
Selective Buchwald–Hartwig arylation of C-amino-1,2,4-triazoles and other coordinating aminoheterocycles enabled by bulky NHC ligands and TPEDO activator†
Alexander V.
Astakhov
 a,
Andrey Yu.
Chernenko
a,
Vadim V.
Kutyrev
a,
Gleb S.
Ranny
a,
Mikhail E.
Minyaev
b,
Victor M.
Chernyshev
*a and
Valentine P.
Ananikov
*ab
a,
Andrey Yu.
Chernenko
a,
Vadim V.
Kutyrev
a,
Gleb S.
Ranny
a,
Mikhail E.
Minyaev
b,
Victor M.
Chernyshev
*a and
Valentine P.
Ananikov
*ab
aPlatov South-Russian State Polytechnic University, (NPI), Prosvescheniya st., 132, Novocherkassk, 346428, Russia. E-mail: chern13@yandex.ru
bZelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, Moscow, 119991, Russia. E-mail: val@ioc.ac.ru
First published on 22nd November 2022
Abstract
C-Amino-1,2,4-triazoles are challenging polynitrogen substrates for metal-catalyzed arylation due to their multidentate character, enhanced coordinating ability and decreased nucleophilicity of the amino group. In the present study, the Buchwald–Hartwig cross-coupling of diverse 3(5)-amino-1,2,4-triazoles with aryl chlorides and bromides delivering (hetero)arylamino-1,2,4-triazoles in good-to-excellent yields under Pd/NHC catalysis was developed. The use of Pd complexes with bulky NHC ligands such as IPr*OMe and TPEDO (1,1,2,2-tetraphenylethane-1,2-diol) as an in situ Pd(II) to Pd(0) reductant enabled the selective arylation of the NH2 group even in acidic NH unprotected substrates and deactivated 1-substituted 5-amino- and 4-substituted 3-amino-1,2,4-triazoles. The reaction mechanism and structure–activity relationships were studied with DFT calculations. A significant effect of the position of the N-substituent in the 1,2,4-triazole ring on the favorable reaction pathways was revealed.
1. Introduction
Palladium-catalyzed Buchwald–Hartwig amination has become an indispensable tool for the construction of (hetero)aromatic amine frames in the pharma industry, agrochemistry and material sciences.1–7 Despite the great progress in the development of efficient catalytic systems for a wide combination of amines and electrophiles, polynitrogen heteroaromatic compounds still remain challenging substrates for Buchwald–Hartwig cross-coupling.8–12 The decreased reactivity of polynitrogen aminoheterocycles can usually be explained by the low nucleophilicity of amino groups combined with the multidentate character of these substrates as well as their arylation products, which are prone to coordinate with metal centers via endocyclic N atoms and thus hinder reductive elimination, oxidative addition, and Pd(II) to Pd(0) reduction at the activation stage.9–131,2,4-Triazole is a highly demanded scaffold in medicinal chemistry14–20 and material sciences.21–24 For example, arylamino-1,2,4-triazoles, such as the anticancer25–28 and anti-SARS-CoV-229–32 drug Bemcentinib (R428), anticancer agents JNJ-770662133,34 and K00546,34,35 and antidepressant JNJ-39393406,36–38 are currently under clinical trials (Fig. 1). In addition, many other 3(5)-arylamino-1,2,4-triazoles have been reported as compounds revealing potential anticancer,39–41 antibacterial,42 anti-HIV,43,44 anti-inflammatory45,46 and antimalarial47,48 activities. Moreover, 3-arylamino-1,2,4-triazoles were recently studied as promising photoactive compounds possessing aggregation-induced emission (AIE)49 and precursors for N-heterocyclic carbene ligands.50–52
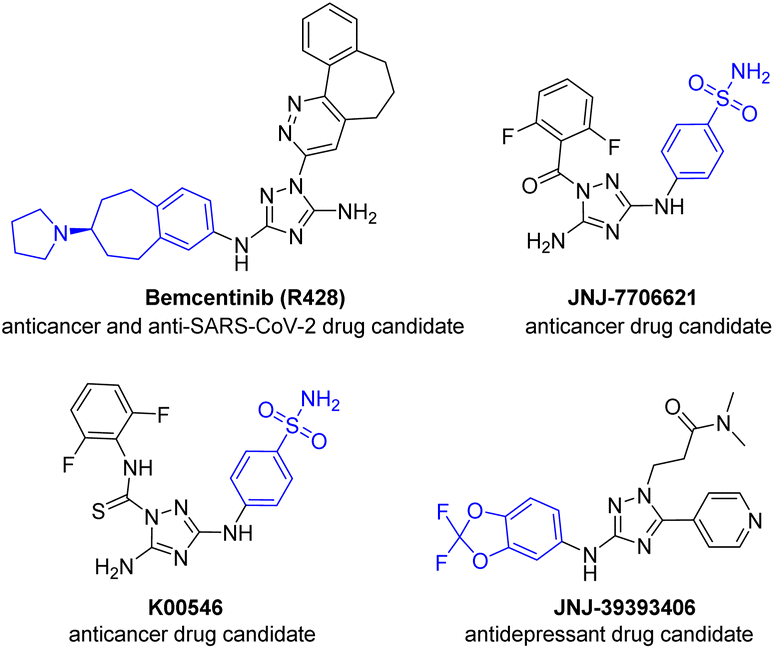 | ||
| Fig. 1 Medicinally important 3(5)-arylamino-1,2,4-triazoles under clinical trials. | ||
Although various methods based on the cyclization of acyclic precursors42,53–57 or amination of 3(5)-bromo-1,2,4-triazoles58–60 have been reported in the literature, arylation of easily available C-amino-1,2,4-triazoles19,61 may be considered one of most efficient pathways to diverse 3(5)-arylamino-1,2,4-triazoles. However, noncatalytic arylation of C-amino-1,2,4-triazoles is limited to activated arylating agents such as nitrochloroarenes,62 whereas metal-catalyzed arylation is encumbered by the strongly coordinating character of these substrates and the low nucleophilicity of the amino group. For example, Cu-catalyzed arylation of aminotriazoles with boronic acids (Chan–Lam reaction) required using over-stoichiometric loadings of Cu salts and a large excess of quite expensive boronic acids,63,64 whereas Pd-catalyzed arylation with aryl halides was only possible in the presence of expensive bulky phosphine ligands at high Pd loadings.65,66
Complexes of palladium with N-heterocyclic carbene ligands (Pd/NHC) were recently reported as efficient catalysts for the selective arylation of various polynitrogen aminoheterocycles. Organ and co-workers described the use of Pd-PEPPSI-IPentCl precatalyst for the (het)arylation of aminopyridines,10 Wu, Qiu and co-workers reported new precatalyst (SIPr)Ph2Pd(cin)Cl efficient in room temperature Buchwald–Hartwig cross-coupling of various (hetero)aryl chlorides with multinitrogen heteroaryl amines,11 and Szostak, Liu and co-workers successfully applied Pd-BIAN-NHC complexes for catalysis of (hetero)arylation of various coordinating aminoheterocycles.12
However, to the best of our knowledge, arylation of C-amino-1,2,4-triazoles under catalysis with Pd/NHC complexes has never been studied before. Herein, we report a facile approach to diverse 3(5)-(hetero)arylamino-1,2,4-triazoles via Pd/NHC-catalyzed (hetero)arylation of 3(5)-amino-1,2,4-triazoles enabled by the bulky NHC-ligand and new efficient Pd(II) to Pd(0) reductant (Scheme 1). The developed catalytic system can also be employed in the N-(hetero)arylation of other coordinating aminoheterocycles.
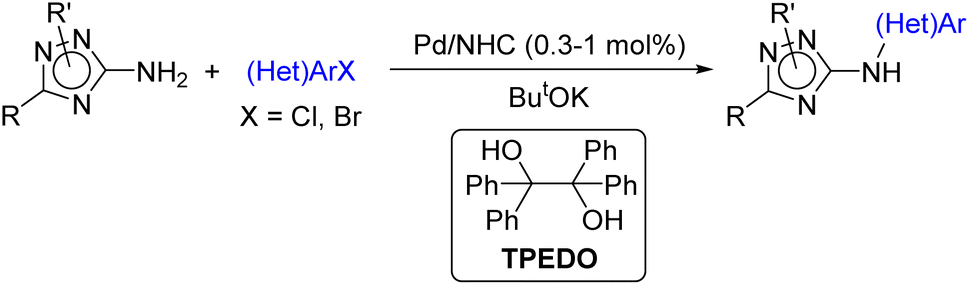 | ||
| Scheme 1 Arylation of diverse 3(5)amino-1,2,4-triazoles (this work). | ||
2. Results and discussion
2.1 Experimental study of arylation of C-amino-1,2,4-triazoles
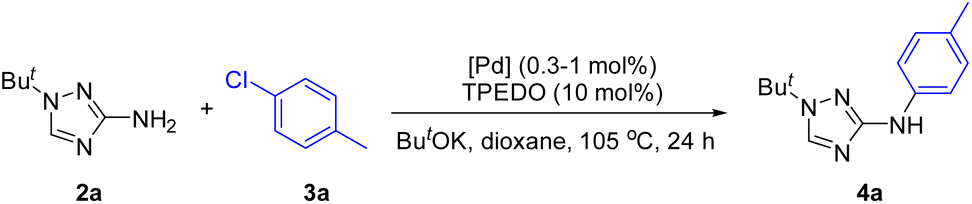
The structures of the Pd/NHC complexes (1a–i) used in the present study are shown in Fig. 2. For comparison, Pd/phosphine catalytic systems consisting of palladium acetate and phosphine ligands presented in Fig. 2 were also evaluated. Arylation of 3-amino-1-tert-butyl-1,2,4-triazole 2a with 4-chlorotoluene 3a in 1,4-dioxane at 105 °C (ButOK as base) was used as a model reaction for the preliminary evaluation of catalytic activities (Table 1 and Tables S1–S4†).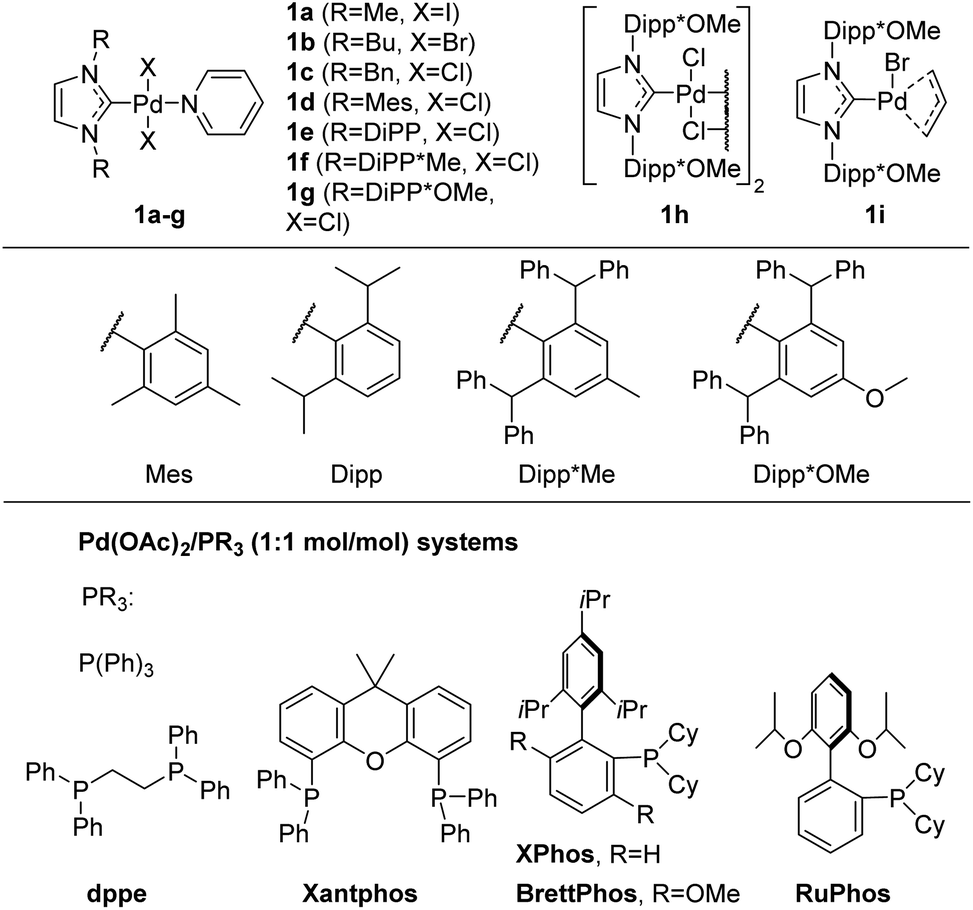 | ||
| Fig. 2 Pd/NHC complexes and phosphine ligands used in this study. | ||
|
|
|||
|---|---|---|---|
| Entry | [Pd] precatalyst (Pd loading, mol%) | Activator | Yield of 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b, % b, % |
| a Reaction conditions: 2a (0.2 mmol), tolyl chloride 3a (0.2 mmol), ButOK (0.5 mmol), palladium precatalyst (0.6–2 μmol, 0.3–1 mol%), TPEDO (0.02 mmol, 10 mol%) if required, dioxane (1 mL), 105 °C, 24 h. b The yields of 4a were determined by GC-MS. | |||
| 1 | 1d (1) | No | 6 |
| 2 | 1e (1) | No | 19 |
| 3 | 1f (1) | No | 68 |
| 4 | 1g (1) | No | 96 |
| 5 | 1h (1) | No | 92 |
| 6 | 1i (1) | No | 99 |
| 7 | 1g (0.5) | No | 94 |
| 8 | 1h (0.5) | No | 89 |
| 9 | 1i (0.5) | No | 99 |
| 10 | 1g (0.3) | No | 75 |
| 11 | 1h (0.3) | No | 85 |
| 12 | 1i (0.3) | No | 95 |
| 13 | 1g (0.3) | TPEDO | 99 |
| 14 | 1h (0.3) | TPEDO | 99 |
| 15 | 1i (0.3) | TPEDO | 89 |
| 16 | 1g (0.2) | TPEDO | 87 |
| 17 | Pd(OAc)2/PPh3 (1) | No | 6 |
| 18 | Pd(OAc)2/dppe (1) | No | 22 |
| 19 | Pd(OAc)2/XantPhos (1) | No | 36 |
| 20 | Pd(OAc)2/XPhos (1) | No | 93 |
| 21 | Pd(OAc)2/XPhos (0.5) | No | 66 |
| 22 | Pd(OAc)2/BrettPhos (1) | No | 49 |
| 23 | Pd(OAc)2/RuPhos (1) | No | 37 |
Initially, the efficiency of various precatalysts was studied at 1 mol% Pd loading. Complexes 1a–c were inactive in the studied conditions (Table S1,† entries 1–3). In addition, fast formation of Pd-black was observed in the reaction mixtures containing precatalysts 1a–c. Apparently, complexes 1a–c suffer facile decomposition via O-NHC,52,67 N-NHC,68 H-NHC and C-NHC coupling reactions.69–72 More stable complexes 1d and 1e with bulkier NHC ligands provided 6% and 19% yields of 4a, respectively (Table 1, entries 1 and 2). The yields of 4a with bromo- and iodoarenes were slightly higher under catalysis with precatalysts 1d and 1e; however, the formation of a side N,N-diarylation product 4a′ was observed (Table S1,† entries 4 and 5, compare results for X = Cl, Br). Complexes 1f–i with the bulkiest NHC ligands, such as Dipp*Me and Dipp*OMe, furnished 68–99% yields of 4a at 1 mol% Pd loading (Table 1, entries 3–6). Close results were obtained when 4-bromo- and 4-iodotoluene were used as arylating agents with precatalysts 1g–i (Table S1†). Obviously, an increase in NHC steric bulkiness enhances the selectivity of arylation with more active aryl bromides and iodides. Remarkably, all the studied Pd/phosphine systems (Fig. 2), except Pd(OAc)2/XPhos, were significantly less efficient (Table 1, entries 17–23).
Encouraged by the results obtained with Pd/NHC complexes 1g–i, we further optimized the reaction conditions to reduce Pd loading. Reducing the Pd loading to 0.5 mol% resulted in a slight decrease in the yield of 4a with complexes 1g and 1h (Table 1, entries 7 and 8), whereas the yield provided by complex 1i remained the same (entry 9). Remarkably, Pd(OAc)2/XPhos furnished a significantly decreased yield of 4a (66%) at 0.5 mol% Pd loading (Table 1, entry 21). Further decreasing Pd loadings to 0.3 mol% (Table 1) resulted in a more appreciable decrease in the 4a yield in the case of precatalysts 1g (75%, Table 1, entry 10) and 1h (85%, Table 1, entry 11) and a smaller decrease in the yield in the case of precatalyst 1i (95%, entry 12). It should be noted that complex 1i contains a sacrificial allylic ligand that serves as internal Pd(II) to Pd(0) reductant in basic media and most likely provides more efficient precatalyst activation.73–75 Apparently, the rate of Pd(II) to Pd(0) reduction has an appreciable effect on the efficiency of Pd/NHC catalytic systems under the studied conditions. Therefore, we explored the possibility of applying external Pd(II) to Pd(0) reductants to enhance the efficiency of Pd/NHC catalytic systems.
