
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyethylene and poly(ethylene-co-vinyl acetate) star polymers by iodine transfer polymerization†
Florian
Baffie
,
Olivier
Boyron
 ,
Muriel
Lansalot
,
Vincent
Monteil
and
Franck
D'Agosto
*
,
Muriel
Lansalot
,
Vincent
Monteil
and
Franck
D'Agosto
*
Univ. Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials (CP2M), 43 Bd du 11 Novembre 1918, 69616 Villeurbanne, France. E-mail: franck.dagosto@univ-lyon1.fr
First published on 4th September 2023
Abstract
Iodine transfer (co)polymerization (ITP) was employed to form polyethylene (PE) and poly(ethylene-co-vinyl acetate) (EVA). ITPs were conducted at 80 bar pressure of ethylene in dimethylcarbonate (DMC) at 70 °C using tri- and tetra-iodofunctionalized chain transfer agents (CTAs) leading to secondary reinitiating radicals. To better comprehend their behaviour, a full study was undertaken to compare the kinetics, the structures of the final polymer and the molar mass control obtained when using analogous iodo and di-iodo CTAs leading to the same secondary reinitiating radials. Well-defined linear PEs carrying one or two iodo chain ends could be formed together with three- and four-arm star PE. The possibility to cleave each arm of the corresponding star structures allowed to show good control of the polymerizations. The transposition to the copolymerization of ethylene and vinyl acetate led to the formation of well-defined four arm star EVA.
Introduction
Star polymers are spatially defined polymers that have attracted and still attract great interest among chemists and material scientists who take advantage of these higher-order and compact architectures to generate unique properties. The development of star polymers has been closely linked to the advances in polymerization techniques, particularly in living polymerizations.1 Several strategies have been used to form star polymers by combining or not controlled/living polymerizations and highly efficient coupling chemistries and among them: arm-first, coupling-onto, and core-first. The arm-first strategy involves the synthesis of linear and well-defined end-functionalized polymer chains and their subsequent crosslinking.2 On the other hand, in the coupling-onto strategy,3 the preformed linear end-functionalized polymer chains are attached onto a multifunctional agent by a coupling reaction.4 The core-first strategy employs a multifunctional initiator to form star polymers by the simultaneous growth of polymer arms by controlled/living polymerizations. Each of these strategies has both inherent advantages and disadvantages. For example, in the arm-first strategy, the number of arms is difficult to control but can be very high. In contrast, with the core-first approach, the number of arms is limited but well defined.5In the particular case of polyethylene (PE), synthetic pathways to produce star polymers have relied on the anionic polymerization of butadiene to produce stars according to the above-mentioned strategies followed by hydrogenation.6,7 Preformed PE chains functionalized with appropriate end groups have been involved in arm-first strategies.8,9 Ma et al.10 originally used polyhomologation of ylides to produce polymethylenes, analogue to polyethylene, from which two polystyrene arms could be grown by reversible-deactivation radical polymerization (RDRP) to yield mikto-arm star polymers. The core-first strategy was implemented by using living ethylene coordination–insertion polymerization with a Pd complex to produce star polyethylenes. The polymerization takes advantage of the formation of a trifunctional active species by combination of the Pd complex with a triacrylate.11–13 This last strategy is straightforward as the star PE is formed directly from polymerization without additional chemistry step. The living nature of the polymerization is however costly in metal (one per chain) and sensitive to polar species and potential polar comonomers.
We recently focused on strategies to produce well-defined polyethylene stars by the core-first method using exclusively RDRPs. Indeed, in the last ten years, RDRPs of ethylene were successful achieved using techniques based on a reversible degenerative (DT) chain transfer. Among the different techniques amenable to control the free radical polymerization of ethylene by DT, reversible addition–fragmentation chain transfer (RAFT),14,15 organotellurium mediated radical polymerization (TeRP)16 and iodine transfer polymerization (ITP)17 have been successfully used to control the free radical growth of the PE chains. Copolymerization with polar monomers such as vinyl acetate (VAc) was also successfully conducted with RAFT,14,15,18 ITP17,19 and cobalt mediated radical polymerization (CoMRP).20–22 As far as we know, there is no example of star PE obtained by RDRP of ethylene using a core-first strategy. Besides, only one example of star copolymers based on ethylene and vinyl acetate (EVA) has been depicted by RAFT using a trifunctional chain transfer agent (CTA).18
Star polymers base on methyl methacrylate have been synthesized by ITP using multifunctional alkyl iodides as CTAs according to a core-first strategy.23,24 In these studies, 3-arm star polymers were synthesized using an alkyl tri-iodide CTA. Considering the successful use of ITP to control ethylene (co)polymerization, a similar strategy was used in the present paper to investigate for the first time a core-first synthesis of star PE starting from ethylene as monomer. An EVA star polymer is also investigated. To achieve this goal, tri and tetrafunctional CTA-S3 and CTA-S4 (Scheme 1), respectively, were considered. Mono- and di-functional CTA-S1 and CTA-S2 (Scheme 1) were also considered for the sake of comparison. As part of a synthetic strategy, these CTAs have significant advantages. Their precursors are readily available and cheap. Furthermore, the ester moiety remaining at the core of the structures can be hydrolyzed to retrieve and characterize the polymer arms.
 | ||
| Scheme 1 Multifunctional alkyl iodides employed to mediate ITP. | ||
Experimental
Materials
Ethylene (Air liquid, 99.95%), 2,2′-azobis(isobutyronitrile) (AIBN, Aldrich, 98%), ethyl 2-bromopropionate (Aldrich, 99%), 2-bromopropionyl bromide (Aldrich, 97%), ethylene glycol (Aldrich, >99%), trimethylolpropane (Aldrich, 97%), pentaerythritol (Aldrich, 98%), sodium iodide (Aldrich, >99.5%), triethylamine (Et3N, Aldrich, >99%), pyridine (Aldrich, >99%), tetrahydrofuran (THF, Fisher, HPLC grade), acetone (Aldrich, >99.5%), dichloromethane (Aldrich, >99.5%), sodium thiosulfate (Na2S2O3, Aldrich, 99%), sodium bicarbonate (NaHCO3, Aldrich, >99%), ammonium chloride (NH4Cl, Aldrich, >99.5%), magnesium sulfate (MgSO4, Aldrich, >99.5%), and dodecane (Aldrich, 99%) were used as received.Dimethyl carbonate (DMC, Aldrich, 99%) was used after bubbling with argon for more than 12 h. Vinyl acetate (VAc, Sigma, >99%) was purified over neutral alumina and was used after bubbling with argon for more than 12 h. Tetrachloroethylene (TCE, ACS Reagent, Aldrich) was purified by passing through silica.
