
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A systematic investigation of the NMR relaxation properties of Fe(III)-EDTA derivatives and their potential as MRI contrast agents†
Rocío
Uzal-Varela
 a,
Fátima
Lucio-Martínez
a,
Alessandro
Nucera
b,
Mauro
Botta
*b,
David
Esteban-Gómez
a,
Laura
Valencia
c,
Aurora
Rodríguez-Rodríguez
*a and
Carlos
Platas-Iglesias
*a
a,
Fátima
Lucio-Martínez
a,
Alessandro
Nucera
b,
Mauro
Botta
*b,
David
Esteban-Gómez
a,
Laura
Valencia
c,
Aurora
Rodríguez-Rodríguez
*a and
Carlos
Platas-Iglesias
*a
aUniversidade da Coruña, Centro de Investigacións Científicas Avanzadas (CICA) and Departamento de Química, Facultade de Ciencias, 15071, A Coruña, Galicia, Spain. E-mail: carlos.platas.iglesias@udc.es; aurora.rodriguez@udc.es
bDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale, Viale T. Michel 11, 15121 Alessandria, Italy. E-mail: mauro.botta@uniupo.it
cDepartamento de Química Inorgánica, Universidade de Vigo, Facultad de Ciencias, 36310 Pontevedra, Spain
First published on 20th January 2023
Abstract
We report a detailed investigation of the potential of Fe(III) complexes with H4EDTA derivatives containing different spacers as magnetic resonance imaging (MRI) contrast agents: trans-cyclohexane-1,2-diamine (t-H4CDTA), cis-cyclohexane-1,2-diamine (c-H4CDTA), propane-1,3-diamine (H4PDTA), benzene-1,2-diamine (H4PhDTA), trans-cyclopentane-1,2-diamine (H4CpDTA) and trans-cyclobutane-1,3-diamine (H4CBuDTA). The Fe(III) complex of the related hexadentate ligand H2CBuDEDPA (6,6′-(((trans-cyclobutane-1,3-diyl)bis(azanediyl))bis(methylene))dipicolinic acid) is also reported for comparative purposes. The X-ray structure of [Fe(1,3-CBuDEDPA)](PF6)·H2O evidences the six-coordination of the ligand to the metal ion and displays a distorted octahedral polyhedron. All complexes show reversible or quasi-reversible cyclic voltammograms in aqueous 0.15 M NaCl due to the Fe(III)/Fe(II) pair, with E1/2 values in the range of +97 to 136 mV (vs. NHE) for the complexes with t-H4CDTA, c-H4CDTA, H4PhDTA and H4CpDTA. The longer spacers of H4PDTA and H4CBuDTA induce a stabilization of the Fe(II) complex (E1/2 = 260 and 294 mV vs. NHE, respectively). The Fe(III) complexes of H4PDTA and H4CBuDTA do not contain inner-sphere water molecules. Thus, their 1H nuclear magnetic relaxation dispersion (NMRD) profiles were analysed using an outer-sphere model. The complexes of c-H4CDTA and H4PhDTA display relaxivities that indicate the presence of a water molecule coordinated to the metal ion. A set of 17O NMR transverse relaxation rates and chemical shifts obtained at different temperatures provided information on the water exchange kinetics. Subsequently, the analysis of the 1H NMRD profiles provided information on the rotational dynamics of the complexes and electronic relaxation. The integrated approach reported here includes a computational DFT and CASSCF study and provides insights into the structural and dynamic parameters affecting the efficiency of these complexes as MRI contrast agents.
Introduction
High-spin Fe(III) complexes with polyamino-polycarboxylate ligands are gaining increasing interest as potential contrast agents (CAs) for magnetic resonance imaging (MRI).1–4 These paramagnetic molecules accelerate the longitudinal relaxation times (T1) of water protons in their vicinity, which can be exploited to improve image contrast and thus aid medical diagnoses.Most CAs that are currently used in clinical practice are complexes containing Gd(III) with a water molecule coordinated to the metal ion.5–8 The chemical exchange of coordinated water molecules with those present in the vicinity of the agent provides an efficient pathway to shorten the longitudinal relaxation times (T1) of their 1H nuclei.9 Furthermore, the T1 values of the water proton nuclei diffusing in the vicinity of the paramagnetic centre are also shortened by the outer-sphere mechanism.10,11 Complexes of the d5 metal ion Mn(II) behave in a similar manner.12,13 The stable and inert complexation of this metal ion has proved difficult.14,15 However, some remarkable successes have been achieved, enabling the initiation of some clinical trials.16 Recently, several research groups showed that Fe(III) complexes can behave as efficient T1 CAs, with efficiencies comparable to those of Gd(III) and Mn(II) analogues with similar molecular size.2,3,17
The efficiency of T1 contrast agents is conveniently assessed by measuring 1H relaxivities (r1p), which provide the relaxation rate enhancement effect originated by the paramagnetic complex at 1 mM concentration.18 A rather large number of parameters affect the observed relaxivities. The Solomon–Bloembergen–Morgan theory of paramagnetic relaxation is generally used to describe the inner-sphere mechanism,19–21 which depends mainly on the distance between the paramagnetic metal ion and 1H nuclei of the coordinated water molecule (rMH), rotational correlation time of the M–H vector (τR), exchange rate of the coordinated water molecule with bulk water, which is the inverse of the mean residence time of a water molecule in the first coordination sphere (kex = 1/τm),22 and electronic relaxation of the paramagnetic centre (Tie, i = 1, 2). Electronic relaxation originates from transient fluctuations of the zero-field-splitting energy (Δ) that result from distortions of the metal coordination sphere characterized by a correlation time τV.23,24 Electron relaxation also affects the outer-sphere mechanism,10,11 which further depends on the distance of closest approach of an outer-sphere water molecule to the metal centre (aMH) and the relative diffusion of bulk water molecules and the paramagnetic centre (DMH). Several of these parameters vary with temperature and introduce additional parameters into the model. Independent information on some of these parameters can be obtained using complementary techniques, most commonly 17O NMR measurements, which provide direct information on water exchange dynamics.25
The systematic studies performed on Gd(III) and Mn(II) complexes over the last 25 years provided a detailed understanding of the factors that affect their relaxivities, which can be controlled in a rational way through ligand design.26 However, very few investigations have been reported for Fe(III) complexes. Our recent study on [Fe(H2O)6]3+ and the Fe(III) complexes with EDTA4− and CDTA4− pointed to remarkable differences between Fe(III) complexes and Mn(II) and Gd(III) analogues.17 More specifically, the inner-sphere relaxivity of Gd(III) and Mn(II) complexes at high magnetic fields (>20 MHz) is dominated by τR, unless water exchange is fast enough so that τR and τM are comparable or τM < τR. However, the high-field relaxivities of Fe(III) complexes also receive significant contribution from T1e, as T1e appears to be shorter for Fe(III) complexes than for Gd(III) and Mn(II) derivatives.
Herein, we report a systematic study of the 1H relaxivities of Fe(III) complexes of the H4EDTA family, in which the spacer connecting the two amine N atoms was varied to modulate the steric compression around the coordinated water molecule and the bite angle of the two amine N atoms. This is expected to affect both the number of coordinated water molecules and their exchange rates.27,28 For instance, longer spacers such as those present in H4PDTA and 1,3-H4CBuDTA (Scheme 1) are likely to hinder the access of water molecules to the metal centre, which may have an impact on the hydration number,29 water exchange30 and the pKa of the coordinated water molecule. This is an important issue to be considered in the case of Fe(III) complexes, as the metal ion is highly acidic and thus forms hydroxo species around neutral pH,31,32 unlike Mn(II) or Gd(III) analogues. The different bite angle of the central diamine group may also affect the geometry of the coordination sphere, and thus electronic relaxation.33–35 The systematic 1H and 17O NMR study reported here sheds light on these factors that control 1H relaxivities. Furthermore, we report cyclic voltammetry experiments to investigate the factors that favour an increased stabilization of the Fe(III) oxidation state, as reduction to Fe(II) may offer an efficient pathway for complex dissociation in vivo. We also describe here the Fe(III) complex of 1,3-H2CBuDEDPA, which lacks coordinated water molecules and was used to investigate the outer-sphere contribution to relaxivity. The X-ray crystal structure of the latter complex is also reported and discussed.
 | ||
| Scheme 1 Ligands discussed in this work. | ||
Results and discussion
Synthesis of the ligands
Ligand H4PDTA is commercially available, while H4PhDTA36,37 and 1,3-H4CBuDTA38 were prepared following the literature procedures. The synthesis of H4CpDTA was achieved by alkylation of trans-cyclopentane-1,2-diamine with tert-butyl bromoacetate in acetonitrile at room temperature, using K2CO3 as a base. Subsequent acid deprotection of the tert-butyl ester groups using aqueous 6 M HCl afforded the ligand in 69% yield over the two steps. The full alkylation of cis-cyclohexane-1,2-diamine with tert-butyl bromoacetate proved to be difficult, but could be achieved with a reasonable yield (39%) under microwave radiation using DIPEA as a base (Experimental section).X-ray crystal structure of [Fe(1,3-CBuDEDPA)](PF6)·H2O
