
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient carbon dot-based photoinitiating systems for 3D-VAT printing†
Dominika
Krok
a,
Wiktoria
Tomal
a,
Alexander J.
Knight
 b,
Alexander I.
Tartakovskii
b,
Nicholas T. H.
Farr
c,
Wiktor
Kasprzyk
a and
Joanna
Ortyl
*ade
b,
Alexander I.
Tartakovskii
b,
Nicholas T. H.
Farr
c,
Wiktor
Kasprzyk
a and
Joanna
Ortyl
*ade
aDepartment of Biotechnology and Physical Chemistry, Faculty of Chemical Engineering and Technology, Cracow University of Technology, Warszawska 24, 30-155 Cracow, Poland. E-mail: jortyl@pk.edu.pl
bDepartment of Physics and Astronomy, University of Sheffield, Sheffield, S3 7RH, UK
cDepartment of Materials Science and Engineering, University of Sheffield, Sheffield S1 3JD, UK
dPhoto HiTech Ltd., Bobrzyńskiego 14, 30-348 Cracow, Poland
ePhoto4Chem Ltd., Lea 114, 30-133 Cracow, Poland
First published on 12th September 2023
Abstract
In this work, different types of carbon dots (CDs) based on citric acid as a precursor were synthesized using an efficient procedure to purify these materials from low molecular by-products and fluorophores. Their structural and optical characteristics were elaborated and compared to commercially available graphene quantum dots. The mechanism of their action in photopolymerization processes was evaluated. Obtained materials proved to perform well in the development of effective photoinitiating systems for 3D printing applications. The morphology and chemical composition of obtained hydrogel printouts were profoundly characterized via SEM, AFM, Nano-FTIR, and s-SNOM.
Introduction
Known as a rising star among carbon nanomaterials, carbon dots (CDs) have attracted considerable interest in various fields in recent years.1 Valuable features including their structure and fascinating spectroscopic and electrochemical properties have led to their numerous applications: in biomedicine,2–4 drug delivery,5 photodynamic therapy,6 photocatalysis7,8 or solar cells.9 In particular, the excellent optical properties of CDs are attracting increasing attention in biomedical10 and photocatalytic applications. This is due to low cytotoxicity,11 good biocompatibility,12 stable chemical inertness,13 efficient light harvesting and excellent light efficiency. Induced electron transfer14 make them promising candidates for various applications in biosensors,15,16 bioimaging,17,18 optoelectronic devices19,20 or solar cells, etc.21 CDs, usually considered as small carbon nanoparticles in suspension or in solid form are divided into three types: graphene quantum dots (GQDs), carbon nanodots (CNDs) and carbonized polymer dots (CPDs). A more detailed classification and description of the different classes of CDs has been described in review papers.22 In terms of synthetic protocols used for CDs fabrication, two approaches are considered i.e. “top-down” and “bottom-up”.23,24 The latter being more frequently used due to its simplicity, cost-effectiveness and high diversity of possible precursors. Although CDs prepared from different precursors and/or using different synthetic conditions often exhibit distinct optical properties,25 some spectroscopic features might be considered as common for all types of CDs. Generally, CDs exhibit one or more strong absorption bands in the short wavelength range of UV/VIS spectrum (from 200 nm to 320 nm), which shows extensive tailing across the entire visible region. As a consequence of above extremely broad absorption, variety of emitting centers present within CDs can be simultaneously excited (directly or via energy transfer) giving rise to excitation-dependent PL emission.26–28Recently, above optical characteristics of CDs have been found useful in the initiation of photopolymerization processes. Despite significant progress in the field of light-induced polymerization reactions, researchers still search for improvements in initiation systems used in these processes.29–32 For example, iodonium salts, as one of the most commonly used initiators, absorb light in a narrow range of electromagnetic spectrum (up to 360 nm).33–35 Therefore, it is necessary to look for molecules, which will change the absorption characteristics of these initiators in order to be compatible with the UV-A and visible range light sources used.36–41 Currently, a number of molecules have been developed that may be suitable for this role.36,41–47 Nevertheless, the fact that not only the appropriate absorption range of the compounds is sought, but also other parameters such as their lack of toxicity, natural origin or their efficiency, make the search for such compounds still relevant. As the spectroscopic properties of CDs are promising, scientists have already made some progress in the development of initiating systems based on these nanometric carbon particles.48 Pioneering work on this matter was done by Matyjaszewski's group, who used heteroatom doped CDs as photocatalysts for reversible addition and fragmentation polymerization (RAFT).49 Then, Kütahya et al., proposed to use CDs in a binary initiating system with iodine or sulfur.50 he aforementioned researchers described the use of carbon-based nanomaterials for radical photopolymerization processes (specifically, RAFT and ATRP processes). These researchers described the use of carbon nanomaterials in radical photopolymerization processes (specifically RAFT and ATRP processes). Consequently, our group, presented for the first time the possibility of using CDs doped with heteroatoms in cationic photopolymerization processes.51 Common initiators used in photopolymerization processes have some shortcomings. One disadvantage of such initiators is inefficient absorption of radiation. In addition, the initiators used are often poorly soluble in monomers and their mixtures, which is a problem especially when higher concentrations of photoinitiators. Carbon dots as photoinitiators are not characterized by the above disadvantages. Photoinitiators in the form of carbon dots are devoid of these disadvantages. They are characterized by excellent spectroscopic properties as well as good solubility. What distinguishes carbon dots from other photoinitiators is low toxicity and even, in some cases, its complete absence. Recently, perovskite carbon dots have proven to be very promising and efficient catalysts.52,53 They have been used to directly initiating photopolymerization processes to produce various polymer compounds. However, these photopolymerizations were easily inhibited in air due to the oxygen quenching effect on carbon radicals. It is worth mentioning, however, that the variety of visible-light-induced photocatalytic processes that perovskite nanocrystals are capable of, coupled with their easy preparation, easily tunable redox potentials, high catalytic efficiency and reusability, create great opportunities for their future applications in green and sustainable organic synthesis. Carbon dots synthesized by us also have a number of positive properties. However, the specific structure of both materials makes them unique to each other.
Herein, we are continuing our research on using CDs in photopolimerization processes, however, focusing on new photoinitiating systems not yet reported in the literature. Thus, we introduce three types of CDs: citric acid-based, as well as ammonia- and ethylenediamine-doped CDs, and compare their effectiveness to commercially available graphene-based CDs as an element of two- or three-component photoinitiating systems dedicated for free radical photopolymerization processes. This approach led to the development of efficient initiating systems and allowed better understanding of the mechanisms according to which CDs performed in these processes. CDs-based photoinitiating systems were studied in free radical polymerization of acrylate monomers. As the proof of concept, CDs-based photoinitiating systems were implemented in two types of 3D-VAT printing processes: DLP and DLW printing, to obtain high-resolution, 3D hydrogel materials.
Experimental
Materials
Citric acid (CA, from Sigma Aldrich), ammonia (NH3·H2O, from Sigma Aldrich) and ethylenediamine (ED2, from Sigma Aldrich) were used to obtain three types of CDs (Fig. 1). The following reagents were used in purification process of the CDs: 1 M NaOH and 1 M HCl aqueous solutions (both from Sigma Aldrich). Commercially available graphene quantum dots, exhibiting blue luminescence (GQDs, from Sigma Aldrich) were used as a reference. | ||
| Fig. 1 Components used in the syntheses of CDs. | ||
The following reagents were used as a part of photoinitiating systems: bis-(4-t-butylphenyl)iodine hexafluorophosphate (IOD, Speedcure 938, Lambson, Wetherby, UK) as a commercial photoinitiator, methyldiethanolamine (MDEA, Sigma Aldrich) as a co-initiator in type II photoinitiating systems. Trimethylolpropane triacrylate (TMPTA, Sigma Aldrich) was used as a model monomer for free radical photopolymerization. In addition, 2-hydroxyethyl acrylate (HEA, Sigma Aldrich) was used to prepare photocurable hydrogels. The structures of above compounds are presented in Fig. S1.†
Synthesis of CDs
A single-pot, bottom-up, solventless procedure for the synthesis of CDs was applied. Three kinds of CDs were obtained through thermal treatment of specific precursors: CA (1 g, 5 mmol) (reaction (1)); CA (1 g, 5 mmol) mixed with ammonia (0.425 g, 25 mmol) (reaction (2)); and CA (1 g, 5 mmol) mixed with ethylenediamine (1.5 g, 25 mmol) (reaction (3)). For each reaction, reagents were placed in a glass vial reactor and kept at 230 °C for 5 h. The reactions were conducted on a heating block with a magnetic stirrer at 100 rpm. The synthesis products were then cooled to room temperature and further processed to purify the CDs. The general synthesis scheme is shown in Fig. S2.†Purification of CDs
Appropriate purification of the CDs was a crucial point in this stage of the studies. Therefore, the solid products obtained after one-pot synthesis were treated with 1 M NaOH solution (15 mL of alkaline solution was used) and then subjected to ultrasound for 30 minutes. The resulting solutions were neutralized with 1 M HCl until the pH of the test solution was neutral (Fig. S3†)The solution containing CDs, fluorophores and impurities was sequentially centrifuged and washed using ultrafiltration units (Vivaspin 20, MWCO 10 kDa) to remove low molecular weight impurities and fluorophores. For verification of dialysis effectiveness, spectroscopic studies of obtained filtrates were conducted (Fig. S4†). The purified aqueous suspensions of CDs were then frozen and lyophilized to obtain a dry product.