A number of potential Pd(II) to Pd(0) reductants76–80 were studied in the conditions of the model reaction between 2a and 3a (Table S2†). Among them, TPEDO (1,1,2,2-tetraphenylethane-1,2-diol) was found to be the most efficient activator (Table S2,† entry 10). The use of TPEDO reduced the loading of complexes 1g and 1h up to 0.3 mol% with the retention of 99% yield of 4a (Table 1, entries 13 and 14). It should be noted that TPEDO and analogous diols were used previously for the reduction of Ni(II) to Ni(0)78,81 complexes and as a source of hydrogen for the hydrogenation of alkenes in the presence of Pd catalysts.82 To the best of our knowledge, the use of TPEDO for Pd/NHC precatalyst activation has not been reported previously.
Further lowering the Pd loading to 0.2 and 0.1 mol% caused a decrease in the 4a yield in the case of all precatalysts even in the presence of TPEDO, which was less intensive than in the absence of the activator (Table 1, entry 16; Table S3†). Kinetic experiments also confirmed the significant promoting effect of TPEDO (Fig. 3 and Fig. S2–S5†). It is interesting to note that the use of TPEDO was inefficient in the case of precatalyst 1i, even though a slight decrease in 4a yield was observed (Table 1, compare entries 15 and 16; Fig. S5†).
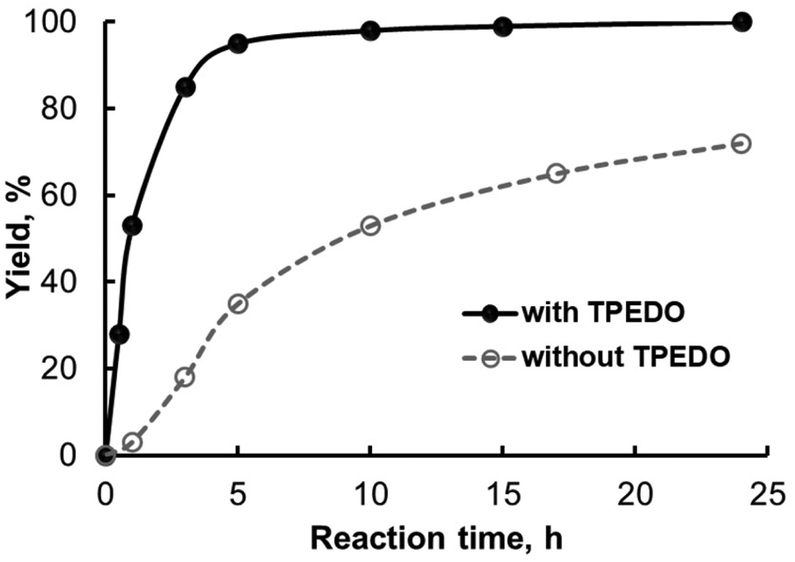 | ||
| Fig. 3 Kinetic curves for the formation of product 4a in the reaction between 2a and 3a. Reaction conditions: 2a (0.2 mmol), 3a (0.2 mmol), ButOK (0.5 mmol), precatalyst 1g (0.6 μmol, 0.3 mol%), TPEDO (0.02 mmol, 10 mol%) if required, dioxane (1 mL), 105 °C. | ||
Variation of the reaction conditions (base, solvent, TPEDO loading) was further studied for more easily available precatalyst 1g (Table S4†). The conditions of the experiment of entry 13 in Table 1 were accepted as optimal.
With the optimized reaction conditions in hand, we evaluated the efficiency of the catalytic system in the arylation of 1-substituted 3-amino-1,2,4-triazoles 2a–g by (hetero)aryl chlorides and bromides (Scheme 2 and Fig. S1†).
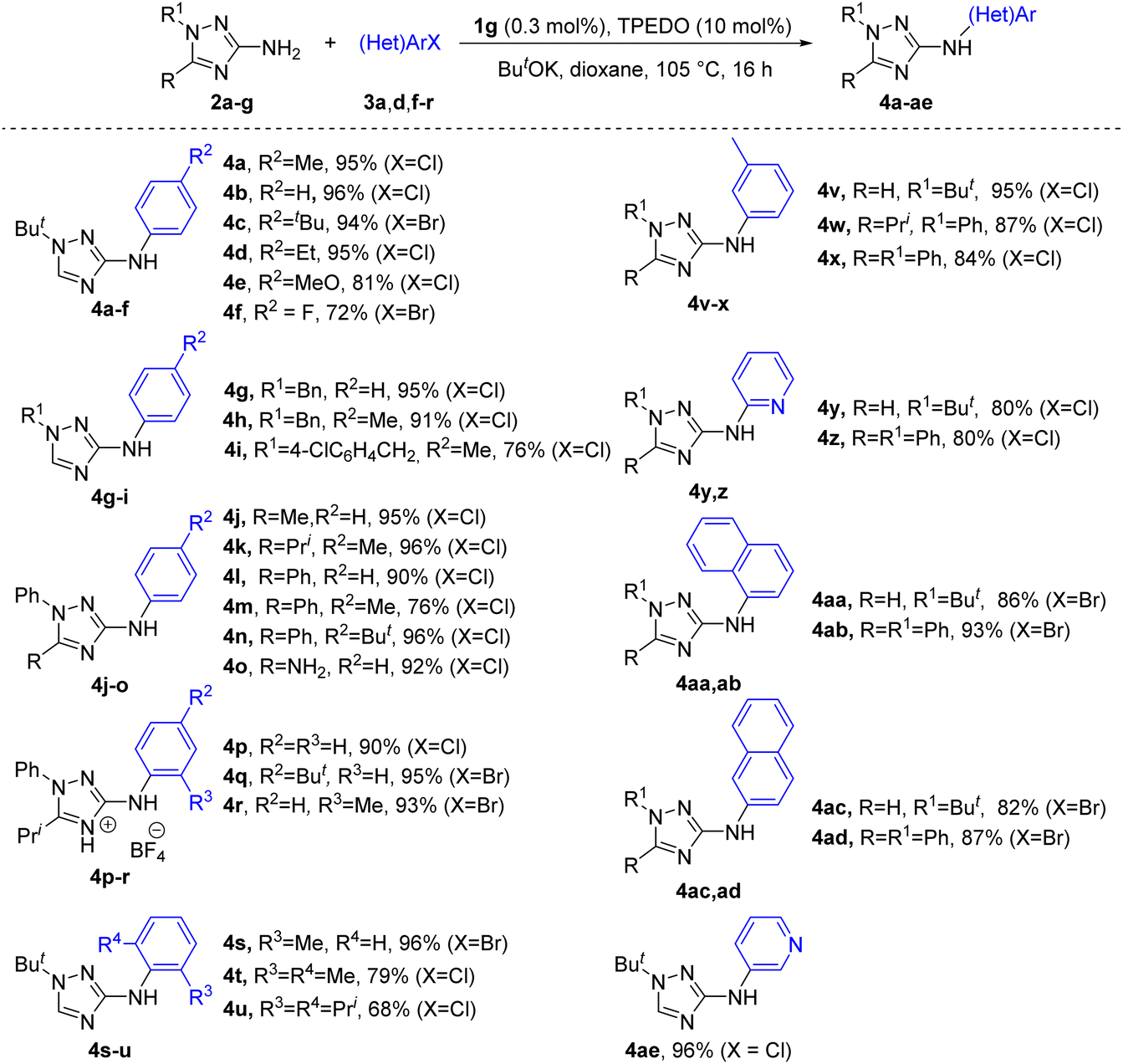 | ||
| Scheme 2 Arylation of 1-substitited 3-amino-1,2,4-triazoles 2a–g. | ||
Diverse 1-substituted 3-(hetero)arylamino-1,2,4-triazoles 4a–ae were obtained in good to excellent yields via the (hetero)arylation of 1-substituted 3-amino-1,2,4-triazoles 2a–g under the optimized conditions (Scheme 2). Even sterically hindered deactivated aryl chlorides provided good yields of products 4t, u. Remarkably, arylation of 3,5-diamino-1-phenyl-1,2,4-triazole 2g (R = NH2, R1 = Ph) afforded monoarylation product 4o owing to the higher nucleophilicity of the 3-NH2 group.61 Compounds 4p–r were isolated as tetrafluoroborate salts due to the high solubility of free bases in organic solvents and difficulties with their purification.
However, attempts at the arylation of 3(5)-amino-1,2,4-triazoles 5b–k with unsubstituted N-atoms of the triazole ring, as well as 1-substituted 5-amino-1,2,4-triazoles 6a–d and 4-substituted 3-amino-1,2,4-triazoles 7a–c (Scheme 3) using the same reaction conditions as in Scheme 2, resulted in low yields of arylated products (∼10–40%). The lowered reactivity of triazoles 5b–k can presumably be explained by the capture of Pd active species by triazole anions formed via deprotonation of the endocyclic NH in basic conditions into less active complexes. This inhibiting effect of acidic endocyclic NH-groups in various azoles is a known phenomenon in metal-catalyzed reactions (see also discussion in further section).8,13 In addition, deprotonation of endocyclic nitrogen should significantly decrease the acidity of the NH2 group and therefore hinder the formation of amide-type intermediates required for further reductive elimination. The decreased reactivity of triazoles 6a–d and 7a–c can be explained by the lowered nucleophilicity of the NH2 group in these compounds, as follows from DFT calculations (see below). We found that deactivated substrates 5, 6 and 7 can be successfully arylated at enhanced to 1 mol% Pd loading (Scheme 3). Remarkably, no formation of endocyclic nitrogen arylation products from substrates 5b–k was detected by GC-MS and NMR analyses of the reaction mixtures.
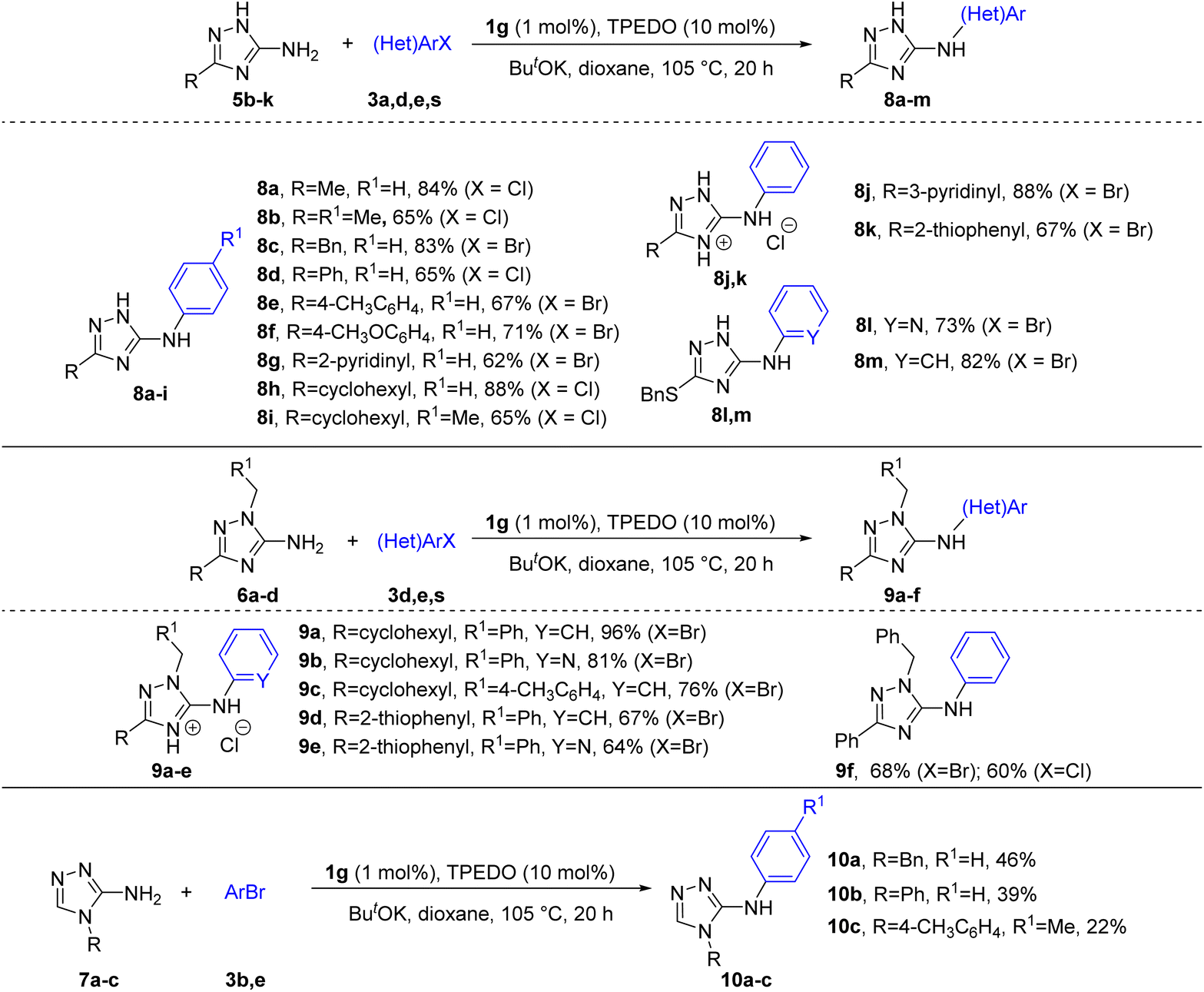 | ||
| Scheme 3 Arylation of C-amino-1,2,4-triazoles 5b–k, 6a–d, 7a–c. | ||
The developed catalytic system is also applicable for the selective N-arylation of other nitrogen heteroaryl amines. Compounds 11a–g were successfully arylated to give the desired products in good to excellent yields (Scheme 4).
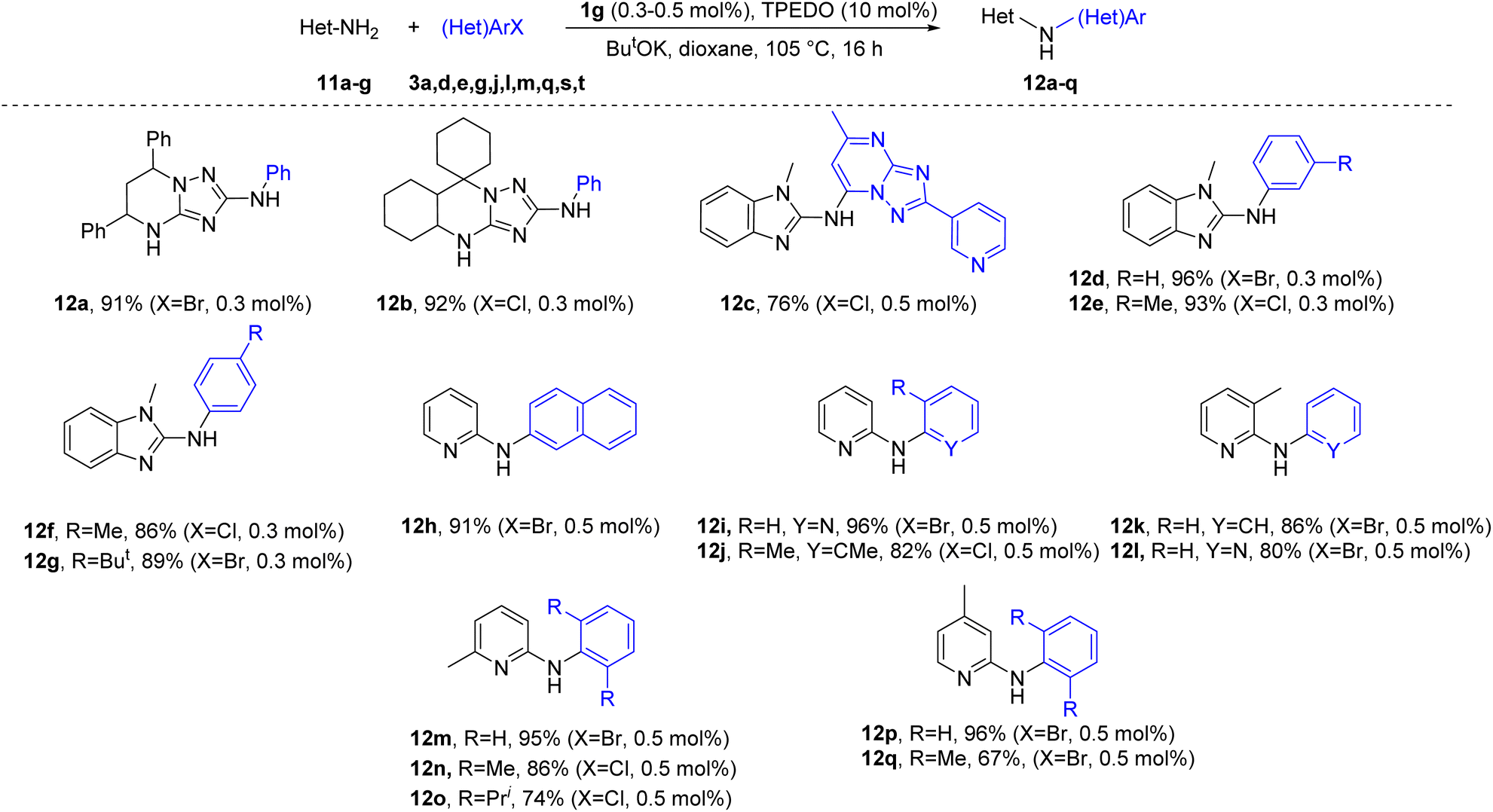 | ||
| Scheme 4 Arylation of aminoheterocycles 11a–g. | ||
The structures of the arylation products 4a–ae, 8a–m, 9a–f, 10a–c, and 12a–q were confirmed by 1H and 13C NMR spectra, including 2D experiments for some compounds and HRMS. Single crystal X-ray analyses were performed for compounds 4h, 4u and 9f (see ESI for details†).