Methods
1H NMR (400 MHz, CDCl3) δ (ppm) = 4.35 (q, J = 6.9 Hz, 1H, I–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –CH3), 4.28–4.17 (m, 2H, O–C2–CH3), 1.82 (d, J = 6.9 Hz, 3H, I–CH–C3), 1.30 (t, J = 7.1 Hz, 3H, O–CH2–C3). 1H NMR (400 MHz, TCE + C6D6) δ (ppm) = 4.25 (q, J = 7.0 Hz, 1H, I–C–CH3), 4.08–3.91 (m, 2H, O–C2–CH3), 1.76 (d, J = 7.0 Hz, 3H, I–CH–C3), 1.07 (t, J = 7.1 Hz, 3H, O–CH2–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 172.0 (Cq,
–CH3), 4.28–4.17 (m, 2H, O–C2–CH3), 1.82 (d, J = 6.9 Hz, 3H, I–CH–C3), 1.30 (t, J = 7.1 Hz, 3H, O–CH2–C3). 1H NMR (400 MHz, TCE + C6D6) δ (ppm) = 4.25 (q, J = 7.0 Hz, 1H, I–C–CH3), 4.08–3.91 (m, 2H, O–C2–CH3), 1.76 (d, J = 7.0 Hz, 3H, I–CH–C3), 1.07 (t, J = 7.1 Hz, 3H, O–CH2–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 172.0 (Cq, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 61.9 (CH2, O–H2–CH3), 23.5 (CH, I–H–CH3), 13.9 (CH3, O–CH2–H3), 13.4 (CH3, I–CH–H3). 13C NMR (101 MHz, TCE + C6D6) δ (ppm) = 171.0 (Cq, O), 61.5 (CH2, O–H2–CH3), 23.8 (CH, I–H–CH3), 14.0 (CH3, O–CH2–H3), 13.0 (CH3, I–CH–H3) HRMS (ESI+): [M + H]+m/z = 228.9720 (calc.), 228.9722 (exp.); [M + Na]+m/z = 250.9539 (calc.), 250.9541 (exp.).
O), 61.9 (CH2, O–H2–CH3), 23.5 (CH, I–H–CH3), 13.9 (CH3, O–CH2–H3), 13.4 (CH3, I–CH–H3). 13C NMR (101 MHz, TCE + C6D6) δ (ppm) = 171.0 (Cq, O), 61.5 (CH2, O–H2–CH3), 23.8 (CH, I–H–CH3), 14.0 (CH3, O–CH2–H3), 13.0 (CH3, I–CH–H3) HRMS (ESI+): [M + H]+m/z = 228.9720 (calc.), 228.9722 (exp.); [M + Na]+m/z = 250.9539 (calc.), 250.9541 (exp.).
The general strategy followed for the synthesis of CTA-S2–4 is depicted in Scheme 2.
 | ||
| Scheme 2 Synthesis of CTA-S2, CTA-S3, and CTA-S4 by a two-step reaction from the corresponding alcohol precursors (% values correspond to yields). | ||
1H NMR (400 MHz, CDCl3) δ (ppm) = 4.41–4.34 (m, 6H, Br–C–CH3 and C2–O), 1.79 (m, 6H, Br–CH–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 170.0 (2Cq, O), 63.1 (2CH2, H2–O), 39.7 (2CH, Br–H–CH3), 21.6 (2CH3, Br–CH–H3). HRMS (ESI+): [M + Na]+m/z = 352.8995 (calc.), 352.8978 (exp.).
Then, to a solution of ethylene glycol bis(2-bromopropionate) (5.00 g, 15.0 mmol, 1.0 eq.) in dry acetone (50 mL) was added a solution of sodium iodide (NaI, 6.30 g, 42.0 mmol, 2.8 eq.) in dry acetone (50 mL). The mixture was stirred at R.T. overnight. The salts were removed by filtration and the filtrate was concentrated under reduced pressure. The crude product was dissolved in dichloromethane (80 mL), washed with a saturated solution of Na2S2O3 (3 × 40 mL) and water (2 × 40 mL). The organic layer was dried over MgSO4, filtered, concentrated in vacuo to afford the desired product as a brown liquid (3.40 g, 65%).
1H NMR (400 MHz, CDCl3) δ (ppm) = 4.48 (q, J = 7.0 Hz, 2H, I–C–CH3), 4.43–4.23 (m, 4H, C2–O), 1.95 (d, J = 7.0 Hz, 6H, I–CH–C3). 1H NMR (400 MHz, TCE + C6D6) δ (ppm) = 4.27 (q, J = 7.0 Hz, 2H, I–C–CH3), 4.07–4.17 (m, 6H, C2–O), 1.77 (d, J = 7.0 Hz, 6H, I–CH–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 171.8 (2Cq, O), 63.0 (2CH2, H2–O), 23.4 (2CH3, I–CH–H3), 12.4 (2CH, I–H–CH3). 13C NMR (101 MHz, TCE + C6D6) δ (ppm) = 171.0 (2Cq, O), 63.1 (2CH2, H2–O), 23.7 (2CH3, I–CH–H3), 12.1 (2CH, I–H–CH3). HRMS (ESI+): [M + Na]+m/z = 448.9717 (calc.), 448.8733 (exp.).
1H NMR (300 MHz, CDCl3) δ (ppm) = 4.37 (q, J = 7.0 Hz, 3H, Br–C–CH3), 4.27–4.01 (m, 2H, C–C2–O), 1.81 (d, J = 7.0 Hz, 9H, Br–CH–C3), 1.56 (q, J = 7.6 Hz, 2H, C2–CH3), 0.92 (t, J = 7.6 Hz, 3H, CH2–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 169.8 (3Cq, O), 64.9 (3CH2, C–H2–O), 41.7 (Cq, –CH2–O), 39.7 (3CH, Br–H–CH3), 23.0 (CH2, H2–CH3), 21.6 (3CH3, Br–CH–H3), 7.5 (CH3, CH2–H3) HRMS (ESI+): [M + H]+m/z = 558.8950 (calc.), 558.8937 (exp.).