Addition of excess KPF6 to an aqueous solution of the [Fe(1,3-CBuDEDPA)]+ complex provided single crystals suitable for X-ray analysis (Fig. 1). Crystals contained the expected cationic complex, a PF6− anion and water molecules, which are involved in hydrogen bonds with the anions and O atoms of the carboxylate groups. The cyclobutane unit adopts a puckered conformation to minimize steric strain,39,40 with C–C–C angles of 88.7° and dihedral C–C–C–C angles of 17.1°. The metal ion is coordinated by the secondary amine N atoms, the pyridine N atoms and two oxygen atoms from the carboxylate groups, resulting in a distorted octahedral coordination environment. The trans angles N(3)–Fe(1)–O(3) and N(2)–Fe(1)–O(1) (153.0°) show significant deviations from linearity, while the N(1)–Fe(1)–N(4) angle [171.36(5)°] is closer to the ideal value for an octahedron.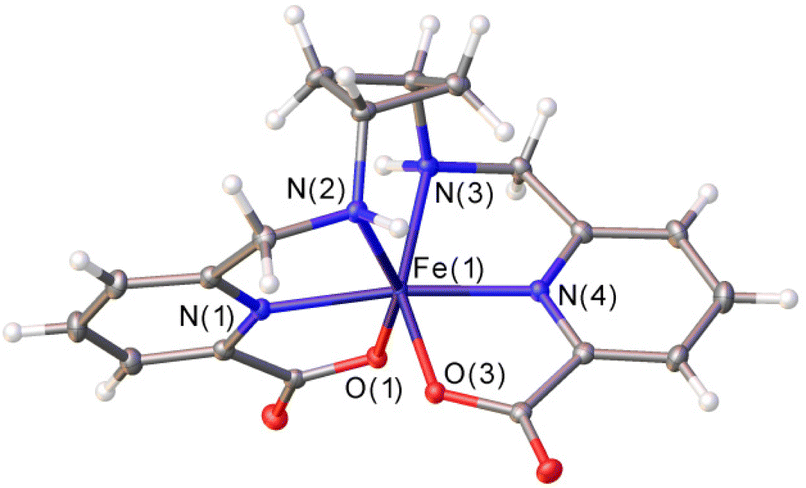 | ||
| Fig. 1 X-ray crystal structure of [Fe(1,3-CBuDEDPA)](PF6)·H2O with ellipsoids plotted at the 50% probability level. Water molecules and anions were omitted for simplicity. Bond distances of the metal coordination sphere (Å): Fe(1)–N(1), 2.0664(13); Fe(1)–N(2), 2.1975(13); Fe(1)–N(3), 2.2015(13); Fe(1)–N(4), 2.0628(13); Fe(1)–O(1), 1.9746(11); Fe(1)–O(3), 1.9692(11). | ||
Cyclic voltammetry studies
The Fe(III) complexes with the EDTA derivatives studied in this work were investigated using cyclic voltammetry experiments in aqueous solutions containing 0.15 M NaCl as supporting electrolyte (Fig. 2). The pH of the solutions was adjusted in the range 5.1–6.8 to avoid the hydrolysis of the water molecule coordinated to Fe(III).31 The redox potential for the Fe(III)/Fe(II) pair should be considered for MRI applications. For instance, reduction of the Fe(III) complex in vivo can provide an efficient pathway for complex dissociation. In our previous work, we showed that the reduction of the complexes with H4CDTA and H4EDTA in the presence of ascorbate was orders of magnitude faster than the half-live of the Fe(III) complexes.17 A similar behaviour was established for Cu(II) complexes relevant for biomedical applications. Indeed, very inert complexes with respect to acid-catalysed dissociation were found to dissociate very quickly upon reduction to Cu(I) by ascorbate.41 Furthermore, Fe(III) reduction may trigger undesirable formation of reactive oxygen species in the presence of H2O2 and ascorbate.42–44 This can be avoided by shifting the electrode potential of the Fe(III) complex out of the window defined by the electrode potentials of the ascorbyl/monohydroascorbate (Asc˙−, H+/Hasc−) and hydrogen peroxide/water, hydroxyl radical (H2O2, H+/HO˙, H2O) couples.45 This window was estimated to be +0.1 V to +0.9 V under physiologically relevant conditions.46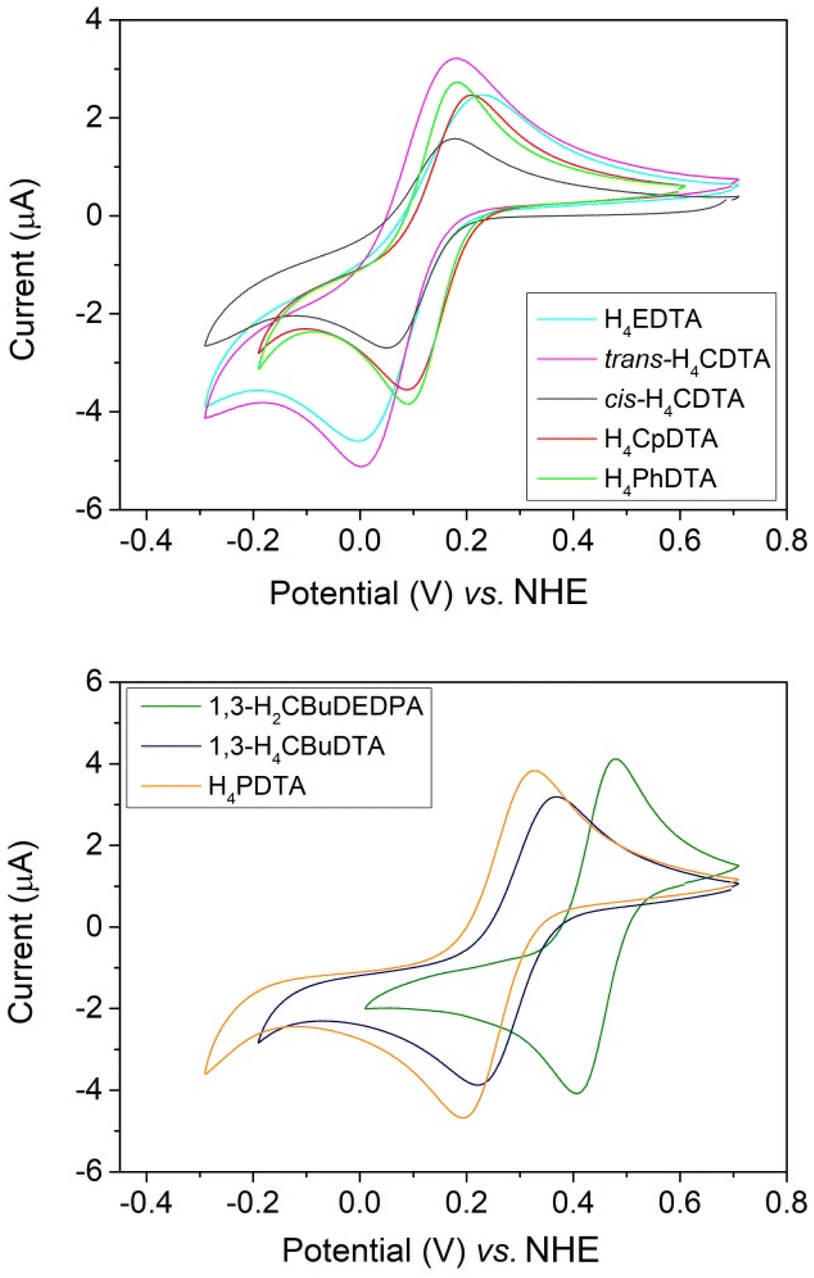 | ||
| Fig. 2 Cyclic voltammograms recorded for Fe(III) complexes (∼2 mM) in 0.15 M NaCl. | ||
All cyclic voltammograms are characterized by well-defined anodic and cathodic waves, characteristic of reversible or quasi-reversible processes. The cyclic voltammogram of [Fe(1,3-CBuDEDPA)]+ displays a peak-to-peak separation (ΔEp) close to the value of 59 mV expected for an electrochemically reversible process.47 Furthermore, ΔEp remains constant when varying the scan rate in the range of 10 to 500 mV s−1. For all other complexes, ΔEp increases with increasing scan rate, which indicates electrochemically quasi-reversible processes. Plots of the peak current versus v1/2, where v is the scan rate, are linear (Fig. S24–S31, ESI†), suggesting diffusion-controlled electrochemical processes.47
The half-wave potentials (E1/2) determined here for [Fe(EDTA)]− and [Fe(t-CDTA)]− agree well with those reported in the literature (−133 and −151 mV vs. SCE,31 which correspond to +108 and +90 vs. NHE).48 The latter two complexes and those of c-CDTA4−, PhDTA4− and CpDTA4− show relatively similar E1/2 values in the range of +97 to +136 mV, indicating that all of these ligands have a similar ability to stabilize Fe(III) (Table 1). However, the complexes with PDTA4− and CBuDTA4− are characterized by more positive E1/2 values of 260 and 294 mV, respectively. This evidences that the elongation of the central spacer of the ligand scaffold results in a stabilization of the Fe(II) complex. Hexadentate ligands like EDTA4− and their derivatives generally form seven-coordinate Fe(III) complexes, where a water molecule completes the metal coordination environment. The inner-sphere water molecule lies approximately on the plane defined by the metal ion, the amine N atoms and the oxygen atoms of two acetate groups.27 The longer spacers present in PDTA4− and CBuDTA4− result in an arrangement of the N2O4 donor set of the ligand that is closer to octahedral, reducing the Oip–Fe–Oip angle (Oip = in-plane oxygen atom of carboxylate groups) and stabilizing Fe(II) due to ligand field effects. The [Fe(1,3-CBuDEDPA)]+ complex displays a distorted octahedral coordination and a softer donor atom set, which results in further stabilization of Fe(II).
| Ligand | E a | E c | ΔEp | E 1/2 | pH |
|---|---|---|---|---|---|
| H4EDTA | 229 | −3 | 232 | 113 | 5.53 |
| t-H4CDTA | 180 | 14 | 166 | 97 | 5.33 |
| c-H4CDTA | 178 | 51 | 127 | 114 | 6.78 |
| H4PhDTA | 180 | 92 | 88 | 136 | 5.62 |
| H4CpDTA | 157 | 97 | 59 | 127 | 5.73 |
| H4PDTA | 324 | 195 | 129 | 260 | 5.87 |
| H4CBuDTA | 368 | 219 | 149 | 294 | 5.68 |
| H2CBuDEDPA | 478 | 407 | 71 | 443 | 5.14 |
All E1/2 values measured for the EDTA derivatives shown in Table 1 fall within the range of typical reducing agents present in vivo. The lowest E1/2 value was determined for [Fe(t-CDTA)]− (E1/2 = +97 mV vs. NHE),48 which is in the lower edge of the redox window under physiological conditions.46 All other complexes have E1/2 values well within the +0.1 V to +0.9 V window.
pH dependence of 1H relaxivity
The relaxivities of the Fe(III) complexes were first measured at a fixed magnetic field strength (1.46 T, which corresponds to a 1H Larmor frequency of 62 MHz) and 298 K (Fig. 3). The relaxivities observed in the pH range of 3–6 allows for classifying these complexes into three different groups: (1) the complexes of PhDTA4−, c-CDTA4− and CpDTA4− are characterized by relaxivities of 2.0–2.3 mM−1 s−1. These values are similar to those reported for the complexes with EDTA4− and t-CDTA4−, which contain one water molecule coordinated to the metal ion (Table 2). (2) A second group of complexes displays low relaxivities of 0.6–0.7 mM−1 s−1, which likely reflects the absence of water molecules in the Fe(III) inner coordination sphere. This is expected for the complex of 1,3-CBuDEDPA2− on the basis of the X-ray structure described above. For the [Fe(PDTA)]− complex, the r1p values suggest that there is a very small inner-sphere contribution to relaxivity, if any. A 17O NMR study reported a fraction of a water-containing species of f = 0.2.28 (3) The complex with 1,3-CBuDTA4− shows an intermediate behaviour that suggests a hydration equilibrium involving q = 1 and q = 0 species (q is the number of coordinated water molecules). A similar situation was observed for the corresponding Mn(II) complex and related systems.38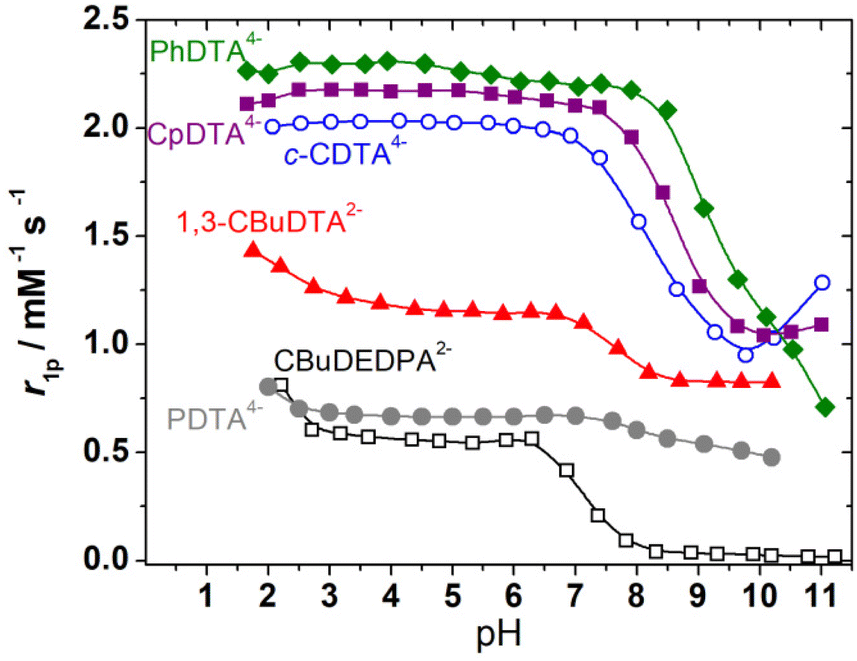 | ||
| Fig. 3 pH dependence of r1p (62 MHz, 1.46 T, 298 K) measured for the complexes investigated in this work. Complex concentrations were in the 3.0–5.6 mM range. | ||
| Ligand | r 1p | pKa | r FeO | A/ħ | ΔG298 | S (PB)d | S (CTP)d | |
|---|---|---|---|---|---|---|---|---|
| a Data from ref. 31. b Complex dissociation and precipitation of Fe(OH)3 prevented pKOH determination. c The lack of δ/λ conformations results in the formation of a single diastereoisomer. d Shape measures obtained for pentagonal bipyramidal S(PB) and capped trigonal prismatic S(CTP) coordination; S = 0 for a coordination environment that matches the reference polyhedron. | ||||||||
| H4EDTA | Δ(δ) | 1.67 | 7.52a | 2.133 | −67.7 | +5.58 | 2.48 | 2.85 |
| Δ(λ) | 2.153 | −63.2 | 1.21 | 4.61 | ||||
| t-H4CDTA | Δ(δ) | 2.11 | 9.54a | 2.147 | −66.3 | −5.47 | 2.31 | 3.10 |
| Δ(λ) | 2.162 | −63.4 | 1.23 | 4.43 | ||||
| c-H4CDTA | Δ(δ) | 2.03 | 8.23 (3) | 2.149 | −65.9 | −11.49 | 2.33 | 3.10 |
| Δ(λ) | 2.169 | −61.4 | 1.15 | 4.83 | ||||
| H4CpDTA | Δ(δ) | 2.12 | 8.57 (2) | 2.136 | −67.6 | −2.71 | 2.54 | 3.03 |
| Δ(λ) | 2.149 | −64.1 | 1.41 | 4.51 | ||||
| 1,3-H4CBuDTA | 1.15 | 7.64(2) | 2.166c | −64.6c | — | 2.17 | 4.78 | |
| H4PhDTA | 2.26 | 2.138c | −67.2c | — | 1.57 | 4.34 | ||
| H4PDTA | 0.66 | — | — | — | — | — | ||
| H2CBuDEDPA | 0.55 | — | — | — | — | — | ||
The r1p values measured for PhDTA4−, CpDTA4− and c-CDTA4− complexes remain constant within experimental error below pH 6 down to pH ∼ 2.5, as observed previously for the EDTA4− and t-CDTA4− analogues.17 However, an increase in relaxivity is observed at low pH for the complexes of 1,3-CBuDTA4−, 1,3-CBuDEDPA2− and PDTA4−. This may be related either to the dissociation of the complexes below pH ∼ 3 or to the formation of a protonated species with higher relaxivity than the non-protonated forms. In the case of PDTA4−, the stability constant of the Fe(III) complex (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 21.6) was found to be considerably lower than that of the EDTA4− analogue (logK = 25.1).49 The speciation diagram of the Fe(III)-PDTA4− system (Fig. S32, ESI†) indicates that complex dissociation is significant below pH ∼ 2, while complex protonation already occurs below pH ∼ 5.50 Thus, the marked relaxivity increase observed below pH 2.5 is most likely associated with complex dissociation. The decrease in relaxivity observed above pH 6 is also in agreement with the speciation diagram.