2.2 Theoretical study of the arylation of C-amino-1,2,4-triazoles
To obtain an insight into the reaction mechanism, we performed a theoretical investigation of the arylation reaction using DFT calculations at the PBE1PBE/def2svp level of theory. Complex (NHC)PdPy (NHC = IPr, Py = pyridine) was selected as a model catalyst. The potassium tert-butoxide dimer was selected as a model base since metal alcoholates in low-polarity solvents are prone to the formation of dimers and oligomers.83The free energy profile of a model reaction between aminotriazole 2a and chlorobenzene is presented in Fig. 4 (detailed consideration of the reaction routes from starting compounds to TS-IIA and TS-IIB is provided in Fig. S7 and S8†). Oxidative addition (OA) of PhCl to the starting complex is preceded by pyridine displacement with chlorobenzene molecules to form intermediate IA, which then undergoes oxidative addition via transition state TS-IIA to form oxidative addition intermediate II, which can then capture a pyridine molecule to form more stable intermediate III. The total energy barrier of the oxidative addition process ΔG‡OA is 21.1 kcal mol−1. It should be noted that OA can also proceed through an alternative pathway without preliminary displacement of a pyridine ligand via intermediates IB and TS-IIB (Fig. 4 and Fig. S7, S8†). However, the alternative pathway is characterized by a significantly higher energy barrier of 27.7 kcal mol−1 and seems less probable. Remarkably, it was reported that oxidative addition of PhI to a similar complex (IPr)Pd(DMF) also required pushing out the DMF coligand from the coordination sphere of palladium upon oxidative addition.84
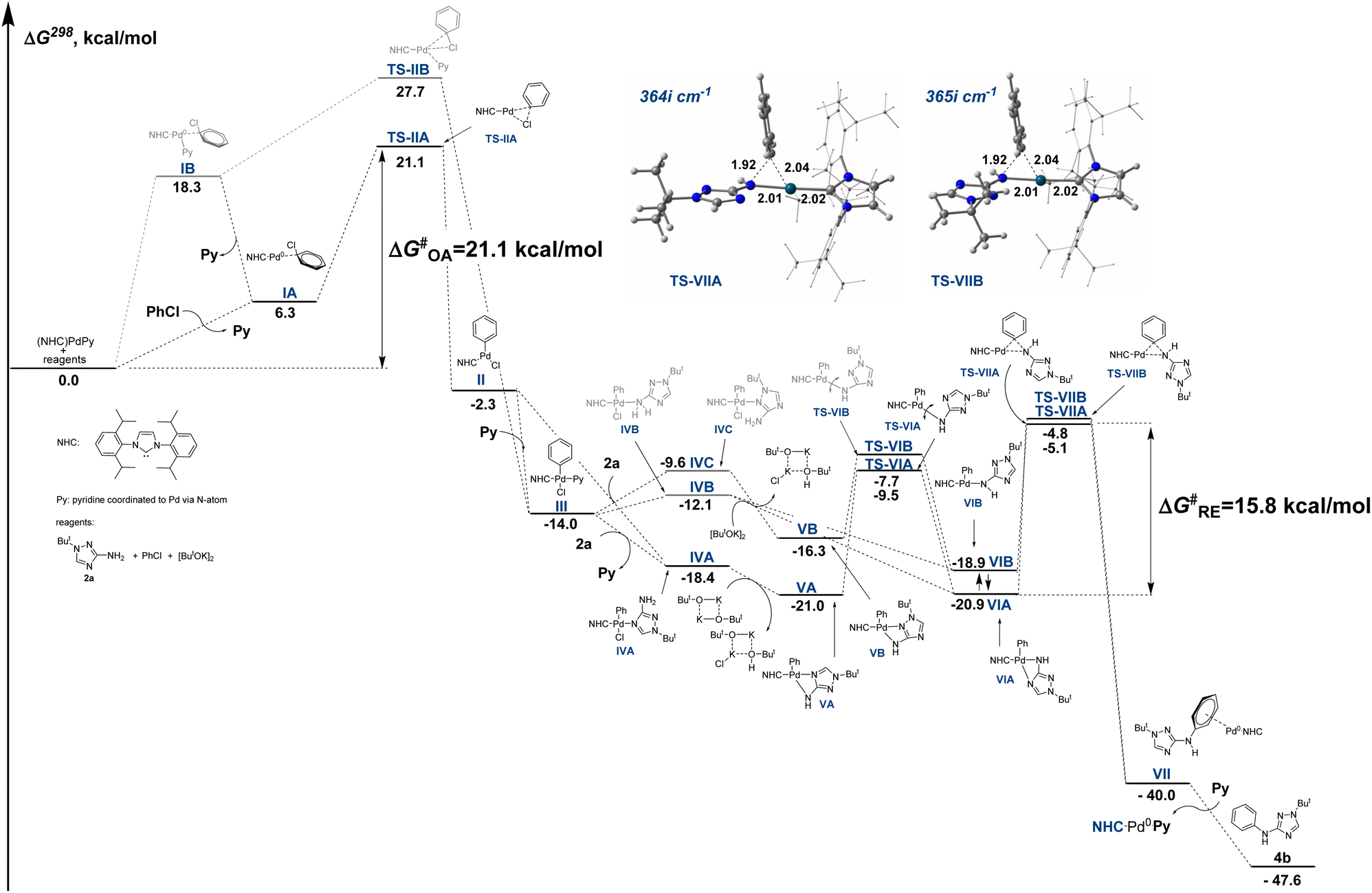 | ||
| Fig. 4 Relative Gibbs free energies of intermediates and transition states of the C–N coupling reaction between aminotriazole 2a and chlorobenzene computed at the PBE1PBE/def2svp level. For details of the reaction routes from starting compounds to TS-IIA and TS-IIB, see Fig. S7 and S8.† | ||
Then, the catalytic process should involve the coordination of the 2a molecule and subsequent deprotonation and reductive elimination steps. However, aminotriazoles are polydentate molecules that can form coordination compounds due to binding with metals by various N atoms, including the NH2 group as well as different N atoms of the triazole ring.21,22 Molecule 2a can coordinate to intermediate II or to intermediate III (with the displacement of the pyridine coligand) to give intermediates IVA, IVB and IVC (Fig. 4 and Fig. S9†). The intermediate IVA is 6.3 and 8.8 kcal mol−1 more stable than intermediates IVB and IVC, respectively (Fig. S6†). In addition, the electrostatic potential of the 2a molecule is localized predominantly on the N4 atom of the triazole ring rather than on the NH2 group or N2 (Fig. S6†), so the formation of the thermodynamically more stable IVA should proceed faster than the formation of intermediates IVB and IVC.
Deprotonation of IVA leads to an intermediate VA, which then rearranges viaTS-VIA into a key intermediate VIA. Deprotonation of the intermediate IVB leads directly to the key intermediate VIA or VIB, whereas deprotonation of IVC leads to an intermediate VB, which then rearranges into an alternative key intermediate VIB. The intermediate VIA is 2.0 kcal mol−1 more stable than VIB. Both deprotonated intermediates VIA and VIB are amenable for the reductive elimination (RE) step to give intermediate VII. The RE from VIA proceeds through a transition state TS-VIIA and is accompanied by a cleavage of a bond between Pd and endocyclic N atom, whereas the RE from VIB proceeds through a transition state TS-VIIB. These transition states differ in conformation of the triazole moiety (Fig. 4). The pathway from VIA to VII through TS-VIIA is only 0.3 kcal mol−1 energetically more profitable than the alternative pathway from VIB through TS-VIIB. Intermediate VII then undergoes displacement of the Pd-coordinated arylated aminotriazole molecule by pyridine and transforms into the starting complex (NHC)PdPy and reaction product 4b.
It should be noted that aminotriazole is used in a large molar excess relative to the starting complex (NHC)PdPy; therefore, replacement of the pyridine coligand by the 2a molecule can lead to new starting active complexes (NHC)PdL, in which L is a Pd-coordinated molecule of aminotriazole. Therefore, the alternative reaction pathway through the more stable complex (NHC)PdL should not be excluded. We studied the alternative reaction pathway (Fig. S9†) starting from the complex (NHC)Pd(L) in which aminotriazole (L) is coordinated to the Pd atom via the N4 atom of the triazole ring (the most stable coordination isomer) and found that this reaction pathway is energetically very similar to the pathway starting from the complex (NHC)PdPy. The main difference is that the displacement of the aminotriazole ligand by the chlorobenzene molecule to give the intermediate IA slightly enhances the barrier of the oxidative addition by 1.8 kcal mol−1 (ΔG‡OA = 22.9 kcal mol−1, Fig. S9†).
According to the results of the DFT calculations (Fig. 4), the oxidative addition of chlorobenzene to the starting complex (NHC)PdPy (ΔG‡OA = 21.1 kcal mol−1) should be the rate-determining step in the arylation of compound 2a. Calculations taking into account solvation effect of 1,4-dioxane using IEFPCM approximation afforded similar results (ΔG‡OA = 20.4 kcal mol−1, ΔG‡RE = 16.7 kcal mol−1, Fig. S10†). It should be noted that the energy barriers of the OA stage may be lower with precatalysts 1f–i containing bulkier IPr* and IPr*OMe ligands because bulkier ligands can facilitate a pyridine or L coligand pushing out and stabilize the OA transition state via attractive noncovalent interactions with aryl chloride.85 Therefore, the effect of aryl halide on the yield of arylation product 4a may be less pronounced under catalysis with precatalysts 1f–i.
On the other hand, the experimentally observed significant effects of the structures of the studied C-amino-1,2,4-triazoles (Schemes 2 and 3), especially the position of a substituent on a nitrogen atom of the triazole ring, on the yields of arylation products suggest that the rate-determining step may depend on the aminotriazole substrate. Therefore, we computed corresponding energy profiles for the arylation of isomeric 3(5)-amino-1(4)-methyl-1,2,4-triazoles 2h, 6e, 7d (Fig. 5 and Fig. S13, S15, S17†). The energy barrier of the OA stage is the same (ΔG‡OA = 21.1 kcal mol−1) for all reactions, as presented in Fig. 5, because the OA stage does not involve the participation of the aminotriazole substrates. However, the computed energy barriers of the reductive elimination stage (ΔG‡RE) differ significantly and amount to 15.9 kcal mol−1, 21.2 kcal mol−1 and 23.7 kcal mol−1 for the arylation of isomeric substrates 2h, 6e and 7d, respectively. In other words, reductive elimination becomes the rate-determining stage in the arylation of compound 7d. In the case of the arylation of compound 6e, the computed energy barriers of the OA and RE stage are very close (21.1 and 21.2 kcal mol−1, respectively). It can be presumed, that the rate determining stage in the arylation of 1-substituted 5-amino-1,2,4-triazoles 6 can depend on the variation of substrate structure and reaction conditions.
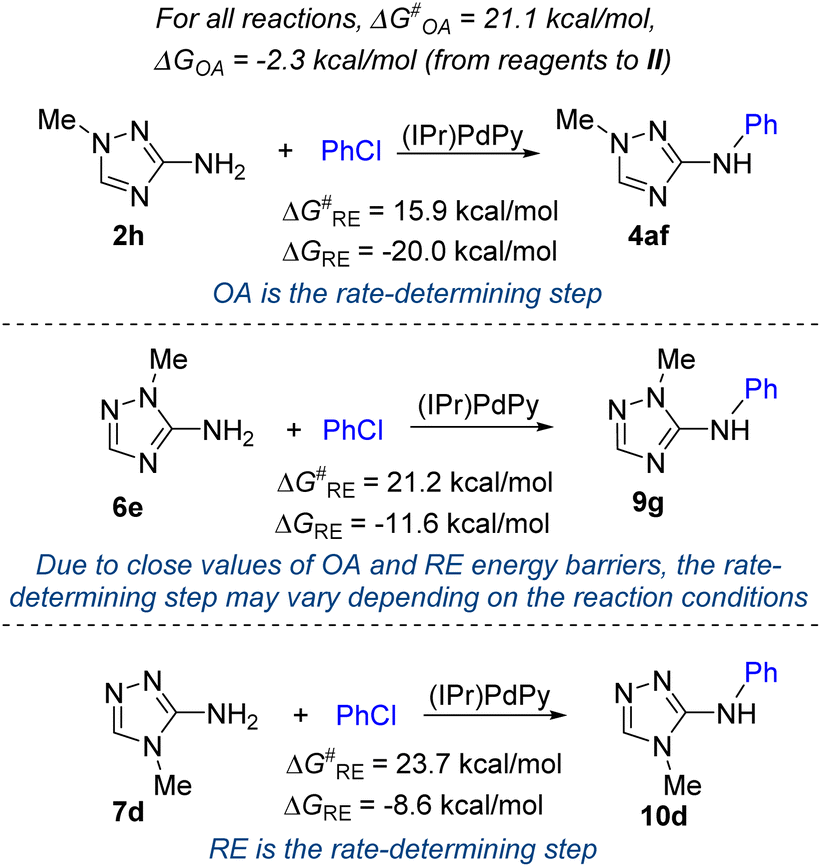 | ||
| Fig. 5 Gibbs free energy barriers (ΔG‡RE) and energies (ΔGRE) of the reductive elimination stage in the arylation of isomeric 1-methyl- and 4-methyl-3(5)-amino-1,2,4-triazoles with chlorobenzene computed at the PBE1PBE/def2svp level. | ||
The computed order of the RE energy barriers (Fig. 5 and Fig. S13, S15, S17, Table S5†) correlates well with the experimentally found reactivity of C-amino-1,2,4-triazoles: 1-substitud 3-amino-1,2,4-triazoles 2 are the most reactive substrates, which require only 0.3 mol% Pd loading (Scheme 2), whereas 1-substituted 5-amino-1,2,4-triazoles 6 and, especially, 4-substituted 3-amino-1,2,4-triazoles 7 are less reactive and require 1 mol% Pd loading (Scheme 3). Remarkably, the observed differences in the reactivity of 1-substituted 3-amino-1,2,4-triazoles 2 and 5-amino-1,2,4-triazoles 6 agree with the differences in nucleophilicity of the NH2-group in isomeric 1-substituted C-amino-1,2,4-triazoles: the 3-NH2 group is significantly more nucleophilic than the 5-NH2 group.61
Arylation of aminotriazoles 5 unsubstituted at the endocyclic nitrogen atoms deserves special discussion. These substrates exist and can react in several tautomeric forms possessing different reactivities (Fig. S19†).86–91 According to previous theoretical and experimental studies, a less reactive 5-amino-1H-tautomeric form can dominate in polar media.86,89,91 In addition, these substrates are quite acidic and can eliminate the proton from the endocyclic NH in strong alkaline media.92,93 The formed triazole N-anions can coordinate to Pd species to give quite stable complexes that can represent the catalyst resting state (for example, XXXIIA, Fig. S20†). Obviously, for these reasons, aminotriazoles 5 unsubstituted at the endocyclic nitrogen atoms demonstrate lower reactivity than 1-substituted 3-amino-1,2,4-triazoles 2 (Schemes 2 and 3). Another question is why these substrates do not form products of arylation at the triazole ring N atoms? The answer to this question is obvious from the consideration of the RE stage (Fig. 6 and Fig. S21†). The lowest energy barrier for the arylation of the NH2 group to give product 8n amounts to 16.0 kcal mol−1, whereas energy barriers for the formation of N1, N2 and N4 arylation products 2i, 6f and 7b amount to 21.2 kcal mol−1, 21.9 kcal mol−1 and 24.0 kcal mol−1, respectively (Fig. 6 and Fig. S21†). Apparently, the kinetics of the RE step determine the selectivity of the arylation of substrates 5.