Then, to a solution of trimethylolpropane tris(2-bromopropionate) (6.99 g, 13.0 mmol, 1.0 eq.) in dry acetone (100 mL) was added a solution of sodium iodide (NaI, 8.36 g, 55.7 mmol, 4.3 eq.) in dry acetone (50 mL). The mixture was stirred at R.T. overnight. The salts were removed by filtration and the filtrate was concentrated under reduced pressure. The crude product was dissolved in dichloromethane (80 mL), washed with a saturated solution of Na2S2O3 (3 × 50 mL) and water (3 × 20 mL). The organic layer was dried over MgSO4, filtered, concentrated in vacuo to afford the desired product as an orange oil (6.36 g, 72%).
1H NMR (400 MHz, CDCl3) δ (ppm) = 4.50 (q, J = 7.0 Hz, 3H, I–C–CH3), 4.29–3.98 (m, 6H, C–C2–O), 1.96 (d, J = 7.0 Hz, 9H, I–CH–C3), 1.59 (q, J = 7.6 Hz, 2H, C2–CH3), 0.94 (t, J = 7.6 Hz, 3H, CH2–C3). 1H NMR (400 MHz, TCE + C6D6) δ (ppm) = 4.27 (q, J = 7.1 Hz, 3H, I–C–CH3), 4.16–3.90 (m, 6H, C–H2–O), 1.77 (d, J = 7.1 Hz, 9H, I–CH–C3), 1.46 (q, J = 7.6 Hz, 2H, C2–CH3), 0.81 (t, J = 7.6 Hz, 3H, CH2–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 171.5 (3Cq, O), 64.8 (3CH2, C–H2–O), 41.9 (Cq, –CH2–O), 23.1 (3CH3, I–CH–H3), 23.0 (CH2, H2–CH3), 12.4 (3CH, I–H–CH3), 7.6 (CH3, CH2–H3). 13C NMR (101 MHz, TCE + C6D6) δ (ppm) = 170.7 (3Cq, O), 65.3 (3CH2, C–H2–O), 42.6 (Cq, –CH2–O), 23.9 (CH2, H2–CH3), 23.8 (3CH3, I–CH–H3), 12.3 (3CH, I–H–CH3), 7.7 (CH3, CH2–H3). HRMS (ESI+): [M + H]+m/z = 680.8703 (calc.), 680.8702 (exp.); [M + Na]+m/z = 702.8521 (calc.), 702.8521 (exp.).
1H NMR (400 MHz, CDCl3) δ (ppm) = 4.39 (q, J = 6.9 Hz, 4H, Br–C–CH3), 4.36–4.18 (m, 8H, C–C2), 1.83 (d, J = 6.9 Hz, 12H, Br–CH–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 169.6 (4Cq, O), 63.1 (4CH2, C–H2), 43.3 (Cq, –CH2), 39.5 (4CH, Br–H–CH3), 21.6 (4CH3, Br–CH–H3). HRMS (ESI+) [M + H]+m/z = 671.8205 (calc.), 671.8202 (exp.).
Then, to a solution of pentaerythritol tetrakis(2-bromopropionate) (3.00 g, 4.4 mmol, 1.0 eq.) in dry acetone (50 mL) was added a solution of sodium iodine (NaI, 5.32 g, 35.5 mmol, 8.0 eq.) in dry acetone (50 mL). The mixture was stirred at R.T. overnight. The salts were removed by filtration, and the solid was washed with dry acetone (40 mL). The filtrate was concentrated under reduced pressure. The crude product was dissolved in dichloromethane (80 mL), washed with a saturated solution of Na2S2O3 (3 × 50 mL) and water (2 × 20 mL). The organic layer was dried over MgSO4, filtered, concentrated in vacuo to afford the desired product as an orange oil (3.33 g, 88%).
1H NMR (400 MHz, CDCl3) δ (ppm) = 4.52 (d, J = 7.0 Hz, 4H, I–C–CH3), 4.43–4.12 (m, 8H, C–C2), 1.97 (d, J = 7.0 Hz, 12H, I–CH–C3). 1H NMR (400 MHz, TCE + C6D6) δ (ppm) = 4.27 (q, J = 7.0 Hz, 4H, I–C–CH3), 4.22–4.07 (m, 8H, C–C2), 1.78 (d, J = 7.0 Hz, 12H, I–CH–C3). 13C NMR (101 MHz, CDCl3) δ (ppm) = 171.3 (4Cq, O), 63.1 (4CH2, C–H2), 43.5 (Cq, –CH2), 23.3 (4CH3, I–CH–H3), 12.0 (4CH, I–H–CH3). 13C NMR (101 MHz, TCE + C6D6) δ (ppm) = 170.6 (4Cq, O), 63.6 (4CH2, C–H2), 44.3 (Cq, –CH2), 23.7 (4CH3, I–CH–H3), 11.9 (4CH, I–H–CH3). HRMS (ESI+) [M + Na]+m/z = 886.7542 (calc.), 886.7545 (exp.).
A reference experiment (radical polymerization, RP) was also conducted in absence of CTA under the exact same conditions for the sake of comparison.
Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TCE/deuterated benzene (C6D6) solutions using a Bruker Avance 400 spectrometer (1H: 400.13 MHz; 13C: 100.61 MHz). The temperature was calibrated with a tube containing 80 vol% of ethylene glycol and 20 vol% of DMSO-d6. 1H NMR spectra were recorded with a 5 mm BBFO+ probe with a z-gradient coil (D1 = 3 s, scans = 256, RO = 20 Hz with O1P = 6.5 and O2P = 4.5). Carbon NMR spectra were recorded with a 10 mm SEX probe, 13C selective with a z-gradient coil. The pulse sequence used includes a decoupling proton with NOE effects, and a 70° spin excitation (D1 = 2 s, scans = 6144, O1P = 110 ppm, O2P = 80 ppm, RO = 20 Hz). The method is said to be “semiquantitative,” and the NMR calculations were carried out between carbons of the same nature (the –CH2–). Chemical shifts are given in parts per million (ppm) with the solvent peak as internal standard.
000 g mol−1 (Polymer Standards Service).
