
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroactive sulfur-rich materials obtained via inverse vulcanization of a diallylsilyl-functionalized ferrocene†
María
Vera-Tuset
a,
Rubén
Mas-Ballesté
 ab,
Isabel
Cuadrado
ab,
Alicia
Moya
*a and
Sonia
Bruña
*ab
ab,
Isabel
Cuadrado
ab,
Alicia
Moya
*a and
Sonia
Bruña
*ab
aDepartamento de Química Inorgánica, Facultad de Ciencias, Calle Francisco Tomás y Valiente, 7, Universidad Autónoma de Madrid, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain. E-mail: sonia.brunna@uam.es; alicia.moya@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Facultad de Ciencias, Calle Francisco Tomás y Valiente, 7, Universidad Autónoma de Madrid, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
First published on 6th February 2024
Abstract
Reaction of elemental sulfur with 1,1′-bis(dimethylallylsilyl)ferrocene (1) in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 or 70:30 wt% at 130 °C results in new polymeric materials as a consequence of an inverse vulcanization process. Through this procedure, allylsilyl-functionalized ferrocene moieties are integrated into a solid matrix structured by the formation of covalent C–S bonds between sulfur atom chains and organometallic co-monomers. The structure and composition of the amorphous materials obtained have been assessed by elemental analysis, TXRF, ATR-IR, solid-state NMR, XPS and SEM. Remarkably, the electrochemical activity of ferrocene fragments is preserved when incorporated into the polysulfide material. Consequently, 1 is the first silicon- and ferrocene-containing molecule being used as a co-monomer to develop redox-responsive polysulfide materials. Furthermore, the interaction of sulfur atoms with Cd(II) or Hg(II) metal centers, present in aqueous solutions, results in the modulation of the electrochemical signal of ferrocene moieties. Therefore, this work presents the synergistic combination of polysulfide chains and ferrocene fragments resulting in electrochemical recognition of harmful metal centers commonly present in polluted hydric resources.
:50 or 70:30 wt% at 130 °C results in new polymeric materials as a consequence of an inverse vulcanization process. Through this procedure, allylsilyl-functionalized ferrocene moieties are integrated into a solid matrix structured by the formation of covalent C–S bonds between sulfur atom chains and organometallic co-monomers. The structure and composition of the amorphous materials obtained have been assessed by elemental analysis, TXRF, ATR-IR, solid-state NMR, XPS and SEM. Remarkably, the electrochemical activity of ferrocene fragments is preserved when incorporated into the polysulfide material. Consequently, 1 is the first silicon- and ferrocene-containing molecule being used as a co-monomer to develop redox-responsive polysulfide materials. Furthermore, the interaction of sulfur atoms with Cd(II) or Hg(II) metal centers, present in aqueous solutions, results in the modulation of the electrochemical signal of ferrocene moieties. Therefore, this work presents the synergistic combination of polysulfide chains and ferrocene fragments resulting in electrochemical recognition of harmful metal centers commonly present in polluted hydric resources.
Introduction
Inverse vulcanization is an emergent route that Pyun and coworkers materialized a decade ago, in 2013,1 as an alternative to obtain polymers with a high sulfur content, also known as polysulfides. While in traditional vulcanization of rubber, found accidentally by Charles Goodyear in 1839, sulfur is introduced in small amounts as a bridge between organic polymer chains,2 inverse vulcanization gives rise to polymers with a much higher sulfur content. Therefore, in the past years, inverse vulcanization has been exploited for reevaluating and enhancing sulfur uses in a sustainable and low-cost process.1,3 Sulfur is a generally benign and exceptionally Earth-abundant element, mainly in volcanic areas. It is produced in high quantities in oil refining and natural gas purification processes. However, its demand is still far from meeting its high levels of production. In this regard, in the last decade, inverse vulcanization reactions have boosted the use of sulfur as a cheap, abundant, and accessible feedstock for high sulfur- content polymeric materials, which is highly desirable.4Inverse vulcanization reactions are carried out at temperatures above the melting point of sulfur (120 °C) when a ring-opening polymerization (ROP) of the S8 ring occurs. Di-radicals (˙(Sn)˙), formed after the ROP, bind to the unsaturated co-monomers (olefinic fragments) in a clean process, in which sulfur acts both as a reactant and as the reaction medium, avoiding the use of solvents.5 Elemental sulfur can self-polymerize, but the resulting polymers are not stable and tend to revert to the S8 ring at room temperature. However, polymers obtained by inverse vulcanization are chemically stable and processable, as the unsaturated co-monomers stabilize the polymer chains.6,7 Despite its novelty, inverse vulcanization has already been experimented with important developments.8 For instance, Hasell and coworkers have recently introduced the use of metal-based catalysts to allow lower temperatures and the use of volatile organic monomers.9,10 Furthermore, acceleration of inverse vulcanization using trialkyl amines as catalysts has been reported.11 Other improvements involve the photoinduced inverse vulcanization,12 which permits catalyst-free reactions at room temperature, or mechanochemical inverse vulcanization,13,14 which in addition allows short reaction times, high atom economy and a greener procedure. A new strategy for the synthesis of high-performance sulfur-based polymers has also been described, which involves anionic hybrid copolymerization of elemental sulfur with acrylate at room temperature, yielding co-polymers with short polysulfide segments.15
Due to the high sulfur content, some polysulfides obtained by inverse vulcanization have shown valuable applications as cathode materials for Li–S batteries,16–19 in infrared thermal imaging and optics,20,21 as flame retardant materials,4 to develop fully renewable adhesive polysulfides,22 or as agents for environmental remediation.23,24 The latter application is based on the preference of sulfur atoms for soft metal centers, such as Hg2+, Ag+, or Pt2+. Among them, the most investigated metal has been mercury,25–28 due to its high toxicity and its ability to generate both important health and environmental problems.
In terms of the co-monomer nature, inverse vulcanization has been investigated with terminal, internal or even exo olefins,29 but it has been examined mainly with linkers of organic nature.29–35 Only a few examples of inorganic co-monomers have been reported so far, primarily based on silane or siloxane linkages,36–39 including complex cyclosiloxanes40 and polyhedral oligomeric silsesquioxanes (POSS),41,42 or based on tin43 as a main group heteroatom-linker. However, to the best of our knowledge, examples of inverse vulcanization with olefin-functionalized co-monomers incorporating transition metals have not yet been reported. Overall, co-monomers of different nature have been commonly used to modulate the material's properties, which focus exclusively on the polysulfide fragments. Therefore, the use of functional linkers that provide the materials with properties non-related to sulfur atoms is still widely disregarded. As a result, there are still important challenges to address, such as the development of methods allowing co-polymerization of linkers containing transition metals, with inherent redox, conductive, or even catalytic properties. Such findings would give more relevance to the co-monomer that binds sulfur, which could act synergistically to obtain co-polymers with improved properties and specific applications.
In this regard, an interesting, robust and redox-responsive transition metal-containing building block is bis(η5-cyclopentadienyl)iron(II) or ferrocene (Fe(η5-C5H5)2). The unique geometry, thermodynamic and kinetic stabilities, versatile reactivity, and redox properties of this iconic molecule have made ferrocene one of the most widely studied organometallic compounds.44,45 Remarkably, ferrocene is readily functionalized, which enables its facile incorporation into more complex molecular systems. Furthermore, covalently incorporating ferrocene into polymers can deeply modify their physical and chemical properties. As a result, ferrocene-based metallopolymers are a distinctive class of functional materials that have gained much attention in recent years.46–50 The possibility to tune the properties of ferrocene-based polymers and macromolecules relies on the redox activity of ferrocene, since this organometallic molecule undergoes fast one-electron oxidation to the positively charged ferrocenium form [Fe(η5-C5H5)2]+. By switching the redox state (FeII/FeIII) of the multiple ferrocenyl units (separately, or in a cooperative fashion) the charge, polarity, color, and hydrophilicity of the molecules may be controlled. Consequently, the exclusive features of metallopolymers having multiple ferrocene units provide specific electrochemical, photo-physical, catalytic, and biological properties with potential for applications as multi-electron reservoirs, electron-transfer mediators, redox sensors and building blocks for electronic devices and smart materials.48,51–54 Other relevant and advanced applications of ferrocene-based derivatives have been extensively reviewed.49,55,56
Recently, we have reported the uses of 1,3-divinyl-1,3-dimethyl-1,3-diferrocenyldisiloxane ([(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)FcMeSi]2O),57 diferrocenylacetylene (FcC
CH)FcMeSi]2O),57 diferrocenylacetylene (FcC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CFc),58 vinylferrocene (FcCHCH2),59 and ethynylferrocene (FcCCH)60 (Fc = Fe(η5-C5H4)(η5-C5H5)) as efficient electroactive ferrocene-based precursors for thiol–ene and thiol–yne radical reactions. Such hydrothiolation reactions allowed the incorporation of the electroactive ferrocene as backbone, pendant, or peripheral moieties into polymers and macromolecules with and without silane connecting groups. The resulting polymers and macromolecules retain the valuable electrochemical activity of ferrocene.
