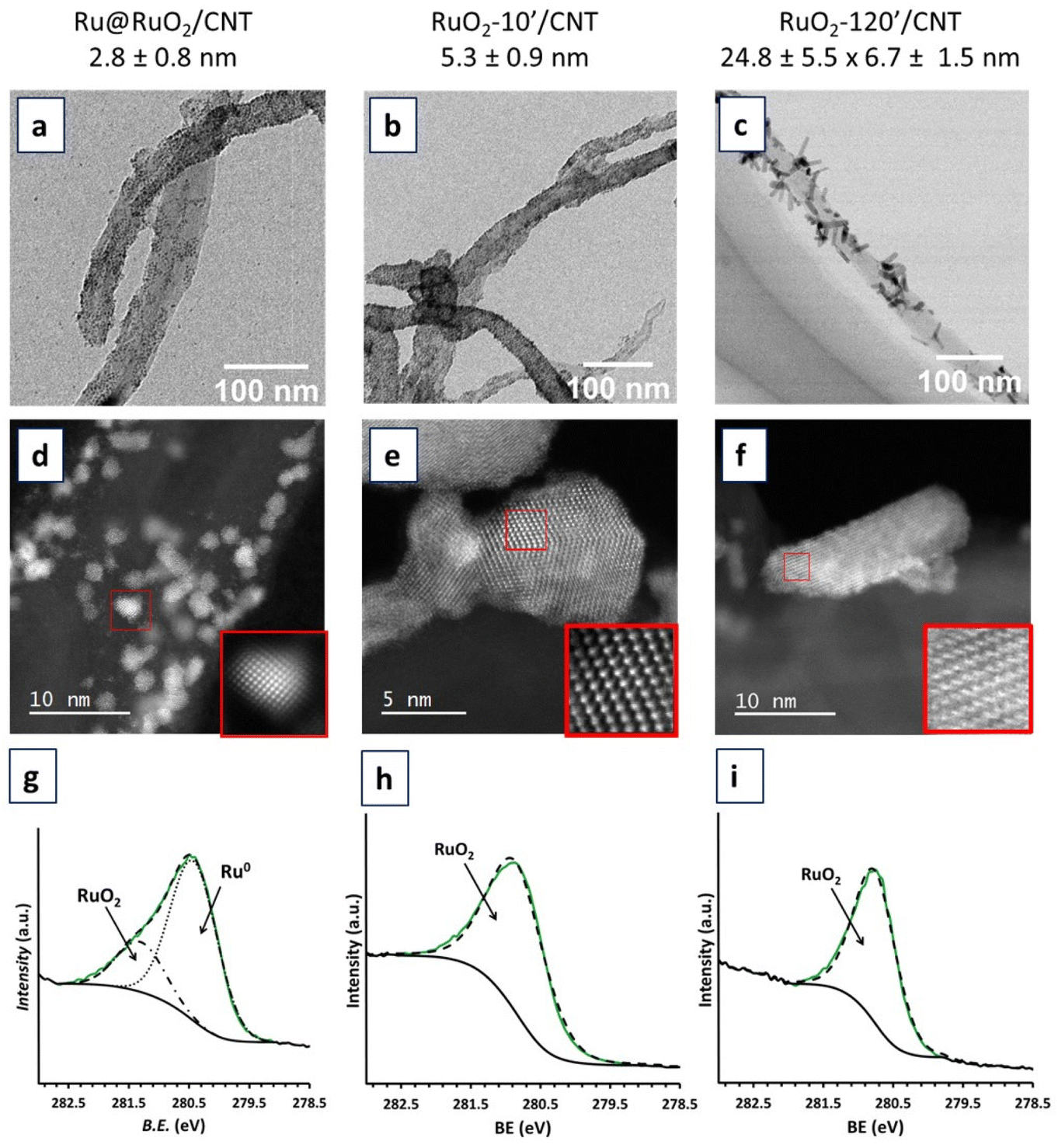 | ||
| Fig. 2 TEM images, HRTEM images and XPS spectra of Ru@RuO2/CNT (a, d and g), RuO2-10′/CNT (b, e and h) and RuO2-120′/CNT (c, f and i) nanomaterials. In the XPS spectra, the RuO2 component is shown by a dotted-dashed black line, the Ru(0) component by a dotted black line, the envelope by a dashed black, the experimental data by a solid green line and the background signal by a solid black line. | ||
TEM and HRTEM analyses showed that the Ru@RuO2/CNT nanomaterial contains well dispersed, well defined and highly crystalline NPs of 2.8 ± 0.8 nm average size at the surface of the carbon support (Fig. 2a and S1†). Careful local EDX analysis of individual NPs in a holey carbon covered copper grid showed the presence of only Ru and small amounts of oxygen in all cases (Fig. S2†). XPS analysis (Fig. 2g) revealed the presence of both Ru(0) and RuO2 in a mixture of ca. 0.76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.24, as shown by the Ru 3d5/2 peaks centered at 280.4 eV and 281.3 eV for metallic Ru and RuO2, respectively. The presence of both Ru(0) and RuO2 in the NPs results from a surface passivation phenomenon that commonly occurs under atmospheric oxygen,9,40–42 yielding core@shell Ru@RuO2 NPs on the CNTs. The crystallinity of the Ru NPs has been confirmed by HRTEM. Electron diffraction patterns obtained after fast Fourier transform (FFT) treatment (Fig. S1†) indicated two families of interplanar distances for individual NPs, namely 0.21, 0.23 and 0.11 nm, that correspond to the (002), (110) and (004) planes of the hexagonal compact crystalline (hcp) structure of bulk Ru(0), and 0.13, 0.12 and 0.25 nm, that correspond to the (022), (111) and (011) planes of the rutile structure of RuO2.