:1 TCE/deuterated benzene (C6D6) solutions using a Bruker Avance 400 spectrometer (1H: 400.13 MHz; 13C: 100.61 MHz). The temperature was calibrated with a tube containing 80 vol% of ethylene glycol and 20 vol% of DMSO-d6. 1H NMR spectra were recorded with a 5 mm BBFO+ probe with a z-gradient coil (D1 = 3 s, scans = 256, RO = 20 Hz with O1P = 6.5 and O2P = 4.5). Carbon NMR spectra were recorded with a 10 mm SEX probe, 13C selective with a z-gradient coil. The pulse sequence used includes a decoupling proton with NOE effects, and a 70° spin excitation (D1 = 2 s, scans = 6144, O1P = 110 ppm, O2P = 80 ppm, RO = 20 Hz). The method is said to be “semiquantitative,” and the NMR calculations were carried out between carbons of the same nature (the –CH2–). Chemical shifts are given in parts per million (ppm) with the solvent peak as internal standard.
000 g mol−1 (Polymer Standards Service).
For THF soluble polymer samples, molar mass measurements were performed using a Viscotek TDA SEC (Malvern Panalytical), including a differential refractive index detector, a viscometer, a light scattering detector and a UV detector. Stabilized THF was used as the mobile phase at a flow rate of 1 mL min−1 at 35 °C. All samples were injected at a concentration of 3 to 6 mg mL−1 after filtration through a 0.45 μm PTFE membrane. The separation was carried out on three Agilent mixed C columns (SDVB, 5 μm, 300 × 7.5 mm) and a guard column. Mn and Đ were determined by means of a conventional calibration curve or by a universal calibration based on certified PS standards (Polymer Standards Service) from 470 to 270000 g mol−1. The molar mass of an unknown PE sample of mass M is calculated based on the measurement of the intrinsic viscosity [η], and the column retention volume, from which the product [η]·M is read on the universal calibration curve constructed with the known PS standards. Knowing independently [η]·M and [η] leads to the calculation of M of the unknown samples. The Omnisec software was used for data acquisition and data analysis.
500 – mass range: 50 to 20000), and ionization source (electrospray (ESI)).
with

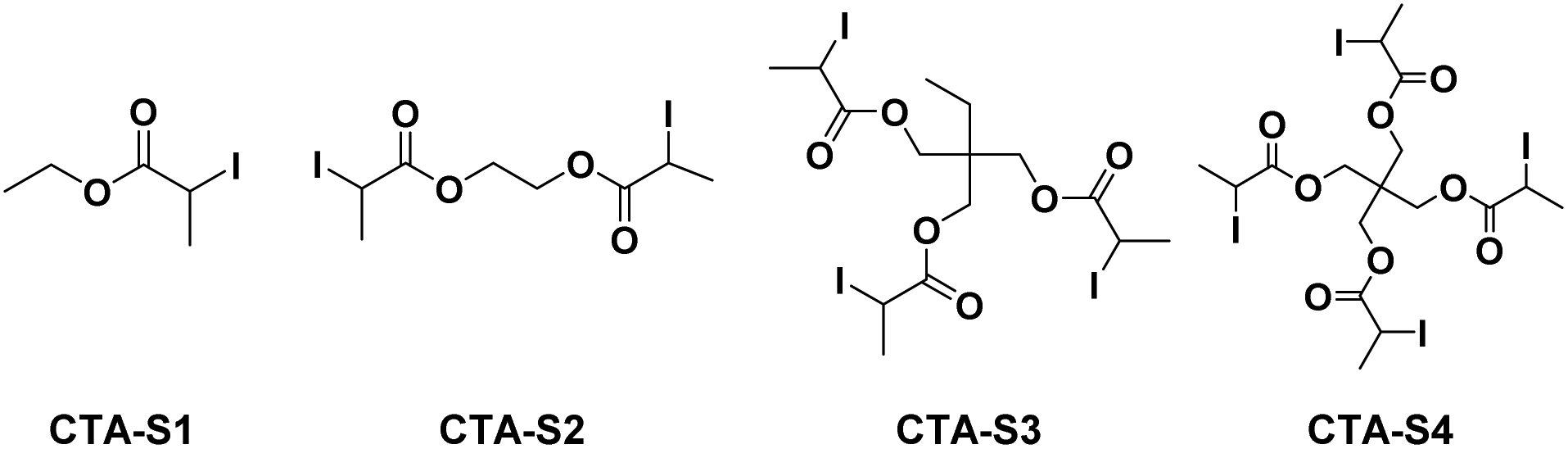
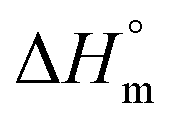 = 293 J g−1 as standard enthalpy of polyethylene.
= 293 J g−1 as standard enthalpy of polyethylene.
Dry nitrogen with a flow rate set at 30 mL min−1 was used as purged gas.
Results and discussion
17,19 were studied and shown to be efficient controlling agents in ITP of ethylene.
CTA-S1-mediated ethylene polymerization was performed under conditions previously used for C6F13I17,19 (70 °C, 80 bar, 50 mL of DMC, 50 mg of AIBN, and [CTA]:[AIBN] = 3:1) and compared to the exact same experiment conducted either in presence of C6F13I or in absence of CTA (free radical polymerization, RP). Fig. 1a shows the CTA conversion and ethylene consumption versus polymerization time. As already observed for C6F13I,17,19 no rate retardation was observed in presence of CTA-S1 compared to the RP. As C6F13I, CTA-S1 is consumed rapidly (conversion > 97% after 5 minutes) and the ethylene consumptions are comparable to those obtained with C6F13I.
 | ||
| Fig. 1 Polymerization conditions: T = 70 °C, P = 80 bar, [CTA]:[AIBN] = 3:1. Ethylene consumption = (mass of dried product) − (mass of AIBN) − (mass of CTA). (a) Ethylene consumption and CTA conversion versus polymerization time for ITP systems compared to the radical polymerization (RP) performed without CTA, and (b) corresponding molar mass evolutions (from HT-SEC). The radical polymerization yielded PE with Mn values of about 8000 g mol−1 and Đ values between 1.5 and 2.1. Molar masses are determined using a conventional PE calibration. | ||
The molar mass evolution versus ethylene consumption is presented in Fig. 1b and the corresponding molar mass distributions for CTA-S1 are given in Fig. S1 (ESI†). The polymerization behaviour with CTA-S1 is very similar to the one with C6F13I with dispersities decreasing upon polymerization to reach values between 1.5 and 1.8 for higher molar masses. In agreement with the theory of RDRP, number average molar masses obtained by HT-SEC (Mn) are relatively close to the theory and follow the expected linear trend.