CFc),58 vinylferrocene (FcCHCH2),59 and ethynylferrocene (FcCCH)60 (Fc = Fe(η5-C5H4)(η5-C5H5)) as efficient electroactive ferrocene-based precursors for thiol–ene and thiol–yne radical reactions. Such hydrothiolation reactions allowed the incorporation of the electroactive ferrocene as backbone, pendant, or peripheral moieties into polymers and macromolecules with and without silane connecting groups. The resulting polymers and macromolecules retain the valuable electrochemical activity of ferrocene.
Based on our previous results concerning hydrothiolation chemistry, we envisioned that alkenylsilane-containing ferrocenes would be interesting precursors for structurally new types of ferrocene- and sulfur-rich metallopolymers through inverse vulcanization. In the present study, we explore the inverse vulcanization reaction of elemental sulfur and the allylsilyl-difunctionalized ferrocene (Fc[Si(CH3)2(–CH2–CHCH2)]2) 1, as co-monomer. Regarding the electrochemical properties of ferrocene and the binding abilities of sulfur, the resulting hybrid electroactive materials could act as sensors for polluting cations with a high environmental significance, such as Cd2+ and Hg2+. Thus, the focus of this work relies on the synergistic combination of the sulfur polymer and the ferrocene-containing co-metallopolymer.
Results and discussion
Synthesis of polysulfide materials based on silicon-containing ferrocenyl units through inverse vulcanization
The key requirements of an inverse vulcanization linker are the presence of unsaturated CC bonds and a suitable, relatively high, boiling point. On the other hand, due to the electron-rich aromatic cyclopentadienyl rings, the ferrocene molecule is readily functionalized via diverse synthetic modifications, which enables its facile incorporation into more complex molecular systems. Based on this, as a first step to achieve the synergistic combination between an electroactive co-monomer and the sulfur chain, a specific linker containing an alkenylsilane-functionalized ferrocene has been designed. Particularly, we decided to focus our attention on a ferrocene bifunctionalized with two reactive allylsilane groups, the 1,1′-bis(dimethylallylsilyl)ferrocene (Fc[Si(CH3)2(–CH2–CHCH2)]2) 1. Furthermore, the presence of two reactive groups in the linker allows the obtention of cross-linked polysulfides, with electroactive ferrocenyl units as an integral part of the main chain.
The precursor 1 was synthesized following the procedure published by Cuadrado and coworkers for similar compounds.61 This route involved a 1,1′-dilithiation reaction of ferrocene with n-butyllithium (n-BuLi), in the presence of TMEDA (N,N,N′,N′-tetramethylethane-1,2-diamine),62 followed by a salt-elimination reaction of Fe(η5-C5H4Li)2·TMEDA with allyl(chloro)dimethylsilane (Scheme 1). Compound 1 was isolated as an air-stable, orange oil, which was purified by column chromatography. The structure of precursor 1 was confirmed by 1H, 13C and 29Si Nuclear Magnetic Resonance (NMR) and Attenuated Total Reflectance InfraRed (ATR-IR) spectroscopies and mass spectrometry (see ESI†). As expected, the 1H NMR spectrum shows three multiplet signals, centered at δ 5.79, 4.88 and 4.84 ppm, and the 13C NMR spectrum presents signals at δ 135.0, 113.2 and 24.9 ppm, all of them corresponding to the allyl groups. In the 29Si NMR spectrum, a single signal appears at δ −3.5 ppm, which is consistent with the value observed for the analogous ferrocene derivative with two vinylsilane groups.61
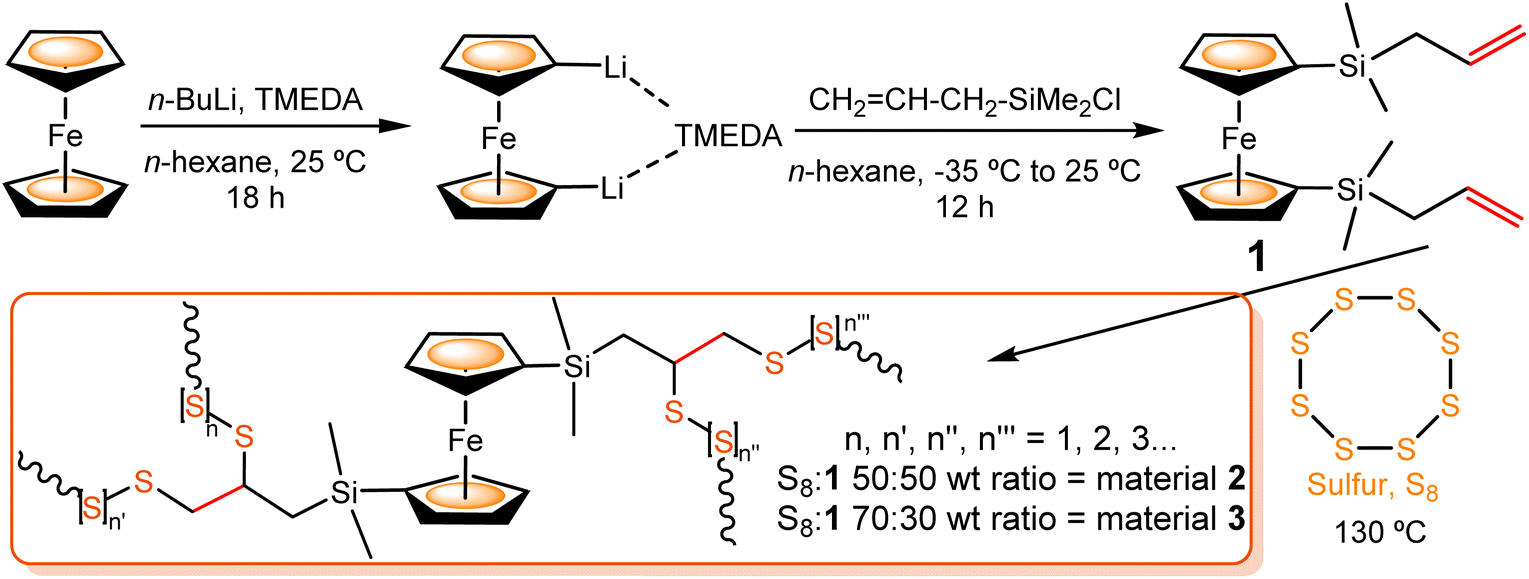 | ||
| Scheme 1 Synthesis of diallylsilyl-functionalized ferrocene co-monomer 1 and polysulfide materials 2 and 3, obtained by inverse vulcanization reaction of elemental sulfur and 1. | ||
To investigate the reactivity of the two allyl groups (covalently linked to the organometallic unit in co-monomer 1) against elemental sulfur, different vulcanization reaction conditions were tested. Specifically, we optimized the reaction conditions using two different S8:1 weight ratios: 50:50 and 70:30 (see Scheme 1 and Table 1).
| Entry | Mass S8 (mg) | Mass 1 (mg) | Time | Material obtained (mass)a | S:Fe ratiob |
|---|---|---|---|---|---|
| a Final mass isolated. b Calculated from TXRF analysis of materials 2 and 3, respectively (see below). c For this control experiment bare ferrocene was used instead of compound 1. | |||||
| 1 | 100 | 100 | 6 h 30 min | 2 (69 mg) | 7:1 |
| 2 | 233 | 100 | 3 h 45 min | 3 (103 mg) | 13:1 |
| 3 | 100 | 0 | 6h | — | — |
| 4 | 0 | 100 | 6h | — | — |
| 5 | 100 | 100 (Fc)c | 6h | — | — |
Considering the melting point of sulfur (∼120 °C), we first tried the milder reaction conditions possible (130 °C), to preserve the integrity of the organometallic moiety (Scheme 1). A first vulcanization reaction with a S8:1 50:50 wt% was explored. After 3 h, the oily solid obtained was washed with CH2Cl2, giving unreacted compound 1 as proved by 1H NMR. The residual yellow solid was found to be unreacted sulfur, soluble in CS2 after several washes. Then, we increased the reaction temperature, but after 3 h at 150 °C a blackish rubber was formed. Its ATR-IR spectrum presented no recognizable signals. The same occurred when the reaction was performed at 130 °C for 20 h. Finally, we investigated the reaction at 130 °C until a red rubber-like material appeared (material 2), after 6 h and 30 min (entry 1). The inverse vulcanization of sulfur with 1 (50:50) was repeated twice at the optimal temperature (130 °C), to investigate its reproducibility. In both cases, rubber-like semisolids were obtained again, showing identical ATR-IR spectra to the one of material 2.
Given the promising results obtained with the 50:50 wt% inverse vulcanization reaction, tested with 1,1′-bis(dimethylallylsilyl)ferrocene 1 as the cross-linked monomer, we subsequently investigated the S8:1 70:30 wt% at 130 °C (see entry 2 in Table 1). After 3 h and 45 min, the formation of a red rubber-like semisolid (material 3) was observed, with identical spectroscopic features to the ones of material 2 (see below). The reproducibility of this reaction was confirmed under the specified conditions.