:0.24, as shown by the Ru 3d5/2 peaks centered at 280.4 eV and 281.3 eV for metallic Ru and RuO2, respectively. The presence of both Ru(0) and RuO2 in the NPs results from a surface passivation phenomenon that commonly occurs under atmospheric oxygen,9,40–42 yielding core@shell Ru@RuO2 NPs on the CNTs. The crystallinity of the Ru NPs has been confirmed by HRTEM. Electron diffraction patterns obtained after fast Fourier transform (FFT) treatment (Fig. S1†) indicated two families of interplanar distances for individual NPs, namely 0.21, 0.23 and 0.11 nm, that correspond to the (002), (110) and (004) planes of the hexagonal compact crystalline (hcp) structure of bulk Ru(0), and 0.13, 0.12 and 0.25 nm, that correspond to the (022), (111) and (011) planes of the rutile structure of RuO2.
HRTEM analysis of the RuO2-10′/CNT nanomaterial (oxidation in air, 300 °C, 10 min) showed an increase of the average size of the Ru NPs, up to 5.3 ± 0.9 nm (Fig. 2b and S3†). This NP growth may be explained by the diffusion of the NPs along the CNTs during the temperature treatment (sintering) as well as the incorporation of oxygen atoms into the structure upon oxidation of the NP core. As can be observed for the RuO2-120′/CNT nanomaterial (Fig. 2c and S4†), the same oxidation treatment for 120 min provoked the coalescence of the NPs and led to the formation of well crystallized Ru-based nanorods (6.7 ± 1.5 nm in width and 24.8 ± 5.5 nm in length). FFT treatment of the HRTEM image or the electron diffraction patterns of RuO2-10′/CNT and RuO2-120′/CNT nanomaterials (Fig. S3 and S5,† respectively) indicate interplanar distances of 0.13, 0.16, 0.17 and 0.25 nm, which agree with the (202), (220), (211) and (101) planes of the rutile structure of RuO2. XPS analyses of the RuO2-10′/CNT and RuO2-120′/CNT nanomaterials (Fig. 2h and i) confirmed the complete oxidation of the as-prepared Ru@RuO2 NPs, the unique peak is observed at 280.8 eV corresponding to the Ru 3d5/2 band of RuO2. This is corroborated by EDX analysis, where the presence of Ru and O elements in high amounts is detected for both samples (Fig. S3 and S4†).
3.3. Electrocatalytic HER studies
The Ru@RuO2/CNT, RuO2-10′/CNT and RuO2-120′/CNT nanomaterials were dispersed in THF and the obtained suspensions were dropcast onto a glassy carbon (GC) rotating disk electrode. The so-obtained Ru@RuO2/CNT/GC, RuO2-10′/CNT/GC, and RuO2-120′/CNT/GC were introduced as working electrodes in a three-electrode cell in 1 M H2SO4 and then subjected to a reductive treatment (20 min chronopotentiometry (CP) at −10 mA cm−2),9,43 leading to the corresponding reduced counterparts: r-Ru@RuO2/CNT/GC, r-RuO2-10′/CNT/GC, and r-RuO2-120′/CNT/GC (“r-” will be used from now on for all hybrid nanomaterials exposed to a reductive potential). Linear sweep voltammetry (LSV) experiments under reductive potentials (from 0.40 V to −0.35 V vs. RHE) were recorded for both the as-synthesized and reduced nanomaterials to evaluate their electrocatalytic performance in the HER. The LSV plots and the main benchmarking parameters are presented in Fig. 3 and Table S1,† respectively.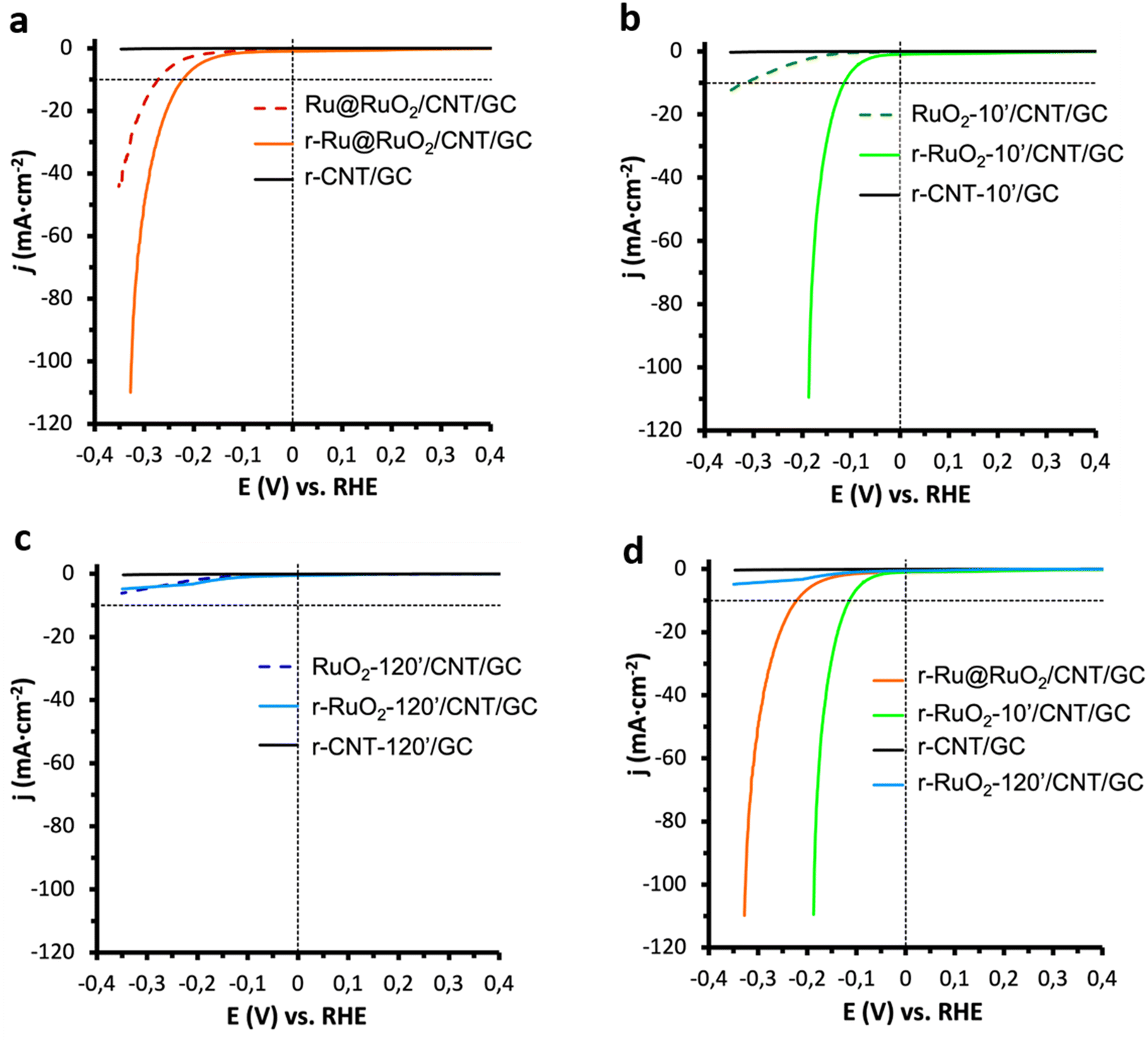 | ||
| Fig. 3 LSVs in 1 M H2SO4 of (a) Ru@RuO2/CNT/GC (dashed red line) and r-Ru@RuO2/CNT/GC (orange line), (b) RuO2-10′/CNT/GC (dashed green line) and r-RuO2-10′/CNT/GC (light green line) and (c) RuO2-120′/CNT/GC (dashed blue line) and r-RuO2-120′/CNT/GC (light blue line), before and after a 20 min reductive chronopotentiometry at j = −10 mA cm−2, respectively; (d) comparison of polarization curves of r-Ru@RuO2/CNT/GC (orange), r-RuO2-10′/CNT/GC (light green line ) and r-RuO2-120′/CNT/GC (light blue line) after a 20 min reductive chronopotentiometry at j = −10 mA cm−2. The LSV curve corresponding to each r-CNT-X/GC (X = 0-, 10- or 120-minutes oxidation at 300 °C) blank (no catalyst present) is shown in black. The thermodynamic HER potential in 1 M H2SO4 and the reference j = −10 mA cm−2 current density are indicated as black dashed lines. | ||