K = 21.6) was found to be considerably lower than that of the EDTA4− analogue (logK = 25.1).49 The speciation diagram of the Fe(III)-PDTA4− system (Fig. S32, ESI†) indicates that complex dissociation is significant below pH ∼ 2, while complex protonation already occurs below pH ∼ 5.50 Thus, the marked relaxivity increase observed below pH 2.5 is most likely associated with complex dissociation. The decrease in relaxivity observed above pH 6 is also in agreement with the speciation diagram.
All complexes display important changes in their relaxivities above pH 6–8. The complexes of PhDTA4−, 1,3-CBuDEDPA2− and PDTA4− show an irreversible decrease in relaxivity together with visible precipitation of Fe(OH)3, which is clear evidence of complex dissociation (logK = 21.8 for the complex with PhDTA4−).51 For the complexes with c-CDTA4−, CpDTA4− and 1,3-CBuDTA4−, the decrease in relaxivity observed above pH ∼ 6 can be attributed to the deprotonation of the coordinated water molecule according to:
| [Fe(L)(H2O)]− ⇄ [Fe(L)(OH)]2− + H+. | (1) |
It is important to note that the formation of the hydroxide complex may also trigger the dimerization of the complex, as demonstrated for the complexes with t-CDTA4− and EDTA4−, and this in turn will affect the pKa of the coordinated water molecule. The fits of the relaxivity data afforded the apparent pKa values of the coordinated water molecule listed in Table 2. These results and the data reported previously for t-CDTA4− and EDTA4− complexes show that the complex with t-CDTA4− displays the highest pKa value, while the complexes with EDTA4− and 1,3-CBuDTA4− present pKa values of 7.5–7.6. As a result, a significant amount of these complexes is expected to hydrolyse at physiological pH. The complexes with CpDTA4− and c-CDTA4− show an intermediate behaviour, with pKa values of 8.57 and 8.23, respectively.
DFT calculations
The Fe(III)-EDTA derivatives studied here were characterized by using DFT calculations (see Computational details below). Following our previous studies, we added a small number of explicit second-sphere water molecules for a better characterization of the Fe(III)-OH2 bond involving the coordinated water molecule, as well as to obtain more accurate 17O hyperfine coupling constants.40,52 This aids in overcoming some limitations of polarized continuum models to account for hydrogen-bonding interactions involving the coordinated water molecules.53,54 The [Fe(EDTA)(H2O)]− complex is known to exist in the solid state and in solution as a mixture of two isomers whose coordination is often described as capped trigonal prismatic (CTP) and pentagonal bipyramidal (PB).55 These two isomers originate from the two sources of helicity associated with: (i) the layout of the four acetate groups, which may rotate clockwise (Λ) or anticlockwise (Δ)56 in a plane perpendicular to the ideal C2 symmetry axis of the complex, and (ii) the conformation of the five-membered chelate ring formed due to the coordination of the two amine N atoms, which can be denoted as δ or λ (Fig. 4).27 The combination of these two sources of helicity yields two enantiomeric pairs of diastereoisomers: the Δ(δ)/Λ(λ) pair, which is considered to give a CTP coordination, and the Δ(λ)/Λ(δ) pair, which gives PB coordination. Of note, the coordination of the trans-cyclobutane-1,3-diamine unit in the [Fe(CBuDTA)(H2O)]− complex results in the formation of a six-membered ring bisected by a mirror plane. Thus, the only source of helicity is associated with the layout of the acetate groups (Δ or Λ). A similar situation holds for [Fe(PhDTA)(H2O)]−, as the coordination of the benzene-1,2-diamine results in the formation of a planar 5-membered chelate ring.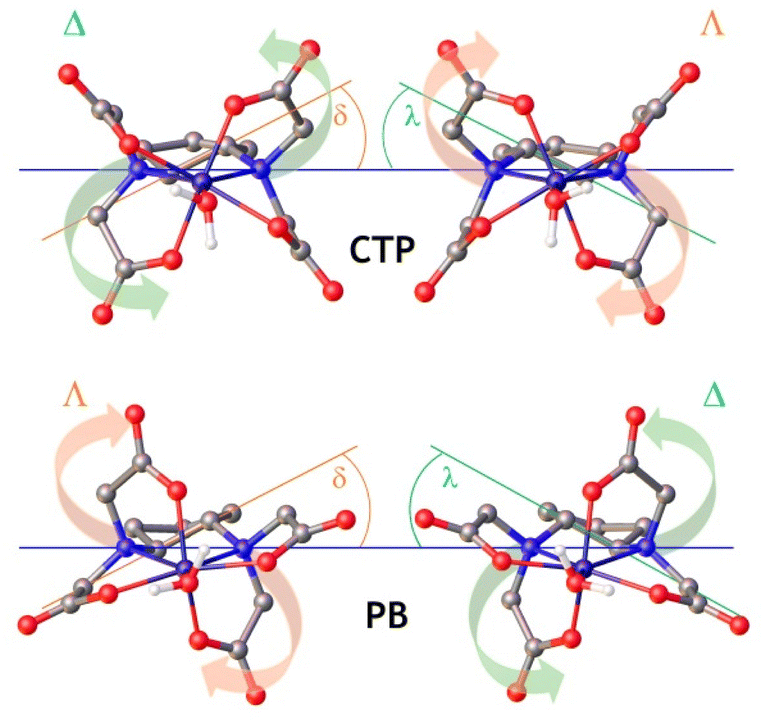 | ||
| Fig. 4 Structures of the capped trigonal prismatic (CTP) and pentagonal bipyramidal (PB) isomers of the [Fe(CpDTA)(H2O)]− complex obtained with DFT calculations, highlighting the different sources of chirality. Hydrogen atoms and second-sphere water molecules omitted for simplicity. | ||
The analysis of the metal coordination environments using shape measures57,58 (Table 2) indicates that both the Δ(δ)/Λ(λ) and Δ(λ)/Λ(δ) sets have coordination polyhedra best described as pentagonal bipyramidal. Indeed, the shape measure takes a value of 0 for a coordination polyhedron that matches perfectly with the reference polyhedron, and increases as the polyhedron is more distorted.59 Nevertheless, the coordination of the metal ion in the Δ(δ)/Λ(λ) pair is more distorted towards a CTP, and we will subsequently refer to this enantiomeric pair as the CTP diastereoisomer.
The [Fe(t-CDTA)(H2O)]− complex displays a CTP coordination in the solid state.27,60 Our calculations indeed predict that the [Fe(t-CDTA)(H2O)]− complex displays a CTP coordination in solution, with the PB isomer displaying a considerably higher energy (ΔG° = +5.5 kJ mol−1, Table 2). This situation is reversed in [Fe(EDTA)(H2O)]−, for which PB coordination is favoured by ∼5.6 kJ mol−1, in agreement with the structures observed in the solid state.61 Both the [Fe(c-CDTA)(H2O)]− and [Fe(CpDTA)(H2O)]− complexes are predicted to have a CTP coordination environment. However, in the latter case, the free energy difference between the PB and CTP isomers is small (∼2.7 kJ mol−1, Table 2).
The pKa values determined for this series of structurally related complexes do not correlate with the Fe–Owater distances (rFeO) calculated with DFT (Table 2), which fall within a rather narrow range of 2.13–2.17 Å. The longest rFeO value was obtained for the [Fe(1,3-CBuDTA)(H2O)]− complex, which appears to be characterised by a number of coordinated water molecules of 0 < q < 1, according to relaxivity measurements. However, this relatively weak Fe–Owater interaction does not correlate with a high pKa value, as would be expected. Indeed, similar pKa values were determined for [Fe(1,3-CBuDEDPA)(H2O)]+ and [Fe(EDTA)(H2O)]−, which possess the longest and shortest Fe–Owater bonds within this family of structurally related complexes, respectively, according to DFT studies. We also notice that the coordinated water molecules are characterized by similar values of the 17O hyperfine coupling constant A/ħ (Table 2), which depends on the difference between α and β electron densities at the nucleus.62 We note that the CTP isomers possess somewhat shorter Fe–Owater distances, which leads to slightly more negative A/ħ values.
1H nuclear magnetic relaxation dispersion (NMRD) profiles
The 1H NMRD profiles of the [Fe(1,3-CBuDEDPA)(H2O)]+ and [Fe(PDTA)(H2O)]− complexes were recorded at different temperatures in the proton Larmor frequency range of 0.01–500 MHz (Fig. 5). The shape of the profiles is similar to those recorded previously for the EDTA4− and CDTA4− analogues, although the r1p values observed for the latter complexes are higher over the whole range of Larmor frequencies investigated. The NMRD profiles were therefore analysed using the outer-sphere model proposed by Freed,10 which depends on the relative diffusion coefficient of the solute and water molecules 298D and its activation energy ED, the distance of closest approach of a freely-diffusing second-sphere water molecule to the metal complex, aFeH, the zero-field splitting (ZFS) energy, 298Δ, and the activation energy EΔ, the correlation time for the modulation of the ZFS, τV, and its activation energy EV. The values of EV were fixed to 1 kJ mol−1 following previous relaxometric studies on Gd(III)18 and Mn(II)63 complexes. Good fits of the experimental data were obtained with the parameters shown in Table 3 by fixing the value of aFeH to 3.7 Å (see also Fig. 5).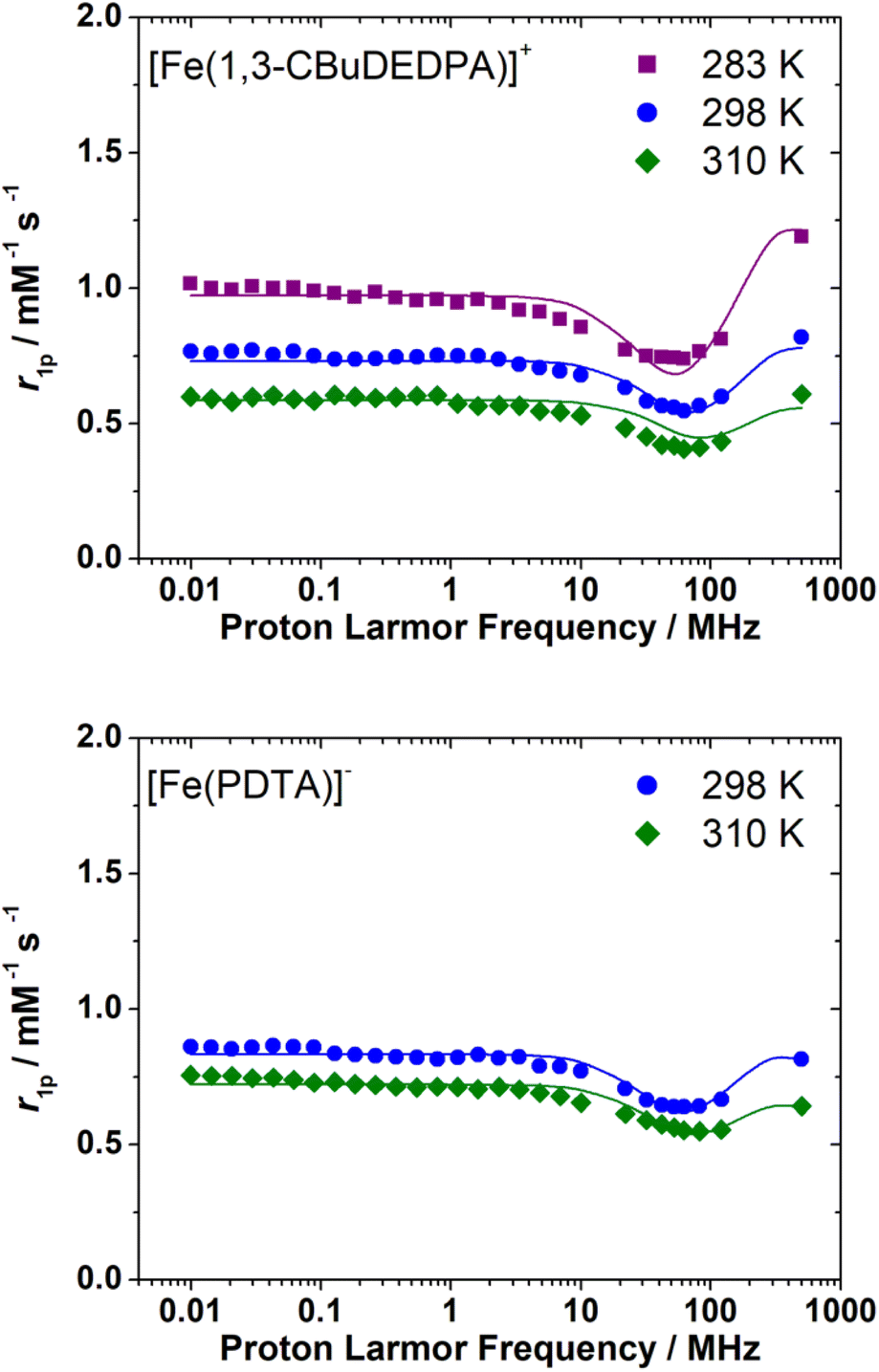 | ||
| Fig. 5 1H NMRD profiles recorded for [Fe(1,3-CBuDEDPA)]+ (3.3 mM, pH 5.5) and [Fe(PDTA)]− (5.8 mM, pH = 5.4) at different temperatures. The solid lines represent the fits of the data, as described in the text. | ||
The fits of the NMRD data provided diffusion coefficients 298D in good agreement with those reported for other small metal complexes.64,65 The fitted 298D values are also close to the self-diffusion coefficient of water in water (2.3 × 10−9 m2 s−1),66 as would be expected due to the fact that water diffuses much faster than the metal complex. The decrease of relaxivity observed upon increasing temperature is related to fast diffusion at high temperature. The values of 298Δ2 are close to those determined previously from the analysis of the 1H NMRD profiles of [Fe(EDTA)(H2O)]− (298Δ2 = 27 × 1020 s−2) and [Fe(t-CDTA)(H2O)]− (298Δ2 = 18 × 1010 s−2).17 An EPR study provides axial (D) and rhombic (E) ZFS parameters for [Fe(EDTA)(H2O)]+34 that correspond to a ZFS energy of Δ2 = 32 × 1020 s−2,67 in excellent agreement with the value obtained from 1H NMRD.