Determination of the morphology of CDs
For the morphological analysis of obtained CDs, a transmission electron microscope (TEM) (TecnaiTF 20 X-TWIN (200 kV)) was used, equipped with a field-terminated emission (FEG) cannon for high total beam current and high stability of operation. The Tecnai TF 20 X-TWIN time-resolved microscope operated at 0.5 nA and the electron beam was focused on a spot with a diameter of 1 nm.Determination of the chemical composition and chemical bonds within CDs
The XPS analyses were carried out in a PHI VersaProbeII Scanning XPS system using monochromatic Al Kα (1486.6 eV) X-rays focused to a 100 μm spot and scanned over the area of 400 μm x 400 μm. The photoelectron take-off angle was 45° and the pass energy in the analyzer was set to 117.50 eV (0.5 eV step) for survey scans and 46.95 eV (0.1 eV step) to obtain high energy resolution spectra for the C 1s, O 1s, Na 1s, Si 2p, N 1s and Cl 2p regions. A dual beam charge compensation with 7 eV Ar+ ions and 1 eV electrons were used to maintain a constant sample surface potential regardless of the sample conductivity. All XPS spectra were charge referenced to the unfunctionalized, saturated carbon (C–C) C 1s peak at 285.0 eV. The operating pressure in the analytical chamber was less than 3 × 10−9 mbar. Deconvolution of spectra was carried out using PHI MultiPak software (v.9.9.3). Spectrum background was subtracted using the Shirley method. The information depth of the XPS analysis, within the geometry of spectrometer can be estimated at about 5 nm.Electrochemical properties of CDs
Voltammetric measurements were carried out to determine the oxidation and reduction potentials of the studied CDs. For this purpose, an electrochemical analyzer M161 and an electrode stand M164, from MTM-ANKO, Poland with a standard three-electrode cell were used. Voltammetry was recorded at a scan rate of 100 mV s−1. The working electrode was a glassy carbon electrode with a diameter of 3 mm (Mineral, Poland), and a platinum wire served as the auxiliary electrode. All potentials were measured with respect to the Ag/AgCl (3M KCl) electrode, which was placed in a double bridge filled with a 3 M KCl solution in the upper part and a solution of the auxiliary electrolyte in a given solvent in the lower part. The two solutions were separated by a plug of cotton wool and separated from the test solution by a thick ceramic frieze. The potential of this reference electrode at 25 °C is +0.209 V relative to a standard hydrogen electrode (SHE). As an auxiliary electrolyte, 0.1 M KCl in distilled water was used. All measurements were carried out in a dry argon atmosphere (5.0 Messer) at 25 °C.Spectroscopic characterization of CDs
Absorption spectra of CDs along with the precursors were analyzed using a SilverNova TEC-X2 spectrophotometer (StellarNet, Inc., Tampa, FL, USA) in the 190–1100 nm range. A deuterium-halogen UV/Vis light source (StellarNet, Inc., Tampa, FL, USA) was used in all experiments in the 190–2500 nm range. The fluorescence emission and excitation spectra of CA-CDs, CA-ED2-CDs and CA-NH3-CDs and GQDs were analyzed using a FluoroMax-4P spectrofluorometer (Horiba, Kyoto, Japan). All spectra were recorded at varying excitation wavelengths in the range of 200–800 nm, using a slit width of 5 nm for both excitation and emission in each measurement. All measurements were carried out in water at room temperature.Real-time FT-IR photopolymerization experiments
The light-induced photopolymerization process involving CDs was analyzed by real-time FT-IR method, using an FT-IR i10 NICOLET™ infrared spectrometer with a horizontal adapter (Thermo Scientific, Waltham, MA, USA). Composition analyzed in these experiments was prepared by dissolving appropriate amounts of the initiating system with various monomers under light-free conditions. Study of polymerization kinetics was conducted using FT-IR method via observation of absorption peaks at specific wavelengths (corresponding to the functional group or bond of the monomers used). Hence, the conversion rate was calculated according to the following formula (eqn (1)):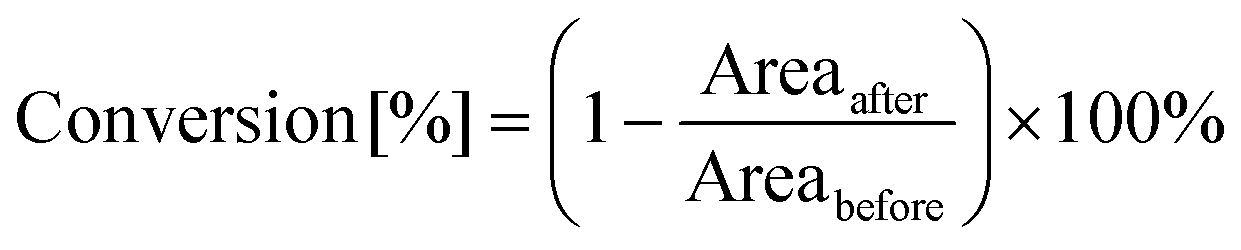 | (1) |
Free-radical photopolymerization of acrylate monomers
For the study of free-radical photopolymerization, compositions consisting of TMPTA together with binary initiating system: IOD (2 wt%) as photoinitiator and CDs (0.2 wt%) as photosensitizers were prepared. MDEA amine (2 wt%) was used as a co-initiator. A few drops of the studied composition were applied to a 1.4 mm thick adapter (a schematic diagram can be found in Fig. S17 in the ESI†) and placed on a horizontal holder in the FT-IR spectrometer. The double bond was monitored continuously at about 6143 cm−1 for 600 s. TMPTA monomer with 2.0 wt% iodonium salt was used as a reference.CDs-based systems for radical photopolymerization of air-resistant polymer hydrogels
Free-radical photopolymerization of HEA in aqueous medium was carried out in order to fabricate hydrogel materials. Firstly, a photo-curable composition consisting of: CDs (0.2 wt%) IOD (2%) and a mixture of HEA monomer and water in a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was prepared. Then, the composition was applied to a 1.4 mm thick adapter (same as in studies of free-radical photopolymerization of acrylate monomers). The photopolymerization process was conducted under UV-LED 405 nm (light intensity of 1400 mA). The conversion of double bonds was followed using the FT-IR spectrometer at about 6125 cm−1. A mixture of HEA/H2O (1/1 wt%) with 2.0 wt% iodonium salt was used as a reference.
:1 was prepared. Then, the composition was applied to a 1.4 mm thick adapter (same as in studies of free-radical photopolymerization of acrylate monomers). The photopolymerization process was conducted under UV-LED 405 nm (light intensity of 1400 mA). The conversion of double bonds was followed using the FT-IR spectrometer at about 6125 cm−1. A mixture of HEA/H2O (1/1 wt%) with 2.0 wt% iodonium salt was used as a reference.