It is important to emphasize the fact that these reactions occur at a relatively low temperature, clearly indicating the high reactivity of organometallic linker 1 through inverse vulcanization reactions. The reactivity of diallylsilyl-functionalized ferrocene 1 against elemental sulfur, in terms of temperature, is much more similar to that of small organic linkers, such as styrene16 or 4-vinylbenzyl chloride63 (130 °C), than to that of inorganic siloxane-based linkers, such as 1,3-diallyl-tetramethyldisiloxane (which requires 180 °C) or 1,1,3,3-tetramethyl-1,3-divinyldisiloxane (160 °C and a Zn catalyst).39 In addition, it is worth noting that these reactions do not require a catalyst, avoiding not only its use but also its removal after the catalyzed reaction.
The ferrocene-containing polysulfide materials 2 and 3 are insoluble in common organic solvents. They were thoroughly washed with CH2Cl2, removing unreacted compound 1 as proved by 1H NMR experiments, and with CS2 to remove unreacted S8.
To prove that the formation of the semisolid materials was due to the inverse vulcanization reaction, three control experiments were performed (Table 1, entries 3–5). All of them were heated at 130 °C for 6 hours. Control 1 (entry 3) involved the heating process of elemental sulfur, which melted. After cooling, it gave a more crystalline solid, but neither a polymer nor an insoluble material. Starting diallylsilyl-functionalized ferrocene 1 was also heated (control 2, entry 4) but no change was observed in its 1H NMR spectrum, showing that compound 1 does not self-polymerize after 6 hours at 130 °C. Finally, a 50:50 wt% reaction between sulfur and ferrocene was investigated (control 3, entry 5). The solid obtained was first washed with CH2Cl2, giving unreacted ferrocene as proved by 1H NMR. The residual yellow solid was sulfur and no material remained after the washing process with CS2. These control experiments prove that the materials obtained after mixing linker 1 and sulfur, under the optimal reaction conditions, correspond to the ensuing products of the inverse vulcanization reaction of S8 and the allyl functionalities present in co-monomer 1 (absent in pure ferrocene). They also prove that materials 2 and 3 are not simple co-solidification products, therefore, new covalent C–S bonds must have been formed.
Characterization of polysulfide materials with ferrocenyl units
Elemental analysis values supported the sulfur-reach nature of both materials obtained by inverse vulcanization reactions (Table S1†), and allowed the estimation of the average number of S atoms per Fc[Si(CH3)2(–CH2–CH–CH2–)]2 unit. As can be seen in Table S1†, material 2 contains an average of 8 sulfur atoms per ferrocenyl unit, while material 3 is estimated to contain 16. Similar results were obtained by total X-ray fluorescence (TXRF) analysis, which allowed to quantify the iron content. This analytical data indicates a S:Fe ratio of 7:1 in material 2 and 13:1 in material 3 (see Fig. S5 and S6†). Consequently, the elemental analysis and the TXRF results proved that at a higher sulfur weight ratio in the reactants, more S atoms bind to 1 forming the final cross-linked polymers. Thus, different starting S8:1 wt% resulted in materials with distinct sulfur content, which was further confirmed by Energy Dispersive X-Ray Spectroscopy (EDX) analysis (see below).
The insoluble and rubber-like materials 2 and 3 could not be ground using a mortar and a pestle, therefore, they were pressed giving uniform films, suitable for ATR-IR spectroscopy. Fig. 1A shows the ATR-IR spectra of materials 2 and 3, together with the spectrum of their ferrocenyl precursor 1. The ferrocenyl and methylsilane peaks appear, as expected, in all the spectra. However, the peaks related to the –CHCH2 group (highlighted in orange in Fig. 1A) can be observed in the spectrum of compound 1 at 1630 cm−1 (ν(CC)) and at 992 and 931 cm−1 (δ (Csp2–H)), but they are absent in the two materials spectra, clearly indicating the reaction of the allyl group through the inverse vulcanization process. The presence of a new peak at around 1100 cm−1 (yellow area) is observed for the obtained materials, which can be assigned to ν(C–S) vibrations, suggesting the formation of new C–S bonds.29,39
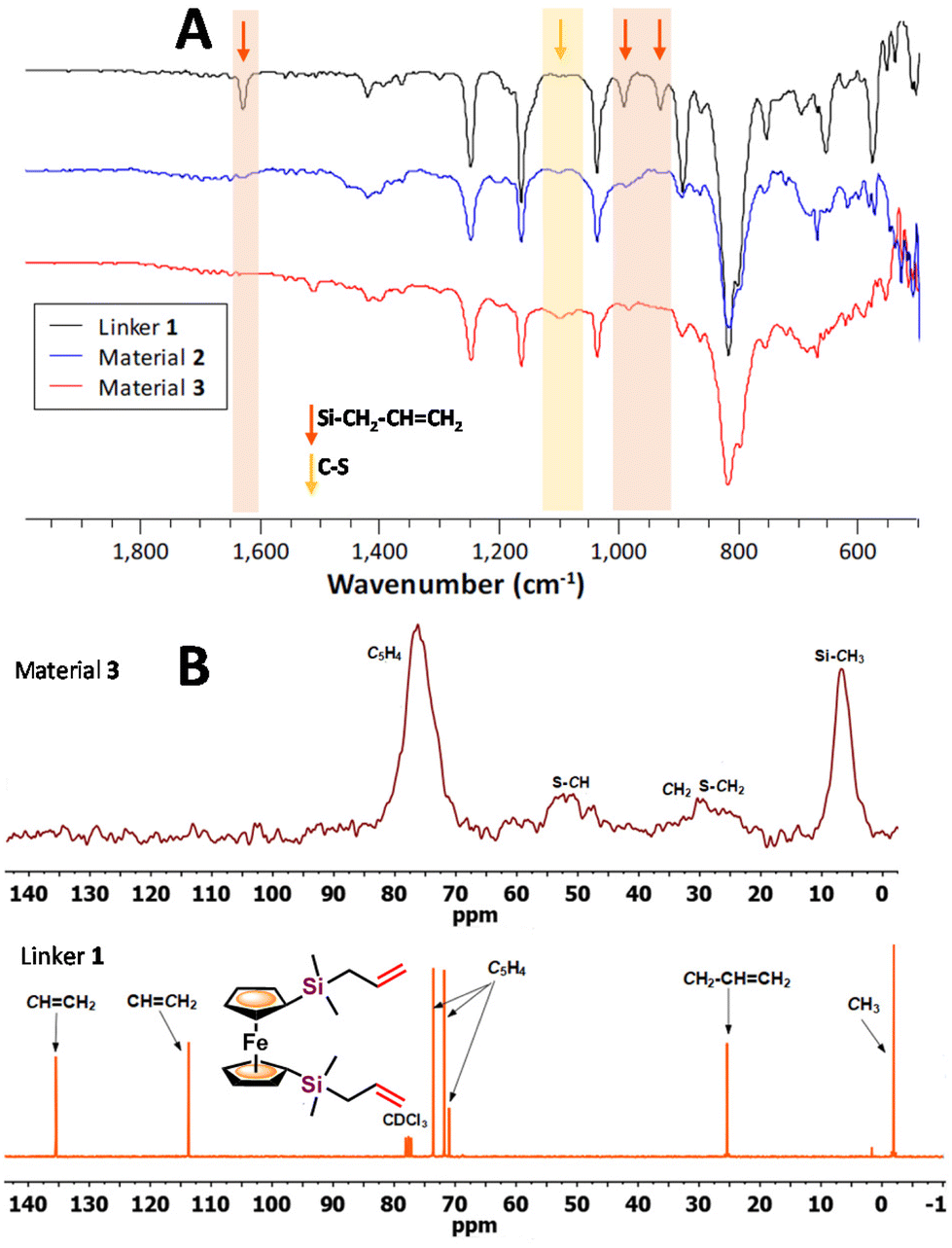 | ||
| Fig. 1 (A) ATR-IR spectrum comparison of starting diallylsilyl-functionalized ferrocene 1 and materials 2 and 3. (B) SS 13C CP/MAS NMR spectrum of material 3 (up) and 13C NMR spectrum in CDCl3 (300 MHz) of starting 1 (down). | ||
The chemical nature of the materials obtained was also investigated through solid-state 13C cross-polarization magic-angle spinning (SS 13C CP/MAS) NMR. Fig. 1B compares the solid-state 13C CP/MAS NMR spectrum of material 3, with the 13C NMR spectrum in CDCl3 of precursor 1. The spectrum of material 2 (see Fig. S7†) displays the same peaks shown for material 3. Some of the signals in the spectra of the materials are comparable to the ones observed in the solution spectrum of precursor 1: at ∼δ 73 ppm for the ferrocenyl rings, ∼δ 24 ppm for the methylene (CH2) units, and ∼δ 0.6 ppm corresponding to the methyl groups. However, no signals are observed in the area between δ 110 and 140 ppm, corresponding to the allyl moieties, and new signals appear at around δ 43–49 ppm assigned to S–CH, and at δ 21–25 ppm for S–CH2 groups, together with the CH2–CHCH2 signal. These observations fully support an effective reaction between compound 1 and elemental sulfur, giving rise to the formation of materials with new C–S bonds and electroactive ferrocenyl units.