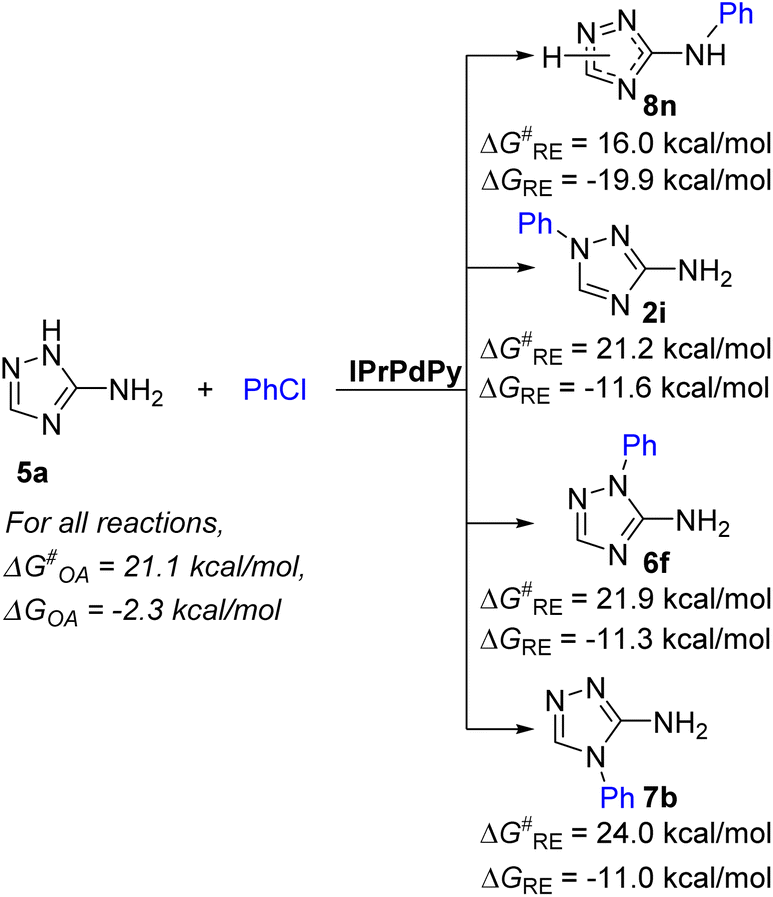 | ||
| Fig. 6 Gibbs free energy barriers (ΔG‡RE) and reaction energies (ΔGRE) of the reductive elimination stages leading to isomeric products in the arylation of 3-amino-1,2,4-triazole 5a computed at the PBE1PBE/def2svp level. | ||
Although theoretical calculations provided some useful insights into the reaction mechanism, one should take into account the limitations in the model applied and limitations in the accuracy of calculations. The present results should be taken as preliminary, and more investigations are required for a better description of reaction pathways. In particular, the discussion on the rate-determining stages and accurate barriers may be further continued.
3. Conclusion
In summary, a new efficient approach for the selective arylation of NH2 groups in diverse C-amino-1,2,4-triazoles and other coordinating polynitrogen heteroaryl amines by (hetero)aryl chlorides and bromides has been developed. The approach relies on the use of a Pd/NHC precatalyst containing a bulky IPr*OMe NHC ligand and 1,1,2,2-tetraphenylethane-1,2-diol (TPEDO) as an in situ activator. The use of TPEDO significantly enhanced the yields of arylation products in comparison with experiments without the use of the Pd(II) reductant. The developed approach provides excellent selectivity for NH2 group arylation even in substrates containing unprotected endocyclic NH moieties.The reactivity of C-amino-1,2,4-triazoles in the arylation decreases in the order 1-substituted 3-amino-1,2,4-triazoles > 3(5)-amino-1,2,4-triazoles unsubstituted at the endocyclic N atoms ∼1-substituted 5-amino-1,2,4-triazoles > 4-substituted 3-amino-1,2,4-triazoles. A theoretical study of the reaction mechanism with the use of DFT calculations revealed a significant effect of the C-amino-1,2,4-triazole structure on the energy barrier of the reductive elimination (RE) stage. The rate of the arylation of more nucleophilic 1-substituted 3-amino-1,2,4-triazoles is most likely limited by the oxidative addition of aryl chloride. In the arylation of less nucleophilic 1-substituted 5-amino-1,2,4-triazoles, the rate determining stage may vary due to closeness of the oxidative addition and reductive elimination energy barriers. However, in the arylation of 4-substituted 3-amino-1,2,4-triazoles, the reductive elimination stage is anticipated to be the rate determining stage. The arylation of 3(5)-amino-1,2,4-triazoles unsubstituted at the endocyclic N atoms, due to their proneness to prototropy, can proceed via a few reaction channels. The decreased reactivity of these substrates may be explained by their high coordinating ability and the formation of coordination complexes with Pd species that are too stable. The observed high selectivity of the arylation of the N atom of the NH2 group is determined by the kinetics of the reductive elimination step, namely, by the significantly lower energy barrier of the reductive elimination of the arylamino-product relative to the energy barriers of the reductive elimination of triazole ring N-arylated products.
4. Experimental section
4.1 General information
1H and 13C{1H} NMR spectra were recorded using a Bruker Avance Neo NMR spectrometer at 300 MHz for 1H and 75 MHz for 13C spectra. 1H and 13C chemical shifts are presented in ppm relative to the residual peak of the solvent signal (δ 7.26 for trace CHCl3 in CDCl3, δ 2.50 for trace DMSO-d5 in DMSO-d6; δ 77.2 for CDCl3, δ 39.5 for DMSO-d6).High-resolution mass spectra were obtained on a Bruker maXis Q TOF spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with an electrospray ionization (ESI) ion source. The measurements were conducted in positive (+) MS ion mode (HV Capillary: 4500 V; Spray Shield: −500 V) with a scan range of m/z 50–1500. External calibration of the mass spectrometer was performed using a low-concentration Agilent tuning mix solution. Direct syringe injection was applied to the analyzed solutions at a flow rate of 3 μL min−1. N2 was applied as the nebulizer gas (0.4 bar) and dry gas (4.0 L min−1, the dry temperature was 250 °C). All the mass spectra were recorded with a 1 Hz frequency and processed using Bruker Data Analysis 4.0 software.
GC-MS analyses were performed using an Agilent 7890A gas chromatograph equipped with an Agilent 5975C mass-selective detector (EI, 70 eV) and an HP-5MS column (30 m × 0.25 mm × 0.25 μm film) using He as the carrier gas at a flow rate of 1.0 mL min−1.
Melting points were determined in a Thiele apparatus in open capillary tubes and were uncorrected.
Precatalysts 1a,941b,951c,961d,971e,971f,981g,76 and starting compounds 1-alkyl-1H-1,2,4-triazol-3-amines (2a–c, g),88,99–105 1-unsubstituted 1H-1,2,4-triazol-3(5)-amines (5b–k),88,99–105 4-benzyl-4H-1,2,4-triazol-3-amines (7a),106 4-phenyl-4H-1,2,4-triazol-3-amines (7b),107 5,7-diaryl-4,5,6,7-tetrahydro-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (11a, b)26 and 4a′,5′,6′,7′,8′,8a’-hexahydro-4′H-spiro[cyclohexane-1,9′-[1,2,4]triazolo[5,1-b]quinazolin]-2′-amine (11c)108 were synthesized via procedures adapted from the literature. Other chemicals were obtained from commercial sources. Solvents were distilled and dried by standard methods prior to use.
:2) and dried at 60 °C in vacuo. Yield 60 mg (66%), light-yellow solid. 1H NMR (CDCl3, 300 MHz) (as a mixture of dimer and monomer): δ 3.38 and 3.53 (two s, 12H, 4CH3O), 4.60 and 4.64 (two s, 4H, 4CH), 5.47 (s, 2H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) Ph2), 6.19 (s, 2H, CPh2), 6.35 (s, 2H, CPh2), 6.47 (s, 2H, CPh2), 6.49–6.50 (m, 3H, Ar), 6.57–6.61 (m, 7H, Ar), 6.71–6.77 (m, 11H, Ar), 6.87–7.11 (m, 40H, Ar), 7.18–7.32 (m, 19H, Ar), 7.41–7.44 (m, 4H, Ar), 7.48–7.50 (m, 4H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ13C NMR (75 MHz, CDCl3) δ 51.2, 51.4, 55.2, 114.6, 116.4, 124.2, 126.0, 126.2, 126.4, 126.6, 128.1, 128.2, 128.3, 128.8, 129.2, 129.4, 129.6, 129.7, 130.7, 130.9, 142.9, 143.5, 143.8, 144.18, 144.24, 144.5, 144.7, 145.1, 146.0, 159.7. Anal. calcd for C138H112Cl4N4O4Pd2, %: C 73.83, H 5.03, N 2.50. Found, %: C 73.90, H 4.99, N 2.54.
Ph2), 6.19 (s, 2H, CPh2), 6.35 (s, 2H, CPh2), 6.47 (s, 2H, CPh2), 6.49–6.50 (m, 3H, Ar), 6.57–6.61 (m, 7H, Ar), 6.71–6.77 (m, 11H, Ar), 6.87–7.11 (m, 40H, Ar), 7.18–7.32 (m, 19H, Ar), 7.41–7.44 (m, 4H, Ar), 7.48–7.50 (m, 4H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ13C NMR (75 MHz, CDCl3) δ 51.2, 51.4, 55.2, 114.6, 116.4, 124.2, 126.0, 126.2, 126.4, 126.6, 128.1, 128.2, 128.3, 128.8, 129.2, 129.4, 129.6, 129.7, 130.7, 130.9, 142.9, 143.5, 143.8, 144.18, 144.24, 144.5, 144.7, 145.1, 146.0, 159.7. Anal. calcd for C138H112Cl4N4O4Pd2, %: C 73.83, H 5.03, N 2.50. Found, %: C 73.90, H 4.99, N 2.54.
4.2 General procedure for the synthesis of compounds 2d–f (see Scheme S2†)
A solution of acyl chloride or acetic anhydride (36 mmol) in dry MeCN (10 mL) was added dropwise while stirring to a solution of S-methylisothiuronium sulfate [(NH2)2CSMe]2SO4 (2.5 g, 9 mmol) and Et3N (4.55 g, 45 mmol) in dry MeCN (20 mL). Then, the resulting mixture was heated under reflux with stirring for 2 h and cooled to room temperature. Then, phenyl hydrazine (1.95 g, 18 mmol) was added to the reaction mixture, and the resulting solution was additionally heated at 80 °C with stirring for 2 h and then evaporated to dryness in vacuo. The oily residue was suspended in a 5% aqueous acetic acid solution (20 mL) and extracted with CHCl3 (3 × 10 mL). The extract was washed with distilled water (2 × 10 mL), dried over anhydrous Na2SO4 and evaporated to dryness in vacuo. The residue obtained was dissolved in a mixture of concentrated HCl (1 mL) and EtOH (20 mL). The solution was heated under reflux within 5 h, evaporated to a small volume (∼5 mL), neutralized with a saturated aqueous solution of NaHCO3 (to pH 8–9) and extracted with CHCl3 (3 × 10 mL). The extract was dried over anhydrous Na2SO4, evaporated to a small volume and subjected to column chromatography on silica gel using CHCl3 as the eluent. Product 2 was finally purified by crystallization from an EtOH–Et2O mixture (1:5).
4.3 General procedure for the synthesis of compounds 6a–d (see Scheme S3†)
A mixture of the appropriate triazole 5c, e, h (3.0 mmol) and MeONa (168 mg, 3.1 mmol) in DMSO (5 mL) was stirred at room temperature for 30 min. Then, the appropriate alkyl halide (3.0 mmol) was added dropwise to the stirred reaction mixture. The mixture was additionally stirred at room temperature within 16 h, diluted with water (20 mL) and extracted with CH2Cl2 (3 × 10 mL). The extract was washed with water (3 × 10 mL), dried over anhydrous Na2SO4 and evaporated in vacuo to a small volume (∼0.5 mL). The residue was diluted with Et2O (3 mL) and cooled to 0–5 °C. The precipitate formed was collected by filtration, washed with a small amount of Et2O, recrystallized from the appropriate solvent and dried in vacuo at 50 °C.4.4 Procedure for the synthesis of 4-(4-methylphenyl)-4H-1,2,4-triazol-3-amine (7c)
A solution of 4-methylaniline (338 mg, 3.15 mmol) in EtOH (5 mL) was added with stirring to the S-methyl-isothiosemicarbazide hydroiodide [NH2NH(NH2)CSMe]I (700 mg, 3 mmol). Then, the resulting mixture was heated under reflux with stirring for 6 h and evaporated to dryness in vacuo. The oily residue was washed with Et2O (3 × 5 mL) and evaporated to dryness in vacuo. The resulting residue was dissolved in concentrated HCOOH (5 mL). The solution was boiled for 2 h, slowly evaporated for 1 h at 140 °C, neutralized with a saturated aqueous NaHCO3 solution (to pH 8–9), and extracted with CHCl3 (5 × 10 ml). The extract was dried over anhydrous Na2SO4, evaporated to a small volume, and precipitated with Et2O. The precipitate formed was collected by filtration, washed with a small amount of Et2O, recrystallized from EtOH–Et2O (1:5) and dried in vacuo at 50 °C.
4.5 Study of N-arylation of compound 2a
A 4 mL screw-capped glass tube equipped with a magnetic stirring bar was charged with the corresponding base (0.5 mmol), aryl halide (0.2 mmol), 1-tert-butyl-1H-1,2,4-triazol-3-amine (2a) (28 mg, 0.2 mmol), appropriate Pd precatalyst (0.2–2 μmol, 0.1–1 mol%), and, if needed, appropriate NHC proligand and 1,1,2,2-tetraphenylethane-1,2-diol (TPEDO, 11 mg, 0.03 mmol, Table 1 and Table S1†). Then, the appropriate solvent (1 mL, Table 1 and Tables S1, S2†) was added to the resulting mixture, and the tube was purged with argon, sealed with a screw cap fitted with a rubber septum, placed in a thermostated oil bath and heated with vigorous stirring at the corresponding temperature for 24 h. Then, the glass tubes were cooled to room temperature, and a solution of AcOH (30 mg, 0.5 mmol) and naphthalene (12.8 mg, 0.1 mmol, an internal standard in 1 mL of acetonitrile) was added by syringe through the septum. An aliquot (5–10 μl) of the reaction mixture was dissolved in 1 ml of CH3CN and analyzed by GC-MS (Fig. S2–S5,†Table 1 and Tables S1–S4†).4.6 General procedure for the synthesis of compounds 4a–ae, 8a–m, 9a–f, 10a–c, and 12a–q
A 7 mL screw-capped glass tube equipped with a magnetic stirring bar was charged with ButOK (224 mg, 2.0 mmol), aryl halide (0.8 mmol), corresponding aminotriazole (2a–g, 5b–k, 6a–d, 7a–c) or compound 11a–g (0.8 mmol), 1,1,2,2-tetraphenylethane-1,2-diol (TPEDO, 30 mg, 0.08 mmol, 10 mol%) and Pd precatalyst 1g (2.9 mg, 2.4 μmol, 0.3 mol% for the arylation compounds 2a–g, 11a–c (see Schemes 2 and 4), or 4–8 μmol, 0.5–1 mol% for the arylation of compounds 5–7, 11d–g (see Schemes 3 and 4). Then, 1,4-dioxane (4 mL) was added to the resulting mixture, and the tube was purged with argon, sealed with a screw cap fitted with a septum, placed in a thermostated oil bath and stirred at 105 °C for 20 h. After cooling to room temperature, the tube was opened, and the reaction mixture was neutralized with AcOH (∼0.2 g) and evaporated to dryness in vacuo.To isolate compounds 4a–o, s–ae, 9f and 12h–q, the residue obtained after evaporation of the reaction mixture was dissolved in CH2Cl2 (10 mL). The solution was washed with water (3 × 10 mL), dried over anhydrous Na2SO4, evaporated to a small volume (0.5–1.0 mL) and subjected to column chromatography on silica gel using CH2Cl2 as the eluent. Products 4a–o, s–ae, 9f and 12h–q were finally purified by recrystallization from the appropriate solvent and dried in vacuo at 50 °C overnight.
