
Selective Buchwald–Hartwig arylation of C-amino-1,2,4-triazoles and other coordinating aminoheterocycles enabled by bulky NHC ligands and TPEDO activator†
 a
Mikhail E.
Minyaev,
b
Victor M.
Chernyshev
*a
and
Valentine P.
Ananikov
*ab
a
Mikhail E.
Minyaev,
b
Victor M.
Chernyshev
*a
and
Valentine P.
Ananikov
*ab
Abstract
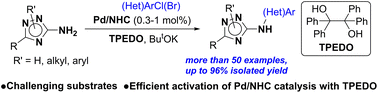
C-Amino-1,2,4-triazoles are challenging polynitrogen substrates for metal-catalyzed arylation due to their multidentate character, enhanced coordinating ability and decreased nucleophilicity of the amino group. In the present study, the Buchwald–Hartwig cross-coupling of diverse 3(5)-amino-1,2,4-triazoles with aryl chlorides and bromides delivering (hetero)arylamino-1,2,4-triazoles in good-to-excellent yields under Pd/NHC catalysis was developed. The use of Pd complexes with bulky NHC ligands such as IPr*OMe and TPEDO (1,1,2,2-tetraphenylethane-1,2-diol) as an in situ Pd(II) to Pd(0) reductant enabled the selective arylation of the NH2 group even in acidic NH unprotected substrates and deactivated 1-substituted 5-amino- and 4-substituted 3-amino-1,2,4-triazoles. The reaction mechanism and structure–activity relationships were studied with DFT calculations. A significant effect of the position of the N-substituent in the 1,2,4-triazole ring on the favorable reaction pathways was revealed.
 Please wait while we load your content...
Please wait while we load your content...