The Ru@RuO2/CNT/GC electrode shows an onset overpotential of 200 mV (η0 = 200 mV) and a current density of 10 mA cm−2 is achieved at an overpotential of 272 mV (η10 = 272 mV) (Fig. 3a, dashed). However, a clear shift of the polarization curve is observed for its reduced counterpart (r-Ru@RuO2/CNT/GC), which shows a decrease in both overpotentials by 50 mV (Fig. 3a, bold) to η0 = 150 mV and η10 = 222 mV. XPS analysis of the r-Ru@RuO2/CNT nanomaterial (Fig. S6b†) shows the total disappearance of the RuO2 phase, leading to a pure Ru(0)-based nanomaterial. Given the already described superior HER activity of the Ru(0) phase over the RuO2 one, the disappearance of the RuO2 phase after the reductive treatment by chronopotentiometry can explain the detected enhancement in HER activity.41,43
The electrocatalytic enhancement after negative bias is even more evident when comparing the HER electro-activity of the RuO2-10′/CNT/GC electrode and that of its reduced counterpart, r-RuO2-10′/CNT/GC (Fig. 3b); η0 and η10 decrease from 130 to 50 mV and from 319 to 115 mV, respectively, and the Tafel slope (Fig. S7†) drops from 289 to 77 mV dec−1. XPS analysis of the r-RuO2-10′/CNT/GC nanomaterial shows an asymmetric XPS peak that includes both RuO2 (Ru 3d5/2 – 281.3 eV) and metallic Ru (Ru 3d5/2 – 280.4 eV) components (Fig. S6c†), with an approximate contribution of 85% and 15%, respectively. The observed activation in the HER may thus arise from the formation of highly active Ru metal atoms, that may eventually form a thin shell on the top of the initial RuO2 core (see Fig. S6a,† bottom). Interestingly, the HER activity of the r-RuO2-10′/CNT/GC electrode, with a RuO2 core and metallic Ru at the surface, outperforms that of the r-Ru@RuO2/CNT/GC electrode, where only metallic Ru is present. Also, the faradaic efficiency of both reduced nanomaterials was evaluated on larger surface-area fluorine doped tin oxide (FTO) electrodes (Fig. S8†). The attained >98% value confirms the production of H2 as the sole reaction happening under turnover conditions.
In contrast, the nanomaterials resulting from a 120 min thermal treatment in both as-synthesized and cathodically polarized versions (RuO2-120′/CNT/GC and r-RuO2-120′/CNT/GC) showed negligible HER electro-activity (Fig. 3c). These results indicate that the presence of large and highly crystalline RuO2 nanorods on the electrode is not favorable for the HER activity, clearly highlighting the influence of the duration of the applied thermal treatment on the electrocatalytic performance. Similar results (poor electro-activity of both as-received and cathodically polarized counterparts) were obtained when the HER performance of bulk commercial RuO2 was assayed under identical electrocatalytic conditions (Fig. S9†). This is also in accordance with previous reports where a decrease in the HER activity of hydrous RuO2 NPs was observed after enhancing their crystallinity through annealing processes.44,45 Given the inactivity of the RuO2-120′/CNT/GC and r-RuO2-120′/CNT/GC samples in the HER, these electrodes have not been studied further.
With the aim at better understanding the reasons behind the activity trends in the HER described in this section, impedance spectroscopy (EIS) measurements (Section 3.4) and DFT calculations (Section 3.5) have been performed (vide infra).