To isolate compounds 8a–i, l, m, 10a–c, and 12a–g the residue obtained after evaporation of the reaction mixture was dissolved in EtOH (7 mL). The solution was passed through a short layer of Celite, evaporated to a small volume (∼1 mL), diluted with water (∼1 mL) and cooled to 0–5 °C. A precipitate formed was collected by filtration and recrystallized from the appropriate solvent and then dried in vacuo at 50 °C.
To isolate hydrochlorides 9a–e, 8j, k and hydrotetrafluoroborates 4p–r, the residue obtained after evaporation and column chromatography of the reaction mixture was dissolved in hot C2H5OH (0.1 mL) and acidified by the addition of concentrated HCl or 40% aqueous HBF4 solution. Then, the resulting acidic solution was diluted with Et2O (1 mL) and cooled to −10 °C. A precipitate formed was collected by filtration, washed with cold ether, recrystallized from the appropriate solvent and dried in vacuo at 80 °C overnight.
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.60 (s, 9H, 3CH3), 2.29 (s, 3H, CH3), 6.55 (s, 1H, NH), 7.09 (d, J = 8.2 Hz, 2H), 7.35–7.38 (m, 2H, Ar), 7.82 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 20.8, 29.4, 58.0, 116.3, 129.6, 129.7, 138.2, 139.0, 160.6. HRMS (ESI) calcd for C13H19N4 [M + H]+m/z 231.1604, found m/z 231.1608.
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.61 (s, 9H, 3CH3), 6.73 (s, 1H, NH), 6.88–6.93 (m, 1H, Ph), 7.26–7.32 (m, 2H, Ph), 7.46–7.50 (m, 2H, Ph), 7.84 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 29.4, 58.0, 116.1, 120.4, 129.2, 138.2, 141.4, 160.3. HRMS (ESI) calcd for C12H17N4 [M + H]+m/z 217.1448, found m/z 217.1453. The spectral data of compound 4b are consistent with the literature data.56
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.22 (t, J = 7.6 Hz, 3H, CH3), 1.61 (s, 9H, 3CH3), 2.60 (q, J = 7.6 Hz, 2H, CH2), 6.85 (s, 1H, NH), 7.11–7.14 (m, 2H, Ar), 7.39–7.42 (m, 2H, Ar), 7.83 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 15.9, 28.2, 29.3, 57.8, 116.2, 128.3, 136.1, 138.0, 139.2, 160.5. HRMS (ESI) calcd for C14H21N4 [M + H]+m/z 245.1761, found m/z 245.1765.
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.60 (s, 9H, 3CH3), 3.78 (s, 3H, CH3), 6.58 (s, 1H, NH), 6.87 (d, J = 9.0 Hz, 2H, Ar), 7.41 (d, J = 9.0 Hz, 2H, Ar), 7.81 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 29.4, 55.8, 57.9, 114.5, 117.7, 135.2, 138.2, 153.9, 160.8. HRMS (ESI) calcd for C13H19N4O [M + H]+m/z 247.1553, found m/z 247.1558.
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.61 (s, 9H, 3CH3), 6.66 (s, 1H, NH), 6.96–7.02 (m, 2H, Ar), 7.40–7.45 (m, 2H, Ar), 7.83 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 29.4, 58.1, 115.6 (d, 2JCF = 22.4 Hz), 117.4 (d, 3JCF = 7.5 Hz), 137.6 (d, 4JCF = 2.1 Hz), 138.3, 157.3 (d, 1JCF = 238.0 Hz), 160.4. HRMS (ESI) calcd for C12H16FN4 [M + H]+m/z 235.1354, found m/z 235.1353.
:5 mixture). 1H NMR (CDCl3, 300 MHz): δ 5.24 (s, 2H, CH2), 6.77 (s, 1H, NH), 6.89–6.94 (m, 1H, Ar), 7.29–7.47 (m, 9H, Ar), 7.75 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 53.7, 116.4, 120.7, 128.2, 128.7, 129.1, 129.2, 135.0, 141.0, 141.7, 161.0. HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1294.
:1 mixture). 1H NMR (CDCl3, 300 MHz): 2.29 (s, 3H, CH3), 5.23 (s, 2H, CH2), 6.61 (s, 1H, NH), 7.08–7.11 (m, 2H, Ar), 7.29–7.42 (m, 7H, Ar), 7.73 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 20.8, 53.6, 116.6, 128.2, 128.6, 129.1, 129.7, 130.1, 135.0, 138.6, 141.7, 161.3. HRMS (ESI) calcd for C16H17N4 [M + H]+m/z 265.1448, found m/z 265.1452.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 2.29 (s, 3H, CH3), 5.19 (s, 2H, CH2), 6.75 (s, 1H, NH), 7.08–7.11 (m, 2H, Ar), 7.23–7.26 (m, 2H, Ar), 7.32–7.37 (m, 4H, Ar), 7.76 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 20.8, 52.8, 116.6, 129.3, 129.4, 129.7, 130.2, 133.6, 134.6, 138.5, 141.7, 161.4. HRMS (ESI) calcd for C16H16ClN4 [M + H]+m/z 299.1058, found m/z 299.1062.
:2 mixture). 1H NMR (CDCl3, 300 MHz): δ 2.50 (s, 3H, CH3), 6.83 (s, 1H, NH), 6.90–6.94 (m, 1H, Ar), 7.27–7.32 (m, 2H, Ar), 7.38–7.44 (m, 1H, Ar), 7.49–7.52 (m, 6H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 13.5, 116.5, 120.7, 124.4, 128.2, 129.2, 129.5, 137.8, 141.0, 150.9, 159.6. HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1292.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.32 (d, J = 6.8 Hz, 6H, 2CH3), 2.28 (s, 3H, CH3), 3.11 (hept, J = 6.8 Hz, 1H, CH), 6.58 (s, 1H, NH), 7.07–7.09 (m, 2H, Ar), 7.37 (d, J = 8.3 Hz, 2H, Ar), 7.43–7.51 (m, 5H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 20.8, 21.8, 25.9, 116.5, 125.5, 128.6, 129.5, 129.6, 129.8, 137.8, 138.6, 159.8 (signals of two aromatic carbons overlap). HRMS (ESI) calcd for C18H21N4 [M + H]+m/z 293.1761, found m/z 293.1770.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 6.89–6.94 (m, 1H, Ar), 7.17 (s, 1H, NH), 7.24–7.42 (m, 11H, Ar), 7.49–7.52 (m, 4H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 116.5, 120.7, 125.4, 128.0, 128.5, 128.7, 129.0, 129.2, 129.4, 130.2, 138.4, 140.9, 152.4, 160.1. HRMS (ESI) calcd for C20H17N4 [M + H]+m/z 113.1448, found m/z 313.1458.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 2.30 (s, 3H, CH3), 6.72 (s, 1H, NH), 7.08–7.11 (m, 2H, Ar), 7.31–7.44 (m, 10H, Ar), 7.48–7.51 (m, 2H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 20.8, 116.7, 125.4, 128.1, 128.4, 128.7, 129.0, 129.4, 129.7, 130.11, 130.13, 138.4, 138.5, 152.4, 160.2. HRMS (ESI) calcd for C21H19N4 [M + H]+m/z 327.1604, found m/z 327.1618.
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.61 (s, 9H, 3CH3), 2.34 (s, 3H, CH3), 6.59 (s, 1H, NH), 6.71–6.74 (m, 1H, Ar), 7.18 (t, J = 7.8 Hz, 1H, Ar), 7.24 (br. s, 1H, Ar), 7.32–7.35 (m, 1H, Ar), 7.83 (s, 1H, CH). 13C{1H} NMR (CDCl3, 75 MHz): δ 21.8, 29.4, 58.0, 113.3, 116.9, 121.3, 129.0, 138.2, 138.9, 141.4, 160.4. HRMS (ESI) calcd for C13H19N4 [M + H]+m/z 231.1604, found m/z 231.1608.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.32 (d, J = 6.8 Hz, 6H, 2CH3), 2.33 (s, 3H, CH3), 3.11 (sept, J = 6.8 Hz, 1H, CH), 6.60 (s, 1H, NH), 6.72–6.74 (m, 1H, Ar), 7.14–7.20 (m, 1H, Ar), 7.22 (br. s, 1H, Ar), 7.34–7.38 (m, 1H, Ar), 7.42–7.54 (m, 5H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 21.8 (three ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 25.9, 113.5, 117.0, 121.5, 125.5, 128.6, 129.1, 129.5, 137.8, 138.9, 141.0, 159.7, 159.8. HRMS (ESI) calcd for C18H21N4 [M + H]+m/z 293.1761, found m/z 293.1766.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 2.17 (s, 3H, CH3), 6.58–6.62 (m, 1H, Ar), 6.73 (s, 1H, NH), 7.01–7.11 (m, 3H, Ar), 7.17–7.29 (m, 6H, Ar), 7.34–7.35 (m, 2H, Ar), 7.36–7.37 (m, 2H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 21.8, 113.7, 117.2, 121.7, 125.4, 128.1, 128.4, 128.7, 129.0, 129.1, 129.4, 130.1, 138.5, 139.0, 140.9, 152.5, 160.1. HRMS (ESI) calcd for C21H19N4 [M + H]+m/z 327.1604, found m/z 327.1616.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 6.84–6.88 (m, 1H, Ar), 7.32–7.44 (m, 8H, Ar + NH), 7.50–7.54 (m, 2H, Ar), 7.63–7.69 (m, 1H, Ar), 8.13–8.17 (m, 1H, Ar), 8.37–8.39 (m, 2H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 110.5, 116.3, 125.5, 128.0, 128.6, 128.7, 129.1, 129.4, 130.2, 138.3, 138.4, 148.2, 152.8, 153.7, 159.0. HRMS (ESI) calcd for C19H16N5 [M + H]+m/z 314.1400, found m/z 314.1414.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.62 (s, 9H, 3CH3), 7.21–7.25 (m, 1H, Ar), 7.92 (s, 1H, CH), 7.99 (s, 1H, Ar), 8.07–8.15 (m, 2H, Ar), 8.66 (s, 1H, NH). 13C{1H} NMR (CDCl3, 75 MHz): δ 29.4, 58.3, 122.5, 123.8, 138.25, 138.33, 138.6, 141.2, 159.9. HRMS (ESI) calcd for C11H16N5 [M + H]+m/z 218.1400, found m/z 218.1407.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 2.27 (s, 3H, CH3), 6.73–6.78 (m, 1H, Ar), 7.15–7.21 (m, 2H, Ar), 7.48–7.52 (m, 2H, Ar), 8.95 (s, 1H, NH), 12.78 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 11.7, 115.5, 118.6, 128.6, 142.4, 150.9, 160.2. HRMS (ESI) calcd for C9H11N4 [M + H]+m/z 175.0978, found m/z 175.0980.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz) (two tautomers): δ 3.87 and 4.00 (two s, 2H, CH2), 6.72–6.77 (m, 1H, Ar), 7.15–7.35 (m, 7H, Ar), 7.48–7.51 (m, 2H, Ar), 9.01 and 9.25 (two s, 1H, NH), 12.29 and 13.01 (two s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 32.0, 115.5, 118.7, 126.6, 128.5, 128.6, 128.7, 137.1, 142.3, 153.5, 160.3. HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1299.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 2.36 (s, 3H, CH3), 6.78–6.83 (m, 1H, Ar), 7.21–7.26 (m, 2H, Ar), 7.30–7.33 (m, 2H, Ar), 7.57–7.60 (m, 2H, Ar), 7.85–7.88 (m, 2H, Ar), 9.42 (s, 1H, NH), 13.57 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 21.0, 115.8, 119.2, 125.7, 128.7, 129.4, 139.1, 142.0, 153.7, 158.8 (one signal of aromatic carbon is broadened and merged into the background or overlapped). HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1301.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz) (two tautomers): δ 3.82 (s, 2H, CH2), 6.75–6.80 (m, 1H, Ar), 7.00–7.11 (m, 2H, Ar), 7.19–7.24 (m, 2H, Ar), 7.57–7.59 (m, 2H, Ar), 7.89–7.92 (m, 2H, Ar), 9.16 and 9.39 (two s, 1H, NH), 12.53 and 13.49 (two s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 55.3, 114.5, 115.6, 118.8, 120.0, 127.4, 128.6, 128.9, 142.3, 152.2, 160.6. HRMS (ESI) calcd for C15H15N4O [M + H]+m/z 267.1240, found m/z 267.1232.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 6.77–6.82 (m, 1H, Ar), 7.21–7.26 (m, 2H, Ar), 7.50–7.54 (m, 1H, Ar), 7.54–7.61 (m, 2H, Ar), 8.00–8.01 (m, 2H, Ar), 8.69–8.70 (m, 1H, Ar), 9.25 (s, 1H, NH), 14.02 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 115.7, 119.1, 121.1, 124.9, 128.7, 137.9, 142.2, 146.3, 149.6, 151.7, 160.9. HRMS (ESI) calcd for C13H12N5 [M + H]+m/z 238.1087, found m/z 238.1095.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 1.19–1.51 (m, 5H, cyclohexyl), 1.63–1.78 (m, 3H, cyclohexyl), 1.91–1.96 (m, 2H, cyclohexyl), 2.62–2.69 (m, 1H, cyclohexyl), 6.72–6.77 (m, 1H, Ar), 7.15–7.21 (m, 2H, Ar), 7.48–7.52 (m, 2H, Ar), 9.12 (s, 1H, NH), 12.93 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 25.3, 25.5, 31.0, 35.5, 115.5, 118.6, 128.6, 142.3, 159.1, 161.0. HRMS (ESI) calcd for C14H19N4 [M + H]+m/z 243.1604, found m/z 243.1597.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 1.16–1.53 (m, 5H, cyclohexyl), 1.64–1.77 (m, 3H, cyclohexyl), 1.91–1.95 (m, 2H, cyclohexyl), 2.20 (s, 3H, CH3), 2.63–2.70 (m, 1H, cyclohexyl), 6.97–7.00 (m, 2H, Ar), 7.38–7.41 (m, 2H, Ar), 8.85 (s, 1H, NH), 12.70 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 20.3, 25.3, 25.5, 30.9, 35.3, 115.5, 127.0, 129.0, 140.0, 158.7, 160.0. HRMS (ESI) calcd for C15H21N4 [M + H]+m/z 257.1761, found m/z 257.1751.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 4.32 (s, 2H, CH2), 6.89–7.01 (m, 2H, Ar), 7.21–7.33 (m, 3H, Ar), 7.36–7.42 (m, 2H, Ar), 7.65–7.71 (m, 1H, Ar), 8.22–8.24 (m, 1H, Ar), 10.69 (s, 1H, NH), 13.07 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 35.0, 110.5, 116.1, 127.1, 128.4, 128.8, 138.2, 138.3, 147.2, 151.7, 152.8, 155.7. HRMS (ESI) calcd for C14H14N5S [M + H]+m/z 284.0964, found m/z 284.0967.
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 4.34 (s, 2H, CH2), 6.96–7.00 (m, 2H, Ar), 7.22–7.39 (m, 9H, Ar + NH) (one NH signal is broadened and merged into the background). 13C{1H} NMR (CDCl3, 75 MHz): δ 37.8, 117.6, 122.0, 127.9, 128.9, 129.0, 129.5, 137.1, 140.4, 153.2, 159.1. HRMS (ESI) calcd for C15H15N4S [M + H]+m/z 283.1012, found m/z 283.1024.