Three-dimensional printing of the hydrogel model structure
Two types of printing techniques were used to prepare three-dimensional materials: digital light processing (DLP) and direct laser writing (DLW). 3D-VAT printouts were obtained using a Lumen X+™ printer (from Cellnk Inc., USA, supplied by Sygnis SA, Poland), which is a specialized polymer printer equipped with a light emitting projector with a wavelength of 405 nm. It is an additive process a laser printer (DK-8-KZ, NEW) under a laser light source @405 nm (spot diameter 50 μm) was also used to produce 3D models. All 3D printouts obtained were analyzed on a DSX1000 digital microscope with a DSX10-SXLOB3X objective with a magnification of 42-420X.Source of light
The light source used for the real-time FTIR experiment was a 405 nm M405LP1 LED (I0 = 22.0 mW cm−2, Thorlabs, Tampa, FL, USA). The LED was powered by a DC2200 driver (Thorlabs, Tampa, FL, USA). The distance between the radiation source and the sample was 210 mm.LV-SEM/AFM/s-SNOM
Low voltage-SEM imaging: FEI Helios Nanolab G3 (FEI Company, USA) and Helios G4 DualBeam (ThermoFisher Scientific, USA) microscopes were employed for surface morphology observations of 3D-VAT printouts. In contrast to common SEM analysis practice, samples did not undergo conductive coating. An accelerating voltage of 1–2 keV, vacuum pressures in the range of 10−6 mbar and a working distance of 4 mm were chosen to avoid sample damage through surface charging. An Everhart–Thornley Detector (ETD) was selected for low magnification of SE images, a Through Lens Detector (TLD) for high magnification SE images and a concentric backscatter (CBS) detector for the collection of backscattered election (BSE) images.The atomic force microscopy (AFM) and scattering-type, scanning near-field microscopy (s-SNOM) data shown in Fig. 12 were collected using a neaSCOPE from attocube systems AG/Neaspec. s-SNOM: The s-SNOM apparatus consisted of an AFM within one arm of an interferometer, and a moveable mirror in the other. A conductive AFM cantilever (Pt/Ir coated ARROW-EFM tip from NanoWorld) was brought into tapping mode operation upon the sample (tapping frequency 77 kHz, tapping amplitude 71 nm), and illumination from a single-wavelength source (MIRcat-QT from Daylight Solutions) outputting at 1490 cm−1 was sent into the interferometer. Under focused illumination, the conductive cantilever tip acts as an optical antenna and a near field is generated at the tip apex (radius around 25 nm). The near field interacts with the sample surface and forms a scattering centre that scatters further incoming photons. The scattered photons were collected at the detector and interfered with photons returning from the movable mirror in the reference arm of the interferometer. This reference mirror was oscillated in order to induce side-band frequency mixing in the optical signal power spectrum, and the optical amplitude and phase data were extracted at the third harmonic of the AFM tapping frequency. The optical amplitude data were normalised to the maximum recorded value. The optical phase data were left unprocessed, and thus the raw values of the phase data in Fig. 12 do not hold physical meaning. Only the contrast between two areas of Fig. 12 should be considered. AFM data: AFM topology data were recorded using the same neaSCOPE from attocube systems AG/Neaspec as used for the s-SNOM measurements. Conductive AFM cantilevers (Pt/Ir coated ARROW-EFM tip from NanoWorld) were used, at a tapping frequency of 77 kHz and a tapping amplitude of 71 nm.
Results and discussion
In this work, CDs were synthesized through thermal treatment of precursors either from sole CA (CA-CDs) or in accompaniment of one of frequently reported nitrogen sources i.e. ammonia and ethylenediamine (CA-NH3-CDs and CA-ED2-CDs, respectively). Both, the reaction conditions and purification protocol applied here were aimed on minimization of the molecular fluorescence contribution to optical properties of obtained CDs. Accordingly, synthesis temperature and time were set to values sufficient for significant carbonization of precursors and fluorophores.54,55 Prior to purification process, brown solidified synthesis products containing CDs were dissolved in 1M NaOH. The use of alkaline solution in this step is justified by the fact that the surface of CDs obtained from CA is covered with carboxyl groups which have to be converted into appropriate salts in order to make the CDs more hydrophilic and, in turn, make the transfer of CDs into aqueous suspension possible. Afterwards, the extent of NaOH was neutralized with HCl and the suspensions were subjected to extensive filtration using centrifugation tubes equipped with dialysis membranes with 10 kDa cut off. Verification of purification efficiency was tested through UV-VIS absorption/fluorescence measurements of filtrates obtained from each run (see Fig. S4†). After sufficient number of purification runs, the final retenates were analyzed using LC-DAD-MS system to exclude the presence of specific low molecular mass fluorophores. Thus, these analyses confirmed that the obtained samples are free from citrazinic acid and IPCA (fluorophores frequently reported as molecular species present in CDs obtained from CA/ammonia and CA/etylenediamine mixtures, respectively)56,57 and other low molecular mass PL emissive species (see Fig S4†). Eventually, the retenates were freeze-dried and the target CDs were obtained as powders and used for further characterization and in photopolymerization experiments.Determination of the chemical composition of CDs and chemical bonds
Surface concentrations of chemical bonds obtained from fitting XPS data for all analyzed samples are listed in Table 1. The C 1s spectra for all samples were fitted with four components (Fig. 2.). First line centered at 285.0 arise from aliphatic carbon C–C and some fraction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C type bonds if present, second line lies at 286.4 eV indicate presence of C–O and/or C–OH and/or C–NH bonds, third line centered at 288.0 eV indicate presence of CO and/or O–C–O and/or N–CO bonds58 and fourth line at 289.0 eV indicate presence of O–CO type bonds.58 The O 1s spectra for all samples were fitted with four components: first line centered at 531.3 eV which indicate O–Si and/or OC type bonds, second line at 532.5 eV from either O–C and/or O–Si bonds, third line positioned at 533.7 eV originates from –OH type bonds and/or adsorbed water and some part of Auger Na KLL line and last line found at 535.8 eV comes from Auger Na KLL line only.59
C type bonds if present, second line lies at 286.4 eV indicate presence of C–O and/or C–OH and/or C–NH bonds, third line centered at 288.0 eV indicate presence of CO and/or O–C–O and/or N–CO bonds58 and fourth line at 289.0 eV indicate presence of O–CO type bonds.58 The O 1s spectra for all samples were fitted with four components: first line centered at 531.3 eV which indicate O–Si and/or OC type bonds, second line at 532.5 eV from either O–C and/or O–Si bonds, third line positioned at 533.7 eV originates from –OH type bonds and/or adsorbed water and some part of Auger Na KLL line and last line found at 535.8 eV comes from Auger Na KLL line only.59
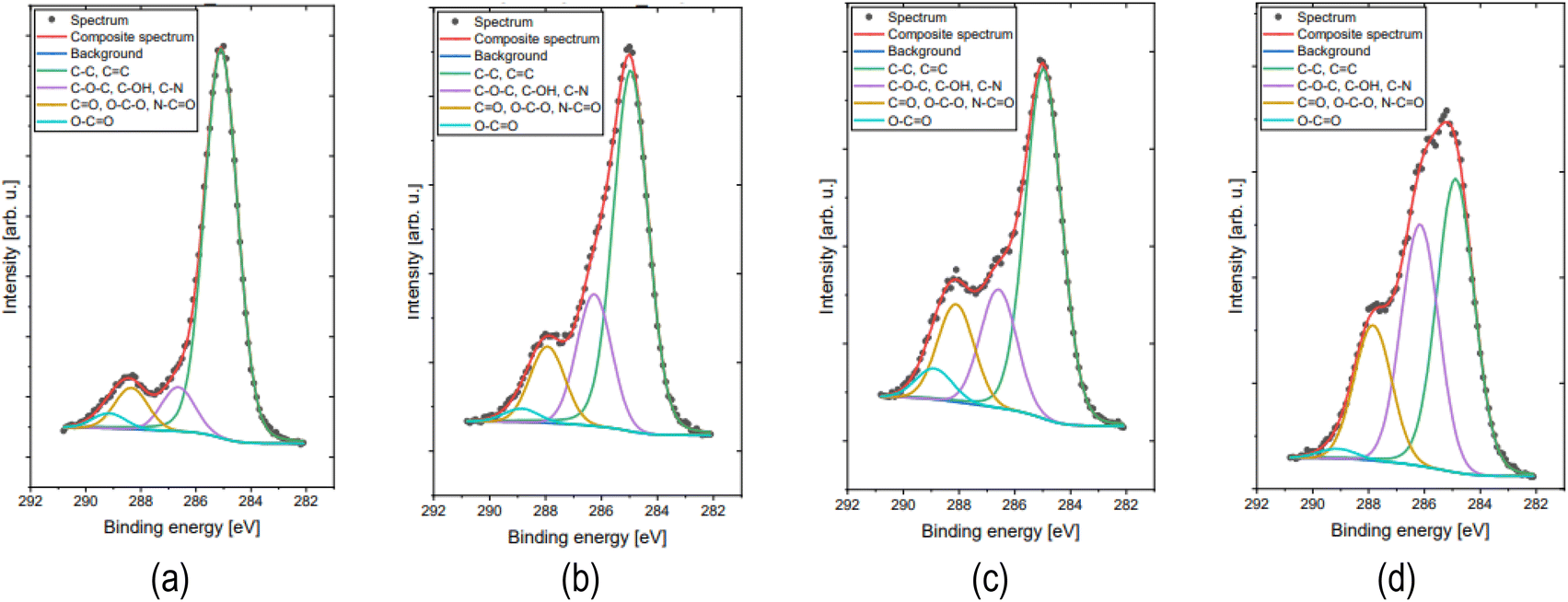 | ||