3.4. Impedance spectroscopy studies
Impedance spectroscopy experiments were first carried out on the as-received CNT supports and their 10′ annealed (air, 300 °C) counterparts. A Randles equivalent circuit was used to fit the experimental data (see Fig. S10† and its inset for a typical Nyquist plot and the equivalent circuit, respectively). Accordingly, capacitance (C) and charge transfer resistance (RCT), as well as series resistance (RS) were obtained. No difference was observed between the as-received and 10′-annealed CNTs (Fig. S11†).Analogous analyses were performed on the r-Ru@RuO2/CNT/GC, RuO2-10′/CNT/GC and r-RuO2-10′/CNT/GC electrodes in order to define the reasons behind their different HER behaviors (Fig. 4).
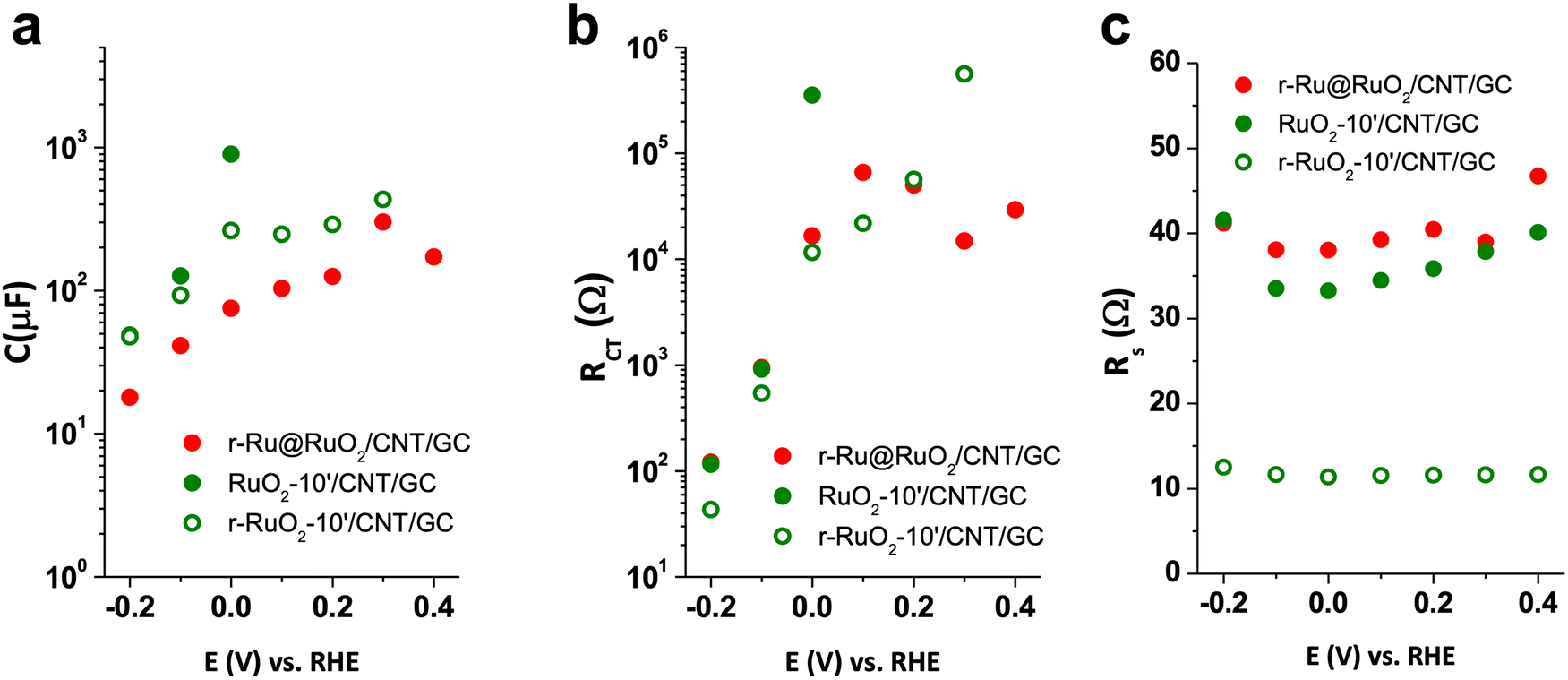 | ||
| Fig. 4 Impedance spectroscopy results C, RCT and RS, fitting the Nyquist plots to a simple Randles circuit (see Fig. S9,† inset) for r-Ru@RuO2/CNT/GC (red circles), RuO2-10′/CNT/GC (green circles) and r-RuO2-10′/CNT/GC (green empty circles). (a) Capacitance (C), (b) charge transfer resistance (RCT) and (c) series resistance (RS). | ||
When comparing the best performing r-RuO2-10′/CNT/GC electrode with its oxidized counterpart, RuO2-10′/CNT/GC, the former presents a lower capacitance and charge transfer resistance before the catalytic region (Fig. 4a and b). This observation is consistent with a decrease in the contribution of the RuO2 phase due to the reductive treatment. Interestingly, the measured uncompensated resistance (series resistance, RS) of this r-RuO2-10′/CNT/GC electrode is four times lower than those for r-Ru@RuO2/CNT/GC and RuO2-10′/CNT/GC, suggesting improved charge transport within the conductive support and further charge extraction due to the thermal oxidation followed by electrochemical reduction (Fig. 4c). However, the three systems present a series resistance (RS) lower than the charge transfer resistance (RCT) in the catalytic region. This observation indicates that the whole process is limited by the catalyst kinetics rather than the cell resistance.