The 1H NMRD profiles recorded for the [Fe(c-CDTA)]−, [Fe(CpDTA)]−, [Fe(PhDTA)]− and [Fe(1,3-CBuDTA)]− complexes are presented in Fig. 6. All measurements were recorded at pH values <7.2 to avoid hydrolysis of the complex and/or complex dissociation. They all have similar shapes and show relatively high relaxivity values that suggest the presence of water molecules coordinated to the metal ion. We notice that the relaxivities measured for [Fe(1,3-CBuDTA)]− are lower than those of [Fe(c-CDTA)]−, [Fe(CpDTA)]− and [Fe(PhDTA)]− at all magnetic field strengths, which again suggests that 0 < q < 1 for the former.
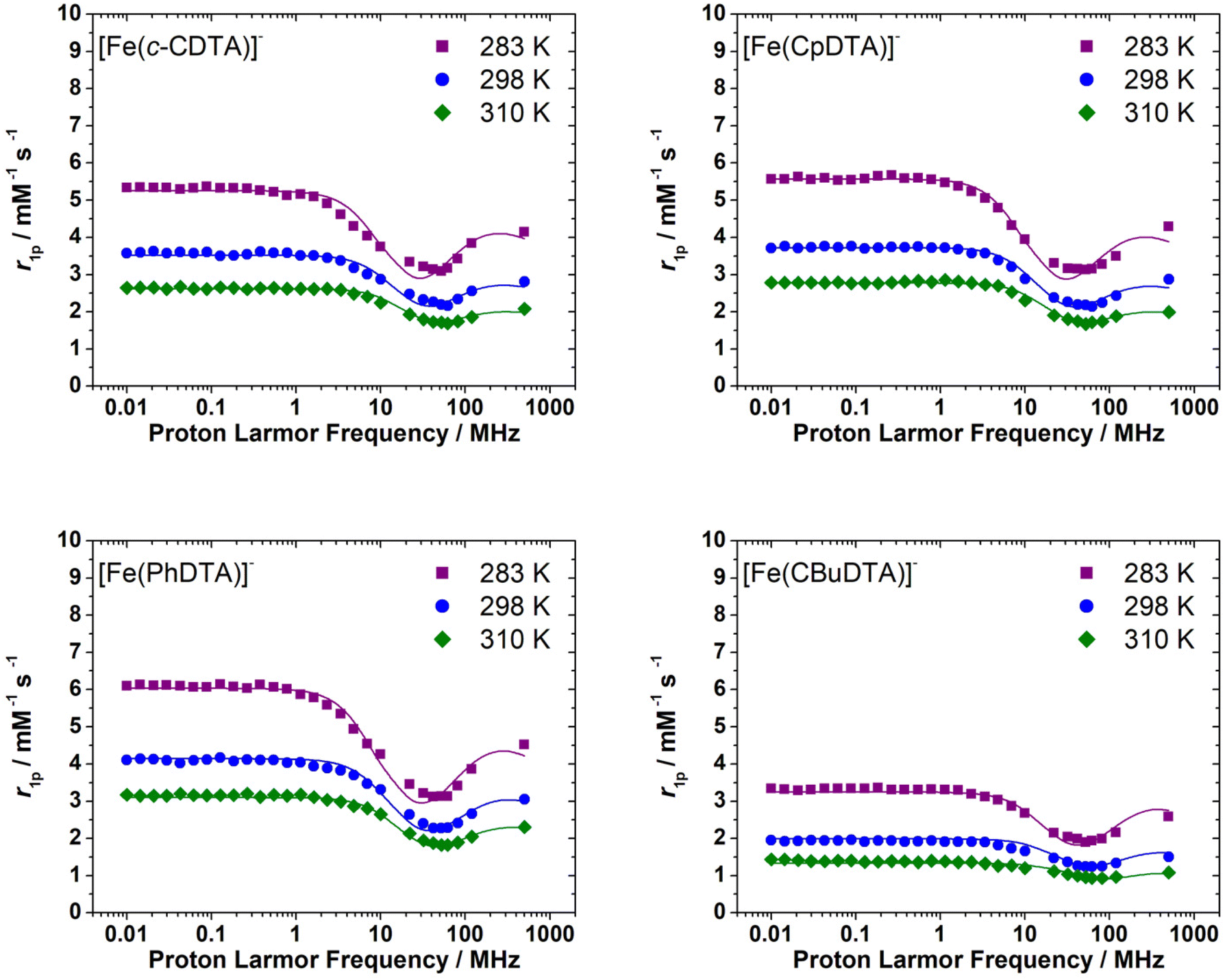 | ||
| Fig. 6 1H NMRD profiles recorded for [Fe(c-CDTA)]− (3.3 mM, pH 6.0), [Fe(CpDTA)]− (5.9 mM, pH 6.1), [Fe(PhDTA)]− (4.9 mM, pH 7.2) and [Fe(1,3-CBuDTA)]− (4.8 mM, pH = 5.5) at different temperatures. The solid lines represent the fits of the data, as described in the text. | ||
Water exchange
A 17O NMR study was performed to gain information on the dynamics of water exchange of this structurally related family of Fe(III) complexes. Reduced transverse relaxation rates (T2r) and chemical shifts (Δωr) were measured from aqueous solutions of the complexes at pH values of ∼5.5 to ensure full complexation of the Fe(III) ion and avoid hydrolysis of the coordinated water molecule. The four complexes investigated here display different trends of 1/T2rversus temperature. In particular, the values of 1/T2r decrease with increasing temperature for [Fe(1,3-CBuDEDPA)(H2O)]+. The temperature dependence of 1/T2r can be rationalized with the following approximated expression: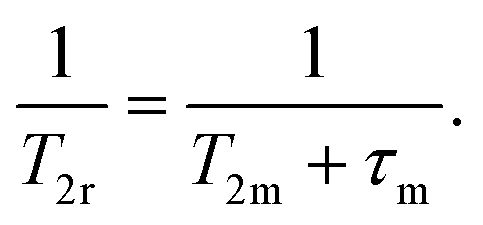 | (2) |
The mean residence time of a water molecule in the inner coordination sphere (τm) decreases with increasing temperature, while the relaxation time of the coordinated water molecule T2m displays the opposite trend. Thus, the temperature dependence of 1/T2r observed for [Fe(1,3-CBuDEDPA)(H2O)]+ is typical of the fast exchange regime, in which T2m dominates the denominator of eqn (2), as τm < T2m. Both [Fe(c-CDTA)]− and [Fe(CpDTA)]− show similar temperature dependence of 1/T2r, which displays a maximum at ∼285 K that corresponds to the temperature at which τm ∼ T2m. This maximum is observed at a higher temperature for [Fe(PhDTA)]− than for [Fe(c-CDTA)]− and [Fe(CpDTA)]−, which indicates that τm is longer for the former. The values of Δωr show inflection points at approximately the temperature in which 1/T2r displays a maximum, as would be expected.18
The 17O NMR data were fitted using the Swift–Connick equations (Fig. 7).68,69 The fits of the data afforded the residence time of the coordinated water molecule (298τOM) and its activation energy (ΔHM), as well as the hyperfine coupling constant A/ħ (Table 4). The latter values are in good agreement with those estimated with DFT (Table 2). In the case of [Fe(1,3-CBuDTA)(H2O)]−, a hydration equilibrium involving the q = 1 and q = 0 species had to be included to obtain satisfactory fits of the 1H NMRD and 17O NMR data:
| [Fe(CBuDTA)(H2O)]− ⇄ [Fe(CBuDTA)]− + H2O. | (3) |
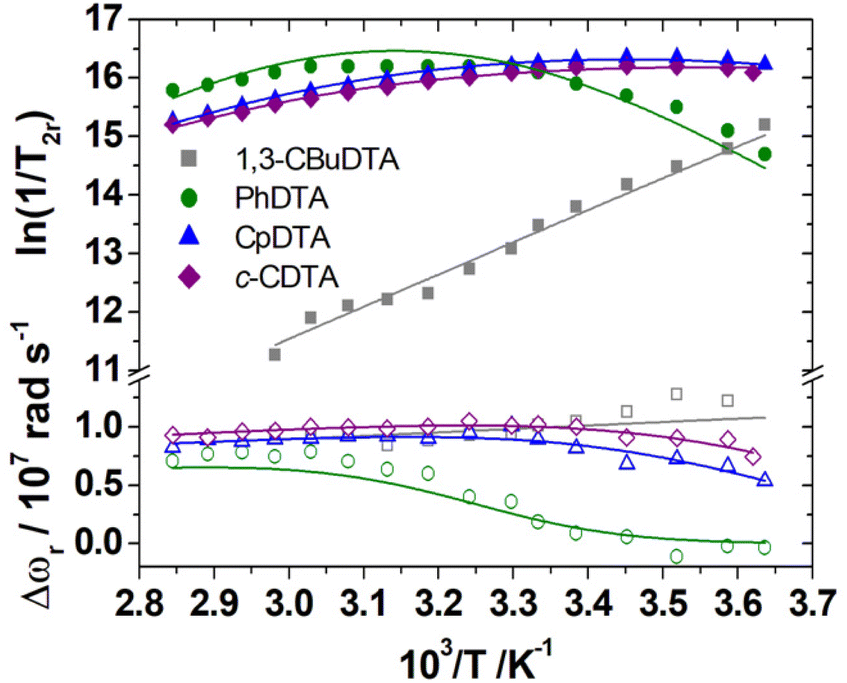 | ||
| Fig. 7 Reduced 17O NMR transverse relaxation rates (1/T2r, filled symbols) and chemical shifts (Δωr, open symbols) measured for [Fe(c-CDTA)]− (3.2 mM, pH 5.9), [Fe(CpDTA)]− (5.9 mM, pH 6.1), [Fe(PhDTA)]− (4.9 mM, pH 7.2) and [Fe(CBuDTA)]− (3.9 mM, pH = 5.5) at 11.75 T. The solid lines represent the fits of the data, as described in the text. | ||
| c-CDTA4− | PhDTA4− | CpDTA4− | 1,3-CBuDTA4− |
t-CDTA4−b |
EDTA4−b |
|
|---|---|---|---|---|---|---|
| a Parameters fixed during the fitting procedure. b Data from ref. 17. | ||||||
| 298 Δ 2/1020 s−2 | 12.5 ± 0.3 | 13.6 ± 0.4 | 12.8 ± 0.3 | 33.2 ± 0.9 | 18.1 | 27.0 |
| E Δ/kJ mol−1 | 9.5 ± 0.4 | 10.0 ± 0.4 | 9.7 ± 0.4 | 9.4 ± 0.4 | 9.8 | 7.8 |
| 298 τ V/ps | 4.9 ± 0.2 | 3.7 ± 0.2 | 4.4 ± 0.2 | 3.2 ± 0.2 | 3.4 | 2.8 |
| E V/kJ mol−1 | 1.0a | 1.0a | 1.0a | 1.0a | 1.0a | 1.0a |
| A O/ħ/106 rad s−1 | −71.2 ± 0.7 | −50.2 ± 3.2 | −65.6 ± 0.7 | −64.6a | −62.8a | −64.8a |
| 298 τ OM/ns | 5.0 ± 0.4 | 57.2 ± 4.4 | 6.5 ± 0.5 | 0.059 ± 0.003 | 36.1 | 0.9 |
| ΔHM/kJ mol−1 | 39.6 ± 1.4 | 60.2 ± 3.0 | 41.0 ± 1.2 | 43.4 ± 2.3 | 51.5 | 30.5 |
| 298 τ R/ps | 45.7 ± 1.2 | 53.3 ± 1.8 | 44.0 ± 1.3 | 40a | 48.4 | 35.1 |
| E R/kJ mol−1 | 21.3 ± 1.2 | 19.3 ± 1.6 | 20.5 ± 1.3 | 20a | 21.1 | 25.2 |
| q 298 | 1a | 1a | 1a | 0.81 | 1a | 1a |
| r FeH/Å | 2.682a | 2.671a | 2.673a | 2.692a | 2.70a | 2.69a |
| a FeH/Å | 3.7a | 3.7a | 3.7a | 3.7a | 3.5a | 3.5a |
| 298 D/10−9 m2 s−1 | 2.5a | 2.5a | 2.5a | 2.5a | 2.24a | 2.24a |
| E D/kJ mol−1 | 17.9a | 17.9a | 17.9a | 17.9a | 20.0a | 20.0a |
The A/ħ value estimated by DFT was used to fit the 17O NMR data of [Fe(1,3-CBuDTA)(H2O)]− in order to reduce the number of fitting parameters (Table 4). This afforded the following thermodynamic parameters for the equilibrium expressed in eqn (3): ΔH° = 42.2 ± 3.4 kJ mol−1 and ΔS° = +129 ± 11 J mol−1 K−1. The positive reaction entropy is typical of dehydration reactions involving both lanthanide70–73 and transition metal ions,28,38 due to water release. The positive reaction enthalpy is probably related to the energy cost associated with the breaking of the Fe-OH2 bond. Thus, the dehydration reaction is entropy-driven, with the dehydration reaction being progressively shifted to the right as the temperature increases. The hydration number at 298 K is q298 = 0.81, decreasing from q = 0.95 at 275 K to q = 0.39 at 335 K.