| Fig. 2 The high energy resolution XPS spectra of C 1s peaks for CDs: (a) CA-CDs; (b) CA-ED2-CDs; (c) CA-NH3-CDs (d) GQDs. | ||
| C | Total C | N | Total N | O | TotalO | Na | Si | Total Si | Cl | ||||||||
| Binding energy [eV] | 285.0 | 286.4 | 288.0 | 289.0 | 399.0 | 400.1 | 531.3 | 532.5 | 533.7 | 1071.6 | 101.9 | 103.2 | 198.4 | ||||
| Oxidation state/Compound | C–C/CC |
C–O–C/C–OH/C–N | CO/N–CO/O–C–O |
O–CO |
C–N | N–CO/–NH3+ |
O–Si/OC |
O–C/O–Si | –OH/H2Oads/Na KLL | Na+ | Silicone/siloxane | SiO2 | Cl− | ||||
| CA-CDs | 58.1 | 6.2 | 5.8 | 2.1 | 72.2 | 0.0 | 0.4 | 0.4 | 10.5 | 7.2 | 3.8 | 21.5 | 3.9 | 0.8 | 0.7 | 1.5 | 0.7 |
| CA-ED2-CDs | 43.0 | 14.7 | 8.5 | 1.5 | 67.7 | 2.6 | 7.4 | 10.0 | 10.6 | 6.4 | 1.2 | 18.2 | 2.4 | 0.8 | 0.7 | 1.5 | 0.1 |
| CA-NH3-CDs | 37.0 | 12.0 | 9.9 | 3.1 | 62.0 | 1.8 | 10.9 | 12.7 | 12.1 | 8.4 | 1.3 | 21.8 | 1.1 | 1.4 | 1.1 | 2.5 | 0.0 |
| GQDs | 28.8 | 22.5 | 12.6 | 0.9 | 64.8 | 0.0 | 15.3 | 15.3 | 10.9 | 5.9 | 0.7 | 17.5 | 0.7 | 0.8 | 0.8 | 1.6 | 0.0 |
The XPS data clearly indicates that all studied CDs exhibit relatively high degree of carbonization. The carbon content is >60% in all cases, which is higher than in CDs reported in literature and obtained at higher temperature and shorter reaction time (300 °C, 30 min)54. This means that the synthetic parameters used in this work were sufficient to produce carbon nanoparticles on one hand and harsh enough to minimize molecular fluorescence on the other.54 The highest content of the total carbon was detected for CA-CDs (∼72%) and the lowest for CA-NH3-CDs (∼62%), which is probably associated with the substitution of C atoms in CDs structure by other elements (N,O). Accordingly, the content of C–C and CC carbon is the highest for CA-based CDs and the residual carbon in CA-CDs exists as a part of hydroxyl, ether, ketone as well as carboxylic acid and anhydrides functionalities (6.2, 5.8 and 2.1%). In contrary, for CDs doped with nitrogen (also commercial GQDs) higher amount of carbon atoms is connected to nitrogen and oxygen species (in comparison to CA-CDs) creating possible defects in graphitic structure of these nanomaterials. It is worth pointing that in commercial CQDs nitrogen exists only as N–CO and –NH3+ species while for N-doped CDs synthesized within this work additionally relevant amount of C–N moieties was recognized. At this point it is important to note that XPS survey spectra show significant quantity of Na+ and Cl− in most of the studied samples (Fig. S7–S10†). The presence of these species is probably associated with the use of NaOH and HCl during purification process, and formation of sodium salts of CDs (via surface carboxylic groups) which is crucial for stabilization of nanoparticle suspension in aqueous environment. The detailed results of the XPS analysis are shown in ESI.†
Morphological analysis using TEM
High-resolution TEM images are very difficult to obtain, due to aggregation of the samples after the freeze-drying process. Nevertheless, in this case TEM was able to confirm the nanometric size of synthesized CDs, achieving dot sizes of about 5 nm. In addition, the spherical morphology of the obtained CDs was confirmed, as demonstrated in Fig. 3. | ||
| Fig. 3 TEM image of (a) CA-ED2-CDs and (b) CA-NH3-CDs. | ||
Determination of the spectroscopic properties of CDs
Owing to the fact, that specific optical properties of CDs remain one of their most intriguing features due to numerous possible applications in different fields of science, in this work a lot of attention has been paid for spectroscopic studies of obtained nanomaterials. Thus, Fig. 4 show the UV-visible absorption spectra of the synthesized CA-based CDs and their precursors i.e. CA, NH3xH2O, and ED2. By analyzing these graphs, it can be seen that the precursors show rather sharp absorption bands in high energy part of the electromagnetic spectra. On the other hand, CDs are characterized by a maximum in the 250–350 nm range and a long “absorbance tail” extending into the visible range which is a common feature observed in CDs. CA-ED2-CDs and especially CA-NH3-CDs exhibited only barely visible signs of specific features appearing as a delicate hills (320–600 nm) over the tail of the main absorption bands, while the absorption tail of CA-CDs is rather smooth and featureless. The differences in the absorption characteristics of studied CDs may be attributed to formation of pyridinic and pyrrolic nitrogen species within the sp2 structures of CD core as well as to nitrogen-derived surface functional groups found in CA-NH3-CDs and CA-ED2-CDs.60 On the contrary, commercially available CDs show strong narrow bands in short-wavelength region and moderately strong but also narrow band around 340 nm. Interestingly, no absorption tailing frequently reported for CDs was observed in this case. The origin of these dramatic differences between CDs synthesized in this work and commercial ones may be explained by differences in materials heterogeneity (substantial heterogeneity of synthesized CDs versus high homogeneity of commercial materials) or in different mechanisms governing absorption and PL emission (multiple types of emission centers versus single type of emission center).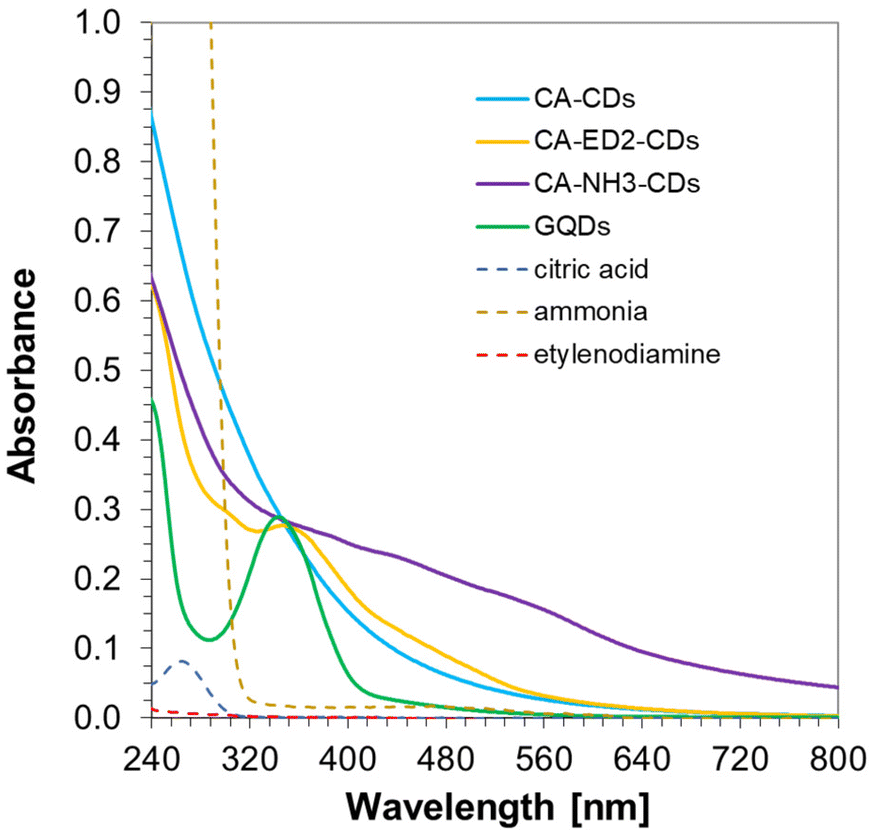 | ||
| Fig. 4 UV-visible absorption spectra of the synthesized citric acid-based CDs and commercial GQDs. Solvent: water. | ||
For PL emission studies all CDs were excited at series of excitation wavelengths. Beyond common spectral characterization, this study was aimed on investigation whether molecular fluorophores contribute to optical properties of obtained CDs. Typically, the PL emission spectrum for molecular fluorophores at different excitation wavelengths does not shift toward longer or shorter wavelengths but is constant. In the case of well-purified CDs, the situation appears different, that is, as the excitation length increases, the emission spectrum of the CDs should shift toward longer wavelengths.61
In, Fig. 5, the fluorescence spectra for CA, CA-ED2-CDs, and CA-NH3-CDs, which were synthesized with the aim of this work and commercial GQDs, have been presented. All synthesized samples show excitation-dependent feature typical for CDs, while commercial product show PL emission characteristics typical for single emission centers. Although, the PL quantum yield measurements have not been conducted in the present study, it can be said with certainty that the GQDs sample show very bright fluorescence in the blue region while CDs exhibit very week PL emission in different regions of electromagnetic spectrum. Interestingly, it seems that CA-CDs show highest homogeneity in emission among synthesized CDs. The shift of PL emission wavelength maxima in this case starts at 465 nm (excitation 320 nm) and gradually increase up to ∼580 nm with markedly decreased PL intensity. For CA-ED2-CDs the situation become more complex as contribution of another emission center starts to be apparent at ∼530 nm. In these CDs both PL centers show excitation-dependent PL emission. Finally, for CA-NH3-CDs several PL centers could be observed giving rise to PL emission across whole visible part of electromagnetic spectrum, confirming that these materials are the most heterogenous among CDs studied here.