:10 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 1.17–1.51 (m, 5H, cyclohexyl), 1.62–1.75 (m, 3H, cyclohexyl), 1.89–1.93 (m, 2H, cyclohexyl), 2.57–2.67 (m, 1H, cyclohexyl), 5.49 (s, 2H, CH2), 7.08–7.13 (m, 1H, Ar), 7.29–7.39 (m, 7H, Ar), 7.50–7.53 (m, 2H, Ar), 10.46 (s, 1H, NH) (one NH signal is broadened and merged into the background). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 25.2, 25.4, 30.5, 35.8, 50.0, 120.0, 123.5, 127.5, 127.8, 128.6, 129.2, 135.8, 138.7, 149.2, 159.1. HRMS (ESI) calcd for C21H25N4 [M-Cl−]+m/z 333.2074, found m/z 333.2081.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.17–1.61 (m, 5H, cyclohexyl), 1.72–1.86 (m, 3H, cyclohexyl), 2.02–2.06 (m, 2H, cyclohexyl), 2.71–2.81 (m, 1H, cyclohexyl), 5.87 (s, 2H, CH2), 7.10–7.14 (m, 1H, Ar), 7.27–7.35 (m, 3H, Ar), 7.62–7.66 (m, 2H, Ar), 7.91–7.97 (m, 1H, Ar), 8.09–8.11 (m, 1H, Ar), 8.53–8.56 (m, 1H, Ar) (two NH signals are broadened and merged into the background). 13C{1H} NMR (CDCl3, 75 MHz): δ 25.8, 31.3, 36.8, 52.0, 116.8, 117.6, 128.4, 128.8, 129.0, 134.8, 138.3, 142.8, 146.8, 150.6, 160.5 (signals of two aliphatic carbons overlap). HRMS (ESI) calcd for C20H24N5 [M − Cl−]+m/z 334.2026, found m/z 334.2040.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.22–1.51 (m, 5H, cyclohexyl), 1.67–1.70 (m, 1H, cyclohexyl), 1.77–1.80 (m, 2H, cyclohexyl), 1.99–2.02 (m, 2H, cyclohexyl), 2.30 (s, 3H, CH3), 2.63–2.68 (m, 1H, cyclohexyl), 5.32 (s, 2H, CH2), 7.01–7.23 (m, 9H, Ar), 10.54 (s, 1H, NH) (one NH signal is broadened and merged into the background). 13C{1H} NMR (CDCl3, 75 MHz): δ 21.3, 25.52, 25.55, 30.4, 35.3, 52.6, 121.2, 125.5, 128.4, 129.6, 130.4, 136.2, 138.6, 146.7, 155.8, 170.8. HRMS (ESI) calcd for C22H27N4 [M − Cl−]+m/z 347.2230, found m/z 347.2240.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 5.26 (s, 2H, CH2), 6.84–6.89 (m, 1H, Ar), 7.20–7.37 (m, 7H, Ar), 7.53–7.56 (m, 2H, Ar), 8.27, 8.69 (two s, 2H, CH + NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 45.5, 116.5, 120.3, 127.1, 127.8, 128.7, 128.8, 136.5, 141.1, 141.5, 150.5. HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1300.
H3), 25.9, 113.5, 117.0, 121.5, 125.5, 128.6, 129.1, 129.5, 137.8, 138.9, 141.0, 159.7, 159.8. HRMS (ESI) calcd for C18H21N4 [M + H]+m/z 293.1761, found m/z 293.1766.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 2.17 (s, 3H, CH3), 6.58–6.62 (m, 1H, Ar), 6.73 (s, 1H, NH), 7.01–7.11 (m, 3H, Ar), 7.17–7.29 (m, 6H, Ar), 7.34–7.35 (m, 2H, Ar), 7.36–7.37 (m, 2H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 21.8, 113.7, 117.2, 121.7, 125.4, 128.1, 128.4, 128.7, 129.0, 129.1, 129.4, 130.1, 138.5, 139.0, 140.9, 152.5, 160.1. HRMS (ESI) calcd for C21H19N4 [M + H]+m/z 327.1604, found m/z 327.1616.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 6.84–6.88 (m, 1H, Ar), 7.32–7.44 (m, 8H, Ar + NH), 7.50–7.54 (m, 2H, Ar), 7.63–7.69 (m, 1H, Ar), 8.13–8.17 (m, 1H, Ar), 8.37–8.39 (m, 2H, Ar). 13C{1H} NMR (CDCl3, 75 MHz): δ 110.5, 116.3, 125.5, 128.0, 128.6, 128.7, 129.1, 129.4, 130.2, 138.3, 138.4, 148.2, 152.8, 153.7, 159.0. HRMS (ESI) calcd for C19H16N5 [M + H]+m/z 314.1400, found m/z 314.1414.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.62 (s, 9H, 3CH3), 7.21–7.25 (m, 1H, Ar), 7.92 (s, 1H, CH), 7.99 (s, 1H, Ar), 8.07–8.15 (m, 2H, Ar), 8.66 (s, 1H, NH). 13C{1H} NMR (CDCl3, 75 MHz): δ 29.4, 58.3, 122.5, 123.8, 138.25, 138.33, 138.6, 141.2, 159.9. HRMS (ESI) calcd for C11H16N5 [M + H]+m/z 218.1400, found m/z 218.1407.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 2.27 (s, 3H, CH3), 6.73–6.78 (m, 1H, Ar), 7.15–7.21 (m, 2H, Ar), 7.48–7.52 (m, 2H, Ar), 8.95 (s, 1H, NH), 12.78 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 11.7, 115.5, 118.6, 128.6, 142.4, 150.9, 160.2. HRMS (ESI) calcd for C9H11N4 [M + H]+m/z 175.0978, found m/z 175.0980.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz) (two tautomers): δ 3.87 and 4.00 (two s, 2H, CH2), 6.72–6.77 (m, 1H, Ar), 7.15–7.35 (m, 7H, Ar), 7.48–7.51 (m, 2H, Ar), 9.01 and 9.25 (two s, 1H, NH), 12.29 and 13.01 (two s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 32.0, 115.5, 118.7, 126.6, 128.5, 128.6, 128.7, 137.1, 142.3, 153.5, 160.3. HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1299.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 2.36 (s, 3H, CH3), 6.78–6.83 (m, 1H, Ar), 7.21–7.26 (m, 2H, Ar), 7.30–7.33 (m, 2H, Ar), 7.57–7.60 (m, 2H, Ar), 7.85–7.88 (m, 2H, Ar), 9.42 (s, 1H, NH), 13.57 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 21.0, 115.8, 119.2, 125.7, 128.7, 129.4, 139.1, 142.0, 153.7, 158.8 (one signal of aromatic carbon is broadened and merged into the background or overlapped). HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1301.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz) (two tautomers): δ 3.82 (s, 2H, CH2), 6.75–6.80 (m, 1H, Ar), 7.00–7.11 (m, 2H, Ar), 7.19–7.24 (m, 2H, Ar), 7.57–7.59 (m, 2H, Ar), 7.89–7.92 (m, 2H, Ar), 9.16 and 9.39 (two s, 1H, NH), 12.53 and 13.49 (two s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 55.3, 114.5, 115.6, 118.8, 120.0, 127.4, 128.6, 128.9, 142.3, 152.2, 160.6. HRMS (ESI) calcd for C15H15N4O [M + H]+m/z 267.1240, found m/z 267.1232.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 6.77–6.82 (m, 1H, Ar), 7.21–7.26 (m, 2H, Ar), 7.50–7.54 (m, 1H, Ar), 7.54–7.61 (m, 2H, Ar), 8.00–8.01 (m, 2H, Ar), 8.69–8.70 (m, 1H, Ar), 9.25 (s, 1H, NH), 14.02 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 115.7, 119.1, 121.1, 124.9, 128.7, 137.9, 142.2, 146.3, 149.6, 151.7, 160.9. HRMS (ESI) calcd for C13H12N5 [M + H]+m/z 238.1087, found m/z 238.1095.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 1.19–1.51 (m, 5H, cyclohexyl), 1.63–1.78 (m, 3H, cyclohexyl), 1.91–1.96 (m, 2H, cyclohexyl), 2.62–2.69 (m, 1H, cyclohexyl), 6.72–6.77 (m, 1H, Ar), 7.15–7.21 (m, 2H, Ar), 7.48–7.52 (m, 2H, Ar), 9.12 (s, 1H, NH), 12.93 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 25.3, 25.5, 31.0, 35.5, 115.5, 118.6, 128.6, 142.3, 159.1, 161.0. HRMS (ESI) calcd for C14H19N4 [M + H]+m/z 243.1604, found m/z 243.1597.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 1.16–1.53 (m, 5H, cyclohexyl), 1.64–1.77 (m, 3H, cyclohexyl), 1.91–1.95 (m, 2H, cyclohexyl), 2.20 (s, 3H, CH3), 2.63–2.70 (m, 1H, cyclohexyl), 6.97–7.00 (m, 2H, Ar), 7.38–7.41 (m, 2H, Ar), 8.85 (s, 1H, NH), 12.70 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 20.3, 25.3, 25.5, 30.9, 35.3, 115.5, 127.0, 129.0, 140.0, 158.7, 160.0. HRMS (ESI) calcd for C15H21N4 [M + H]+m/z 257.1761, found m/z 257.1751.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 4.32 (s, 2H, CH2), 6.89–7.01 (m, 2H, Ar), 7.21–7.33 (m, 3H, Ar), 7.36–7.42 (m, 2H, Ar), 7.65–7.71 (m, 1H, Ar), 8.22–8.24 (m, 1H, Ar), 10.69 (s, 1H, NH), 13.07 (s, 1H, NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 35.0, 110.5, 116.1, 127.1, 128.4, 128.8, 138.2, 138.3, 147.2, 151.7, 152.8, 155.7. HRMS (ESI) calcd for C14H14N5S [M + H]+m/z 284.0964, found m/z 284.0967.
:1 mixture). 1H NMR (CDCl3, 300 MHz): δ 4.34 (s, 2H, CH2), 6.96–7.00 (m, 2H, Ar), 7.22–7.39 (m, 9H, Ar + NH) (one NH signal is broadened and merged into the background). 13C{1H} NMR (CDCl3, 75 MHz): δ 37.8, 117.6, 122.0, 127.9, 128.9, 129.0, 129.5, 137.1, 140.4, 153.2, 159.1. HRMS (ESI) calcd for C15H15N4S [M + H]+m/z 283.1012, found m/z 283.1024.
:10 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 1.17–1.51 (m, 5H, cyclohexyl), 1.62–1.75 (m, 3H, cyclohexyl), 1.89–1.93 (m, 2H, cyclohexyl), 2.57–2.67 (m, 1H, cyclohexyl), 5.49 (s, 2H, CH2), 7.08–7.13 (m, 1H, Ar), 7.29–7.39 (m, 7H, Ar), 7.50–7.53 (m, 2H, Ar), 10.46 (s, 1H, NH) (one NH signal is broadened and merged into the background). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 25.2, 25.4, 30.5, 35.8, 50.0, 120.0, 123.5, 127.5, 127.8, 128.6, 129.2, 135.8, 138.7, 149.2, 159.1. HRMS (ESI) calcd for C21H25N4 [M-Cl−]+m/z 333.2074, found m/z 333.2081.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.17–1.61 (m, 5H, cyclohexyl), 1.72–1.86 (m, 3H, cyclohexyl), 2.02–2.06 (m, 2H, cyclohexyl), 2.71–2.81 (m, 1H, cyclohexyl), 5.87 (s, 2H, CH2), 7.10–7.14 (m, 1H, Ar), 7.27–7.35 (m, 3H, Ar), 7.62–7.66 (m, 2H, Ar), 7.91–7.97 (m, 1H, Ar), 8.09–8.11 (m, 1H, Ar), 8.53–8.56 (m, 1H, Ar) (two NH signals are broadened and merged into the background). 13C{1H} NMR (CDCl3, 75 MHz): δ 25.8, 31.3, 36.8, 52.0, 116.8, 117.6, 128.4, 128.8, 129.0, 134.8, 138.3, 142.8, 146.8, 150.6, 160.5 (signals of two aliphatic carbons overlap). HRMS (ESI) calcd for C20H24N5 [M − Cl−]+m/z 334.2026, found m/z 334.2040.
:10 mixture). 1H NMR (CDCl3, 300 MHz): δ 1.22–1.51 (m, 5H, cyclohexyl), 1.67–1.70 (m, 1H, cyclohexyl), 1.77–1.80 (m, 2H, cyclohexyl), 1.99–2.02 (m, 2H, cyclohexyl), 2.30 (s, 3H, CH3), 2.63–2.68 (m, 1H, cyclohexyl), 5.32 (s, 2H, CH2), 7.01–7.23 (m, 9H, Ar), 10.54 (s, 1H, NH) (one NH signal is broadened and merged into the background). 13C{1H} NMR (CDCl3, 75 MHz): δ 21.3, 25.52, 25.55, 30.4, 35.3, 52.6, 121.2, 125.5, 128.4, 129.6, 130.4, 136.2, 138.6, 146.7, 155.8, 170.8. HRMS (ESI) calcd for C22H27N4 [M − Cl−]+m/z 347.2230, found m/z 347.2240.
:1 mixture). 1H NMR (DMSO-d6, 300 MHz): δ 5.26 (s, 2H, CH2), 6.84–6.89 (m, 1H, Ar), 7.20–7.37 (m, 7H, Ar), 7.53–7.56 (m, 2H, Ar), 8.27, 8.69 (two s, 2H, CH + NH). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 45.5, 116.5, 120.3, 127.1, 127.8, 128.7, 128.8, 136.5, 141.1, 141.5, 150.5. HRMS (ESI) calcd for C15H15N4 [M + H]+m/z 251.1291, found m/z 251.1300.
4.7 X-ray crystal structure determination of compounds 4h, 4u and 9f
Details of the X-ray structure determinations are presented in ESI, section S3.†Crystallographic data for 4h, 4u and 9f have been deposited with the Cambridge Crystallographic Data Center. CCDC 2194495 (4h), CCDC 2194496 (4u) and CCDC 2194497 (9f) contain the supplementary crystallographic data for this paper.
5. Computational details
All geometry optimizations were performed by the PBE1PBE method113,114 using the def2svp basis set115 in the Gaussian 09116 software package. The PBE1PBE method is one of the cost-effective and reliable DFT methods often used for describing chemical reactions involving transition metal complexes.117–120 The calculations were performed both in vacuum and in dioxane medium (in the IEF PCM approximation).121,122 The IPr ligand was used in the calculations instead of bulkier IPr*OMe to reduce computational time. Although IPr provides lower catalytic efficiency, the general regularities of the arylation reaction were similar for both ligands according to the experimental data. For optimized molecular structures, the thermodynamic parameters were calculated at a temperature of 298 K. In the vibrational spectrum of each transition state, there is one imaginary frequency corresponding to the vibration along the reaction coordinate. Molecular structures and MESP density isosurfaces were visualized using Chemcraft software.123Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (RSF grant 22-23-00380). The authors also thank the Shared Research Center “Nanotechnologies” of the Platov South-Russian State Polytechnic University and the Department of Structural Studies of Zelinsky Institute of Organic Chemistry for analytical services.References
- P. Ruiz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, Buchwald-Hartwig reaction: an overview, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, The Buchwald–Hartwig amination after 25 years, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- P. A. Forero-Cortés and A. M. Haydl, The 25th anniversary of the Buchwald–Hartwig amination: development, applications, and outlook, Org. Process Res. Dev., 2019, 23, 1478–1483 CrossRef.
- B. T. Ingoglia, C. C. Wagen and S. L. Buchwald, Biaryl monophosphine ligands in palladium-catalyzed C–N coupling: An updated User's guide, Tetrahedron, 2019, 75, 4199–4211 CrossRef CAS PubMed.
- I. P. Beletskaya and A. D. Averin, Metal-catalyzed reactions for the C(sp2)7N bond formation: Achievements of recent years, Russ. Chem. Rev., 2021, 90, 1359–1366 CrossRef.
- B. Seifinoferest, A. Tanbakouchian, B. Larijani and M. Mahdavi, Ullmann-Goldberg and Buchwald-Hartwig C−N Cross Couplings: Synthetic Methods to Pharmaceutically Potential N-Heterocycles, Asian J. Org. Chem., 2021, 10, 1319–1344 CrossRef CAS.
- M. Su, N. Hoshiya and S. L. Buchwald, Palladium-Catalyzed Amination of Unprotected Five-Membered Heterocyclic Bromides, Org. Lett., 2014, 16, 832–835 CrossRef CAS.
- E. P. K. Olsen, P. L. Arrechea and S. L. Buchwald, Mechanistic Insight Leads to a Ligand Which Facilitates the Palladium-Catalyzed Formation of 2-(Hetero)Arylaminooxazoles and 4-(Hetero)Arylaminothiazoles, Angew. Chem., Int. Ed., 2017, 56, 10569–10572 CrossRef CAS.
- A. Khadra, S. Mayer and M. G. Organ, Pd-PEPPSI-IPentCl: A Useful Catalyst for the Coupling of 2-Aminopyridine Derivatives, Chem. – Eur. J., 2017, 23, 3206–3212 CrossRef CAS.
- J.-S. Ouyang, S. Liu, B. Pan, Y. Zhang, H. Liang, B. Chen, X. He, W. T. K. Chan, A. S. C. Chan, T.-Y. Sun, Y.-D. Wu and L. Qiu, A Bulky and Electron-Rich N-Heterocyclic Carbene–Palladium Complex (SIPr)Ph2Pd(cin)Cl: Highly Efficient and Versatile for the Buchwald–Hartwig Amination of (Hetero)aryl Chlorides with (Hetero)aryl Amines at Room Temperature, ACS Catal., 2021, 11, 9252–9261 CrossRef CAS.
- D.-H. Li, X.-B. Lan, A.-X. Song, M. M. Rahman, C. Xu, F.-D. Huang, R. Szostak, M. Szostak and F.-S. Liu, Buchwald-Hartwig Amination of Coordinating Heterocycles Enabled by Large-but-Flexible Pd-BIAN-NHC Catalysts, Chem. – Eur. J., 2022, 28, e202103341 CAS.