The 29Si CP/MAS NMR spectra of both materials (Fig. S8 and S10†) show only one signal, at δ −2.6 ppm for material 2 and δ −2.5 ppm for material 3. These values correspond to silicon atoms bonded to alkyl groups. They are only slightly shifted downfield, about δ 1 ppm, compared to the same signal of precursor 1 (δ −3.5 ppm). This small difference reflects that the inverse vulcanization reaction does not induce a significant modification in the electronic environment of the silicon atoms within the –Si–CH2–CHCH2 functionalities when transformed into new –Si–CH2–CH(S)n–CH2–(S)n′ units.
X-ray Photoelectron Spectroscopy (XPS) was used to investigate the chemical composition of the two inverse vulcanized polymer materials 2 and 3. The XPS survey spectra (Fig. 2A) of both materials show signals corresponding to C, S, Fe, Si and O atoms. The main difference observed is in the relative intensity of the sulfur signal but no significant difference in the binding energy of all the atoms is detected. This indicates variations in the sulfur-to-ferrocene ratios between both samples, with material 3 (70:30 initial wt%) presenting a higher S/Fe ratio compared to material 2 (50:50 initial wt%) (2.5 and 1.7, respectively).
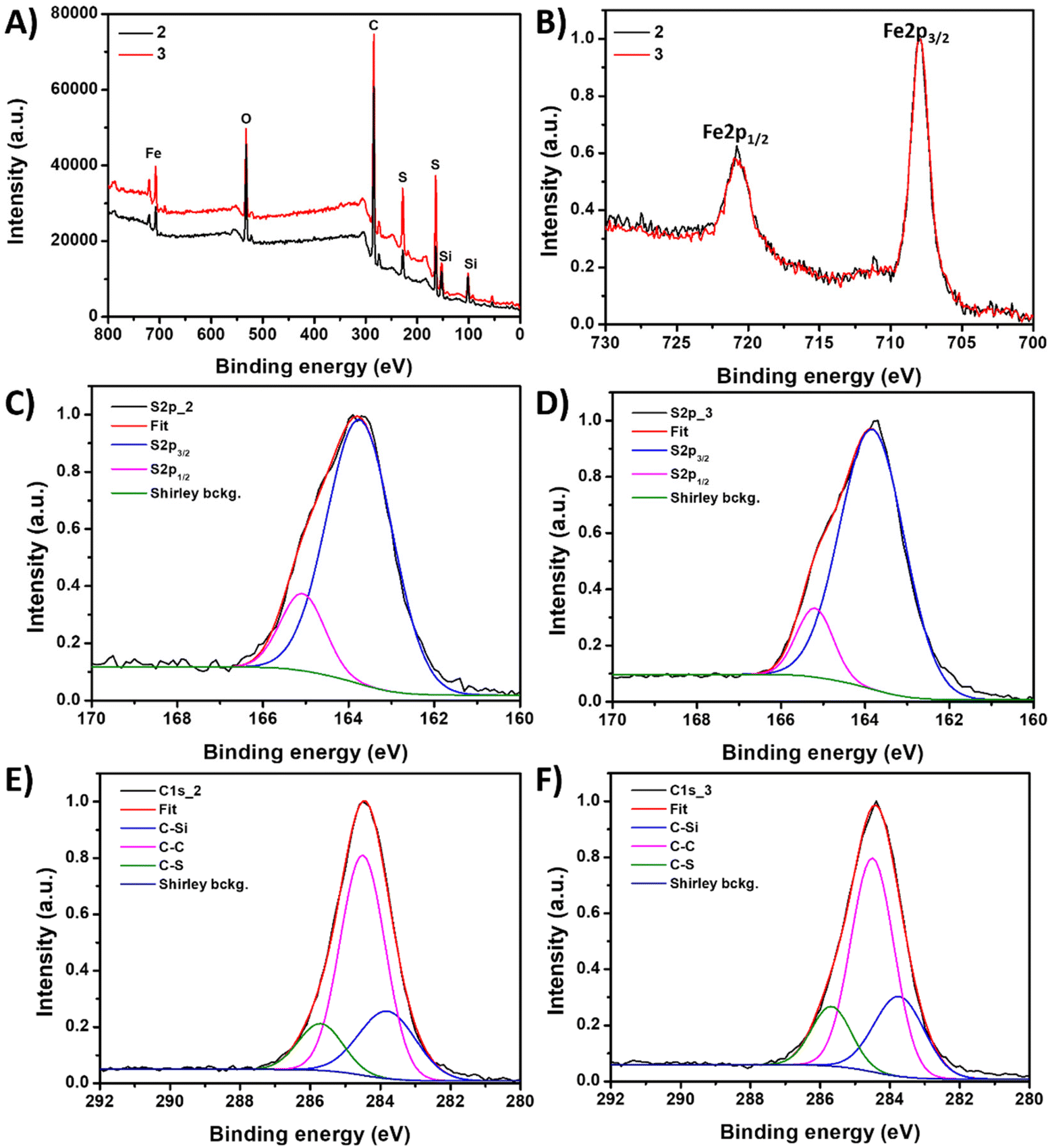 | ||
| Fig. 2 (A) XPS spectra of material 2 (black line) and 3 (red line) samples. (B) XPS spectra of both samples in the Fe 2p region. (C and D) Fit spectra of the S 2p region for materials 2 and 3, respectively. (E and F) Fit spectra of the C 1s region for materials 2 and 3, respectively. | ||
Furthermore, a fitting analysis of the XPS spectra was performed to obtain essential insight into the oxidation states of the atoms, especially S and Fe atoms. The analysis of the S 2p region (Fig. 2C and D) displays two signals centered at 163.7 and 165.1 eV, which are commonly attributed in the literature to S–S and S–C environments, respectively.13,39 This interpretation aligns with previous studies that assigned these signals to sulfur atoms incorporated in hydrocarbon chains, specifically C–S–S–C and C–S–C groups.64 Notably, the absence of signals at binding energies higher than 166 eV confirms the absence of oxidized sulfur species, such as sulfonates or sulfates in any of the samples. In the Fe 2p region, two contributions appear at 720.6 eV and 707.9 eV (Fig. 2B). These signals correspond to the 2p1/2 and 2p3/2 orbitals of Fe(II)65 and confirm that the ferrocene units were not oxidized during the material synthesis, preserving their original redox properties. Additionally, the XPS analysis indicates that the Fe from the ferrocene units is not chemically connected to the sulfur atoms.
The fit of the C 1s region (Fig. 2E and F) has three main contributions at 283.7, 284.5 and 285.7 eV which are ascribed to C–Si, C–C and C–S bonds,66 respectively, consistent with the material's composition. Therefore, the XPS results agree with the solid NMR experiments and suggest that the sulfur has successfully reacted with the diallylsilyl-functionalized ferrocene co-monomer and chemically connected with carbon to form C–S bonds, avoiding oxidation during the inverse vulcanization process. Similarly, the ferrocene units were preserved in the polymer structure, retaining their redox properties.
Scanning Electron Microscopy (SEM) was employed to investigate the morphology of the materials 2 and 3 obtained through the process of inverse vulcanization. Independently of the reactant proportion, the resulting materials display a rubber-like appearance, consistent with the texture of crosslinked network structures observed in similar systems.67
To gain a closer view of the microscale morphology of the material, cryogenic grinding was performed under liquid N2, breaking the material into small grains. SEM images (Fig. 3B and C) revealed a heterogeneous morphology, comprising grains with smooth top surfaces. Importantly, the morphology of sulfur after a heat treatment, identical to the material's synthesis except for the addition of the co-monomer (Fig. 3A), was not observed in the resulting materials.