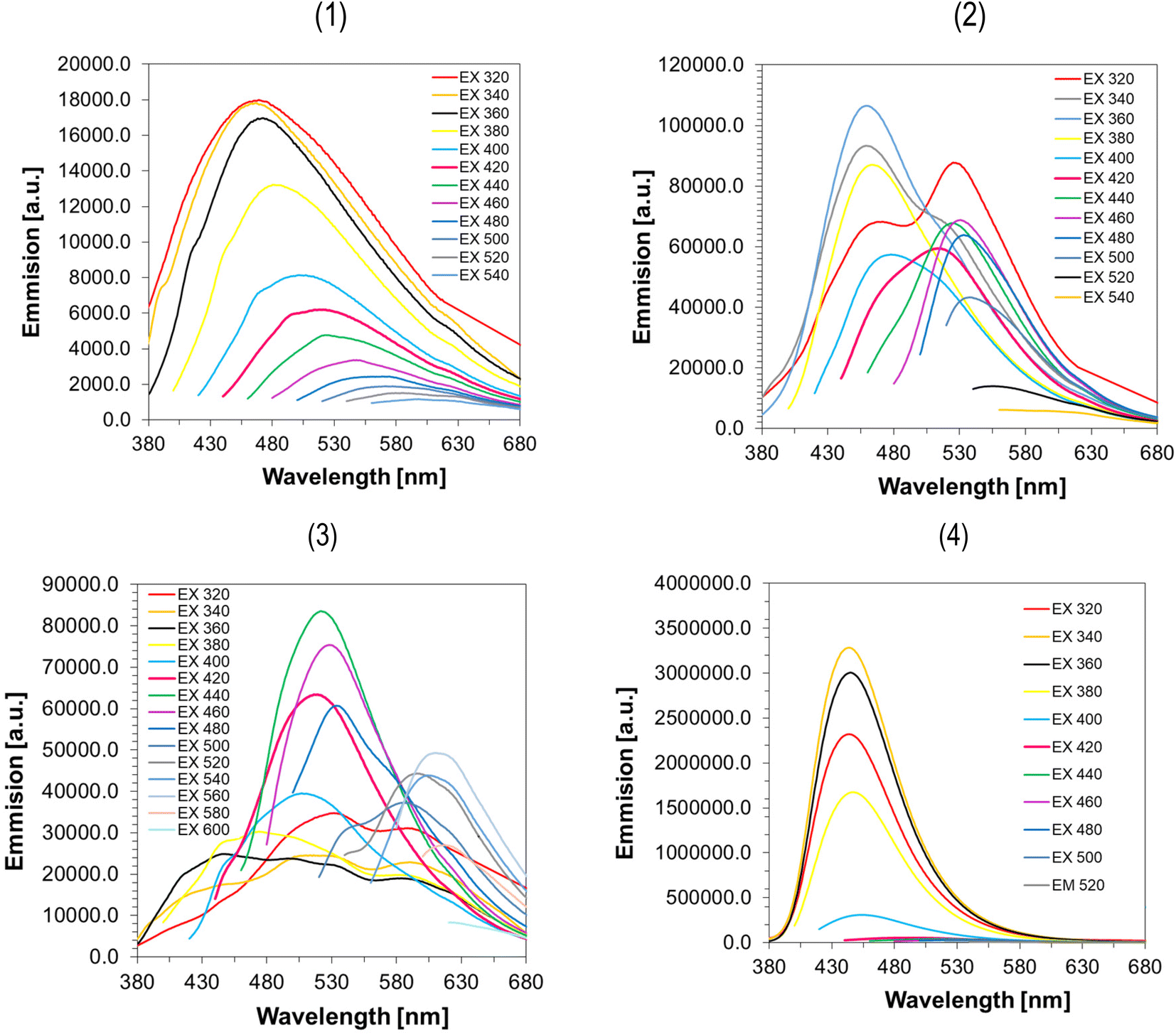 | ||
| Fig. 5 Fluorescence spectra of aqueous solutions of (1) CA-CDs (2) CA-ED2-CDs (3) CA-NH3-CDS (4) GQDs determined with the use of various excitation wavelengths. | ||
Real time FT-IR experiments
The use of CDs in free-radical photopolymerization processes first took place in Jiang's laboratory. CDs acted as photosensitizers in the Reversible Addition–Fragmentation Chain Transfer (RAFT) photopolymerization process. Another researcher working on the use of CDs in radical polymerization was Kutahya.50 In this case, initiating systems based on CDs were enriched with iodonium or sulfonium salts and copper was used as a catalyst for the reaction. The use of CDs in cationic polymerization reactions had not been reported in the literature until our group reported it in 2021. It concerned the use of CDs as sensitizers of iodonium salt in radical photopolymerization processes and as photocatalysts for cationic polymerization. This represented the most environmentally friendly approach to date using carbon nanomaterials in this field of photopolymerization. This time we would like to switch the performance of CDs doped with ethylenediamine and ammonia as effective components in systems that initiate radical polymerization. In addition, we are also investigating a commercially available material containing a fluorophore in this direction. For this purpose, system consisting of CDs (0.2 wt%) and an iodonium salt (2.0 wt%) to initiating the free radical photopolymerization reaction of the acrylate monomer TMPTA (Fig. S15†). A mixture of TMPTA monomer and iodonium salt (2.0 wt%) without CDs was used as a reference.
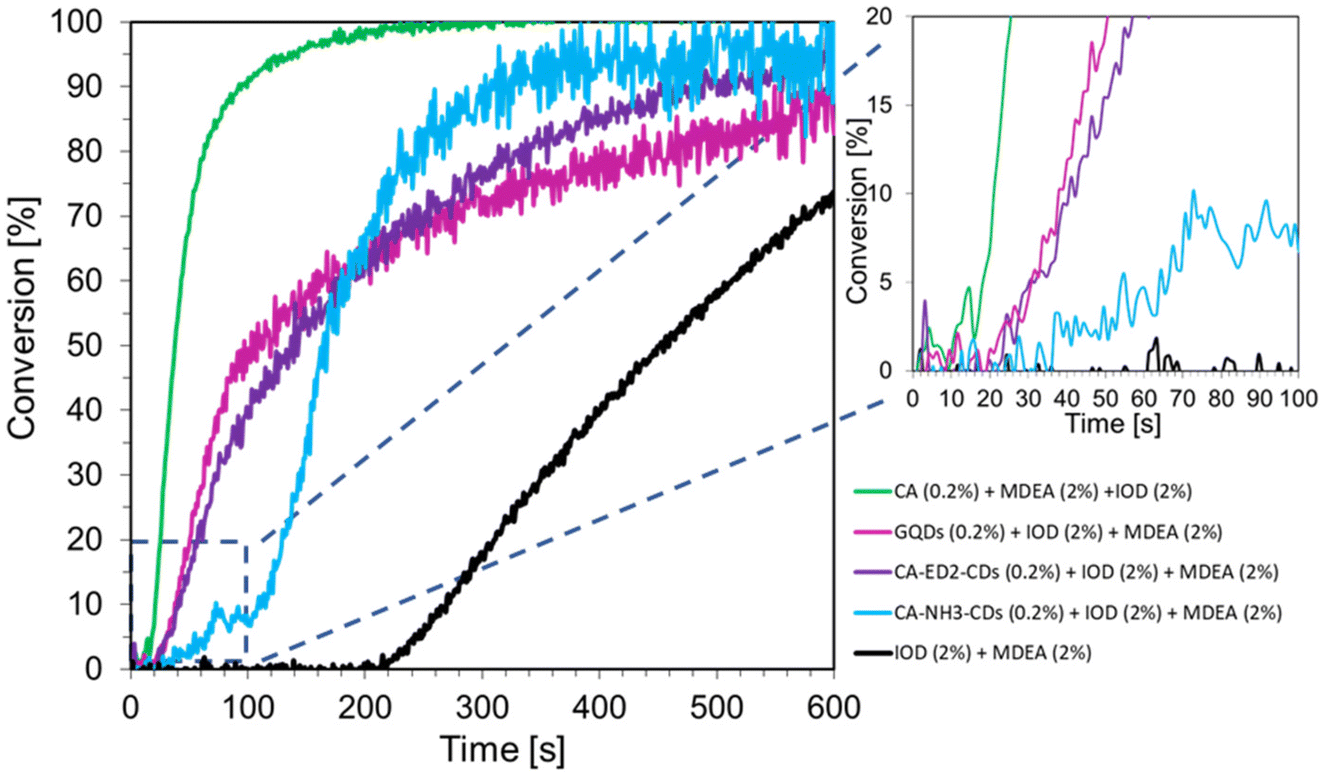
The reaction was carried out under aerobic conditions for 600 s, at 25 °C. The conversion time profiles are shown in Fig. 6. The results clearly indicate that the used photoinitiating systems based on CDs and iodonium salt initiate free-radical polymerization reactions of the TMPTA acrylate monomer. Nitrogen-doped CDs affect the degree of monomer conversion and the slope of the kinetic curve. The best results in two-components systems were obtained with ethylenediamine-doped dots. This may be related to the electrochemical properties of CDs. In addition, a system consisting of CDs (0.2 wt%) and MDEA amine (2 wt%) to initiate the free radical photopolymerization reaction of the acrylate monomer TMPTA. In this case, the CDs with ED2 also showed the best performance, however, the conversion rates obtained are much lower than before.