The values of the mean residence times of water molecules in the inner coordination sphere (298τOM) follow the qualitative trends described above. The [Fe(CBuDTA)(H2O)]− complex displays an extremely fast water exchange rate, with a 298τOM value of only 59 ps. This is likely related to the weak coordination of the water molecule to the metal ion and to the presence of a hydration equilibrium, which favours a fast exchange following a dissociative mechanism.74 The volumes of activation determined for different Fe(III)-EDTA4− derivatives, including t-CDTA4− and PhDTA4−, are positive, indicating dissociatively activated water exchange mechanisms.75 The 298τOM values obtained for [Fe(c-CDTA)(H2O)]− and [Fe(CpDTA)(H2O)]− are two orders of magnitude shorter than that of [Fe(CBuDTA)(H2O)]−, while [Fe(PhDTA)(H2O)]− displays the longest 298τOM value among the EDTA4− derivatives investigated here. The value of 298τOM determined here for [Fe(PhDTA)(H2O)]− (57.2 ns) is slightly shorter than that reported in the literature (83 ns).75,76 Overall, the water exchange rates of these EDTA4− derivatives cover a range of three orders of magnitude from about 60 ps to 60 ns. We note that the [Fe(c-CDTA)(H2O)]− complex displays slightly faster water exchange than the [Fe(t-CDTA)(H2O)]− analogue, as observed for the corresponding Mn(II) complexes.37
Rotational dynamics and electronic relaxation
The relaxivity data were fit to the standard Solomon–Bloembergen–Morgan theory of paramagnetic relaxation.19–21 The values of the rotational correlation times (298τR) and the corresponding activation energies ER are very reasonable considering the size of the complexes, which provides confidence in the reliability of the fits. Similar values for these parameters were obtained previously from NMRD studies of Mn(II) complexes of similar size.63,77,78For the fits of the data, the values of 298D, ED and aFeH were fixed to those obtained for the PDTA4− analogue (Table 3). The distance between the water protons of the coordinated water molecule and the metal ion were fixed to the values obtained with DFT calculations. Satisfactory fits of the data could be obtained only by assuming that the ZFS energy 298Δ displays an Arrhenius dependence with absolute temperature with an activation energy EΔ. The mechanism responsible for electron spin relaxation may have contributions from both transient and static ZFS contributions.24,79–81 The transient ZFS mechanism is due to modulation of the ZFS due to transient distortions of the metal coordination environment, with an associated correlation time τV. The static contribution arises from the time average in the molecular frame of the ZFS of the complex, and it is modulated by the rotational correlation time τR.82 The correlation times τV obtained from the fit of the NMRD data are one order of magnitude lower than τR (Table 4), which in turn are very reasonable considering the size of the complexes. The values of τV are compatible with the transient modulation of the ZFS by distortions of the coordination geometry induced by vibrations, which was estimated to be in the range of ∼0.1 to a few ps for Gd(III) and Mn(II) complexes.23,67,83 This suggests that the transient mechanism dominates electronic relaxation for these complexes. One can reasonably assume that increasing the temperature induces a more significant fluctuation of the ZFS energy (larger spread of Δ values), which is accounted for by the activation energy EΔ.
The family of EDTA4− derivatives investigated here show very similar values of 298Δ2 and activation parameters of EΔ ∼ 10 kJ mol−1. These results indicate that the slight changes of the coordination environment, associated with the different nature of the central spacer, have a minor impact on the electronic relaxation. To gain additional insight into electronic relaxation, we computed the ZFS parameters for this family of complexes using complete active space self-consistent field (CASSCF) calculations, incorporating dynamic correlation with perturbation theory (NEVPT2, see Computational details). These calculations provided the common D and E ZFS parameters using effective Hamiltonian theory,84 as described previously for Mn(II) complexes.67,85 These complexes are characterized by similar ZFS parameters, in line with the results obtained from NMRD studies.
The values of D calculated for the Δ(δ)/Λ(λ) enantiomeric pair are negative, although the prediction of the sign of D is challenging for situations in which E/D is close to the rhombic limit of 1/3, as is the case here.86 A similar E/D value of 0.31 was obtained for the EDTA4− complex using EPR measurements, with |D| = 0.83 cm−1.87 The values of D calculated for the Δ(λ)/Λ(δ) isomers, which display PB coordination environments, are conversely positive. The sign of D is related to the splitting of the Kramers doublets arising from S = 5/2 electronic ground state, as observed previously for Mn(II) complexes.86 If two of the three Kramers doublets are below the centre of gravity, D is positive (Fig. 8), while the reverse situation is observed when two Kramers doublets are above the centre of gravity. Regardless of the sign of D, all complexes investigated here present very similar overall splitting of the three Kramer doublets. For axially symmetric systems, the energy difference between the highest- and lowest-energy Kramers doublets equals 6D. In the present case, this energy difference ranges from 2.05 to 2.65 cm−1, which yields values of |D| of 0.3–0.4 cm−1. The splitting of the Kramers doublets in Mn(II) complexes with polyamino-polycarboxylate ligands was found to be one order of magnitude smaller,85,88 which explains the slow electronic relaxation observed for Mn(II) complexes compared to Fe(III) analogues. The splitting of the Ms = ±5/2, Ms = ±3/2 and Ms = ±1/2 that generates the three Kramers doublets is induced by spin–orbit coupling with excited quartet states. The lowest-energy excited quartet state was found to lie ∼21000 cm−1 above the sextet ground state for Mn(II) complexes,85 while our calculations reduced this energy to ∼17600 cm−1 for the Fe(III) complexes investigated here. Thus, the larger ZFS energies calculated for Fe(III) complexes compared to Mn(II) analogues are related to smaller sextet-quartet energy differences in the former.
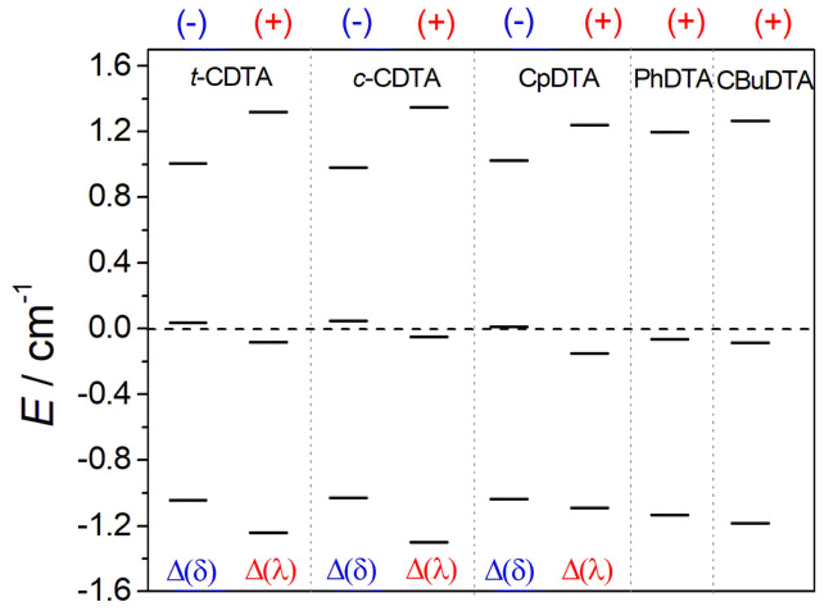 | ||
| Fig. 8 Splitting of the Kramers doublets obtained with CASSCF/NEVPT2 calculations and the signs of D. | ||
The values of the ZFS energy Δ were estimated using the following expression, which provides the energy of the static ZFS:9,89
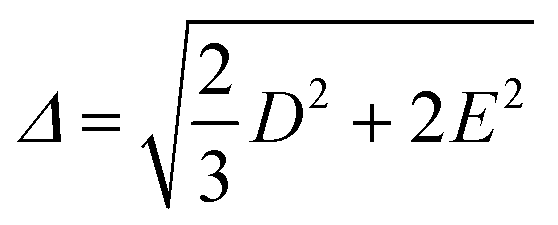 | (4) |
| Ligand | D/cm−1 | E/D | Δ/1010 rad s−1 | Δ 2/1020 s−2 | |
|---|---|---|---|---|---|
| a The lack of δ/λ conformations results in the formation of a single diastereoisomer. | |||||
| t-H4CDTA | Δ(δ) | −0.300 | 0.289 | 5.2 | 27 |
| Δ(λ) | +0.380 | 0.266 | 6.4 | 41 | |
| c-H4CDTA | Δ(δ) | −0.299 | 0.269 | 5.1 | 26 |
| Δ(λ) | +0.389 | 0.288 | 6.7 | 45 | |
| H4CpDTA | Δ(δ) | −0.297 | 0.310 | 5.2 | 27 |
| Δ(λ) | +0.366 | 0.185 | 5.9 | 35 | |
| H4PhDTA | +0.345 | 0.273 | 5.9 | 34 | |
| 1,3-H4CBuDTA | +0.369 | 0.251 | 6.2 | 38 | |
Conclusions
We have analysed in this work some of the physicochemical properties of Fe(III) complexes with H4EDTA derivatives that are relevant for their application as MRI contrast agents. The results obtained here confirm that Fe(III) complexes are very promising MRI contrast agent candidates. However, several challenges still need to be addressed to obtain probes with optimal properties. For instance:(1) Improve the redox stability of the Fe(III) complexes to avoid their reduction in vivo to the Fe(II) analogues. While the reduction of Fe(III) in vivo may be exploited to design redox-responsive agents, it is also likely that metal ion reduction results in the undesirable generation of reactive oxygen species (ROS) by catalytic decomposition of H2O2. Thus, it may be beneficial to shift the electrode potential of the Fe(III)/Fe(II) pair outside the range for redox reactivity under physiological conditions.
(2) Attaining relaxivities comparable to those of the classical Gd(III) MRI contrast agents used in clinical practice requires the presence of water molecules coordinated to the Fe(III) ion. Inner-sphere water molecules in Fe(III) complexes are often hydrolysed to form hydroxo-complexes around physiological pH, which results in a decrease in relaxivity. Thus, an ideal Fe(III)-based contrast agent should contain a coordinated water molecule with a pKa value well above 7.4. We have shown that t-H4CDTA displays the highest pKa value among the complexes studied here, and that the pKa does not correlate with the Fe–Owater bond distances.