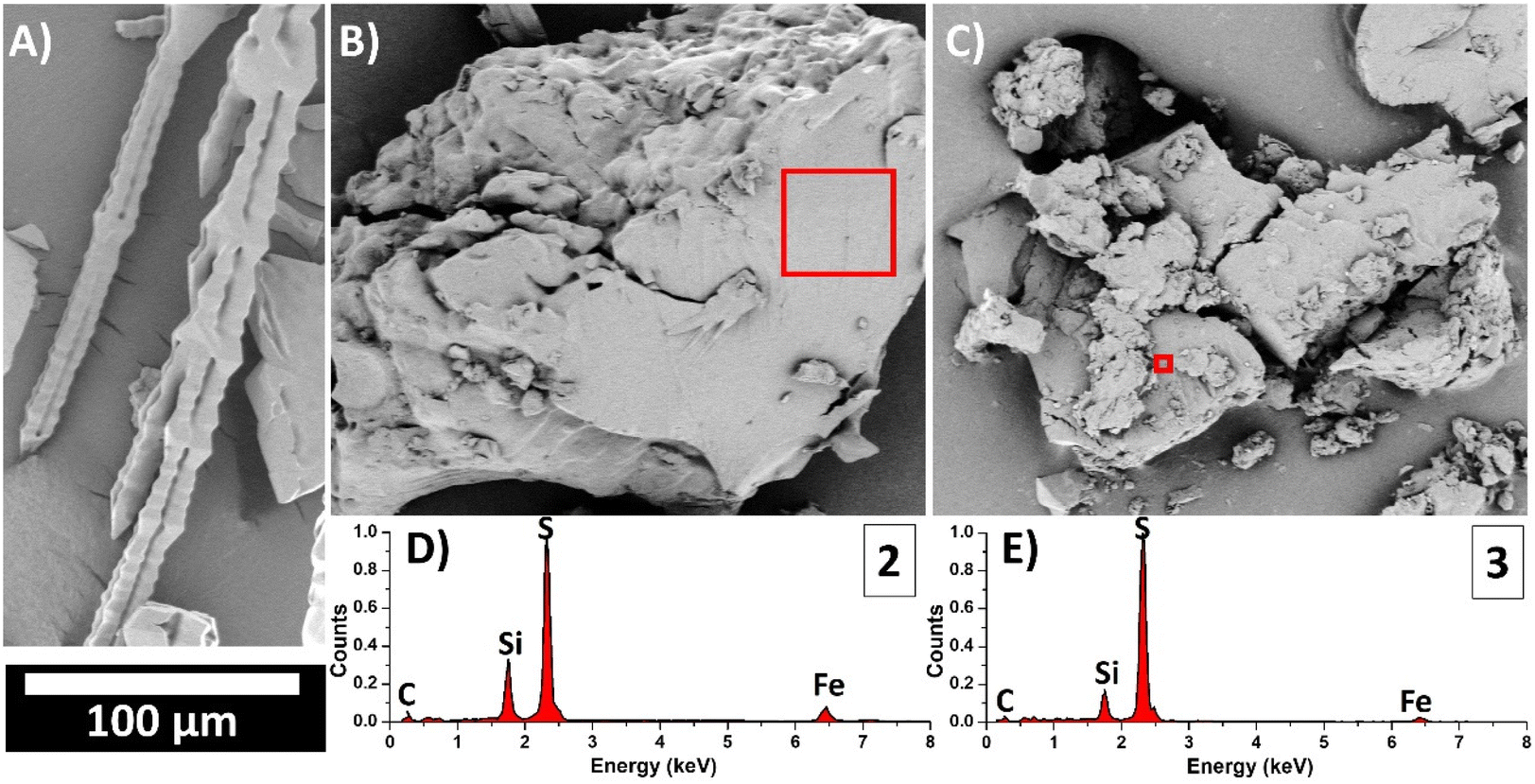 | ||
| Fig. 3 SEM images of (A) heat-treated sulfur, (B) material 2 and (C) material 3 samples. Red boxes represent the areas where EDX spectra of (D) material 2 and (E) material 3 samples were acquired. The bar scale is the same for the three SEM images. | ||
To semi-quantitatively explore the chemical composition of the materials, SEM was combined with Energy Dispersive X-Ray Spectroscopy (SEM-EDX). Multiple regions of interest were analyzed (see Table S2†), enabling a detailed analysis of the material's homogeneity. The results indicated relative atomic concentrations of S/Fe of 6.9 ± 0.8% and 15.8 ± 0.6% (mean ± SD) for materials 2 (S8:1 50:50 initial wt%) and 3 (S8:1 70:30 initial wt%), respectively. EDX spectra for each sample are also included in Fig. 3D and E and correspond to the areas highlighted with red boxes in Fig. 3B and C. The comparison of these spectra demonstrates an increase in the intensity signal of sulfur and a decrease in the signals of C, Si and Fe from the material 2 to the material 3. Therefore, the EDX analysis confirmed the expected S/Fe ratios for both samples (see Table S2†), validating the accuracy of the synthesis procedure and the elemental analysis and TXRF data.
The thermal stability of the ferrocenyl-functionalized polysulfides was investigated by thermogravimetric analysis (TGA). Both materials are stable up to around 200 °C, for experiments performed under a nitrogen atmosphere (Fig. S12 and S13†). This temperature is similar to the one reported for diverse polysulfides obtained by inverse vulcanization reactions, with different linkers.17,30,39 As expected, increasing sulfur content in the materials decreases the residual weight at 900 °C, which is higher (10.8%) for the material obtained with a 50:50 wt% and a high content of ferrocenyl species than for the S8:1 70:30 (8.3%).
Electrochemical studies
Before the study of the redox properties of materials 2 and 3, we investigated the solution electrochemical behavior of 1,1′-bis(dimethylallylsilyl)ferrocene (1) by cyclic voltammetry (CV) and square wave (SWV) voltammetry. The data were recorded in a 10−1 M acetonitrile solution of the supporting electrolyte (tetra-n-butylammonium hexafluorophosphate, [n-Bu4N][PF6]) and 10−3 M of 1. The working electrode was a gold-disk.The cyclic voltammetric response of diallylsilyl-functionalized ferrocene 1 (see Fig. S14†) exhibits a single well-resolved oxidation at E1/2 = 450 mV vs. a saturated calomel electrode (SCE), assigned to the Fe+II/Fe+III redox system. In addition, the plot of peak current versus v1/2 (v = scan rate) is linear (Fig. S15†), indicating that the redox process is diffusion-controlled.68,69 The voltammetric features (Ipc/Ipa = 1.00; the peak-to-peak separation values, ΔEp, 74 mV at slow scan rates, and Ep independent of the scan rate) show that the oxidation of ferrocenyl-containing compound 1 is electrochemically reversible,68,69 resulting in the production of the stable and soluble species [Fc[Si(CH3)2(–CH2–CHCH2)]2]+. After 20 scans, reversibility remains unaffected.
Then, we focused on the electrochemical behavior of materials 2 and 3, obtained by inverse vulcanization reactions. In principle, sulfur-rich ferrocene-containing polymers 2 and 3 are particularly interesting for the modification of electrodes, as they contain thioether and polysulfide bonds of the –Si–(CH2)2–S–(S)n– type, potentially suitable for chemisorption processes. In addition, the modification of electrode surfaces with electroactive species is a field of high activity due to the applications of modified electrodes in important areas of research such as biosensor development, molecular recognition, electrocatalysis, and molecular electronics.68,70 Common procedures to fabricate conducting modified electrodes consist of chemisorption, electrodeposition or evaporatively cast film formation. However, these methodologies require the immersion of the working electrode into an organic solution containing the dissolved modifier, or the evaporation of the modifier solution into the electrode.71 Consequently, carbon paste-based electrodes modified with ferrocene-containing polysulfides 2 and 3 were prepared. The use of chemically modified carbon paste electrodes has attracted increasing attention due to their valuable advantages, such as chemical inertness, robustness, renewability, stable response, no need for internal solution, and suitability for electrocatalysis and sensitive and selective detection of environmental contaminants, pharmaceutical compounds, and biological species.72 Notably, they are non-toxic and environmentally friendly electrodes.
Accordingly, a hollow electrode was properly filled with a homogenized hybrid paste, formed by the corresponding ferrocene-based sulfur-rich electroactive material, high-purity graphite powder and one drop of mineral oil as the binder. An initial assessment of the electrochemical features of materials 2 and 3 was obtained using a 10−1 M CH3CN solution of [n-Bu4N][PF6], as the solvent/electrolyte medium.
Table 2 summarizes the electrochemical parameters obtained by CV and SWV for both materials, Fig. 4 shows their cyclic voltammograms and Fig. S16† their square wave voltammograms. The cyclic voltammetric responses of both materials exhibit a pair of broad peaks at E1/2 = 661 mV (2) and E1/2 = 703 mV (3) vs. SCE, assigned to the Fe+II/Fe+III redox system. As can be observed, the peak-to-peak separation values, (ΔEp = 587 in the case of material 2, and 307 for material 3), are in both cases considerably greater than the one observed in solution for their precursor 1 (74 mV). This significant difference can be attributed to the kinetic limitations in the charge transport through the films formed by this type of modified electrodes.73 Electrodes modified with materials 2 and 3 showed stable and reproducible electrochemical responses, at least, after five months of the first measurement.