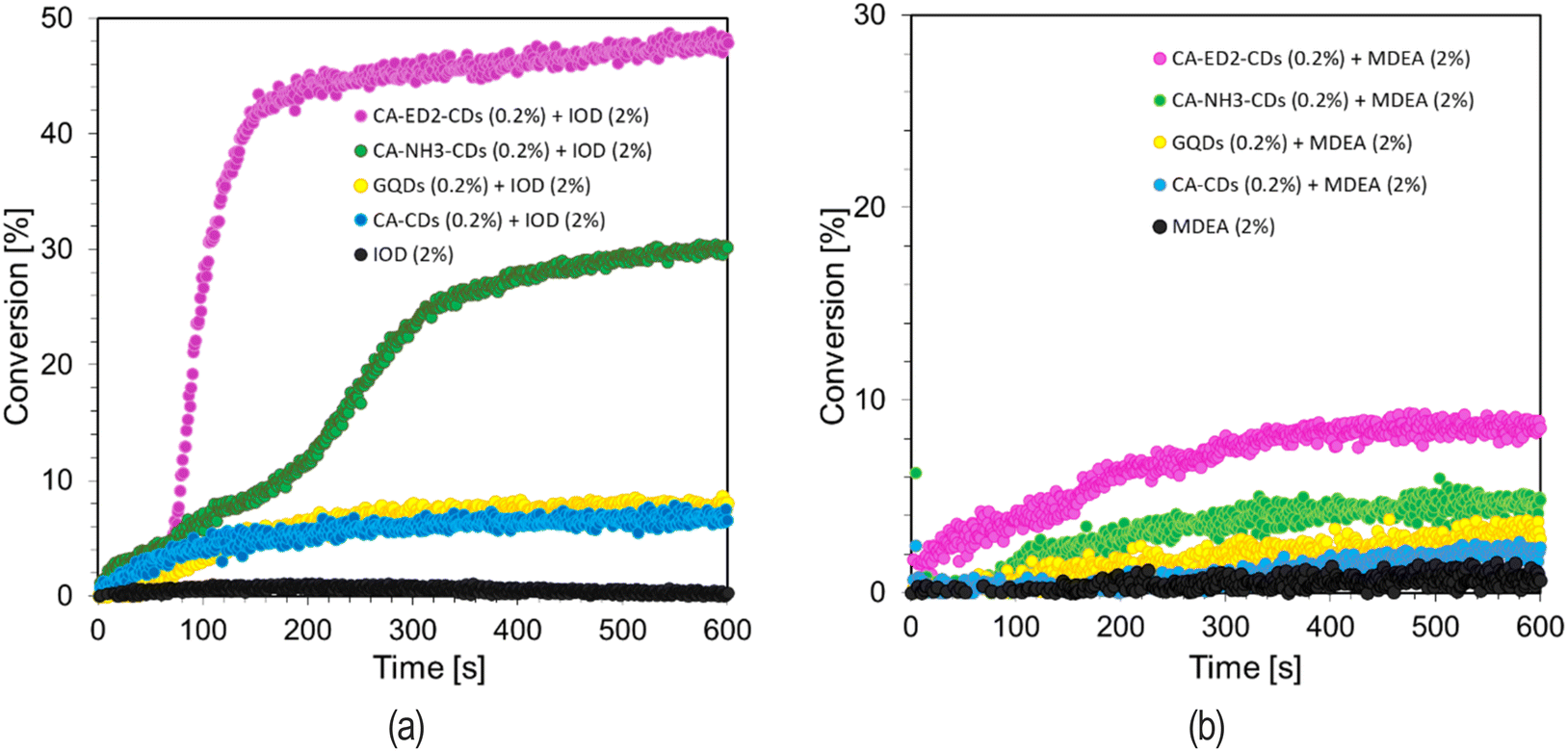 | ||
| Fig. 6 Radical photopolymerization profiles (acrylate function conversion vs. irradiation time) initiated by a bimolecular photoinitiating system based on (a) IOD (2.0 wt%) and CDs (0.2 wt%), (b) MDEA (2 wt%) and CDs (0.2 wt%) under irradiation at 405 nm. | ||
To compare the performance of two-component systems with three-component systems, compositions consisting of CDs (0.2 wt%), iodonium salt (IOD 2.0 wt%) and amine (2 wt%) were prepared for the initiation of radical photopolymerization processes. Similar TMPTA monomer conversion rates were obtained for compositions containing different CDs. The slope of the kinetic curve for CDs with ethylenediamine is the smallest, which indicates that these dots work most efficiently in the described system. To compare the performance of two-component systems with three-component systems, compositions consisting of CDs (0.2 wt%), iodonium salt (IOD 2.0 wt%) and amine (2 wt%) were prepared for the initiation of radical photopolymerization processes (Fig. 7). Similar TMPTA monomer conversion rates were obtained for compositions containing different CDs. The slope of the kinetic curve for CDs with ethylenediamine is the smallest, which indicates that these dots work most efficiently in the described system. In addition, for some curves (CA-CDs (0.2%) + IOD (2%) + MDEA (2%); CA-ED2-CDs (0.2%) + IOD (2%) + MDEA (2%); CA-NH3-CDs (0.2%) + IOD (2%) + MDEA (2%)), large noise in the ∼100–600 s range is noticeable. This is due to the disappearance of the band responsible for the CC double bond (achieving maximum conversion), which in turn reflects a large scatter in the directly read area of the band and calculated conversion. This effect is not noticeable in the case of the other two kinetic curves (GQDs (0.2%) + IOD (2%) + MDEA (2%); IOD (2%) + MDEA (2%)), which is due to the lower conversion obtained, so the band responsible for double bonds (6.143 cm−1) did not disappear and its areas could be read with high accuracy.