(3) Complex stability/inertness is also a key issue, as the agent should remain intact until excreted from the body. We have shown here that the complexes of H4PhDTA and H4PDTA experience dissociation even in weakly basic solutions.
(4) The shape of the NMRD profiles of the complexes investigated here are similar to those of the parent H4EDTA and t-H4CDTA complexes, and show a maximum around 7 T. This makes Fe(III) complexes very good candidates as high-field MRI contrast agents. At high magnetic fields, the inner-sphere relaxivity is affected by both rotation and electronic relaxation. The results presented in this contribution show that electronic relaxation is quite insensitive to the nature of the central spacer in this family of H4EDTA derivatives. However, additional studies are required using other ligand families to understand whether electronic relaxation can be tuned by varying the metal coordination environment.
In conclusion, we have shown that Fe(III) complexes show very interesting properties that make then very attractive as MRI contrast agents. We hope that the guidelines reported here will aid coordination chemists in the design of new complexes with improved properties.
Experimental and computational section
General considerations
All solvents and reagents used were purchased from commercial sources, and were of reagent grade quality and used as supplied without further purification. High-resolution electrospray-ionization time-of-flight ESI-TOF mass spectra were recorded in the positive and negative mode using an LTQ-Orbitrap Discovery Mass Spectrometers coupled to a Thermo Accela HPLC. Medium performance liquid chromatography (MPLC) was carried out using a Puriflash XS 420 InterChim Chromatographer instrument equipped with a UV-DAD detector in normal or reverse phase, depending on the product solubility. Aqueous solutions were lyophilized using a Biobase BK-FD10 Series apparatus. 1H and 13C NMR spectra of the ligands and their precursors were recorded at 298 K on a Bruker AVANCE III 300, a Bruker AVANCE 400 or a Bruker AVANCE 500 spectrometer. Microwave-assisted reactions were carried out in an Anton Paar Monowave 300 reactor operating at 2455 MHz in a 10 mL sealed reaction vials with stirring. The system operated at 0–850 W power. The samples were irradiated with the appropriate power to achieve the temperature of 100 °C, utilizing the “as fast as possible” heating mode (hold time: 2 min, approximately). The reaction mixture temperature was monitored via built-in IR sensor.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). MS (ESI+, CH3CN/H2O): 355.1112 m/z; calculated for [C13H20N2O8]Na+ 355.1112.
O). MS (ESI+, CH3CN/H2O): 347.1449 m/z; calculated for [C14H22N2O8]H+ 347.1449.
O).
O). MS (ESI+, CH3CN/H2O): 355.1112 m/z; calculated for [C13H20N2O8]Na+ 355.1112.
O). MS (ESI+, CH3CN/H2O): 347.1449 m/z; calculated for [C14H22N2O8]H+ 347.1449.
O).
General procedure for the synthesis of the iron complexes
Complexes were synthetized as described in the literature.17 The ligand was dissolved in 3 mL of Milli-Q water, and the pH was adjusted to 1 with 1 M HCl if necessary. Then, a solution of FeCl3·6H2O in 2 mL of water was added (ratio 1:1.05 ligand to metal). The mixture was left stirring for 18 h at room temperature. After this time, the pH was adjusted to 6 with either 1 M NaOH or KOH solution to promote the precipitation of the free Fe(III). Finally, the solution was centrifuged, filtered and lyophilized.
Crystal structure determination
Crystallographic data were collected at 100 K using a Bruker D8 Venture diffractometer with a Photon 100 CMOS detector and Mo-Kα radiation (λ = 0.71073 Å) generated by an Incoatec high brilliance microfocus source equipped with Incoatec Helios multilayer optics. The software APEX391 was used for collecting frames of data, indexing reflections, and the determination of lattice parameters, SAINT92 for integration of intensity of reflections, and SADABS93 for scaling and empirical absorption correction. The structure was solved by dual-space methods using the program SHELXT.94 All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2 using the program SHELXL-2014.95 Hydrogen atoms were inserted at calculated positions and constrained with isotropic thermal parameters. CCDC 2225832 contains the supplementary crystallographic data [Fe(CBuDEDPA)]PF6.† Table S1† contains the crystallographic data and the structure refinement parameters.1H NMRD and 17O NMR measurements
1/T11H Nuclear Magnetic Relaxation Dispersion (NMRD) profiles were acquired with a Fast-Field Cycling (FFC) Stelar SmarTracer relaxometer (Stelar s.r.l., Mede, PV, Italy) over a continuum of proton Larmor frequencies from 9.97 × 10−3 to 10 MHz, with an uncertainty from 1/T1 of ca. 1%. Data in the range of 20–120 MHz proton Larmor frequency were measured with a High Field Relaxometer (Stelar) equipped with the HTS-110 3T Metrology Cryogen-free Superconducting Magnet. The analyses were carried out by using the standard inversion recovery sequence (20 experiments, 2 scans) with a typical 90° pulse width of 3.5 μs, and the reproducibility of the data was within ± 0.5%. The temperature was controlled with a Stelar VTC-91 heater airflow equipped with a copper-constantan thermocouple (uncertainty of ± 0.1 K).17O measurements were recorded on a Bruker Avance III spectrometer (11.7 T) equipped with a 5 mm probe and standard temperature control unit. Aqueous solutions of the complexes were enriched to reach 2.0% of the 17O isotope (Cambridge Isotope). The transverse relaxation rates were calculated from the signal width at a half-height. The concentration of the Fe(III) complexes was assessed by 1H-NMR measurements (Bruker Avance III Spectrometer equipped with a wide bore 11.7 Tesla magnet), by using the well-established bulk magnetic susceptibility method.96
Electrochemical measurements
Cyclic voltammetry experiments were carried out using a three-electrode configuration with an Autolab PGSTAT302M potentiostat-galvanostat. The working electrode was a glassy carbon disc (Metrohm 61204600), whose surface was polished before each measurement using α-Al2O3 (0.3 μm) and washed with distilled water. A Ag/AgCl reference electrode filled with 3 M KCl (Metrohm 6.0726.100) was used as the reference electrode, while a Pt wire was used as the counter electrode. All potentials were converted to the NHE scale for the sake of clarity, using the relationship E(NHE) = E(Ag/AgCl) + 210 mV.97 All potentials are provided vs. the NHE reference electrode. The solutions of the complexes (∼2.0 × 10−3 M) containing 0.15 M NaCl as supporting electrolyte were deoxygenated by bubbling N2 prior each measurement.Computational details
The geometries of the Fe(III) complexes were optimized with the Gaussian 16 program package (revision C.01)98 using the wB97XD functional, which is a long-range corrected hybrid density functional incorporating atom–atom dispersion corrections,99 and the Def2-TZVPP basis set.100 The integration grid was set with the integral = ultrafine keyword. Solvent effects were considered using a polarizable continuum model101 with the default options implemented in Gaussian using scrf = (pcm, solvent = water). Frequency calculations were used to confirm that the optimized structures correspond to local energy minima on the potential energy surface.The ORCA program package (version 5.0.3)102,103 was used to calculate 17O hyperfine coupling tensors and ZFS parameters. Hyperfine coupling constants were obtained using DFT with the TPSSh functional,104 which was found to perform well for this specific problem.105 The Def2-TZVPP basis set was employed together with auxiliary basis sets generated with the Autoaux106 procedure to accelerate the calculations with the resolution of identity and chain of spheres (RIJCOSX) method.107–109 The hyperfine coupling constant tensor contains contributions from the isotropic Fermi contact term and the anisotropic spin–dipolar and spin–orbit coupling (SOC) contributions. The latter was considered using the spin–orbit mean-field (SOMF) method.110,111 The SOC contribution to 17O hyperfine coupling constants was found to be negligible. The unrestricted natural orbitals generated from these calculations were used as starting orbitals for complete active space self-consistent field (CASSCF) calculations,112,113 in which dynamic correlation was incorporated using the strongly contracted variant of N-Electron Valence State Perturbation Theory (SC-NEVPT2)114,115 and SOC effects were introduced using quasi-degenerate perturbation theory (QDPT).116,117 These calculations used the Def2-QZVPP basis set and the Def2/JK auxiliary basis set together with the resolution of identity (RI-JK) method.100,118,119 The active space of the state-averaged CASSCF calculations consisted of five electrons distributed over the five metal-based 3d orbitals CAS(5,5), including one sextet, 24 quartet and 75 doublet roots. All ORCA calculations incorporated water solvent effects with the SMD solvation model.120
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R.-R. (Grant PID2019-108352RJ-I00) and C. P.-I. and D. E.-G. thank Ministerio de Ciencia e Innovación (Grant PID2019-104626GB-I00) and Xunta de Galicia (Grant ED431B 2020/52) for generous financial support. The authors are indebted to Centro de Supercomputación of Galicia (CESGA) for providing the computer facilities. R. U.-V. thanks Xunta de Galicia (Grant ED481A-2018/314) for funding her Ph.D. contract. L. V. is indebted to CACTI (Universidade de Vigo) for X-Ray measurements.References
- P. Boehm-Sturm, A. Haeckel, R. Hauptmann, S. Mueller, C. K. Kuhl and E. A. Schellenberger, Low-Molecular-Weight Iron Chelates May Be an Alternative to Gadolinium-based Contrast Agents for T1-weighted Contrast-enhanced MR Imaging, Radiology, 2018, 286, 537–546 CrossRef PubMed.
- E. M. Snyder, D. Asik, S. M. Abozeid, A. Burgio, G. Bateman, S. G. Turowski, J. A. Spernyak and J. R. Morrow, A Class of FeIII Macrocyclic Complexes with Alcohol Donor Groups as Effective T1 MRI Contrast Agents, Angew. Chem., Int. Ed., 2020, 59, 2414–2419 CrossRef CAS PubMed.
- H. Wang, V. C. Jordan, I. A. Ramsay, M. Sojoodi, B. C. Fuchs, K. K. Tanabe, P. Caravan and E. M. Gale, Molecular Magnetic Resonance Imaging Using a Redox-Active Iron Complex, J. Am. Chem. Soc., 2019, 141, 5916–5925 CrossRef CAS PubMed.
- D. Asik, R. Smolinski, S. M. Abozeid, T. B. Mitchell, S. G. Turowski, J. A. Spernyak and J. R. Morrow, Modulating the Properties of Fe(III) Macrocyclic MRI Contrast Agents by Appending Sulfonate or Hydroxyl Groups, Molecules, 2020, 25, 2291 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- H. Li and T. J. Meade, Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances, J. Am. Chem. Soc., 2019, 141, 17025–17041 CrossRef CAS PubMed.
- L. Helm, J. R. Morrow, C. J. Bond, F. Carniato, M. Botta, M. Braun, Z. Baranyai, R. Pujales-Paradela, M. Regueiro-Figueroa, D. Esteban-Gómez, C. Platas-Iglesias and T. J. Scholl, in New Developments in NMR, ed. V. C. Pierre and M. J. Allen, Royal Society of Chemistry, Cambridge, 2017, pp. 121–242 Search PubMed.
- K. Chan and W. Wong, Small molecular gadolinium(III) complexes as MRI contrast agents for diagnostic imaging, Coord. Chem. Rev., 2007, 251, 2428–2451 CrossRef CAS.
- L. Helm, Relaxivity in paramagnetic systems: Theory and mechanisms, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 45–64 CrossRef CAS.
- J. H. Freed, Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. II. Finite jumps and independent T1 processes, J. Chem. Phys., 1978, 68, 4034–4037 CrossRef CAS.
- P. H. Fries, C. Gateau and M. Mazzanti, Practical Route to Relative Diffusion Coefficients and Electronic Relaxation Rates of Paramagnetic Metal Complexes in Solution by Model-Independent Outer-Sphere NMRD. Potentiality for MRI Contrast Agents, J. Am. Chem. Soc., 2005, 127, 15801–15814 CrossRef CAS PubMed.
- M. Botta, F. Carniato, D. Esteban-Gómez, C. Platas-Iglesias and L. Tei, Mn(II) compounds as an alternative to Gd-based MRI probes, Future Med. Chem., 2019, 11, 1461–1483 CrossRef CAS PubMed.
- B. Drahoš, J. Kotek, P. Hermann, I. Lukeš and É. Tóth, Mn2+ Complexes with Pyridine-Containing 15-Membered Macrocycles: Thermodynamic, Kinetic, Crystallographic, and 1H/17O Relaxation Studies, Inorg. Chem., 2010, 49, 3224–3238 CrossRef PubMed.