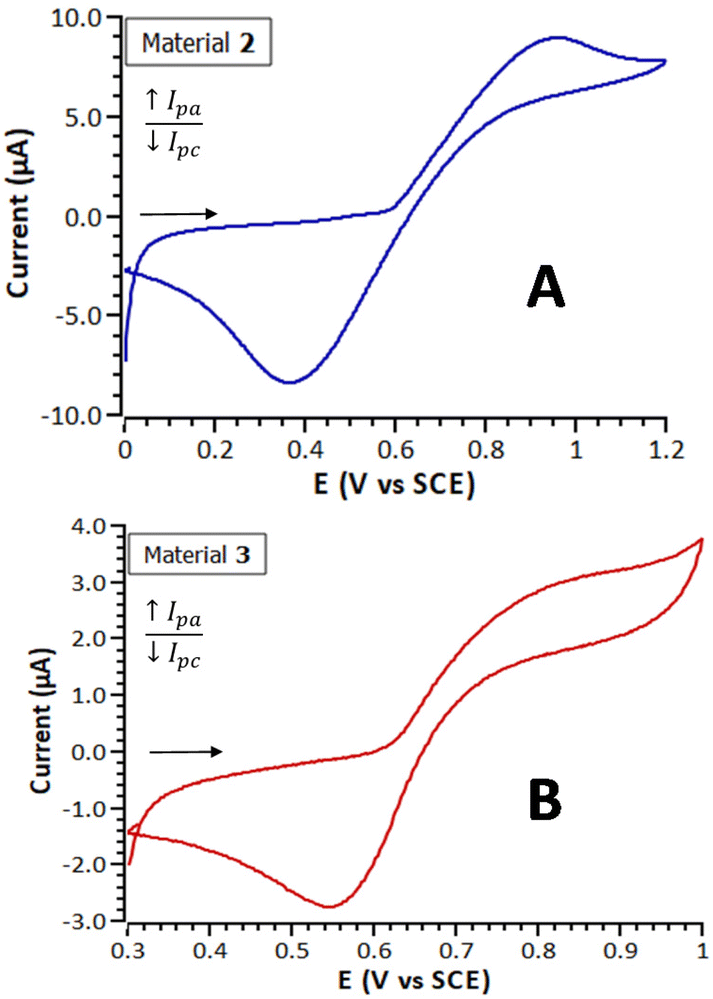 | ||
| Fig. 4 Cyclic voltammograms (first cycle shown of the 10 cycles measured in each case) of materials 2 (A) and 3 (B) measured with a carbon paste working electrode, in a 10−1 M CH3CN solution of [n-Bu4N][PF6], at 50 mV s−1. | ||
| Material | SWV | CV | |||
|---|---|---|---|---|---|
| E 1/2 (mV) | E pa | E pc | E 1/2 = (Epa + Epc)/2 (mV) | ΔEp = Epa − Epc (mV) | |
| a Potentials measured against SCE, using a carbon paste working electrode, a 10−1 M CH3CN solution of [n-Bu4N][PF6] as solvent/electrolyte medium, and under a scan rate of 50 mV s−1. | |||||
| 2 | 669 | 955 | 368 | 661 | 587 |
| 3 | 679 | 857 | 550 | 703 | 307 |
In the SWV voltammograms of materials 2 and 3 (Fig. S16†) two voltammetric waves can be observed. Under these measured conditions, the SWV experiments show a better resolution of the redox processes than the corresponding CV voltammograms, in which only broad peaks are detected. This observation indicates that not all the electroactive ferrocenyl units within the polymer chains of materials 2 and 3 present the same electrochemical environment, as expected for a heterogeneous crosslinked structure of this type. The peak splitting phenomenon has been previously observed, for example in gold electrode surfaces modified with films of ferrocenylalkanethiolate self-assembled monolayers, and it is attributed to two different structural domains.74
Cation recognition studies of carbon paste electrodes modified with ferrocene-containing polysulfide 2
In the field of electrochemical detection, ferrocene-based polymers and macromolecules play an important role because they are useful for electrochemical sensing cations, anions, and neutral molecules by a change in the oxidation potential of the ferrocene moiety.75,76 In addition, modified carbon paste electrodes are particularly useful for the analysis of metal ions using voltammetric methods.A remarkable structural feature of the sulfur- and metallocenyl-rich materials 2 and 3 is that they could combine, in a synergistic manner, the potential cation-binding ability of the sulfur atoms with the redox activity of the ferrocenyl moieties. The electroactive groups can be affected by the presence of the cations interacting with the sulfur atoms, and consequently, materials 2 and 3 constitute potential candidates for sensing heavy- and transition-metal ions.
As proof of concept, we explored the electrochemical behavior of material 2, which presents the highest content of the electroactive ferrocenyl species. To investigate its applicability to the detection of heavy metal pollutants in contaminated hydric resources, we performed SWV experiments (Fig. 5) with material 2 in the presence of water solutions of Zn2+, Cd2+ and Hg2+. SWV was selected since this technique is more sensitive than other electrochemical methods, and it is particularly useful when electrode processes are coupled with adsorption phenomena.77
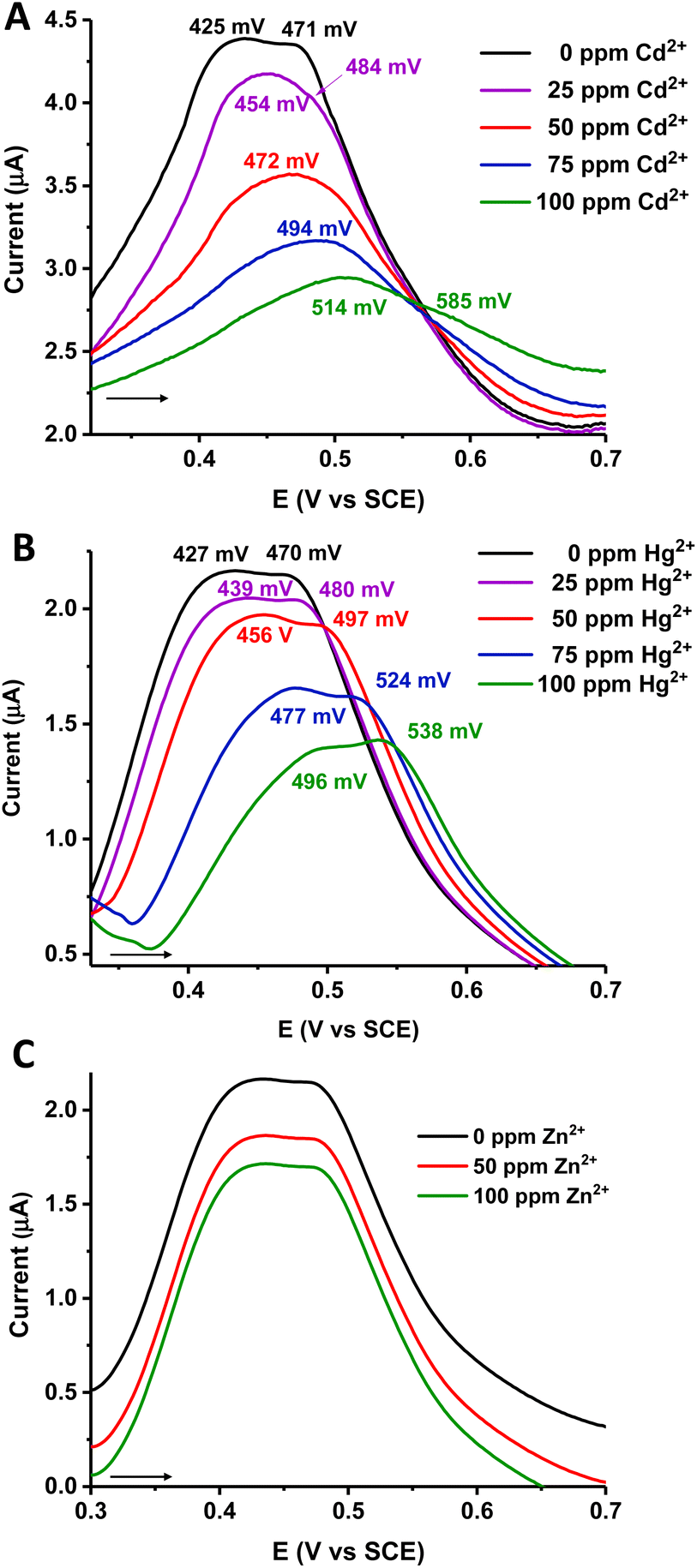 | ||
| Fig. 5 Evolution of the SWV of the carbon paste working electrode prepared with ferrocenyl-functionalized material 2, recorded in a 0.1 M water solution of [NH4][PF6], when increasing amounts of Cd2+ (A), Hg2+ (B) or Zn2+ (C) cations were added. | ||
To a water solution of [NH4][PF6] (0.4 mL, 0.1 M), increasing amounts (from 25 ppm to a final 100 ppm concentration) of the corresponding cation were added in a stepwise manner. The electrochemical responses of each cation addition were measured with the carbon paste working electrode modified with material 2. As can be seen in Fig. 5, in the absence of any cation, modified electrode 2 gives two differentiated peaks in the SWV, when recorded in water/[NH4][PF6], with E1/2 values of ∼426 and 470 mV. After the progressive addition of Cd2+ (Fig. 5A) and Hg2+ (Fig. 5B) a clear sequential anodic shift of the oxidation waves is observed, indicating that material 2 can electrochemically recognize both Hg2+ and Cd2+ cations. Furthermore, as the positive charges of the corresponding cation increase near the redox-active sites in 2, a destabilization process of the ferrocenyl moieties occurs in the material, and consequently, they are oxidized at a more positive potential, as expected.57,78
In the case of Cd2+ (Fig. 5A), the more cathodic peak progressively shifts from 425 mV (for 0 ppm of Cd2+) to 514 mV (after cation addition until a final 100 ppm concentration), resulting in a total anodic shift of 89 mV. The more anodic peak, appearing at 471 mV (for 0 ppm of Cd2+) decreases in intensity, concerning the more cathodic wave, as the Cd2+ is added. After a final 100 ppm cation concentration, this second peak appears at 585 mV, 114 mV shifted from its starting peak potential. Additions over 100 ppm of Cd2+ gave no recognized changes in the SWV response. On the other hand, when the Hg2+ cation is progressively added to the water/[NH4][PF6] solution, both starting peaks of material 2 (at 427 mV and 470 mV) are shifted to more positive potentials. The maximum perturbation of the SWV peaks is again obtained at a Hg2+ cation concentration of 100 ppm (Fig. 5B), with the more cationic wave shifting to 496 mV (shift = 69 mV) and the more anodic to 538 mV (shift = 68 mV).