3.5. DFT calculations
To get further insight into the origin of the high catalytic activity of the NPs in r-RuO2-10′/CNT/GC, we performed DFT (PBE-D2)26,29 periodic calculations (see the Computational details in Section 2.3 for more information). The NPs in r-RuO2-10′/CNT/GC were represented with three different slab models, covering different amounts of Ru over a crystalline RuO2 surface (Fig. 5). A Ru surface, as a model of NPs in r-Ru@RuO2/CNT/GC, and a Pt surface, as a reference material, were also considered for comparison. The nomenclature of the computational models is Ru and Pt for the metallic systems, and RuO2@RuN for the models of NPs in r-RuO2-10′/CNT/GC (N indicates the number of Ru atoms over the RuO2 surface). | ||
| Fig. 5 Studied adsorption sites for (a) the adsorption of one single H atom (θ = 1/N) and (b) the adsorption of one additional H atom to an H monolayer (θ = (N + 1)/N). Site name indicates the number of Ru atoms (1, 2 or 3) interacting with H and the involved centers. Outermost atom color labeling indicates the atomic Bader charges without adsorbed hydrogen. The adsorption energy for each site is reported in the ESI.† | ||
According to Nørskov and co-workers, the H adsorption energy can be used as a descriptor of the electrocatalytic activity in the HER of a specific material.39,46,47 Therefore, we computed the adsorption energy of H for the five models at three different hydrogen coverages (θ), namely 1 hydrogen on the surface (θ = 1/N), 1 hydrogen per Ru surface atom (θ = 1), and one additional hydrogen atom exceeding the 1 hydrogen per Ru center ratio (θ = (N + 1)/N). Several different sites were considered in each case (Fig. 5).
For Ru and Pt surfaces with well-ordered close packed planes, we analyzed three different sites: (1) above a tetrahedral (Td) hole (3Td); (2) above an octahedral (Oh) hole (3Oh) and (3) over a metal atom of the surface (1Top). Alternatively, for RuO2@Ru7, RuO2@Ru10 and RuO2@Ru20 we explored several sites covering the most representative environments, including the interaction of H with 3, 2 and 1 Ru centers (Fig. 5). Table 1 reports the highest adsorption energies of the first H atom (θ = 1/N), the mean value of the adsorption energy of one H per Ru surface site (θ = 1) and the adsorption energy range of additional hydrogen atoms after the adsorption of one hydrogen atom per Ru site (θ = (N + 1)/N). Note that in this latter case there are also several potential adsorption sites, from which we considered a selected representative set (Fig. 5b). The adsorption energies of each site with the optimized structures of all considered species can be found in the ESI (Fig. S12–S16†).
| Model | θ = 1/N | θ = 1a | θ = (N + 1)/Nb |
|---|---|---|---|
| a Highest adsorption energy at this H coverage. b Range of adsorption energies for this coverage. c H adsorption energies computed with a RuO2@Ru20 model containing a 8 RuO2 layer thick core (Fig. S17†). | |||
| Pt | −58.0a | −50.3 | −32.0 to −34.9 |
| Ru | −71.1a | −66.8 | +14.1 to +33.8 |
| RuO2@Ru7 | −76.6 to −49.7b | −66.3 | −23.5 to −31.2 |
| RuO2@Ru10 | −76.1 to −31.4b | −59.7 | −27.5 to −32.2 |
| RuO2@Ru20 | −70.7 to −30.5b | −57.9 | −31.9 to −51.3 |
| −70.3 to −38.5c | −56.6c | −20.5 to −55.3c | |
At low coverages (θ = 1/N), H adsorbs strongly on Ru (Eads(H) = −71.1 kJ mol−1), with a marked preference for 3Oh sites. The adsorption of H on Pt is weaker (Eads(H) = −58.0 kJ mol−1) and shows no preference for any of the three sites studied. At higher coverages (θ = 1), the same trends are observed, the adsorption of hydrogen on Ru is strong and the adsorption on Pt is weaker (−66.8 vs. −50.3 kJ mol−1) and without a clear preferred site. In both cases, however, the adsorption energies are markedly higher (in absolute value) than those defining a good catalyst according to the Norskøv descriptor (Eads(H) = −23 kJ mol−1).39 Remarkably, the adsorption of additional H atoms to the monolayer is unfavorable on Ru, but it is still favorable on Pt, with adsorption energies of about −32.0 kJ mol−1. These data suggest that H adsorbs too strongly on Ru at low coverages, thus preventing H2 evolution. Moreover, at high H coverages (exceeding 1 H per surface Ru center), H2 evolution on crystalline Ru is hampered by the unfavorable H adsorption. In contrast, while at low coverages H2 evolution on Pt is also unfavorable due to a strong H adsorption, H adsorption at high coverages is weak and close to the ideal value,39 and thus H2 evolution is strongly favored on Pt.
The outermost Ru centers of RuO2@Ru7, RuO2@Ru10 and RuO2@Ru20, which are models of r-RuO2-10′/CNT/GC, show a high degree of amorphization (Fig. 5). This is attributed to the roughness of the (110) RuO2 surface, which prevents the formation of Ru close packed planes. Consequently, exposed atoms present a lower coordination and the surface does not show well-defined Oh, Td and Top sites. The adsorption energy of H on RuO2@Ru7, RuO2@Ru10 and RuO2@Ru20 at low coverage (θ = 1/N) ranges from −76.6 to −30.5 kJ mol−1, depending on the number of Ru centers interacting with the adsorbed hydrogen and their charges (vide infra). In all cases, the adsorption energy of the sites presenting the strongest interaction with H of each model is similar to the values computed for Ru, but there are also several sites where H adsorbs more weakly. Indeed, the H adsorption energy tends to increase (in absolute value) upon increasing the number of metal centers that the adsorbed H is interacting with, and on decreasing the positive charge of the Ru centers interacting with H. As a consequence, the final adsorption energies are probably the compromise of three factors: (1) the lower metal coordination in amorphous RuO2@Ru7, RuO2@Ru10 and RuO2@Ru20 models that leads to stronger H adsorption; (2) the layer amorphization that hampers the adsorption of H on the ideal Oh sites and (3) the partial oxidation of the external Ru atoms by RuO2, the latter two factors make the Ru–H interaction weaker.