- M. A. Düfert, K. L. Billingsley and S. L. Buchwald, Suzuki-Miyaura Cross-Coupling of Unprotected, Nitrogen-Rich Heterocycles: Substrate Scope and Mechanistic Investigation, J. Am. Chem. Soc., 2013, 135, 12877–12885 CrossRef.
- R. Aggarwal and G. Sumran, An insight on medicinal attributes of 1,2,4-triazoles, Eur. J. Med. Chem., 2020, 205, 112652 CrossRef CAS PubMed.
- S. A. El-Sebaey, Recent Advances in 1,2,4-Triazole Scaffolds as Antiviral Agents, ChemistrySelect, 2020, 5, 11654–11680 CrossRef CAS.
- A. Abdelli, S. Azzouni, R. Plais, A. Gaucher, M. L. Efrit and D. Prim, Recent advances in the chemistry of 1,2,4-triazoles: Synthesis, reactivity and biological activities, Tetrahedron Lett., 2021, 86, 153518 CrossRef CAS.
- M. Strzelecka and P. Świątek, 1,2,4-Triazoles as Important Antibacterial Agents, Pharmaceuticals, 2021, 14, 224 CrossRef CAS PubMed.
- W. Zafar, S. H. Sumrra and Z. H. Chohan, A review: Pharmacological aspects of metal based 1,2,4-triazole derived Schiff bases, Eur. J. Med. Chem., 2021, 222, 113602 CrossRef CAS PubMed.
- S. Nasri, M. Bayat and K. Kochia, Strategies for synthesis of 1,2,4-triazole-containing scaffolds using 3-amino-1,2,4-triazole, Mol. Diversity, 2022, 26, 717–739 CrossRef CAS.
- S. H. Sumrra, W. Zafar, M. Imran and Z. H. Chohan, A review on the biomedical efficacy of transition metal triazole compounds, J. Coord. Chem., 2022, 75, 293–334 CrossRef CAS.
- J. G. Haasnoot, Mononuclear, oligonuclear and polynuclear metal coordination compounds with 1,2,4-triazole derivatives as ligands, Coord. Chem. Rev., 2000, 200–202, 131–185 CrossRef CAS.
- G. Aromí, L. A. Barrios, O. Roubeau and P. Gamez, Triazoles and tetrazoles: Prime ligands to generate remarkable coordination materials, Coord. Chem. Rev., 2011, 255, 485–546 CrossRef.
- L. G. Lavrenova and O. G. Shakirova, Spin Crossover and Thermochromism of Iron(II) Coordination Compounds with 1,2,4-Triazoles and Tris(pyrazol-1-yl)methanes, Eur. J. Inorg. Chem., 2013, 2013, 670–682 CrossRef CAS.
- H. L. C. Feltham, A. S. Barltrop and S. Brooker, Spin crossover in iron(II) complexes of 3,4,5-tri-substituted-1,2,4-triazole (Rdpt), 3,5-di-substituted-1,2,4-triazolate (dpt−), and related ligands, Coord. Chem. Rev., 2017, 344, 26–53 CrossRef CAS.
- D. Zdżalik-Bielecka, K. Kozik, A. Poświata, K. Jastrzębski, M. Jakubik and M. Miączyńska, Bemcentinib and Gilteritinib Inhibit Cell Growth and Impair the Endo-Lysosomal and Autophagy Systems in an AXL-Independent Manner, Mol. Cancer Res., 2022, 20, 446–455 CrossRef.
- J. Lorens, C. E. Arce-Lara, E. Arriola, P. Brunsvig, E. C. Costa, M. Domine, K. H. Dragnev, E. Felip, R. G. Campelo, M. Krebs, S. P. Aix, J. F. Spicer, J. M. T. Perez, N. Vinolas, R. J. Holt, A. Brown and M. J. Chisamore, Phase II open-label, multi-centre study of bemcentinib (BGB324), a first-in-class selective AXL inhibitor, in combination with pembrolizumab in patients with advanced NSCLC, J. Clin. Oncol., 2018, 36, 3078–3078 Search PubMed.
- A. Hoel, T. Osman, F. Hoel, H. Elsaid, T. Chen, L. Landolt, J. Babickova, K. J. Tronstad, J. B. Lorens, G. Gausdal, H.-P. Marti and J. Furriol, Axl-inhibitor bemcentinib alleviates mitochondrial dysfunction in the unilateral ureter obstruction murine model, J. Cell. Mol. Med., 2021, 25, 7407–7417 CrossRef CAS.
- M. S. Beg, A. M. Lowy, P. J. O'Dwyer, G. S. Jameson, E. H. Borazanci, H. Patel, C. Massey, J. Schoelermann, J. Lorens, F. Fattah, K. Crane, E. F. Williams, J. Clark, D. D. V. Hoff and R. A. Brekken, A randomized clinical trial of chemotherapy with gemcitabine/cisplatin/nabpaclitaxel with or without the AXL inhibitor bemcentinib (BGB324) for patients with advanced pancreatic cancer, J. Clin. Oncol., 2019, 37, TPS473 Search PubMed.
- M. Dittmar, J. S. Lee, K. Whig, E. Segrist, M. Li, B. Kamalia, L. Castellana, K. Ayyanathan, F. L. Cardenas-Diaz, E. E. Morrisey, R. Truitt, W. Yang, K. Jurado, K. Samby, H. Ramage, D. C. Schultz and S. Cherry, Drug repurposing screens reveal cell-type-specific entry pathways and FDA-approved drugs active against SARS-Cov-2, Cell Rep., 2021, 35, 108959 CrossRef CAS.
- J. A. Encinar and J. A. Menendez, Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2′-O-Methylation of Viral RNA, Viruses, 2020, 12, 525 CrossRef CAS.
- M. Goyal, N. Tewatia, H. Vashisht, R. Jain and S. Kumar, Novel corona virus (COVID-19); Global efforts and effective investigational medicines: A review, J. Infect. Public Health, 2021, 14, 910–921 CrossRef PubMed.
- N. R. Jena, Drug targets, mechanisms of drug action, and therapeutics against SARS-CoV-2, Chem. Phys. Impact, 2021, 2, 100011 CrossRef.
- Q. Wang, L. Su, N. Liu, L. Zhang, W. Xu and H. Fang, Cyclin dependent kinase 1 inhibitors: a review of recent progress, Curr. Med. Chem., 2011, 18, 2025–2043 CrossRef CAS.
- R. Lin, P. J. Connolly, S. Huang, S. K. Wetter, Y. Lu, W. V. Murray, S. L. Emanuel, R. H. Gruninger, A. R. Fuentes-Pesquera, C. A. Rugg, S. A. Middleton and L. K. Jolliffe, 1-Acyl-1H-[1,2,4]triazole-3,5-diamine Analogues as Novel and Potent Anticancer Cyclin-Dependent Kinase Inhibitors: Synthesis and Evaluation of Biological Activities, J. Med. Chem., 2005, 48, 4208–4211 CrossRef CAS.
- O. Fedorov, K. Huber, A. Eisenreich, P. Filippakopoulos, O. King, A. N. Bullock, D. Szklarczyk, L. J. Jensen, D. Fabbro, J. Trappe, U. Rauch, F. Bracher and S. Knapp, Specific CLK Inhibitors from a Novel Chemotype for Regulation of Alternative Splicing, Chem. Biol., 2011, 18, 67–76 CrossRef CAS.
- M. Davidson, L. Levi, J. Park, I. Nastas, L. Ford, S. Rassnick, C. Canuso, J. M. Davis and M. Weiser, The effects of JNJ-39393406 a positive allosteric nicotine modulator on mood and cognition in patients with unipolar depression: A double-blind, add-on, placebo-controlled trial, Eur. Neuropsychopharmacol., 2021, 51, 33–42 CrossRef CAS PubMed.
- G. Winterer, J. Gallinat, J. Brinkmeyer, F. Musso, J. Kornhuber, N. Thuerauf, D. Rujescu, R. Favis, Y. Sun, M. A. Franc, S. Ouwerkerk-Mahadevan, L. Janssens, M. Timmers and J. R. Streffer, Allosteric alpha-7 nicotinic receptor modulation and P50 sensory gating in schizophrenia: A proof-of-mechanism study, Neuropharmacology, 2013, 64, 197–204 CrossRef CAS.
- M. N. S. Gendy, C. Ibrahim, M. E. Sloan and B. Le Foll, in Substance Use Disorders: From Etiology to Treatment, ed. M. A. Nader and Y. L. Hurd, Springer International Publishing, Cham, 2020, pp. 395–420 Search PubMed.
- S.-Q. Yin, J.-J. Wang, C.-M. Zhang and Z.-P. Liu, The development of MetAP-2 inhibitors in cancer treatment, Curr. Med. Chem., 2012, 19, 1021–1035 CrossRef CAS PubMed.
- J. P. Marino, P. W. Fisher, G. A. Hofmann, R. B. Kirkpatrick, C. A. Janson, R. K. Johnson, C. Ma, M. Mattern, T. D. Meek, M. D. Ryan, C. Schulz, W. W. Smith, D. G. Tew, T. A. Tomazek, D. F. Veber, W. C. Xiong, Y. Yamamoto, K. Yamashita, G. Yang and S. K. Thompson, Highly Potent Inhibitors of Methionine Aminopeptidase-2 Based on a 1,2,4-Triazole Pharmacophore, J. Med. Chem., 2007, 50, 3777–3785 CrossRef CAS PubMed.
- X. Ouyang, X. Chen, E. L. Piatnitski, A. S. Kiselyov, H.-Y. He, Y. Mao, V. Pattaropong, Y. Yu, K. H. Kim, J. Kincaid, L. Smith, W. C. Wong, S. P. Lee, D. L. Milligan, A. Malikzay, J. Fleming, J. Gerlak, D. Deevi, J. F. Doody, H.-H. Chiang, S. N. Patel, Y. Wang, R. L. Rolser, P. Kussie, M. Labelle and M. C. Tuma, Synthesis and structure–activity relationships of 1,2,4-triazoles as a novel class of potent tubulin polymerization inhibitors, Bioorg. Med. Chem. Lett., 2005, 15, 5154–5159 CrossRef CAS PubMed.
- Ş. Demirayak, K. Benkli and K. Güven, Synthesis and antimicrobial activities of some 3-arylamino-5-[2-(substituted 1-imidazolyl)ethyl]-1,2,4-triazole derivatives, Eur. J. Med. Chem., 2000, 35, 1037–1040 CrossRef PubMed.
- N. Neamati, A. Mazumder, S. Sunder, J. Owen, R. Schultz and Y. Pommier, 2-Mercaptobenzenesulphonamides as Novel Inhibitors of Human Immunodeficiency virus Type 1 Integrase and Replication, Antiviral Chem. Chemother., 1997, 8, 485–495 CrossRef CAS.
- G. Barbaro, A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, Highly active antiretroviral therapy: current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome, Curr. Pharm. Des., 2005, 11, 1805–1843 CrossRef CAS.
- N. Mehta, M. Kaur, M. Singh, S. Chand, B. Vyas, P. Silakari, M. S. Bahia and O. Silakari, Purinergic receptor P2X7: A novel target for anti-inflammatory therapy, Bioorg. Med. Chem., 2014, 22, 54–88 CrossRef CAS.
- S. D. Guile, L. Alcaraz, T. N. Birkinshaw, K. C. Bowers, M. R. Ebden, M. Furber and M. J. Stocks, Antagonists of the P2X7 Receptor. From Lead Identification to Drug Development, J. Med. Chem., 2009, 52, 3123–3141 CrossRef CAS.
- R. M. R. J. da Silva, M. O. Gandi, J. S. Mendonça, A. S. Carvalho, J. P. Coutinho, A. C. C. Aguiar, A. U. Krettli and N. Boechat, New hybrid trifluoromethylquinolines as antiplasmodium agents, Bioorg. Med. Chem., 2019, 27, 1002–1008 CrossRef CAS PubMed.
- L. C. S. Pinheiro, L. M. Feitosa, M. O. Gandi, F. F. Silveira and N. Boechat, The Development of Novel Compounds Against Malaria: Quinolines, Triazolpyridines, Pyrazolopyridines and Pyrazolopyrimidines, Molecules, 2019, 24, 4095 CrossRef PubMed.
- W. Guo, G. Liu, L. Deng, W. Mei, X. Zou, Y. Zhong, X. Zhuo, X. Fan and L. Zheng, Metal- and Oxidant-Free Green Three-Component Desulfurization and Deamination Condensation Approach to Fully Substituted 1H-1,2,4-Triazol-3-amines and Their Photophysical Properties, J. Org. Chem., 2021, 86, 17986–18003 CrossRef CAS PubMed.
- E. Amit, I. Berg and E. Gross, Self-Assembled Monolayers of Nitron: Self-Activated and Chemically Addressable N-Heterocyclic Carbene Monolayers with Triazolone Structural Motif, Chem. – Eur. J., 2020, 26, 13046–13052 CrossRef CAS PubMed.
- M. Sevim, S. B. Kavukcu, A. Kınal, O. Şahin and H. Türkmen, C–C coupling formation using nitron complexes, Dalton Trans., 2020, 49, 16903–16915 RSC.
- A. Y. Chernenko, A. V. Astakhov, V. V. Kutyrev, E. G. Gordeev, J. V. Burykina, M. E. Minyaev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Stabilization of the Pd–NHC framework with 1,2,4-triazol-5-ylidene ligands toward decomposition in alkaline media, Inorg. Chem. Front., 2021, 8, 3382–3401 RSC.
- H. Beyzaei, Z. Khosravi, R. Aryan and B. Ghasemi, A green one-pot synthesis of 3(5)-substituted 1,2,4-triazol-5(3)-amines as potential antimicrobial agents, J. Iran. Chem. Soc., 2019, 16, 2565–2573 CrossRef CAS.
- A. I. Velter, F. P. Bischoff, D. Berthelot, M. De Cleyn, D. Oehlrich, L. Jaroskova, G. Macdonald, G. Minne, S. Pieters, F. Rombouts, S. Van Brandt, Y. Van Roosbroeck, M. Surkyn, A. A. Trabanco, G. Tresadern, T. Wu, H. Borghys, M. Mercken, C. Masungi and H. Gijsen, Anilinotriazoles as potent gamma secretase modulators, Bioorg. Med. Chem. Lett., 2014, 24, 5805–5813 CrossRef CAS PubMed.
- A. Zarguil, S. Boukhris, M. L. El Efrit, A. Souizi and E. M. Essassi, Easy access to triazoles, triazolopyrimidines, benzimidazoles and imidazoles from imidates, Tetrahedron Lett., 2008, 49, 5883–5886 CrossRef CAS.
- R. Noël, X. Song, R. Jiang, M. J. Chalmers, P. R. Griffin and T. M. Kamenecka, Efficient Methodology for the Synthesis of 3-Amino-1,2,4-triazoles, J. Org. Chem., 2009, 74, 7595–7597 CrossRef PubMed.
- W.-P. Yen, F.-C. Kung and F. F. Wong, 1,3-Dipolar Cycloaddition of Carbodiimides and Nitrilimines: Synthesis and Mechanistic Study of 5-Amino-1,2,4-triazoles, Eur. J. Org. Chem., 2016, 2328–2335 CrossRef CAS.
- Y. Fan, Y. Xia, J. Tang, F. Ziarelli, F. Qu, P. Rocchi, J. L. Iovanna and L. Peng, An Efficient Mixed-Ligand Pd Catalytic System to Promote C-N Coupling for the Synthesis of N-Arylaminotriazole Nucleosides, Chem. – Eur. J., 2012, 18, 2221–2225 CrossRef CAS PubMed.
- M. Cong, Y. Fan, J.-M. Raimundo, J. Tang and L. Peng, Pd(dba)2 vs Pd2(dba)3: An in-Depth Comparison of Catalytic Reactivity and Mechanism via Mixed-Ligand Promoted C–N and C–S Coupling Reactions, Org. Lett., 2014, 16, 4074–4077 CrossRef CAS.
- Y. Fan, Y. Xia, J. Tang, P. Rocchi, F. Qu, J. Iovanna and L. Peng, Ligand-Mediated Highly Effective and Selective C−N Coupling for Synthesizing Bioactive N-Aryltriazole Acyclonucleosides, Org. Lett., 2010, 12, 5712–5715 CrossRef CAS PubMed.
- V. M. Chernyshev, A. G. Vlasova, A. V. Astakhov, S. V. Shishkina and O. V. Shishkin, Reactivity of C-Amino-1,2,4-triazoles toward Electrophiles: A Combined Computational and Experimental Study of Alkylation by Halogen Alkanes, J. Org. Chem., 2015, 80, 375–385 CrossRef CAS PubMed.