In general, the responses of modified electrode 2 are similar against both cations, only with slightly higher shifts in the case of the Cd2+ addition. Sulfur atoms, which are the majority in the polymer chains, are expected to be harder bases than sulfide units, in a considerable minority. This fact could explain that material 2 can recognize not only the softest Hg2+ cation, but also the Cd2+. Interestingly, as proved by the initial SWV responses (at 0 ppm for both cation series) recorded in Fig. 5A and B, the observed Cd2+ and Hg2+ interactions with material 2 are reversible. Thus, after thorough washing of the modified electrode in water, this can be further reused.
In pronounced contrast, we found that addition of up to 100 ppm of the Zn2+ cation, the hardest of the series, leaves the SWV electrochemical response of modified electrode 2 essentially unchanged (see Fig. 5C). Therefore, the synergistic effect between sulfur atoms, which are the ones that probably interact with the soft Cd2+ and Hg2+ cations, and the electroactive ferrocenyl moieties that form part of the structure of material 2, allows the qualitative electrochemical recognition of two different cations in water solutions. Consequently, carbon paste electrodes modified with ferrocene-containing polysulfides 2 and 3 are promising in the development of electrochemical sensors.
Conclusions
In the present study, the inverse vulcanization reaction of elemental sulfur and allylsilyl difunctionalized ferrocene 1 (Fc[Si(CH3)2(–CH2–CHCH2)]2), as co-monomer, was explored for the first time. The high reactivity of allylsilyl fragments in 1 allowed its quantitative incorporation into a polysulfide matrix through an inverse vulcanization procedure, under mild conditions. Analytical, spectroscopic and microscopic techniques confirm the successful covalent hybridization of ferrocene fragments with elemental sulfur preserving the molar ratios of the starting precursors. This approach allows the interesting strategy of introducing physico-chemical properties into sulfur-based materials that are not specific to polysulfide fragments. In particular, redox-responsive materials are isolated as a consequence of including electroactive ferrocene moieties. In addition, electrochemical properties can be combined with the recognition phenomena shown by sulfur chains towards soft metal centers. Thus, the results reported here suggest that a synergetic combination of the ferrocene and polysulfide properties can be exploited in the design of a new generation of electrochemical sensors, which would be of environmental interest due to their potential to detect heavy metal pollutants in contaminated waters. From a broader perspective, this work demonstrates the feasibility of incorporating redox-active metallocene building blocks into sulfur-based polymers by inverse vulcanization procedures, opening the door to a wide range of functional materials with applications related to metal centers, such as sensing, optoelectronics or catalysis.
Author contributions
Conceptualization, S.B. and I.C.; synthesis, M. V.-T and S.B.; characterization, M. V.-T, R.M.-B, S.B. and A.M.; electrochemical studies, S.B. and I.C.; cation recognition studies, S.B., I.C. and R.M.-B; writing, original draft preparation and editing, S.B., A.M., I.C., and R.M.-B; supervision, S.B. and A.M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministerio de Ciencia e Innovación (MICINN) of Spain for its financial support through projects PID2021-125207NB-C31, PID2019–110637RB-I00 and PID2022–141016OB-I00.Notes and references
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524, DOI:10.1038/NCHEM.1624.
- C. Guise-Richardson, Technol. Cult., 2010, 51, 357–387 CrossRef.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502, DOI:10.1002/anie.201604615.
- K.-S. Kang, K. A. Iyer and J. Pyun, Chem. – Eur. J., 2022, 28, e202200115, DOI:10.1002/chem.202200115.
- J. Bao, K. P. Martin, E. Cho, K.-S. Kang, R. S. Glass, V. Coropceanu, J. Bredas, Jr., W. O. Parker, J. T. Njardarson and J. Pyun, J. Am. Chem. Soc., 2023, 145, 12386–12397, DOI:10.1021/jacs.3c03604.
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125, DOI:10.1016/j.progpolymsci.2016.04.003.
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22, DOI:10.1021/jacs.1c09329.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105, 10.1039/c9py00636b.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10(647) DOI:10.1038/s41467-019-08430-8.
- B. Zhang, H. Gao, P. Yan, S. Petcher and T. Hasell, Mater. Chem. Front., 2020, 4, 669–675, 10.1039/c9qm00606k.
- J. H. Hwang, J. M. Lee, J. H. Seo, G. Y. Noh, W. Byun, S. Kim, W. Lee, S. Park, D.-G. Kim and Y. S. Kim, Green Chem., 2023, 25, 4641–4646, 10.1039/d3gc01102j.
- J. Jia, J. Liu, Z.-Q. Wang, T. Liu, P. Yan, X.-Q. Gong, C. Zhao, L. Chen, C. Miao, W. Zhao, S. Cai, X.-C. Wang, A. I. Cooper, X. Wu, T. Hasell and Z. Quan, Nat. Chem., 2022, 14, 1249–1257, DOI:10.1038/s41557-022-01049-1.
- P. Yan, W. Zhao, F. McBride, D. Cai, J. Dale, V. Hanna and T. Hasell, Nat. Commun., 2022, 13(4824) DOI:10.1038/s41467-022-32344-7.
- L. He, J. Yang, H. Jiang, H. Zhao and H. Xia, Ind. Eng. Chem. Res., 2023, 62, 9587–9594, DOI:10.1021/acs.iecr.3c00801.
- H. Yang, J. Huang, Y. Song, H. Yao, W. Huang, X. Xue, L. Jiang, Q. Jiang, B. Jiang and G. Zhang, J. Am. Chem. Soc., 2023, 145, 14539–14547, DOI:10.1021/jacs.3c04746.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116, DOI:10.1002/pola.28266.
- A. Hoefling, Y. J. Lee and P. Theato, Macromol. Chem. Phys., 2017, 218, 1600303, DOI:10.1002/macp.201600303.
- R. Liu, D. Lin, Q. Wang, Y. Chen and D. Wang, ACS Sustainable Chem. Eng., 2023, 11, 8517–8523, DOI:10.1021/acssuschemeng.3c00988.
- S. Ren, P. Sang, W. Guo and Y. Fu, Polym. Chem., 2022, 13, 5676–5690, 10.1039/d2py00823h.
- T. S. Kleine, R. S. Glass, D. L. Lichtenberger, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2020, 9, 245–259, DOI:10.1021/acsmacrolett.9b00948.
- D. A. Boyd, C. C. Baker, J. D. Myers, V. Q. Nguyen, G. A. Drake, C. C. McClain, F. H. Kung, S. R. Bowman, W. Kim and J. S. Sanghera, Chem. Commun., 2017, 53, 259–262, 10.1039/c6cc08307b.
- K. B. Sayer, V. L. Miller, Z. Merrill, A. E. Davis and C. L. Jenkins, Polym. Chem., 2023, 14, 3091–3098, 10.1039/d3py00390f.
- F. G. Müller, L. S. Lisboa and J. M. Chalker, Adv. Sustainable Syst., 2023, 7, 2300010, DOI:10.1002/adsu.202300010.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718, DOI:10.1002/anie.201508708.
- H. Lin, Y. Lai and Y. Liu, ACS Sustainable Chem. Eng., 2019, 7, 4515–4522, DOI:10.1021/acssuschemeng.8b06815.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386, 10.1039/c6cc00938g.
- M. Mann, B. Zhang, S. J. Tonkin, C. T. Gibson, Z. Jia, T. Hasell and J. M. Chalker, Polym. Chem., 2022, 13, 1320–1327, 10.1039/d1py01416a.
- N. A. Lundquist, Y. Yin, M. Mann, S. J. Tonkin, A. D. Slattery, G. G. Andersson, C. T. Gibson and J. M. Chalker, Polym. Chem., 2022, 13, 5659–5665, 10.1039/d2py00903j.
- Y. Onose, Y. Ito, J. Kuwabara and T. Kanbara, Polym. Chem., 2022, 13, 5486–5493, 10.1039/d2py00774f.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2016, 49, 767–773, DOI:10.1021/acs.macromol.5b02791.
- H. Berk, B. Balci, S. Ertan, M. Kaya and A. Cihaner, Mater. Today Commun., 2019, 19, 336–341, DOI:10.1016/j.mtcomm.2019.02.014.