Fig. 2 shows the 1H NMR spectrum of a PE produced in the presence of CTA-S1 after 3 hours of polymerization. The iodine end-capped PE structure is shown by signal of the methylene hydrogens adjacent to the iodine atom (a, –CH2I at 2.95 ppm). The use of CTA-S1 offers the possibility to see both chain-ends (d, –OC(O)CH(CH3)– at 2.30 ppm and f, CH3CH2OC(O)– at 4.00 ppm). A comparison of the signal integration from the methylene protons a with that of signal f indicates a quantitative chain-end functionality. Nevertheless, it is worth mentioning that in systems governed by reversible transfer and conducted in the presence of azoinitiator, a fraction of dead chains corresponding at least to the fraction of chains initiated by the free radical initiator should form. In the present case, despite the rather low CTA/AIBN ratio, this fraction of chains seems minimal. The unexpected very high chain end fidelity can be related to an error associated with the determination by NMR of the integration values, although this error should remain low. This can also be due to a different decomposition rate of AIBN and initiating efficiency under high pressure (80 bar) and the conditions employed here. A good match between experimental and expected Mn values was observed after 6 hours (Mn(NMR) = 2500 g mol−1vs. Mn(theo.) = 2350 g mol−1). The discrepancy after 45 minutes (Mn(NMR) = 950 g mol−1vs. Mn(theo.) = 500 g mol−1) is probably due to the larger dispersities obtained (>2) at the beginning of the polymerization and the transition between the pre-equilibrium and the main equilibrium governed by the reversible DT.17 These data nevertheless illustrate well the successful ITP of ethylene mediated by CTA-S1.
 | ||
| Fig. 2 1H NMR spectrum (in TCE/C6D6 at 90 °C) of the PE synthesized at 70 °C, 80 bar in the presence of CTA-S1 after 3 h. “+” NMR solvent impurities. AIBN stands for the methyls from the chain-ends of the PE chains initiated by AIBN. The water is coming from the NMR solvent. | ||
Besides, when setting the integral of the methylene hydrogens adjacent to the iodine chain-end to 2, the multiplet from proton d at 2.30 ppm shows an integral around half of the expected value (0.6 instead of 1). This is surprising as the integrals of a and f are very nicely matching. The integral value of d is stable throughout the polymerization, thus ruling out the possibility of H-abstraction of hydrogen d during polymerization. A quantitative 1H NMR analysis with a D1 of 50 s (instead of 3 s used routinely) was performed to ensure that protons have relaxed fully between pulses. However, the integral value of d remained the same. The influence of temperature analysis was also studied (70, 90 °C in TCE/C6D6 and 110 °C in o-dichlorobenzene (DCB)/d-DCB) without significantly impacting the values of the corresponding integrals. The presence of an asymmetric carbon at the α chain-end (–OC(O)CH(CH3)CH2–) led us to consider whether the integral difference was not due to the presence of diastereoisomers. Indeed, the methylene hydrogens c (–OC(O)CH(CH3)CH2–) are diastereotopic, and the corresponding two protons are non-equivalent. This is shown by the presence of two resonances at 1.60 and 1.40 ppm for the corresponding methylene (Fig. 2).27,28 Therefore, we assumed that the proton d could also be split into two resonances, one at 2.30 ppm and the other between 1.50 and 1.75 ppm. The integrals between 1.50 and 1.75 ppm are indeed consistent with this assumption: 2Hb + 1Hc + 0.5Hd = 3.5H. Such peculiar behavior for similar protons d in a poly(vinylidene fluoride) chain has indeed been already observed.29
Fig. 3 shows the ethylene consumption versus polymerization time for the respective systems. Again, there is no rate retardation compared to the system without CTA. Like CTA-S1, CTA-S2–4 were consumed almost instantly.
 | ||
| Fig. 3 Polymerization conditions: T = 70 °C, P = 80 bar, [iodine]:[AIBN] = 3:1. Ethylene consumption = (mass of dried product) − (mass of AIBN) − (mass of CTA). (a) Ethylene consumption versus polymerization time for ITP systems compared to the radical polymerization (RP) performed without CTA, and (b) corresponding molar mass evolutions (from HT-SEC). Molar masses are determined by a conventional PE calibration for CTA-S1, CTA-S2 and by a universal calibration based on PS calibration for CTA-S3 and CTA-S4. | ||
The molar mass evolution versus ethylene consumption is presented in Fig. 3b, and the corresponding molar mass distributions are given in Fig. 4. A PE calibration was used for the linear PE (CTA-S1, CTA-S2), whereas a universal calibration based on PS samples was preferred for the star polymers (CTA-S3 and CTA-S4). Molar mass distributions remain narrow, and a clear shift is observed toward higher Mn with time, indicating a successful polymerization control. As previously observed, dispersities decrease with time to reach values between 1.6 and 1.9 for higher molar masses. Mn(HT-SEC) remained relatively close to the theory and followed the targeted linear trend (Fig. 3b).
 | ||
| Fig. 4 Molar mass distributions of PE obtained by HT-SEC during ITP of ethylene mediated by (a) CTA-S2, (b) CTA-S3, and (c) CTA-S4 at 70 °C and 80 bar of ethylene pressure and after the indicated polymerization times. | ||
Fig. 5 shows the spectra of CTA-S4 and of the PE produced in the presence of CTA-S4 after 1.5 h and 6 h of polymerization. The characteristic resonances of CTA-S4 (d′, –OC(O)CH(CH3)I at 4.25 ppm and e′, –OC(O)CH(CH3)I at 1.80 ppm), disappear upon ethylene polymerization (Fig. 5a and b). The iodine end-capped PE structure is proven by the presence of methylene hydrogens adjacent to the iodine atom (a, –CH2I at 2.95 ppm). As for CTA-S1, the use of CTA-S4 offers the possibility to see both extremities of the PE arms (f, –CH2OC(O)– at 4.10 ppm and d, –OC(O)CH(CH3)– at 2.35 ppm). A comparison of signal intensity from the methylene hydrogens a and f indicates only a minimal loss of iodine chain-end functionality (<5%) (Fig. 5c). A good match between Mn(NMR) and Mn(theo.) was observed (Fig. S2†). For example, after 6 hours, Mn(NMR) = 8950 g mol−1vs. Mn(theo.) = 9050 g mol−1. This illustrates the successful ITP of ethylene mediated by CTA-S4.