- Z. L. Chioato, T. M. Klapötke, F. Mieskes, J. Stierstorfer and M. Weyrauther, (Picrylamino)-1,2,4-triazole Derivatives – Thermally Stable Explosives, Eur. J. Inorg. Chem., 2016, 2016, 956–962 CrossRef CAS.
- W. Li, Y. Fan, Y. Xia, P. Rocchi, R. Zhu, F. Qu, J. Neyts, J. L. Iovanna and L. Peng, Cu-Mediated Selective N-Arylation of Aminotriazole Acyclonucleosides, Helv. Chim. Acta, 2009, 92, 1503–1513 CrossRef CAS.
- Y. Liu, Y. Xia, Y. Fan, A. Maggiani, P. Rocchi, F. Qu, J. L. Iovanna and L. Peng, N-Aryltriazole ribonucleosides with potent antiproliferative activity against drug-resistant pancreatic cancer, Bioorg. Med. Chem. Lett., 2010, 20, 2503–2507 CrossRef CAS PubMed.
- T. A. Moss, M. S. Addie, T. Nowak and M. J. Waring, Room-Temperature Palladium-Catalyzed Coupling of Heteroaryl Amines with Aryl or Heteroaryl Bromides, Synlett, 2012, 285–289 CrossRef CAS.
- J. Shen and H. Zhang, Synthesis of 1-substituted 3-amino-1H-1,2,4-triazoles from ethyl N-(5-phenyl-1,2,4-oxadiazol-3-yl)formimidate, Tetrahedron, 2015, 71, 6164–6169 CrossRef CAS.
- V. M. Chernyshev, O. V. Khazipov, M. A. Shevchenko, A. Y. Chernenko, A. V. Astakhov, D. B. Eremin, D. V. Pasyukov, A. S. Kashin and V. P. Ananikov, Revealing the unusual role of bases in activation/deactivation of catalytic systems: O–NHC coupling in M/NHC catalysis, Chem. Sci., 2018, 9, 5564–5577 RSC.
- V. M. Chernyshev, O. V. Khazipov, M. A. Shevchenko, D. V. Pasyukov, J. V. Burykina, M. E. Minyaev, D. B. Eremin and V. P. Ananikov, Discovery of the N–NHC Coupling Process under the Conditions of Pd/NHC- and Ni/NHC-Catalyzed Buchwald–Hartwig Amination, Organometallics, 2022, 41, 1519–1531 CrossRef CAS.
- V. M. Chernyshev, E. A. Denisova, D. B. Eremin and V. P. Ananikov, The key role of R–NHC coupling (R=C, H, heteroatom) and M–NHC bond cleavage in the evolution of M/NHC complexes and formation of catalytically active species, Chem. Sci., 2020, 11, 6957–6977 RSC.
- A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, A New Mode of Operation of Pd-NHC Systems Studied in a Catalytic Mizoroki–Heck Reaction, Organometallics, 2017, 36, 1981–1992 CrossRef CAS.
- E. G. Gordeev, D. B. Eremin, V. M. Chernyshev and V. P. Ananikov, Influence of R–NHC Coupling on the Outcome of R–X Oxidative Addition to Pd/NHC Complexes (R=Me, Ph, Vinyl, Ethynyl), Organometallics, 2018, 37, 787–796 CrossRef CAS.
- D. O. Prima, M. Madiyeva, J. V. Burykina, M. E. Minyaev, D. A. Boiko and V. P. Ananikov, Evidence for “cocktail”-type catalysis in Buchwald–Hartwig reaction. A mechanistic study, Catal. Sci. Technol., 2021, 11, 7171–7188 RSC.
- M. S. Viciu, R. F. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Activation and Reactivity of (NHC)Pd(allyl)Cl (NHC=N-Heterocyclic Carbene) Complexes in Cross-Coupling Reactions, Organometallics, 2002, 21, 5470–5472 CrossRef CAS.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, Modified (NHC)Pd(allyl)Cl (NHC=N-Heterocyclic Carbene) Complexes for Room-Temperature Suzuki−Miyaura and Buchwald−Hartwig Reactions, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS.
- P. R. Melvin, D. Balcells, N. Hazari and A. Nova, Understanding Precatalyst Activation in Cross-Coupling Reactions: Alcohol Facilitated Reduction from Pd(II) to Pd(0) in Precatalysts of the Type (η3-allyl)Pd(L)(Cl) and (η3-indenyl)Pd(L)(Cl), ACS Catal., 2015, 5, 5596–5606 CrossRef CAS.
- O. V. Khazipov, M. A. Shevchenko, D. V. Pasyukov, A. Y. Chernenko, A. V. Astakhov, V. A. Tafeenko, V. M. Chernyshev and V. P. Ananikov, Preventing Pd–NHC bond cleavage and switching from nano-scale to molecular catalytic systems: amines and temperature as catalyst activators, Catal. Sci. Technol., 2020, 10, 1228–1247 RSC.
- M. Sayah and M. G. Organ, Potassium Isopropoxide: For Sulfination It is the Only Base You Need!, Chem. – Eur. J., 2013, 19, 16196–16199 CrossRef CAS PubMed.
- N. Ishida, Y. Kamae and M. Murakami, Preparation of Ni(cod)2 Using Light as the Source of Energy, Organometallics, 2019, 38, 1413–1416 CrossRef CAS.
- H. Hu, A. M. Gilliam, F. Qu and K. H. Shaughnessy, Enolizable Ketones as Activators of Palladium(II) Precatalysts in Amine Arylation Reactions, ACS Catal., 2020, 10, 4127–4135 CrossRef CAS.
- S.-T. Kim and M.-H. Baik, Reductive activation of PdII-precatalysts via decarboxylation of pivalate in direct C–H arylation reactions, Chem. Commun., 2020, 56, 13868–13871 RSC.
- N. Ishida, Y. Masuda, F. Sun, Y. Kamae and M. Murakami, A Strained Vicinal Diol as a Reductant for Coupling of Organyl Halides, Chem. Lett., 2019, 48, 1042–1045 CrossRef CAS.
- N. Ishida, Y. Kamae, K. Ishizu, Y. Kamino, H. Naruse and M. Murakami, Sustainable System for Hydrogenation Exploiting Energy Derived from Solar Light, J. Am. Chem. Soc., 2021, 143, 2217–2220 CrossRef CAS PubMed.
- J. Klett, Structural Motifs of Alkali Metal Superbases in Non-coordinating Solvents, Chem. – Eur. J., 2021, 27, 888–904 CrossRef CAS PubMed.
- A. Y. Kostyukovich, A. M. Tsedilin, E. D. Sushchenko, D. B. Eremin, A. S. Kashin, M. A. Topchiy, A. F. Asachenko, M. S. Nechaev and V. P. Ananikov, In situ transformations of Pd/NHC complexes with N-heterocyclic carbene ligands of different nature into colloidal Pd nanoparticles, Inorg. Chem. Front., 2019, 6, 482–492 RSC.
- T. Szilvási and T. Veszprémi, Internal Catalytic Effect of Bulky NHC Ligands in Suzuki–Miyaura Cross-Coupling Reaction, ACS Catal., 2013, 3, 1984–1991 CrossRef.
- O. G. Parchment, I. H. Hillier, D. V. S. Green, N. A. Burton, J. O. Morley and H. F. Schaefer, A theoretical study, using ab initio methods, of tautomerism in 3-amino-1,2,4-triazole in the gas phase and in aqueous solution, J. Chem. Soc., Perkin Trans. 2, 1992, 1681–1684 RSC.
- M. H. Palmer and D. Christen, An ab initio study of the structure, tautomerism and molecular properties of the C- and N-amino-1,2,4-triazoles, J. Mol. Struct., 2004, 705, 177–187 CrossRef CAS.
- A. V. Dolzhenko, G. Pastorin, A. V. Dolzhenko and W. K. Chui, An aqueous medium synthesis and tautomerism study of 3(5)-amino-1,2,4-triazoles, Tetrahedron Lett., 2009, 50, 2124–2128 CrossRef CAS.
- M. Pagacz-Kostrzewa, R. Bronisz and M. Wierzejewska, Theoretical and matrix isolation FTIR studies of 3-amino-1,2,4-triazole and its isomers, Chem. Phys. Lett., 2009, 473, 238–246 CrossRef CAS.
- M. Pagacz-Kostrzewa, A. Bil and M. Wierzejewska, UV-induced proton transfer in 3-amino-1,2,4-triazole, J. Photochem. Photobiol., A, 2017, 335, 124–129 CrossRef CAS.
- S. Meng, Y. Zhao, J. Xue and X. Zheng, Environment-dependent conformation investigation of 3-amino-1,2,4-triazole (3-AT): Raman Spectroscopy and density functional theory, Spectrochim. Acta, Part A, 2018, 190, 478–485 CrossRef CAS PubMed.
- G. F. Kröger and W. Freiberg, Ionisationskonstanten von 1, 2, 4-Triazolen [1], Z. Chem., 1965, 5, 381–382 CrossRef.
- G. I. Koldobskii and V. A. Ostrovskii, Acid-base properties of five-membered nitrogen-containing heterocycles (review), Chem. Heterocycl. Compd., 1988, 24, 469–480 CrossRef.
- D. M. Khramov, E. L. Rosen, J. A. V. Er, P. D. Vu, V. M. Lynch and C. W. Bielawski, N-Heterocyclic carbenes: deducing σ- and π-contributions in Rh-catalyzed hydroboration and Pd-catalyzed coupling reactions, Tetrahedron, 2008, 64, 6853–6862 CrossRef CAS.
- L. G. Pezük, B. Şen, F. E. Hahn and H. Türkmen, Heterobimetallic Complexes Bridged by Imidazol{[4,5-f][1,10]-phenanthrolin}-2-ylidene: Synthesis and Catalytic Activity in Tandem Reactions, Organometallics, 2019, 38, 593–601 CrossRef.
- K.-T. Chan, Y.-H. Tsai, W.-S. Lin, J.-R. Wu, S.-J. Chen, F.-X. Liao, C.-H. Hu and H. M. Lee, Palladium Complexes with Carbene and Phosphine Ligands: Synthesis, Structural Characterization, and Direct Arylation Reactions between Aryl Halides and Alkynes, Organometallics, 2010, 29, 463–472 CrossRef CAS.
- M. Teci, E. Brenner, D. Matt and L. Toupet, N-Heterocyclic Carbenes Functioning as Monoligating Clamps, Eur. J. Inorg. Chem., 2013, 2013, 2841–2848 CrossRef CAS.
- S. G. Guillet, V. A. Voloshkin, M. Saab, M. Beliš, K. Van Hecke, F. Nahra and S. P. Nolan, Understanding existing and designing novel synthetic routes to Pd-PEPPSI-NHC and Pd-PEPPSI-PR3 pre-catalysts, Chem. Commun., 2020, 56, 5953–5956 RSC.
- A. A. Abdel-Hafez, H. A. H. Elsherief, M. Jo, M. Kurokawa, K. Shiraki, T. Kawahata, T. Otake, N. Nakamura and M. Hattori, Synthesis and Evaluation of Anti-HIV-1 and Anti-HSV-1 Activities of 4H-[1,2,4]-Triazolo [1,5-a]pyrimidin-5-one Derivatives, Arzneimittelforschung, 2002, 52, 833–839 CAS.
- V. M. Chernyshev, E. V. Tarasova, A. V. Chernysheva and V. A. Taranushich, Synthesis of 3-pyridyl-substituted 5-amino-1,2,4-triazoles from aminoguanidine and pyridinecarboxylic acids, Russ. J. Appl. Chem., 2011, 84, 1890–1896 CrossRef CAS.
- E. V. Tarasova, V. M. Chernyshev, A. V. Chernysheva and R. S. Abagyan, Thermodynamic and kinetic aspects of a single-reactor synthesis of 5-amino-3-methyl-1,2,4-triazole hydrochloride from aminoguanidine and acetic acid, Russ. J. Appl. Chem., 2011, 84, 400–406 CrossRef CAS.
- N. V. Palysaeva, K. P. Kumpan, M. I. Struchkova, I. L. Dalinger, A. V. Kormanov, N. S. Aleksandrova, V. M. Chernyshev, D. F. Pyreu, K. Y. Suponitsky and A. B. Sheremetev, A Direct Approach to a 6-Hetarylamino[1,2,4]triazolo[4,3-b][1,2,4,5]tetrazine Library, Org. Lett., 2014, 16, 406–409 CrossRef CAS PubMed.
- R. Romagnoli, F. Prencipe, P. Oliva, S. Baraldi, P. G. Baraldi, A. Brancale, S. Ferla, E. Hamel, R. Bortolozzi and G. Viola, 3-Aryl/Heteroaryl-5-amino-1-(3′,4′,5′-trimethoxybenzoyl)-1,2,4-triazoles as antimicrotubule agents. Design, synthesis, antiproliferative activity and inhibition of tubulin polymerization, Bioorg. Chem., 2018, 80, 361–374 CrossRef CAS PubMed.
- S. Platte, M. Korff, L. Imberg, I. Balicioglu, C. Erbacher, J. M. Will, C. G. Daniliuc, U. Karst and D. V. Kalinin, Microscale Parallel Synthesis of Acylated Aminotriazoles Enabling the Development of Factor XIIa and Thrombin Inhibitors, ChemMedChem, 2021, 16, 3672–3690 CrossRef CAS PubMed.
- L. E. A. Godfrey and F. Kurzer, Heterocyclic compounds from urea derivatives. Part I. A new synthesis of 3-amino-5-mercapto(and -hydroxy)-1,2,4-triazoles, J. Chem. Soc., 1960, 3437–3444 RSC.
- A. Er-Rhaimini and R. Mornet, Synthesis and photochemical degradation of N-arylmethyl derivatives of the herbicide 3-amino-1,2,4-triazole, J. Heterocycl. Chem., 1992, 29, 1561–1566 CrossRef CAS.
- F. Kurzer and K. Douraghi-Zadeh, Heterocyclic compounds from urea derivatives. Part VII. Addition-products of diaminoguanidine and carbodi-imides, and their cyclisation, J. Chem. Soc., 1965, 3912–3922 RSC.
- A. I. Matesanz, J. Perles and P. Souza, New palladium and platinum complexes with bioactive 3,5-diacetyl-1,2,4-triazol bis(4-cyclohexyl thiosemicarbazone) ligand: chemistry, antiproliferative activity and preliminary toxicity studies, Dalton Trans., 2012, 41, 12538–12547 RSC.
- Q. Cao, W. I. Nicholson, A. C. Jones and D. L. Browne, Robust Buchwald–Hartwig amination enabled by ball-milling, Org. Biomol. Chem., 2019, 17, 1722–1726 RSC.
- G. V. Boyd and S. R. Dando, The action of cyanamide on 1,3,4-oxadiazolium and pyrylium salts, J. Chem. Soc. C, 1971, 3873–3875 RSC.
- S. N. Santos, G. Alves de Souza, T. M. Pereira, D. P. Franco, C. de Nigris Del Cistia, C. M. R. Sant'Anna, R. B. Lacerda and A. E. Kümmerle, Regioselective microwave synthesis and derivatization of 1,5-diaryl-3-amino-1,2,4-triazoles and a study of their cholinesterase inhibition properties, RSC Adv., 2019, 9, 20356–20369 RSC.
- J. Shen, B. Wong, C. Gu and H. Zhang, Negishi Approach to 1,5-Disubstituted 3-Amino-1H-1,2,4-triazoles, Org. Lett., 2015, 17, 4678–4681 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- P. Hirva, M. Haukka, M. Jakonen and M. A. Moreno, DFT tests for group 8 transition metal carbonyl complexes, J. Mol. Model., 2008, 14, 171–181 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Density functional theory for transition metals and transition metal chemistry, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- M. Steinmetz and S. Grimme, Benchmark Study of the Performance of Density Functional Theory for Bond Activations with (Ni,Pd)-Based Transition-Metal Catalysts, ChemistryOpen, 2013, 2, 115–124 CrossRef CAS PubMed.
- A. V. Astakhov, S. B. Soliev and V. M. Chernyshev, Metal-ligand bond dissociation energies in the Ni, Pd, and Pt complexes with N-heterocyclic carbenes: effect of the oxidation state of the metal (0,+2), Russ. Chem. Bull., 2020, 69, 2073–2081 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. Klamt, C. Moya and J. Palomar, A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach, J. Chem. Theory Comput., 2015, 11, 4220–4225 CrossRef CAS PubMed.
- G. A. Zhurko, Chemcraft - graphical program for visualization of quantum chemistry computations, Version 1.8 (buid 622b); Zhurko, G. A., Ivanovo, Russia, 2005, https://chemcraftprog.com Accessed on May 28, 2022 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and DFT calculations data, single crystal X-ray data and copies of NMR spectra. CCDC 2194495, 2194496 and 2194497. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi01832b |
| This journal is © the Partner Organisations 2023 |