- P. Cieslik, P. Comba, B. Dittmar, D. Ndiaye, É. Tóth, G. Velmurugan and H. Wadepohl, Exceptional Manganese(II) Stability and Manganese(II)/Zinc(II) Selectivity with Rigid Polydentate Ligands, Angew. Chem., Int. Ed., 2022, 61, e202115580 CrossRef CAS PubMed.
- D. Ndiaye, M. Sy, A. Pallier, S. Même, I. Silva, S. Lacerda, A. M. Nonat, L. J. Charbonnière and É. Tóth, Unprecedented Kinetic Inertness for a Mn2+–Bispidine Chelate: A Novel Structural Entry for Mn2+ –Based Imaging Agents, Angew. Chem., Int. Ed., 2020, 59, 11958–11963 CrossRef CAS PubMed.
- D. J. Erstad, I. A. Ramsay, V. C. Jordan, M. Sojoodi, B. C. Fuchs, K. K. Tanabe, P. Caravan and E. M. Gale, Tumor Contrast Enhancement and Whole-Body Elimination of the Manganese-Based Magnetic Resonance Imaging Contrast Agent Mn-PyC3A:, Invest. Radiol., 2019, 54, 697–703 CrossRef CAS PubMed.
- Z. Baranyai, F. Carniato, A. Nucera, D. Horváth, L. Tei, C. Platas-Iglesias and M. Botta, Defining the conditions for the development of the emerging class of FeIII-based MRI contrast agents, Chem. Sci., 2021, 12, 11138–11145 RSC.
- D. H. Powell, O. M. N. Dhubhghaill, D. Pubanz, L. Helm, Y. S. Lebedev, W. Schlaepfer and A. E. Merbach, Structural and Dynamic Parameters Obtained from 17O NMR, EPR, and NMRD Studies of Monomeric and Dimeric Gd3+ Complexes of Interest in Magnetic Resonance Imaging: An Integrated and Theoretically Self-Consistent Approach 1, J. Am. Chem. Soc., 1996, 118, 9333–9346 CrossRef CAS.
- I. Solomon, Relaxation Processes in a System of Two Spins, Phys. Rev., 1955, 99, 559–565 CrossRef CAS.
- N. Bloembergen, Proton Relaxation Times in Paramagnetic Solutions, J. Chem. Phys., 1957, 27, 572–573 CrossRef CAS.
- N. Bloembergen and L. O. Morgan, Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation, J. Chem. Phys., 1961, 34, 842–850 CrossRef CAS.
- L. Helm and A. E. Merbach, Inorganic and Bioinorganic Solvent Exchange Mechanisms, Chem. Rev., 2005, 105, 1923–1960 CrossRef CAS PubMed.
- P. H. Fries, Computing Electronic Spin Relaxation for Gd3+-Based Contrast Agents - Practical Implementation, Eur. J. Inorg. Chem., 2012, 2012, 2156–2166 CrossRef CAS.
- S. Rast, A. Borel, L. Helm, E. Belorizky, P. H. Fries and A. E. Merbach, EPR Spectroscopy of MRI-Related Gd(III) Complexes: Simultaneous Analysis of Multiple Frequency and Temperature Spectra, Including Static and Transient Crystal Field Effects, J. Am. Chem. Soc., 2001, 123, 2637–2644 CrossRef CAS PubMed.
- J. A. Peters, The reliability of parameters obtained by fitting of 1H NMRD profiles and 17O NMR data of potential Gd3+-based MRI contrast agents: Fitting of 1H NMRD profiles and 17O NMR data, Contrast Media Mol. Imaging, 2016, 11, 160–168 CrossRef CAS PubMed.
- T. J. Clough, L. Jiang, K.-L. Wong and N. J. Long, Ligand design strategies to increase stability of gadolinium-based magnetic resonance imaging contrast agents, Nat. Commun., 2019, 10, 1420 CrossRef PubMed.
- J. Maigut, R. Meier, A. Zahl and R. van Eldik, Effect of Chelate Dynamics on Water Exchange Reactions of Paramagnetic Aminopolycarboxylate Complexes, Inorg. Chem., 2008, 47, 5702–5719 CrossRef CAS PubMed.
- J. Maigut, R. Meier, A. Zahl and R. van Eldik, Triggering Water Exchange Mechanisms via Chelate Architecture. Shielding of Transition Metal Centers by Aminopolycarboxylate Spectator Ligands, J. Am. Chem. Soc., 2008, 130, 14556–14569 CrossRef CAS PubMed.
- A. Rodríguez-Rodríguez, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, M. Fekete, M. Botta, R. Tripier and C. Platas-Iglesias, Lanthanide(III) Complexes with Ligands Derived from a Cyclen Framework Containing Pyridinecarboxylate Pendants. The Effect of Steric Hindrance on the Hydration Number, Inorg. Chem., 2012, 51, 2509–2521 CrossRef PubMed.
- R. Ruloff, É. Tóth, R. Scopelliti, R. Tripier, H. Handel and A. E. Merbach, Accelerating water exchange for GdIII chelates by steric compression around the water binding site, Chem. Commun., 2002, 2630–2631 RSC.
- A. Brausam, J. Maigut, R. Meier, P. Á. Szilágyi, H.-J. Buschmann, W. Massa, Z. Homonnay and R. van Eldik, Detailed Spectroscopic, Thermodynamic, and Kinetic Studies on the Protolytic Equilibria of FeIIIcydta and the Activation of Hydrogen Peroxide, Inorg. Chem., 2009, 48, 7864–7884 CrossRef CAS PubMed.
- P. Kocot, K. Szaciłowski and Z. Stasicka, Photochemistry of the [FeIII(edta)(H2O)]− and [FeIII(edta)(OH)]2− complexes in presence of environmentally relevant species, J. Photochem. Photobiol., A, 2007, 188, 128–134 CrossRef CAS.
- M. Azarkh and E. J. J. Groenen, Simulation of multi-frequency EPR spectra for a distribution of the zero-field splitting, J. Magn. Reson., 2015, 255, 106–113 CrossRef CAS PubMed.
- G. Mathies, H. Blok, J. A. J. M. Disselhorst, P. Gast, H. van der Meer, D. M. Miedema, R. M. Almeida, J. J. G. Moura, W. R. Hagen and E. J. J. Groenen, Continuous-wave EPR at 275 GHz: Application to high-spin Fe3+ systems, J. Magn. Reson., 2011, 210, 126–132 CrossRef CAS PubMed.
- G. Mathies, P. Gast, N. D. Chasteen, A. N. Luck, A. B. Mason and E. J. J. Groenen, Exploring the Fe(III) binding sites of human serum transferrin with EPR at 275 GHz, JBIC, J. Biol. Inorg. Chem., 2015, 20, 487–496 CrossRef CAS PubMed.
- Y. Liu, X. Dong, J. Sun, C. Zhong, B. Li, X. You, B. Liu and Z. Liu, Two-photon fluorescent probe for cadmium imaging in cells, Analyst, 2012, 137, 1837 RSC.
- K. Pota, Z. Garda, F. K. Kálmán, J. L. Barriada, D. Esteban-Gómez, C. Platas-Iglesias, I. Tóth, E. Brücher and G. Tircsó, Taking the next step toward inert Mn2+ complexes of open-chain ligands: the case of the rigid PhDTA ligand, New J. Chem., 2018, 42, 8001–8011 RSC.
- R. Uzal-Varela, D. Lalli, I. Brandariz, A. Rodríguez-Rodríguez, C. Platas-Iglesias, M. Botta and D. Esteban-Gómez, Rigid versions of PDTA4− incorporating a 1,3-diaminocyclobutyl spacer for Mn2+ complexation: stability, water exchange dynamics and relaxivity, Dalton Trans., 2021, 50, 16290–16303 RSC.
- V. Dragojlovic, Conformational analysis of cycloalkanes, ChemTexts, 2015, 1, 14 CrossRef.
- O. Porcar-Tost, A. Pallier, D. Esteban-Gómez, O. Illa, C. Platas-Iglesias, É. Tóth and R. M. Ortuño, Stability, relaxometric and computational studies on Mn2+ complexes with ligands containing a cyclobutane scaffold, Dalton Trans., 2021, 50, 1076–1085 RSC.
- R. Uzal-Varela, V. Patinec, R. Tripier, L. Valencia, M. Maneiro, M. Canle, C. Platas-Iglesias, D. Esteban-Gómez and E. Iglesias, On the dissociation pathways of copper complexes relevant as PET imaging agents, J. Inorg. Biochem., 2022, 236, 111951 CrossRef CAS PubMed.
- J. C. Joyner, J. Reichfield and J. A. Cowan, Factors Influencing the DNA Nuclease Activity of Iron, Cobalt, Nickel, and Copper Chelates, J. Am. Chem. Soc., 2011, 133, 15613–15626 CrossRef CAS PubMed.
- J. C. Joyner and J. A. Cowan, Targeted Cleavage of HIV RRE RNA by Rev-Coupled Transition Metal Chelates, J. Am. Chem. Soc., 2011, 133, 9912–9922 CrossRef CAS PubMed.
- D. P. Jones and H. Sies, The Redox Code, Antioxid. Redox Signal., 2015, 23, 734–746 CrossRef CAS PubMed.
- M. Merkofer, R. Kissner, R. C. Hider, U. T. Brunk and W. H. Koppenol, Fenton Chemistry and Iron Chelation under Physiologically Relevant Conditions: Electrochemistry and Kinetics, Chem. Res. Toxicol., 2006, 19, 1263–1269 Search PubMed.
- W. H. Koppenol and R. H. Hider, Iron and redox cycling. Do's and don'ts, Free Radicals Biol. Med., 2019, 133, 3–10 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- M. Song, K. E. Daniels, A. Kiani, S. Rashid-Nadimi and M. D. Dickey, Interfacial Tension Modulation of Liquid Metal via Electrochemical Oxidation, Adv. Intell. Syst., 2021, 3, 2100024 CrossRef.
- A. E. Martell, R. J. Motekaitis, D. Chen, R. D. Hancock and D. McManus, Selection of new Fe(III)/Fe(II) chelating agents as catalysts for the oxidation of hydrogen sulfide to sulfur by air, Can. J. Chem., 1996, 74, 1872–1879 CrossRef CAS.
- J. M. Wilson and R. F. Carbonaro, Capillary electrophoresis study of iron(II) and iron(III) polyaminocarboxylate complex speciation, Environ. Chem., 2011, 8, 295–303 CrossRef CAS.
- J. Sanchiz, S. Domínguez, A. Mederos, F. Brito and J. M. Arrieta, Tetramethyl Carboxylic Acids Derived from o-Phenylenediamines as Sequestering Agents for Iron(III): Thermodynamic Studies. X-ray Crystal Structure of Sodium Aqua(4-chloro-1,2-phenylenediamine-N,N,N ‘, N‘-tetraacetato)ferrate(III)−Water (1/1.5), Inorg. Chem., 1997, 36, 4108–4114 CrossRef CAS.
- V. Patinec, G. A. Rolla, M. Botta, R. Tripier, D. Esteban-Gómez and C. Platas-Iglesias, Hyperfine Coupling Constants on Inner-Sphere Water Molecules of a Triazacyclononane-based Mn(II) Complex and Related Systems Relevant as MRI Contrast Agents, Inorg. Chem., 2013, 52, 11173–11184 CrossRef CAS PubMed.
- D. Riccardi, H.-B. Guo, J. M. Parks, B. Gu, L. Liang and J. C. Smith, Cluster-Continuum Calculations of Hydration Free Energies of Anions and Group 12 Divalent Cations, J. Chem. Theory Comput., 2013, 9, 555–569 CrossRef CAS PubMed.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard III, Calculation of Solvation Free Energies of Charged Solutes Using Mixed Cluster/Continuum Models, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS PubMed.
- F. Neese and E. I. Solomon, Detailed Spectroscopic and Theoretical Studies on [Fe(EDTA)(O2)]3− : Electronic Structure of the Side-on Ferric−Peroxide Bond and Its Relevance to Reactivity, J. Am. Chem. Soc., 1998, 120, 12829–12848 CrossRef CAS.
- J. Maigut, R. Meier, A. Zahl and R. van Eldik, Elucidation of the Solution Structure and Water-Exchange Mechanism of Paramagnetic [FeII(edta)(H2O)]2−, Inorg. Chem., 2007, 46, 5361–5371 CrossRef CAS PubMed.
- D. Casanova, P. Alemany, J. M. Bofill and S. Alvarez, Shape and Symmetry of Heptacoordinate Transition-Metal Complexes: Structural Trends, Chem. – Eur. J., 2003, 9, 1281–1295 CrossRef CAS PubMed.
- D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, Minimal Distortion Pathways in Polyhedral Rearrangements, J. Am. Chem. Soc., 2004, 126, 1755–1763 CrossRef CAS PubMed.
- S. Alvarez, Polyhedra in (inorganic) chemistry, Dalton Trans., 2005, 2209 RSC.