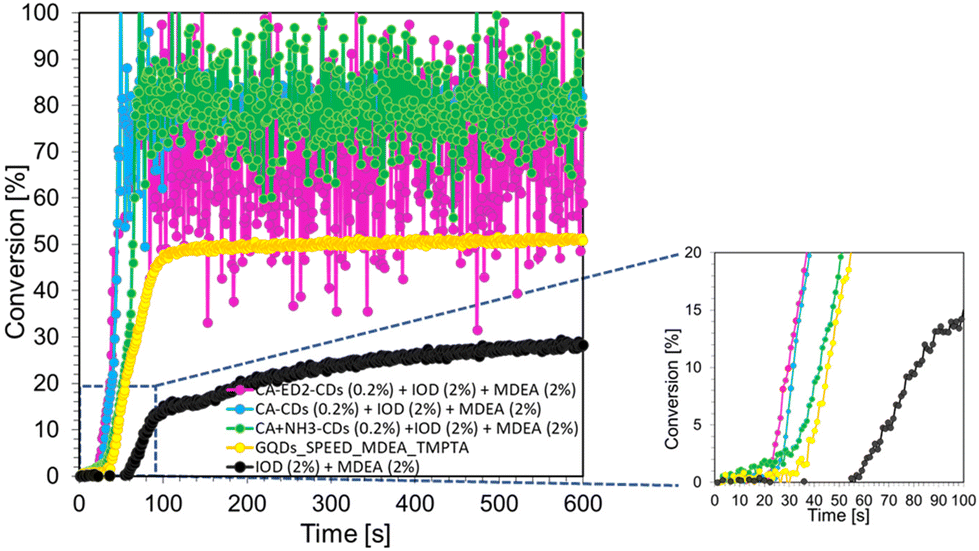 | ||
| Fig. 7 Radical photopolymerization profiles (acrylate function conversion vs. irradiation time) initiated by a three-molecular photoinitiating system based on IOD (2.0 wt%), MDEA (2 wt%) and CDs (0.2 wt%) under irradiation at 405 nm. | ||
In order to better compare the properties of CDs in initiating systems, the slope of kinetic curves and induction times were determined. The slope was determined as dconversion/dt. It is clear, that the composition with commercial CDs showed the longest induction time. A similar induction time was shown by the composition with the addition of the dots doped with ammonia. The best initiating effect was shown by CDs prepared from sole citric acid and those doped with ethylenediamine (CA-ED2-CDs) (Table 2).
| Carbon dots | Initiating system | dα/dt [s−1] | Induction time [s] |
|---|---|---|---|
| CA-ED2-CDs | MDEA + IOD | 1.95 | 19 |
| CA-CDs | 1.62 | 25 | |
| CA-NH3-CDs | 1.21 | 37 | |
| GQDs | 1.24 | 38 | |
| — | 0.46 | 58 |
O. Particular attention should be paid to the possibility of the hydrogel acting as both a donor and an acceptor of hydrogen bonds, depending on its supramolecular structure. Considering how important polymer hydrogels are nowadays and widely used, it was decided to develop compositions with CDs. For this purpose, previously proven initiating systems and hydroxy-functional acrylic monomer dedicated to the preparation of hydrogel materials were used. The results showed that the highest potential in the three-component initiating systems reveled CA-based CDs (Fig. S16†).
As presented in Fig. 8 free-radical photopolymerization of HEA/water composition, initiated with three-component photoinitiating systems, results in formation of hydrogel material, with high final monomer conversion. The best results in all kinetic parameters: CC conversion, short induction time and high slope of the kinetic curve was obtained for IOD (2.0 wt%), MDEA (2.0 wt%) and CA (0.2 wt%) system (Fig. 9).
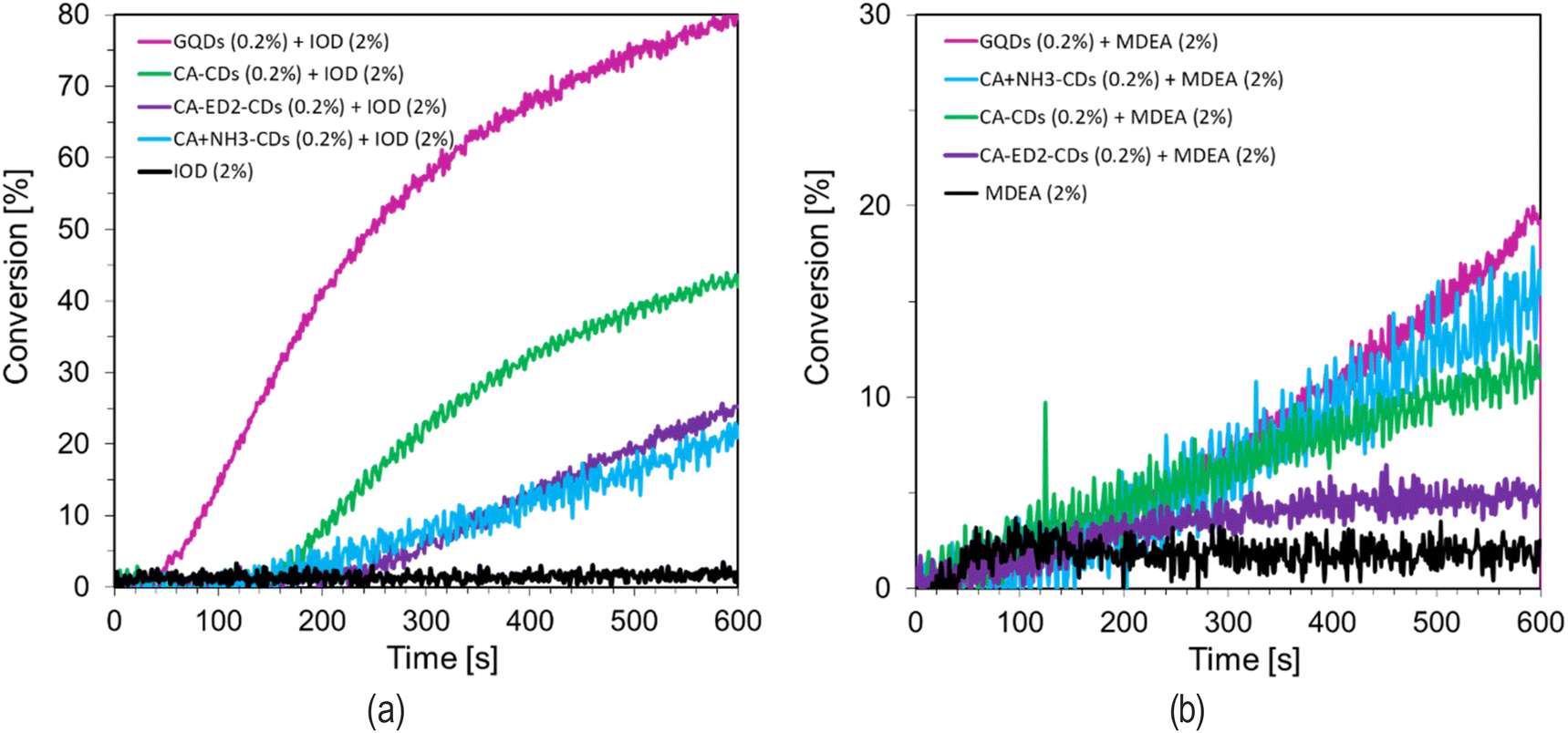 | ||
| Fig. 8 Free-radical photopolymerization profiles (acrylate function conversion vs. irradiation time) during the manufacturing of hydrogels initiated by a bimolecular photoinitiating system based on (a) IOD (2.0 wt%) and CDs (0.2 wt%), (b) MDEA (2.0 wt%) and CDs (0.2 wt%) under irradiation at 405nm. | ||
 | ||
| Fig. 9 Free-radical photopolymerization profiles (acrylate function conversion vs. irradiation time) during the manufacturing of hydrogels initiated by a three-molecular photoinitiating system based on IOD (2.0 wt%), MDEA (2.0 wt%) and CDs (0.2 wt%) under irradiation at 405 nm. | ||
Calculated kinetic parameters for the process of light manufacturing of hydrogel materials are summarized in Table 3.
| Carbon dots | Initiating system | dα/dt [s−1] | Induction time [s] |
|---|---|---|---|
| CA-CDs | MDEA + IOD | 2.31 | 16 |
| GQDs | 0.89 | 29 | |
| CA-ED2-CDs | 0.66 | 28 | |
| CA-NH3-CDs | 0.62 | 88 | |
| — | 0.24 | 220 |
These initiating systems (with iodonium salt) operate according to a mechanism of photo-oxidation mechanism, in which the iodonium salt, as a strong electron acceptor, undergoes reduction by electron transfer, while the CDs, after absorbing light, become electron donors in this process. However, in the course of the study, the action of the CDs was also checked in photoinitiating systems reacting according to the photoreduction mechanism. In such a system, a so-called co-initiator molecule is needed (this role is successfully performed by tertiary amines), which will be the electron donor for the excited acceptor molecule of the – CDs. Popular co-initiators are amines, for example methyl diethanolamine (MDEA), which has a low oxidation potential (864 mV) and was therefore used in the present study (Fig. 10).