The average adsorption energies of one H atom per exposed Ru center (θ = 1) are quite similar for the three RuO2@Ru7, RuO2@Ru10 and RuO2@Ru20 models (−66.3, −59.7 and −57.9 kJ mol−1, respectively) and close to the value on crystalline Ru (−66.8 kJ mol−1), thus suggesting that H2 evolution does not arise from coverages below θ = 1. Indeed, the HER takes place when additional H atoms are added to a coverage of θ = 1. In this scenario and in contrast to what is computed for crystalline Ru, the adsorption of one additional H atom on the three models of r-RuO2-10′/CNT/GC is favorable and usually weak, with values ranging between −23.5 and −51.3 kJ mol−1. Indeed, while there are still some sites showing strong adsorption energies, most of the considered centers present values that are similar to those obtained for Pt or even closer to the ideal value. Remarkably, enlarging the RuO2 core for the RuO2@Ru20 model (see Fig. S17†) does not modify the H adsorption energies (Table 1), thus suggesting that the thickness of the RuO2 core has very little influence on the material catalytic activity. Overall, calculations are in agreement with the higher HER catalytic activity of r-RuO2-10′/CNT/GC when compared to r-Ru@RuO2/CNT/GC, and the calculations suggest that this originates from two factors: (1) the amorphization of the external Ru atoms, that increases the number of hydrogen atoms that can be adsorbed and (2) the partial oxidation of the surface Ru centers by RuO2, that globally weakens the H adsorption (less negative values).
4. Concluding remarks
In the literature, the performances of the state-of-the-art CNT-supported Ru NP-based HER electrocatalysts are usually discussed on the basis of the as-synthesized Ru/RuO2 mixtures.16,17 The interconversion of the two Ru phases that can occur under HER conditions and their respective (or synergistic) role in the ultimate HER performance have not been previously deeply studied, nor the effect of the structural properties (crystalline/amorphous) of these composite materials. In order to clarify these issues, in our study we have combined the organometallic approach for the synthesis of Ru NPs with thermal and electrochemical processes. This allowed us to have at disposal a series of CNT-supported Ru-containing NPs of assorted composition, disposition of the Ru phases and crystalline order, for a comparison of their catalytic activities in the HER (Scheme 1). Our results show that the relative disposition of the Ru/RuO2 phases, their interconversion under reductive turnover conditions and the degree of structural order in the studied materials affect the HER electrocatalytic performance. | ||
| Scheme 1 Proposed scenario for the oxidation/reduction processes occurring at the surface of CNT-supported Ru-based NPs studied in this work. CP = Chronopotentiometry. In A–E the CNT support has been omitted for the sake of clarity. | ||
The decomposition of [Ru(COD)(COT)] under H2 (3 bar, R.T.) in the presence of CNTs followed by air exposure led to a CNT-supported Ru-based nanomaterial with small Ru@RuO2 core–shell NPs well-dispersed onto the CNTs (Scheme 1, A). Subsequent oxidative thermal treatment (ambient air, 300 °C) of this nanomaterial provided CNT-supported RuO2 NPs (Scheme 1, B) and CNT-supported RuO2 nanorods (Scheme 1, C), after a 10 or 120 min treatment, respectively. Reductive chronopotentiometry under HER electrocatalytic conditions (−10 mA cm−2) in acidic medium (1 M H2SO4) allowed the reduction of the RuO2 shell of the Ru@RuO2 NPs in the as-synthesized nanomaterial A, yielding CNT-supported fully metallic Ru NPs (Scheme 1, D). An identical CP reductive treatment on the fully oxidized RuO2 NPs in nanomaterial B led to RuO2@Ru core–shell NPs (Scheme 1, E), while the RuO2 nanorods in C remained the same (Scheme 1, C). Surface reduction in C is prevented by the high reorganizational energy of the highly crystalline RuO2 surface, giving an inactive nanomaterial in the HER. Interestingly, when applied to the Ru@RuO2 NPs in A or the RuO2 NPs in B, the CP reductive treatment (Scheme 1, A → D and B → E) yielded the superior HER electrocatalysts D and E, respectively. These results highlighted the predominant role of the metallic Ru sites at the NP surface over the RuO2 ones in the HER.
Even more remarkably, the nanomaterial E that contains RuO2@Ru core–shell NPs showed a superior HER activity than its counterpart D, containing fully metallic Ru NPs. In order to rationalize this difference in HER electroactivity, a DFT analysis of the H-adsorption energy values was performed. Periodic slab models were used to mimic the hybrid Ru–RuO2 nanomaterials in comparison with pure Ru and Pt metal surfaces, with different H coverages. The calculated data of H adsorption energies reproduce the experimental catalytic activity trends and reinforce their use as descriptors for analyzing their HER activity. When the Ru shell is only one layer thick or below, surface amorphization prevents the adsorption of H at the octahedral sites, which weakens the Ru–H interaction and generates additional sites with adsorption energies close to those of Pt. Moreover, the presence of a RuO2 core weakens the H adsorption energies, bringing them closer to the ideal value. Additionally, the presence of a Ru layer on top of the RuO2 core yields improved charge transport properties, as demonstrated by EIS measurements. Overall, the r-RuO2-10′/CNT/GC nanomaterial E, which contains RuO2@Ru NPs, shows higher electrocatalytic activity when compared to D (pure Ru metal) likely due to two factors: its surface partial oxidation and, more importantly, its amorphization. This amorphization may also explain the high HER electro-activity reported for very small Ru NPs, which are quite amorphous and have numerous surface sites and defects.9,10 This hypothesis is supported by H adsorption calculations on Ru NPs that highlighted the presence of these less coordinated H atoms.9,48,49
All in all, the results obtained using controlled CNT-supported Ru-based nanomaterials have put forward the correlation between the oxidation state of Ru (and its evolution under reductive turnover conditions), the degree of structural order at the NP surface and the H adsorption energies as the key activity descriptors. This work provides valuable guidelines to rationally design Ru-based electrocatalysts of higher performance for the HER.