- Y. Zhang, N. G. Pavlopoulos, T. S. Kleine, M. Karayilan, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 7–12, DOI:10.1002/pola.29266.
- B. Zhang, S. Petcher and T. Hasell, Chem. Commun., 2019, 55, 10681–10684, 10.1039/c9cc04380b.
- S. Park, D. Lee, H. Cho, J. Lim and K. Char, ACS Macro Lett., 2019, 8, 1670–1675, DOI:10.1021/acsmacrolett.9b00827.
- N. Q. Tufts, N. C. Chiu, E. N. Musa, T. C. Gallagher, D. B. Fast and K. C. Stylianou, Chem. – Eur. J., 2023, 29, e202203177, DOI:10.1002/chem.202203177.
- J. M. Scheiger, M. Hoffmann, P. Falkenstein, Z. Wang, M. Rutschmann, V. W. Scheiger, A. Grimm, K. Urbschat, T. Sengpiel, J. Matysik, M. Wilhelm, P. A. Levkin and P. Theato, Angew. Chem., Int. Ed., 2022, 61, e202114896, DOI:10.1002/anie.202114896.
- A. P. Grimm, J. M. Scheiger, P. W. Roesky and P. Theato, Polym. Chem., 2022, 13, 5852–5860, 10.1039/d2py00773h.
- D. Wang, Z. Tang, R. Huang, H. Li, C. Zhang and B. Guo, Macromolecules, 2022, 55, 8485–8494, DOI:10.1021/acs.macromol.2c01678.
- K. W. Park, Z. Zujovic and E. M. Leitao, Macromolecules, 2022, 55, 2280–2289, DOI:10.1021/acs.macromol.1c02558.
- K. W. Park, E. A. Tafili, F. Fan, Z. Zujovic and E. M. Leitao, Polym. Chem., 2022, 13, 4717–4726, 10.1039/d2py00581f.
- R. Anyszka, M. Kozanecki, A. Czaderna, M. Olejniczak, J. Sielski, M. Sicinski, M. Imiela, J. Wreczycki, D. Pietrzak, T. Gozdek, M. Okraska, M. I. Szynkowska, P. Malinowski and D. M. Bielinski, J. Sulfur Chem., 2019, 40, 587–597, DOI:10.1080/17415993.2019.1648470.
- H. Berk, M. Kaya and A. Cihaner, Polym. Chem., 2022, 13, 5152–5158, 10.1039/d2py00761d.
- D. A. Boyd, V. Q. Nguyen, C. C. McClain, F. H. Kung, C. C. Baker, J. D. Myers, M. P. Hunt, W. Kim and J. S. Sanghera, ACS Macro Lett., 2019, 8, 113–116, DOI:10.1021/acsmacrolett.8b00923.
- P. Štěpnička, Dalton Trans., 2022, 51, 8085–8102, 10.1039/d2dt00903j.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29, DOI:10.1002/ejic.201600983.
- D. Schmitt, S. M. Abdel-Hafez, M. Tummeley, V. Schunemann, M. Schneider, V. Presser and M. Gallei, Macromolecules, 2023, 56, 7086–7101, DOI:10.1021/acs.macromol.3c01257.
- T. Zhai, K. Ambrose, A. Nyayachavadi, K. G. Walter, S. Rondeau-Gagne and J. I. Feldblyum, Chem. Commun., 2021, 58, 238–241, 10.1039/d1cc05022b.
- M. Gallei and C. Ruettiger, Chem. – Eur. J., 2018, 24, 10006–10021, DOI:10.1002/chem.201800412.
- R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407, 10.1039/c6cs00155f.
- R. Pietschnig, Chem. Soc. Rev., 2016, 45, 5216–5231, 10.1039/c6cs00196c.
- G. Roy, R. Gupta, S. R. Sahoo, S. Saha, D. Asthana and P. C. Mondal, Coord. Chem. Rev., 2022, 473, 214816, DOI:10.1016/j.ccr.2022.214816.
- X. Su, H. J. Kulik, T. F. Jamison and T. A. Hatton, Adv. Funct. Mater., 2016, 26, 3394–3404, DOI:10.1002/adfm.201600079.
- R. Chen, J. Feng, J. Jeon, T. Sheehan, C. Rüttiger, M. Gallei, D. Shukla and X. Su, Adv. Funct. Mater., 2021, 31, 2009307, DOI:10.1002/adfm.202009307.
- P. B. Medina, V. A. Contreras, F. Hartmann, D. Schmitt, A. Klimek, J. Elbert, M. Gallei and X. Su, ACS Appl. Mater. Interfaces, 2023, 15, 22112–22122, DOI:10.1021/acsami.3c01670.
- Y. Wang, D. Astruc and A. S. Abd-El-Aziz, Chem. Soc. Rev., 2019, 48, 558–636, 10.1039/c7cs00656j.
- Z. Wei, H. Duan, G. Weng and J. He, J. Mater. Chem. C, 2020, 8, 15956–11598, 10.1039/d0tc03810e.
- S. Bruña, I. Martínez-Montero, A. M. González-Vadillo, C. Martín-Fernández, M. M. Montero-Campillo, O. Mó and I. Cuadrado, Macromolecules, 2015, 48, 6955–6969, DOI:10.1021/acs.macromol.5b01683.
- S. Bruña, A. Valverde-González, M. M. Montero-Campillo, O. Mó and I. Cuadrado, Dalton Trans., 2022, 51, 15412–15424, 10.1039/d2dt02378d.
- I. Martínez-Montero, S. Bruña, A. M. González-Vadillo and I. Cuadrado, Macromolecules, 2014, 47, 1301–1315, DOI:10.1021/ma4025202.
- A. Enríquez, A. M. González-Vadillo, I. Martínez-Montero, S. Bruña, L. Leemans and I. Cuadrado, Organometallics, 2014, 33, 7307–7317, DOI:10.1021/om501110w.
- B. García, C. M. Casado, I. Cuadrado, B. Alonso, M. Morán and J. Losada, Organometallics, 1999, 18, 2349–2356, DOI:10.1021/om990109j.
- I. Manners, J. Inorg. Organomet. Polym., 1993, 3, 185–196, DOI:10.1007/BF00683928.
- I. Gomez, A. F. De Anastro, O. Leonet, J. Alberto Blazquez, H. Grande, J. Pyun and D. Mecerreyes, Macromol. Rapid Commun., 2018, 39, 1800529, DOI:10.1002/marc.201800529.
- F. Wang, H. Luo, Q. Wang, J. Wang and J. Xu, Molecules, 2009, 14, 4737–4746, DOI:10.3390/molecules14114737.
- C. M. Woodbridge, D. L. Pugmire, R. C. Johnson, N. M. Boag and M. A. Langell, J. Phys. Chem. B, 2000, 104, 3085–3093, DOI:10.1021/jp993235.
- D. Chen, C. Gan, X. Fan, L. Zhang, W. Li, M. Zhu and X. Quan, Materials, 2019, 12, 2800, DOI:10.3390/ma12172800.
- N. A. Lundquist, A. D. Tikoalu, M. J. H. Worthington, R. Shapter, S. J. Tonkin, F. Stojcevski, M. Mann, C. T. Gibson, J. R. Gascooke, A. Karton, L. C. Henderson, L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2020, 26, 10035–10044, DOI:10.1002/chem.202001841.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd ed, John Wiley & Sons, New York, 2001 Search PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206, DOI:10.1021/acs.jchemed.7b00361.
- R. W. Murray, Molecular Design of Electrode Surfaces, in Techniques of Chemistry, ed. R. W. Murray, John Wiley & Sons, New York, 1992, Vol. 22, pp. 1–45 Search PubMed.
- R. A. Durst, A. J. Baumner, R. W. Murray, R. P. Buck and C. P. Andrieux, Pure Appl. Chem., 1997, 69, 1317–1323, DOI:10.1351/pac199769061317.
- S. Tajik, H. Beitollahi, F. G. Nejad, M. Safaei, K. Zhang, Q. Van Le, R. S. Varma, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 21561–21581, 10.1039/d0ra03672b.
- H. D. Abruña, Coord. Chem. Rev., 1988, 86, 135–189, DOI:10.1016/0010-8545(88)85013-6.
- H. Tian, Y. Dai, H. Shao and H. Yu, J. Phys. Chem. C, 2013, 117, 1006–1012, DOI:10.1021/jp310012v.
- A. Pal, S. R. Bhatta and A. Thakur, Coord. Chem. Rev., 2021, 431, 213685, DOI:10.1016/j.ccr.2020.213685.
- R. Sun, L. Wang, H. Yu. Zain-ul-Abdin, Y. Chen, J. Huang and R. Tong, Organometallics, 2014, 33, 4560–4573, DOI:10.1021/om5000453.
- V. Mirceski, S. Skrzypek and L. Stojanov, Chemtexts, 2018, 4, 17, DOI:10.1007/s40828-018-0073-0.
- C. Arivazhagan, R. Borthakur and S. Ghosh, Organometallics, 2015, 34, 1147–1155, DOI:10.1021/om500948c.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01283b |
| This journal is © The Royal Society of Chemistry 2024 |