 | ||
| Fig. 5 1H NMR spectra (in TCE/C6D6 at 90 °C) of (a) CTA-S4, and PE synthesized in the presence of CTA-S4 after (b) 1.5 h and (c) 3 h. The end group –CH3 stems from intramolecular and intermolecular chain transfer inherent in ethylene radical polymerization. “o” transfer to polymerization solvent DMC. AIBN stands for the methyls from the chain-ends of the PE chains initiated by AIBN. The water is coming from the NMR solvent. | ||
Comparably to what was observed with CTA-S1, the multiplet from proton d at 2.35 ppm presents an integral around half of the expected value (2 instead of 4) (Fig. 5c). And as observed for CTA-S1, the integral of proton d is stable throughout the polymerization. Considering the very good agreement between integrals of protons a and f, consistent with successful control of the growth of 4 PE arms from CTA-S4, we again assigned this lower integral value to the effect of the diastereotopic protons of the adjacent methylene discussed in the case of CTA-S1.
1H NMR analyses of PEs synthesized in the presence of CTA-S2, CTA-S3 and CTA-S4 exhibit the same features and led to very similar behaviour and conclusions (see Fig. S2 in ESI†).
Moreover, two multiplets of small intensity at 2.05 and 2.45 ppm can be observed at short polymerization times in each case (Fig. 5b, after 1 h 30 min for CTA-S4). The resonances of the protons a at 2.95 ppm are also superimposed with other triplets. Their intensities decrease over time, and they disappear after 6 hours of polymerization (Fig. 5c).
To elucidate the origin of these signals, the copolymer obtained after 1 h 30 min in the presence of CTA-S4 was purified by precipitation in methanol. The polymer (200 mg) was dissolved in 20 mL of toluene at 90 °C and stirred for two hours. The solution was subsequently poured in 150 mL of methanol, and a white solid precipitated. The solid was filtered and dried under vacuum to afford a white powder (175 mg). After evaporation of the filtrate, a white wax was obtained (25 mg). The solid (Fig. 6c) and the filtrate residue (Fig. 6b) were analyzed by 1H NMR. The two multiplets at 2.05 and 2.45 ppm are no longer visible in the solid obtained from precipitation, confirming that these resonances are impurities of low molar mass. On the other hand, these signals are more intense in the filtrate residue. These observations are very similar and consistent with the ones made when synthesizing telechelic PEs using fluorinated di-iodo CTA.19 In this case, the origin of these signals arose from (i) the formation of asymmetric oligomers during the degenerative chain transfer preequilibrium, and (ii) the impact of the first additions of ethylene units on the chemical shifts of the protons of the α- and ω-chain ends in the formed products. In the present case, the same explanation can be given. The disappearance of the signals at 2.05 and 2.45 ppm and the very clean triplet for proton a obtained after 6 hours of polymerization (Fig. 6c) shows that the PE arms are indeed growing on all the branches of the targeted star.
 | ||
| Fig. 6 1H NMR spectra (in TCE/C6D6 at 90 °C) of PE synthesized at 70 °C, 80 bar in the presence of CTA-S4 after 1 h 30 min, (a) before reprecipitation, and after precipitation: (b) after evaporation of the filtrate and (c) solid part. The end group –CH3 stems from intramolecular and intermolecular chain transfer inherent in ethylene radical polymerization. AIBN stands for the methyls from the chain-ends of the PE chains initiated by AIBN. The water is coming from the NMR solvent. | ||
SEC separates the chains in a polymer sample based on their hydrodynamic sizes (radius of gyration, Rg). A branched polymer and its analogue linear polymer of equal Rg elute thus out at the same rate. Therefore, SEC cannot differentiate both polymers. For the same molar mass, the Rg of a linear polymer is higher than the one of a branched polymer.30 The branching content can be obtained from either the viscometer or the light scattering detector. However, the molar masses of the PE used in this study are too low to consider measurement with a light scattering detector (isotropic scattering).30–32
Zimm and Stockmayer proposed different formulae to calculate the number of arms in a polymer, depending on the architecture (well-defined star, brush, comb). For polymers with branches randomly distributed, the number of branches (Bn) is correlated to the branching factor (g) (eqn (1)).33
 | (1) |
In the case of a well-defined star, it can be simplified to give eqn (2).
 | (2) |
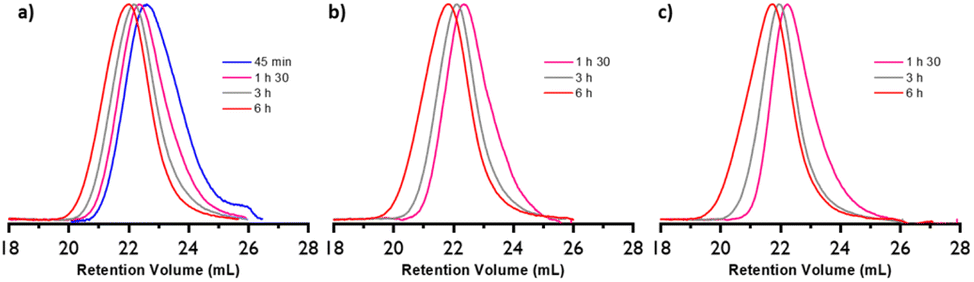
The branching factor g can be expressed with the viscosity branching factor (g′) and the structure factor (ε) (eqn (3)).34
| g′ = gε | (3) |
 | (4) |
The intrinsic viscosity can be measured directly by the viscometer of the HT-SEC. Therefore, we used this approach to determine the number of arms of each polymer and compare it with the expected values.