Author contributions
Conceptualization, N. R., X. S.-M., K. P., and X. S.; investigation, N. R., D. F., L. G., S. C., G. M., J. C., V. C., J. H.-D., L. F., C. M., and V. C.; writing – original draft preparation, R. B., L. F., N. R., X. S.-M., X. S. and J. G.-A.; writing – review and editing, S. G., M. S., L. R.-S., J. C., R. B., K. P., N. R., L. F., J. G.-A., C. M. and X. S.; supervision, X. S.-M., L. R.-S., M. S., J. G.-A., R. B., X. S., N. R., and S. G.; and funding acquisition, X. S.-M., K. P., S. G., L. F., J. G.-A., and X. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the MICINN/FEDER projects PID2019-104171RB-I00, PID2020-112715GB-I00, PID2021-128197NA-I00 and TED2021-129237B-I00, the Red Española de Supercomputación (RES) through QHS-2021-3-0016 project, the CNRS, the Univ. Toulouse III – Paul Sabatier and the GDRI HC3A Franco-Catalan action. J. C. thanks the UAB for a PhD grant. G. M. and D. A. F. thank MICINN for their FPU PhD grants. X. S. thanks ICREA for the ICREA Academia prize 2020. L. F. is indebted to the Ramón y Cajal Program (RYC2018-025394-I fellowship). S. G. acknowledges Universitat Jaume I for financial support through the project UJI-B2019-20. C. A. M. acknowledges the Generalitat Valenciana for the APOSTD/2021/251 fellowship. The authors also acknowledge Guillaume Sauthier for XPS analyses.References
- X. Li, H. Zhang, Q. Hu, W. Zhou, J. Shao, X. Jiang, C. Feng, H. Yang and C. He, Amorphous NiFe Oxide-based Nanoreactors for Efficient Electrocatalytic Water Oxidation, Angew. Chem., Int. Ed., 2023, 62, e202300478 CrossRef CAS PubMed.
- Q. Hu, Z. Wang, X. Huang, Y. Qin, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, A unique space confined strategy to construct defective metal oxides within porous nanofibers for electrocatalysis, Energy Environ. Sci., 2020, 13, 5097–5103 RSC.
- Q. Hu, Z. Han, X. Wang, G. Li, Z. Wang, X. Huang, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Facile Synthesis of Sub-Nanometric Copper Clusters by Double Confinement Enables Selective Reduction of Carbon Dioxide to Methane, Angew. Chem., Int. Ed., 2020, 59, 19054–19059 CrossRef CAS PubMed.
- N. S. Lewis, Research opportunities to advance solar energy utilization, Science, 2016, 351, aad1920 CrossRef PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- S. Fukuzumi and Y. Yamada, Catalytic activity of metal-based nanoparticles for photocatalytic water oxidation and reduction, J. Mater. Chem., 2012, 22, 24284–24296 RSC.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Subnanometric Ru clusters with upshifted D band center improve performance for alkaline hydrogen evolution reaction, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- J. Creus, S. Drouet, S. Suriñach, P. Lecante, V. Collière, R. Poteau, K. Philippot, J. García-Antón and X. Sala, Ligand-Capped Ru Nanoparticles as Efficient Electrocatalyst for the Hydrogen Evolution Reaction, ACS Catal., 2018, 8, 11094–11102 CrossRef CAS.
- J. Creus, J. de Tovar, N. Romero, J. García-Antón, K. Philippot, R. Bofill and X. Sala, Ruthenium Nanoparticles for Catalytic Water Splitting, ChemSusChem, 2019, 12, 2493–2514 CrossRef CAS PubMed.
- Q. Wang, M. Ming, S. Niu, Y. Zhang, G. Fan and J. S. Hu, Scalable Solid-State Synthesis of Highly Dispersed Uncapped Metal (Rh, Ru, Ir) Nanoparticles for Efficient Hydrogen Evolution, Adv. Energy Mater., 2018, 8, 1801698 CrossRef.
- N. Karousis, N. Tagmatarchis and D. Tasis, Current progress on the chemical modification of carbon nanotubes, Chem. Rev., 2010, 110, 5366–5397 CrossRef CAS PubMed.
- J. Sheng and Y. Li, ACS Appl. Mater. Interfaces, 2022, 14, 20455–20462 CrossRef CAS PubMed.
- X. Zhang, Y. Ye, H. Wang and S. Yao, Deposition of platinum-ruthenium nano-particles on multi-walled carbon nano-tubes studied by gamma-irradiation, Radiat. Phys. Chem., 2010, 79, 1058–1062 CrossRef CAS.
- B. Santosh, Z. Wu, R. Sai Siddhartha, G. Krishna Prasad, S. S. Ramamurthy, S. Mitra and R. B. Dandamudi, Ruthenium decorated carbon nanoink as highly active electrocatalyst in hydrogen evolution reaction, Int. J. Hydrogen Energy, 2016, 41, 23007–23014 CrossRef CAS.
- M. Zhang, J. Chen, H. Li, P. Cai, Y. Li and Z. Wen, Ru-RuO2/CNT hybrids as high-activity pH-universal electrocatalysts for water splitting within 0.73 V in an asymmetric-electrolyte electrolyzer, Nano Energy, 2019, 61, 576–583 CrossRef CAS.
- D. H. Kweon, M. S. Okyay, S. J. Kim, J. P. Jeon, H. J. Noh, N. Park, J. Mahmood and J. B. Baek, Ruthenium anchored on carbon nanotube electrocatalyst for hydrogen production with enhanced Faradaic efficiency, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- M. Cheng, H. Geng, Y. Yang, Y. Zhang and C. C. Li, Optimization of the Hydrogen-Adsorption Free Energy of Ru-Based Catalysts towards High-Efficiency Hydrogen Evolution Reaction at all pH, Chem. – Eur. J., 2019, 25, 8579–8584 CrossRef CAS PubMed.
- R. Ding, L. Lin, C. Pei, X. Yu, Q. Sun and H. S. Park, Hierarchical Architectures Based on Ru Nanoparticles/Oxygen-Rich-Carbon Nanotubes for Efficient Hydrogen Evolution, Chem. – Eur. J., 2021, 27, 11150–11157 CrossRef CAS PubMed.