- S. Seibig and R. van Eldik, Structural information on trans-1,2-diaminocyclohexane-N,N′-tetraacetateferrate(IIl) in the solid and aqueous phase, Inorg. Chim. Acta, 1998, 279, 37–43 CrossRef CAS.
- R. Meier and F. W. Heinemann, Structures of the spontaneously resolved six-coordinate potassium chloro-(ethylenediaminetriacetato acetic acid) iron(III) monohydrate and the seven-coordinate potassium (ethylenediaminetetracetato) iron(III) sesquihydrate, Inorg. Chim. Acta, 2002, 337, 317–327 CrossRef CAS.
- F. Neese, Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- G. A. Rolla, C. Platas-Iglesias, M. Botta, L. Tei and L. Helm, 1H and 17O NMR Relaxometric and Computational Study on Macrocyclic Mn(II) Complexes, Inorg. Chem., 2013, 52, 3268–3279 CrossRef CAS PubMed.
- L. Vander Elst, A. Sessoye, S. Laurent and R. N. Muller, Can the Theoretical Fitting of the Proton-Nuclear-Magnetic-Relaxation-Dispersion (Proton NMRD) Curves of Paramagnetic Complexes Be Improved by Independent Measurement of Their Self-Diffusion Coefficients?, Helv. Chim. Acta, 2005, 88, 574–587 CrossRef CAS.
- A. Roca-Sabio, C. S. Bonnet, M. Mato-Iglesias, D. Esteban-Gómez, É. Tóth, A. de Blas, T. Rodríguez-Blas and C. Platas-Iglesias, Lanthanide Complexes Based on a Diazapyridinophane Platform Containing Picolinate Pendants, Inorg. Chem., 2012, 51, 10893–10903 CrossRef CAS PubMed.
- R. Mills, Self-diffusion in normal and heavy water in the range 1-45.deg., J. Phys. Chem., 1973, 77, 685–688 CrossRef CAS.
- C. Platas-Iglesias, D. Esteban-Gómez, L. Helm and M. Regueiro-Figueroa, Transient versus Static Electron Spin Relaxation in Mn2+ Complexes Relevant as MRI Contrast Agents, J. Phys. Chem. A, 2016, 120, 6467–6476 CrossRef CAS PubMed.
- T. J. Swift and R. E. Connick, NMR–Relaxation Mechanisms of O17 in Aqueous Solutions of Paramagnetic Cations and the Lifetime of Water Molecules in the First Coordination Sphere, J. Chem. Phys., 1962, 37, 307–320 CrossRef CAS.
- T. J. Swift and R. E. Connick, Erratum: NMR–Relaxation Mechanisms of 17O in Aqueous Solutions of Paramagnetic Cations and the Lifetime of Water Molecules in the First Coordination Sphere, J. Chem. Phys., 1964, 41, 2553–2554 CrossRef CAS.
- N. Graeppi, D. H. Powell, G. Laurenczy, L. Zékány and A. E. Merbach, Coordination equilibria and water exchange kinetics of lanthanide(III) propylenediaminetetraacetates and other magnetic resonance imaging related complexes, Inorg. Chim. Acta, 1995, 235, 311–326 CrossRef CAS.
- C. Platas-Iglesias, D. M. Corsi, L. V. Elst, R. N. Muller, D. Imbert, J.-C. G. Bünzli, É. Tóth, T. Maschmeyer and J. A. Peters, Stability, structure and dynamics of cationic lanthanide(III) complexes of N,N′-bis(propylamide)ethylenediamine-N,N′-diacetic acid, Dalton Trans., 2003, 727 RSC.
- E. Balogh, M. Mato-Iglesias, C. Platas-Iglesias, É. Tóth, K. Djanashvili, J. A. Peters, A. de Blas and T. Rodríguez-Blas, Pyridine- and Phosphonate-Containing Ligands for Stable Ln Complexation. Extremely Fast Water Exchange on the GdIII Chelates, Inorg. Chem., 2006, 45, 8719–8728 CrossRef CAS PubMed.
- R. Janicki and A. Mondry, Structural and thermodynamic aspects of hydration of Gd(iii) systems, Dalton Trans., 2019, 48, 3380–3391 RSC.
- É. Tóth, O. M. N. Dhubhghaill, G. Besson, L. Helm and A. E. Merbach, Coordination equilibrium- a clue for fast water exchange on potential magnetic resonance imaging contrast agents?, Magn. Reson. Chem., 1999, 701–708 CrossRef.
- T. Schneppensieper, S. Seibig, A. Zahl, P. Tregloan and R. van Eldik, Influence of Chelate Effects on the Water-Exchange Mechanism of Polyaminecarboxylate Complexes of Iron(III), Inorg. Chem., 2001, 40, 3670–3676 CrossRef CAS PubMed.
- M. Mizuno, S. Funahashi, N. Nakasuka and M. Tanaka, Water exchange of the (o-phenylenediamine-N,N,N′,N′-tetraacetato)ferrate(III) complex in aqueous solution as studied by variable-temperature, -pressure, and -frequency oxygen-17 NMR techniques, Inorg. Chem., 1991, 30, 1550–1553 CrossRef CAS.
- A. Forgács, R. Pujales-Paradela, M. Regueiro-Figueroa, L. Valencia, D. Esteban-Gómez, M. Botta and C. Platas-Iglesias, Developing the family of picolinate ligands for Mn2+ complexation, Dalton Trans., 2017, 46, 1546–1558 RSC.
- S. Laine, C. S. Bonnet, F. K. Kálmán, Z. Garda, A. Pallier, F. Caillé, F. Suzenet, G. Tircsó and É. Tóth, Mn2+ complexes of open-chain ligands with a pyridine backbone: less donor atoms lead to higher kinetic inertness, New J. Chem., 2018, 42, 8012–8020 RSC.
- J. Kowalewski, C. Luchinat, T. Nilsson and G. Parigi, Nuclear Spin Relaxation in Paramagnetic Systems: Electron Spin Relaxation Effects under Near-Redfield Limit Conditions and Beyond, J. Phys. Chem. A, 2002, 106, 7376–7382 CrossRef CAS.
- S. Rast, P. H. Fries and E. Belorizky, Static zero field splitting effects on the electronic relaxation of paramagnetic metal ion complexes in solution, J. Chem. Phys., 2000, 113, 8724–8735 CrossRef CAS.
- E. Strandberg and P.-O. Westlund, Paramagnetic Proton Nuclear Spin Relaxation Theory of Low-Symmetry Complexes for Electron Spin Quantum NumberS=52, J. Magn. Reson., 1999, 137, 333–344 CrossRef CAS PubMed.
- E. Belorizky and P. H. Fries, Simple analytical approximation of the longitudinal electronic relaxation rate of Gd(iii) complexes in solutions, Phys. Chem. Chem. Phys., 2004, 6, 2341 RSC.
- S. Khan, R. Pollet, R. Vuilleumier, J. Kowalewski and M. Odelius, An ab initio CASSCF study of zero field splitting fluctuations in the octet ground state of aqueous [Gd(iii)(HPDO3A)(H 2 O)], J. Chem. Phys., 2017, 147, 244306 CrossRef PubMed.
- L. Lang and F. Neese, Spin-dependent properties in the framework of the dynamic correlation dressed complete active space method, J. Chem. Phys., 2019, 150, 104104 CrossRef PubMed.
- R. Uzal-Varela, L. Valencia, D. Lalli, M. Maneiro, D. Esteban-Gómez, C. Platas-Iglesias, M. Botta and A. Rodríguez-Rodríguez, Understanding the Effect of the Electron Spin Relaxation on the Relaxivities of Mn(II) Complexes with Triazacyclononane Derivatives, Inorg. Chem., 2021, 60, 15055–15068 CrossRef CAS PubMed.
- S. Zein and F. Neese, Ab Initio and Coupled-Perturbed Density Functional Theory Estimation of Zero-Field Splittings in MnII Transition Metal Complexes, J. Phys. Chem. A, 2008, 112, 7976–7983 CrossRef CAS PubMed.
- R. Aasa, Powder Line Shapes in the Electron Paramagnetic Resonance Spectra of High–Spin Ferric Complexes, J. Chem. Phys., 1970, 52, 3919–3930 CrossRef CAS.
- S. Khan, A. Kubica-Misztal, D. Kruk, J. Kowalewski and M. Odelius, Systematic theoretical investigation of the zero-field splitting in Gd(III) complexes: Wave function and density functional approaches, J. Chem. Phys., 2015, 142, 034304 CrossRef PubMed.
- P. H. Fries and E. Belorizky, Determination of the Static Zero-Field Splitting of Gd3+ Complexes in Solution from the Shifts of the Central Magnetic Fields of Their EPR Spectra, ChemPhysChem, 2012, 13, 2074–2081 CrossRef CAS PubMed.
- C. Platas-Iglesias, M. Mato-Iglesias, K. Djanashvili, R. N. Muller, L. V. Elst, J. A. Peters, A. de Blas and T. Rodríguez-Blas, Lanthanide Chelates Containing Pyridine Units with Potential Application as Contrast Agents in Magnetic Resonance Imaging, Chem. – Eur. J., 2004, 10, 3579–3590 CrossRef CAS PubMed.
- APEX3 Version 2016.1, Bruker AXS Inc., 2016 Search PubMed.
- SAINT Version 8.37A, Bruker AXS Inc., 2015 Search PubMed.
- G. M. Sheldrick, SADABS Version 2014/5, Bruker AXS Inc. Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- D. M. Corsi, C. Platas-Iglesias, H. van Bekkum and J. A. Peters, Determination of paramagnetic lanthanide(III) concentrations from bulk magnetic susceptibility shifts in NMR spectra, Magn. Reson. Chem., 2001, 39, 723–726 CrossRef CAS.
- E. P. Friis, J. E. T. Andersen, L. L. Madsen, N. Bonander, P. Moller and J. Ulstrup, Dynamics of Pseudomonas aeruginosa azurin and its Cys3Ser mutant at single-crystal gold surfaces investigated by cyclic voltammetry and atomic force microscopy, Electrochim. Acta, 1998, 43, 1114–1122 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- S. Kossmann, B. Kirchner and F. Neese, Performance of modern density functional theory for the prediction of hyperfine structure: meta-GGA and double hybrid functionals, Mol. Phys., 2007, 105, 2049–2071 CrossRef CAS.
- G. L. Stoychev, A. A. Auer and F. Neese, Automatic Generation of Auxiliary Basis Sets, J. Chem. Theory Comput., 2017, 13, 554–562 CrossRef CAS PubMed.
- F. Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed.
- S. Kossmann and F. Neese, Comparison of two efficient approximate Hartee–Fock approaches, Chem. Phys. Lett., 2009, 481, 240–243 CrossRef CAS.
- R. Izsák and F. Neese, An overlap fitted chain of spheres exchange method, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, A mean-field spin-orbit method applicable to correlated wavefunctions, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- F. Neese, Efficient and accurate approximations to the molecular spin-orbit coupling operator and their use in molecular g-tensor calculations, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- P.-Å. Malmqvist and B. O. Roos, The CASSCF state interaction method, Chem. Phys. Lett., 1989, 155, 189–194 CrossRef CAS.
- C. Kollmar, K. Sivalingam, B. Helmich-Paris, C. Angeli and F. Neese, A perturbation–based super–CI approach for the orbital optimization of a CASSCF wave function, J. Comput. Chem., 2019, 40, 1463–1470 CrossRef CAS PubMed.
- C. Angeli, S. Borini, M. Cestari and R. Cimiraglia, A quasidegenerate formulation of the second order n-electron valence state perturbation theory approach, J. Chem. Phys., 2004, 121, 4043–4049 CrossRef CAS PubMed.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, Introduction of n -electron valence states for multireference perturbation theory, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- D. Maganas, S. Sottini, P. Kyritsis, E. J. J. Groenen and F. Neese, Theoretical Analysis of the Spin Hamiltonian Parameters in Co(II)S 4 Complexes, Using Density Functional Theory and Correlated ab initio Methods, Inorg. Chem., 2011, 50, 8741–8754 CrossRef CAS PubMed.
- M. Atanasov, D. Aravena, E. Suturina, E. Bill, D. Maganas and F. Neese, First principles approach to the electronic structure, magnetic anisotropy and spin relaxation in mononuclear 3d-transition metal single molecule magnets, Coord. Chem. Rev., 2015, 289–290, 177–214 CrossRef CAS.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Weigend, Hartree–Fock exchange fitting basis sets for H to Rn, J. Comput. Chem., 2008, 29, 167–175 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, high-resolution MS, cyclic voltammograms, plots of the linear dependence of anodic and cathodic peak currents with the square root of the scan rate, crystal data and structure refinement details and optimized geometries obtained with DFT. CCDC 2225832. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02665a |
| This journal is © the Partner Organisations 2023 |