Nevertheless, PEs synthesized by a radical process are low-density polyethylenes (LDPE). LDPEs are already branched structures and contain both long and short-chain branching (LCB and SCB, respectively). This branching nature adds to the difficulty in characterizing the star nature of our polymers. Under our conditions, the formed PE chains are much less branched than the conventional LDPE as the softer polymerization conditions employed here (80 bar, 70 °C) induces less transfer reactions.14–17
To calculate the number of arms when CTA-S3 and CTA-S4 were employed, the polymers synthesized in the presence of CTA-S1 and CTA-S2 were used as linear equivalents in eqn (4). The number of arms calculated for PE synthesized in the presence of CTA-S3 was on average 2.8 after 3 h of reaction. For PE synthesized in the presence of CTA-S4, the average value was 4.1 after 3 h of reaction. The consistency observed with the expected values of 3 and 4 when CTA-S3 and CTA-S4 are used, respectively, showed that the targeted 3- and 4-armed star PEs were obtained.
Eventually, the PEs synthesized in the presence of the different CTAs present the advantage of having ester functions that can be selectively cleaved to retrieve and assess the homogeneity of PE arms.
The PEs obtained after 6 hours of polymerization were hydrolyzed with tBuOK and subsequently analyzed by SEC (Fig. 7). Fig. 7a shows that, as expected, this treatment is not affecting the molar mass of the PE obtained with CTA-S1. On the other hand, for the PE synthesized in the presence of CTA-S2, CTA-S3, and CTA-S4, the molar mass distributions shifted toward lower molar masses (Fig. 7b–d). As expected, the shift is more pronounced when the number of arms in the targeted star polymer increases. As the [iodine]:[AIBN] ratio was kept the same for all the experiments, the targeted molar masses of each PE arm are identical after the same polymerization time. This is confirmed by the overlay of the molar mass distributions in Fig. 7e. Moreover, using a PE calibration, Mn(SEC) values of 2150, 1950, 2000, 1950 g mol−1 (Đ of 1.92, 1.97, 2.19, 2.29, respectively) were measured after treatment of PE obtained with CTA-S1, CTA-S2, CTA-S3, and CTA-S4, respectively. All these data confirmed the successful synthesis of star PEs.
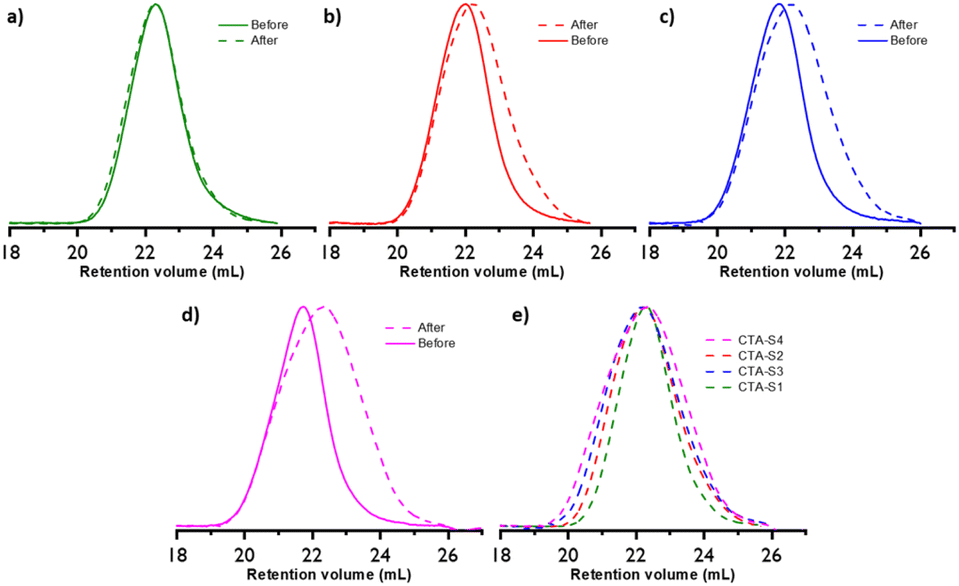 | ||
| Fig. 7 Molar mass distributions of PE before and after hydrolysis by tBuOK of PE obtained in the presence of (a) CTA-S1, (b) CTA-S2, (c) CTA-S3, and (d) CTA-S4. (e) Molar mass distributions comparison after hydrolysis. | ||
:[AIBN] = 3:1) to target a VAc content of 25 mol%. The kinetics of the corresponding experiments are presented in Table 1.
| Run | Time (h) | Monomer cons.b (g) | M n(theo.)c (g mol−1) | VAc contentd (mol%) | M n(NMR)e (g mol−1) | M n(SEC)f (g mol−1) | Đ | T m (°C) | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|
|
a AIBN (0.3 mmol), [iodine]:[AIBN] = 3:1 at 70 °C and 80 bar in 25 mL of DMC with 25 mL of VAc.
b Monomer consumption = (mass of dried product) − (mass of AIBN) − (mass of CTA).
c
M
n(theo.) = (monomer cons. [g])/(CTA [mol]) + M(CTA).
d Calculated by 1H NMR by comparing the CH2 signals of the ethylene unit and the CH of the vinyl acetate unit.
e Calculated by comparing the 1H NMR signals of the EVA chain and the iodo chain-end.
f Determined by THF-SEC with a PS calibration.
g Determined by DSC.
|
|||||||||
| 1 | 1.5 | 2.7 | 12500 |
26 | 14500 |
14200 |
1.5 | 0.6 | −30.6 |
| 2 | 3 | 5.9 | 27000 |
25 | 34300 |
22400 |
1.4 | 0.1 | −29.3 |
| 3 | 6 | 11.7 | 51000 |
25 | 58500 |
36200 |
1.6 | −1.7 | −30.4 |
Molar masses measured by 1H NMR (Fig. 8a) showed an excellent consistency with the expected values (i.e., for run 3 in Table 1, Mn(NMR) = 58500 g mol−1 and Mn(theo.) = 51000 g mol−1). The polymerization system yields copolymers with unimodal molar mass distributions shifting toward higher molar masses for higher monomer consumptions (Fig. 8b). As already discussed,19Mn(SEC) and Đ for the P(E-co-VAc) should be taken with caution. Indeed, different arms and monomer contents might lead to different elution behavior during the SEC experiment. We thus considered NMR spectroscopy being more reliable for a quantitative comparison of the Mn values of the copolymers. Still, good agreement was observed between Mn(SEC), Mn(NMR), and Mn(theo.) (Table 1). In conclusion, the ITcoP of ethylene and VAc mediated by CTA-S4 shows a good control. The star copolymers exhibit glass transition and melting temperatures comparable to those obtained previously for linear analogues with the same VAc content.19
 | ||
| Fig. 8 (a) 1H NMR spectrum and (b) molar mass distributions obtained by THF-SEC of P(E-co-VAc) obtained during the ITcoP of ethylene and VAc mediated by CTA-S4 (runs 4–6) with a VAc content of 25 mol%. | ||
The EVA star polymers are more challenging to characterize by hydrolysis similarly to their PE analogues since the hydrolysis of the EVA core resulted in the hydrolysis of VAc units, complicating the chromatograms comparisons.