- M. Fan, X. Chen, M. Zhang, L. Cui, X. He and X. Zou, Highly dispersed Ru nanoclusters anchored on B,N co-doped carbon nanotubes for water splitting, Inorg. Chem. Front., 2022, 9, 968–976 RSC.
- X. Sun, W. Li, J. Chen, X. Yang, B. Wu, Z. Wang, B. Li and H. Zhang, Boron-induced activation of Ru nanoparticles anchored on carbon nanotubes for the enhanced pH-independent hydrogen evolution reaction, J. Colloid Interface Sci., 2022, 616, 338–346 CrossRef CAS PubMed.
- Z. Liu, X. Yang, G. Hu and L. Feng, Ru Nanoclusters Coupled on Co/N-Doped Carbon Nanotubes Efficiently Catalyzed the Hydrogen Evolution Reaction, ACS Sustainable Chem. Eng., 2020, 8, 9136–9144 CrossRef CAS.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, Organometallic approach for the synthesis of nanostructures, New J. Chem., 2013, 37, 3374–3401 RSC.
- K. Philippot, P. Lignier and B. Chaudret, Organometallic ruthenium nanoparticles and catalysis, Top. Organomet. Chem., 2014, 48, 319–370 CrossRef.
- G. Attard and C. Barnes, Surfaces, Oxford University Press, 1998 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmü, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11184 CrossRef CAS PubMed.
- S. Grimme, Accurate description of van der Waals complexes by density functional theory including empirical corrections, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- P. E. Blöchl, C. J. Först and J. Schimpl, The Projector Augmented Wave Method: ab-initio molecular dynamics with full wave functions, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillonin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. Heras-Domingo, M. Sodupe and X. Solans-Monfort, Interaction between Ruthenium Oxide Surfaces and Water Molecules. Effect of Surface Morphology and Water Coverage, J. Phys. Chem. C, 2019, 123, 7786–7798 CrossRef CAS.
- D. González, B. Camino, J. Heras-Domingo, A. Rimola, L. Rodríguez-Santiago, X. Solans-Monfort and M. Sodupe, BCN-M: A Free Computational Tool for Generating Wulff-like Nanoparticle Models with Controlled Stoichiometry, J. Phys. Chem. C, 2020, 124, 1227–1237 CrossRef.
- D. González, J. Heras-Domingo, M. Sodupe, L. Rodríguez-Santiago and X. Solans-Monfort, Importance of the oxyl character on the IrO2 surface dependent catalytic activity for the oxygen evolution reaction, J. Catal., 2021, 396, 192–201 CrossRef.
- D. González, M. Sodupe, L. Rodríguez-Santiago and X. Solans-Monfort, Metal coordination determines the catalytic activity of IrO2 nanoparticles for the oxygen evolution reaction, J. Catal., 2022, 412, 78–86 CrossRef.
- R. F. W. Bader, A Quantum Theory of Molecular Structure and Its Applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- L. Mallón, C. Cerezo-Navarrete, N. Romero, M. Puche, J. García-Antón, R. Bofill, K. Philippot, L. M. Martínez-Prieto and X. Sala, Ru nanoparticles supported on alginate-derived graphene as hybrid electrodes for the hydrogen evolution reaction, New J. Chem., 2022, 46, 49–56 RSC.
- J. Creus, L. Mallón, N. Romero, R. Bofill, A. Moya, J. L. G. Fierro, R. Mas-Ballesté, X. Sala, K. Philippot and J. García-Antón, Ruthenium Nanoparticles Supported on Carbon Microfibers for Hydrogen Evolution Electrocatalysis, Eur. J. Inorg. Chem., 2019, 2019, 2071–2077 CrossRef CAS.
- I. Álvarez-Prada, D. Peral, M. Song, J. Muñoz, N. Romero, L. Escriche, A. Acharjya, A. Thomas, R. Schomäcker, M. Schwarze, X. Sala, M. Tasbihi and J. García-Antón, Ruthenium nanoparticles supported on carbon-based nanoallotropes as co-catalyst to enhance the photocatalytic hydrogen evolution activity of carbon nitride, Renewable Energy, 2021, 168, 668–675 CrossRef.
- S. Drouet, J. Creus, V. Collière, C. Amiens, J. García-Antón, X. Sala and K. Philippot, A porous Ru nanomaterial as an efficient electrocatalyst for the hydrogen evolution reaction under acidic and neutral conditions, Chem. Commun., 2017, 53, 11713–11716 RSC.
- J. Lee, S. A. Sher Shah, P. J. Yoo and B. Lim, Hydrous RuO2 nanoparticles as highly active electrocatalysts for hydrogen evolution reaction, Chem. Phys. Lett., 2017, 673, 89–92 CrossRef CAS.
- H. S. Park, J. Yang, M. K. Cho, Y. Lee, S. Cho, S. D. Yim, B. S. Kim, J. H. Jang and H. K. Song, RuO2 nanocluster as a 4-in-1 electrocatalyst for hydrogen and oxygen electrochemistry, Nano Energy, 2019, 55, 49–58 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Computational high-throughput screening of electrocatalytic materials for hydrogen evolution, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- A. Moya, J. Creus, N. Romero, J. Alemán, X. Solans-Monfort, K. Philippot, J. García-Antón, X. Sala and R. Mas-Ballesté, Organocatalytic vs. Ru-based electrochemical hydrogenation of nitrobenzene in competition with the hydrogen evolution reaction, Dalton Trans., 2020, 49, 6446–6456 RSC.
- A. Comas-Vives, K. Furman, D. Gajan, M. C. Akatay, A. Lesage, F. H. Ribeiro and C. Copéret, Predictive morphology, stoichiometry and structure of surface species in supported Ru nanoparticles under H2 and CO atmospheres from combined experimental and DFT studies, Phys. Chem. Chem. Phys., 2016, 18, 1969–1979 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi00698k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2023 |