Conclusions
In conclusion, successful ITPs of ethylene were conducted with CTA-S1–S4. In each cases a good control over the polymerization was shown and the final PE structures analyzed by 1H NMR matched the expected ones. This was confirmed by analyzing the cleaved arms in the three and four-arm structures. The star nature of the PE obtained when CTA-S3 and CTA-S4 were used to mediate ITP was further shown by intrinsic viscosity measurements allowing to calculate a number of arms that is consistent with the expected one. These good results prompted us to investigate the synthesis of EVA stars by successfully conducting ethylene and vinyl acetate ITPcoP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. B. acknowledges the French Ministère de l'Enseignement Supérieur de la Recherche et de l'Innovation (MESRI) for funding.Notes and references
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369 RSC.
- J. Kim, H. Y. Jung and M. J. Park, Macromolecules, 2020, 53, 746–763 CrossRef CAS.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed.
- T. Tsoukatos and N. Hadjichristidis, J. Polym. Sci. Part A: Polym. Chem., 2002, 40, 2575–2582 CrossRef CAS.
- Y. Zhang, H. Li, Z. Xu, W. Bu, C. Liu, J.-Y. Dong and Y. Hu, Polym. Chem., 2014, 5, 3963–3967 RSC.
- D. Khedaioui, B. Burcher, D. Gajan, D. Montarnal, F. D'Agosto and C. Boisson, Polym. Chem., 2020, 11, 3884–3891 RSC.
- Y. Xue, S.-S. Zhang, K. Cui, J. Huang, Q.-L. Zhao, P. Lan, S.-K. Cao and Z. Ma, RSC Adv., 2015, 5, 7090–7097 RSC.
- K. Zhang, Z. Ye and R. Subramanian, Macromolecules, 2009, 42, 2313–2316 CrossRef CAS.
- P. Liu, E. Landry, Z. Ye, H. Joly, W.-J. Wang and B.-G. Li, Macromolecules, 2011, 44, 4125–4139 CrossRef CAS.
- X. Xia, Z. Ye, S. Morgan and J. Lu, Macromolecules, 2010, 43, 4889–4901 CrossRef CAS.
- C. Dommanget, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2014, 53, 6683–6686 CrossRef CAS PubMed.
- A. Wolpers, C. Bergerbit, B. Ebeling, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2019, 58, 14295–14302 CrossRef CAS PubMed.
- Y. Nakamura, B. Ebeling, A. Wolpers, V. Monteil, F. D'Agosto and S. Yamago, Angew. Chem., Int. Ed., 2018, 57, 305–309 CrossRef CAS PubMed.
- A. Wolpers, F. Baffie, L. Verrieux, L. Perrin, V. Monteil and F. D'Agosto, Angew. Chem., Int. Ed., 2020, 59, 19304–19310 CrossRef CAS PubMed.
- A. Wolpers, F. Baffie, V. Monteil and F. D'Agosto, Macromol. Rapid Commun., 2021, 42, 2100270 CrossRef CAS PubMed.
- F. Baffie, M. Lansalot, V. Monteil and F. D'Agosto, Polym. Chem., 2022, 13, 2469–2476 RSC.
- A. Kermagoret, A. Debuigne, C. Jérôme and C. Detrembleur, Nat. Chem., 2014, 6, 179–187 CrossRef CAS PubMed.
- J. Demarteau, A. Kermagoret, C. Jérôme, C. Detrembleur and A. Debuigne, Controlled Radical Polymerization: Materials, American Chemical Society, 2015, vol. 1188, pp. 47–61 Search PubMed.
- J. Demarteau, P. B. V. Scholten, A. Kermagoret, J. De Winter, M. A. R. Meier, V. Monteil, A. Debuigne and C. Detrembleur, Macromolecules, 2019, 52, 9053–9063 CrossRef CAS.
- L. Lei, M. Tanishima, A. Goto and H. Kaji, Polymers, 2014, 6, 860–872 CrossRef.
- M. Tanishima, A. Goto, L. Lei, A. Ohtsuki, H. Kaji, A. Nomura, Y. Tsujii, Y. Yamaguchi, H. Komatsu and M. Miyamoto, Polymers, 2014, 6, 311–326 CrossRef.
- Y. Liu and Z. Fan, Des. Monomers Polym., 2015, 18, 251–261 CrossRef CAS.
- J. Bernard, A. Favier, L. Zhang, A. Nilasaroya, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2005, 38, 5475–5484 CrossRef CAS.
- H. Shimamura, T. Sunazuka, T. Izuhara, T. Hirose, K. Shiomi and S. Ōmura, Org. Lett., 2007, 9, 65–67 CrossRef CAS PubMed.
- V. Morozova, J. Skotnitzki, K. Moriya, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 5516–5519 CrossRef CAS PubMed.
- M. Guerre, G. Lopez, T. Soulestin, C. Totée, B. Améduri, G. Silly and V. Ladmiral, Macromol. Chem. Phys., 2016, 217, 2275–2285 CrossRef CAS.
- A. S. Kulkarni and G. Beaucage, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1395–1405 CrossRef CAS.
- M. Gaborieau and P. Castignolles, Anal. Bioanal. Chem., 2011, 399, 1413–1423 CrossRef CAS PubMed.
- P. Liu, W. Liu, W.-J. Wang, B.-G. Li and S. Zhu, Macromol. React. Eng., 2017, 11, 1600012 CrossRef.
- B. H. Zimm and W. H. Stockmayer, J. Chem. Phys., 1949, 17, 1301–1314 CrossRef CAS.
- P. J. Flory and T. G. Fox, J. Am. Chem. Soc., 1951, 73, 1904–1908 CrossRef CAS.
- W.-J. Wang, S. Kharchenko, K. Migler and S. Zhu, Polymer, 2004, 45, 6495–6505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00849e |
| This journal is © The Royal Society of Chemistry 2023 |