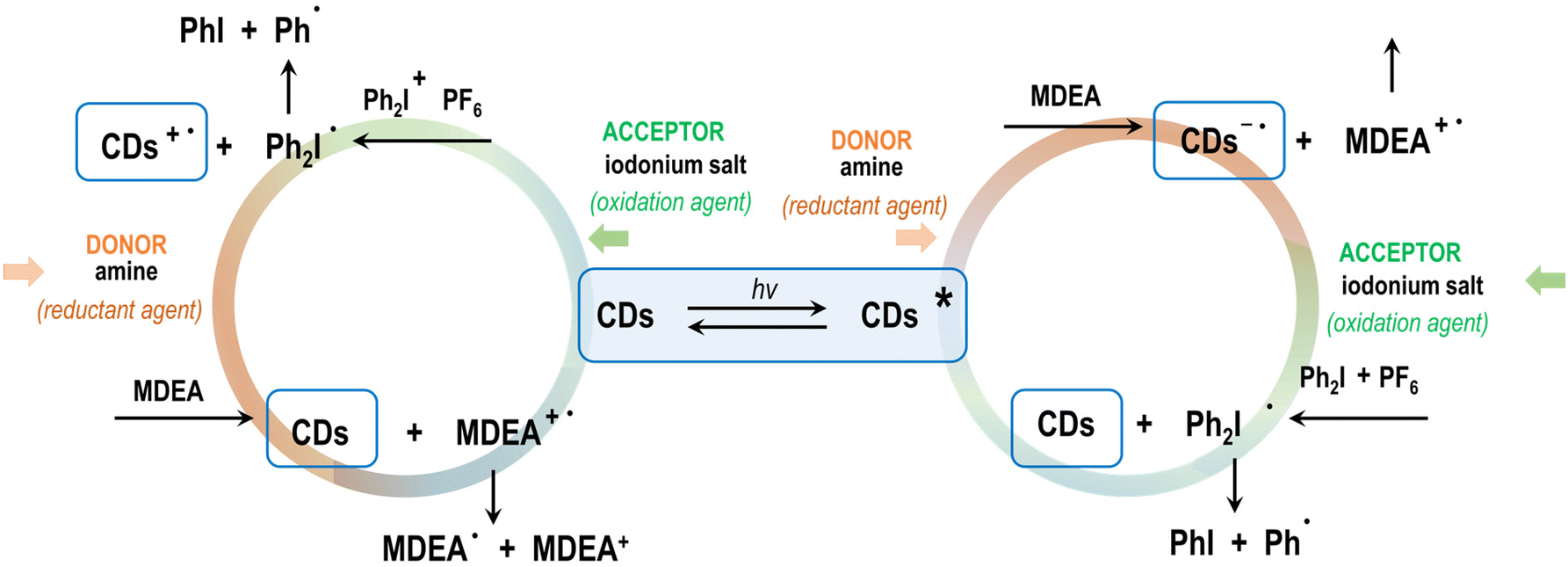 | ||
| Fig. 10 A catalytic cycle using the CDs under study. | ||
3D printing experiments
Today, 3D printing is a topic that unites many researchers, since it allows to create three-dimensional materials in a controlled manner from many raw materials and with a previously defined geometry. We decided to test various CDs as part of initialization systems for the manufacturing of 3D materials. Our previous work51 showed that CDs prepared from sole citric acid were effective for 3D printing in the presence of appropriate monomers and an initiator. So far, the present work has demonstrated that ethylenediamine-doped CDs have the greatest potential to effectively initiate the photopolymerization process in 3D-VAT printing. Accordingly, a composition consisting of the acrylate monomers TMPTA/PEGDA (1/1 wt%) and the initiating system IOD/CA-ED2-CDs(2.0/0.2 wt%) was prepared for 3D printing under a LED light source @405 nm using a Lumen X+™ printer. A resulting printout is shown in Fig. 11. From above mentioned composition, print with a significant thickness of ∼2 mm and very high spatial resolution was obtained.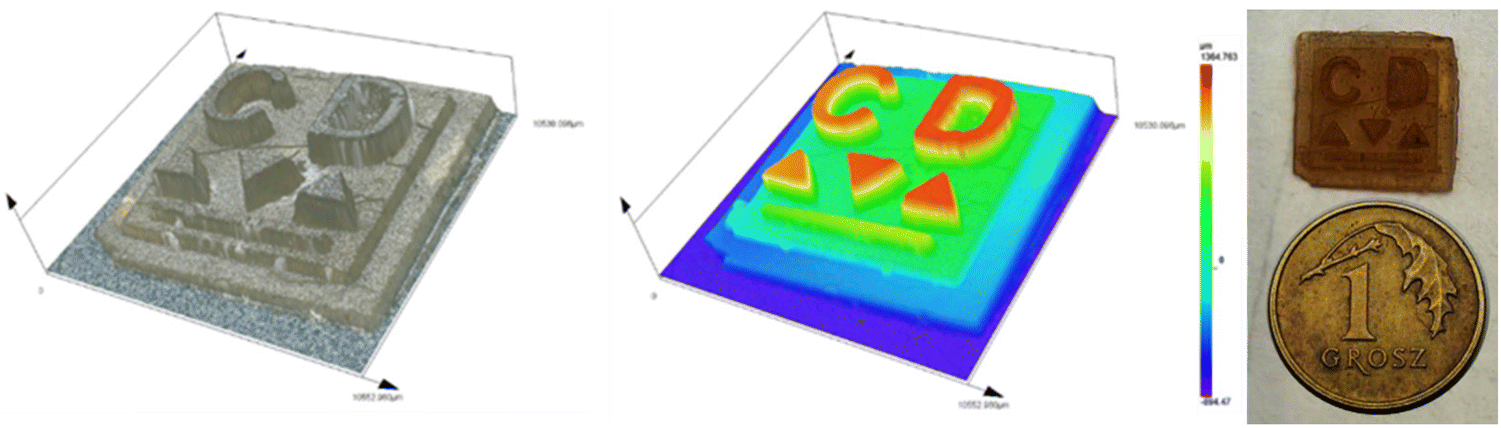 | ||
| Fig. 11 3D printout made by the cationic photopolymerization of a TMPTA:PEGDA (1/1 wt%) monomers in the presence of a two-component photoinitiating system based on CA-ED2-CDs (0.2 wt%) and IOD (2.0 wt%). | ||
Morphological analysis of 3D printed hydrogels using SEM/AFM/Nano-FTIR/s-SNOM
To evaluate the surface morphology and topography of the 3D printed 3D-VAT printouts Low Voltage Scanning Electron Microscopy (LV-SEM) was performed. From the low magnification ETD image presented (Fig. 12A), at the >10 μm scale, the surface appears to be uniform with little variation observed across the material's surface. High resolution SE imaging (Fig. 12B) presents similar findings of limited surface variation at the micron sale. However, from BSE imaging it is notable that what appears to be CB structures are identifiable. Due to the increased penetration depth of BSE imaging, it is assumed that the carbon dot structures are embedded within the material and are present sub-surface. The BSE emission image when centered on a proposed carbon dot particle (Fig. 12C) indicates the presence of some surface elemental variation most notably at the sub-micron scale. | ||
| Fig. 12 (A) Low magnification secondary electron (SE) image of a 3D-VAT printout taken using an Everhart–Thornley Detector (ETD). (B) High resolution SE image of a 3D-VAT printout taken using a Through Lens Detector (TLD). (C) Backscattered election (BSE) image taken using a concentric backscatter (CBS) detector. (D): AFM height topography of a carbon dot at the surface of a hydrogel sample. (E) AFM mechanical phase data taken simultaneously with the data in (D). AFM phase data is sensitive to a number of surface properties (hardness, adhesion, etc.) and is often difficult to interpret. In this case, we simply note that the AFM phase contrast observed in (E) allows for easy distinction between areas of the hydrogel (high AFM phase) and the carbon dot surface (low AFM phase). (F): s-SNOM phase data taken simultaneously with the data in (D), with incident illumination at 1490 cm−1. The s-SNOM data was demodulated at the 3rd harmonic of the AFM tapping frequency to reduce the influence of background effects. The hydrogel and the carbon dot particle have different optical responses under the incident illumination, and so s-SNOM phase contrast is observed between the different regions of the AFM scan. Corresponding s-SNOM amplitude data is shown in Fig. S22 of the ESI.† | ||
Further surface characterization of the hydrogel samples performed with AFM and s-SNOM techniques revealed that, occasionally, carbon dot particles can be found at or emerging from the surface of the hydrogel. Fig. 12D presents the surface topography of an 8 μm by 6.8 μm region of hydrogel as measured through AFM, which is in keeping with the surface characterization data presented in Fig. 12A–C. It is not obvious from the topography data in Fig. 12D alone which features of the sample surface relate to carbon material. However, the carbon dot particles can be identified through the mechanical properties of their surface: Fig. 15E presents the AFM phase data from the scan shown in Fig. 12D, with AFM phase being sensitive to various mechanical surface properties of the sample material such as hardness and adhesion. A strong phase contrast is observed between the soft hydrogel and the harder carbon dot material, allowing for the identification of a carbon dot particle that is only partially covered by the hydrogel. Additionally, Fig. 12F presents s-SNOM optical phase data taken during the scan shown in Fig. 12D, using illumination at 1490 cm−1. s-SNOM measurements are sensitive to optical properties such as refractive index and absorption, and the differences in these properties between the hydrogel and carbon dot materials creates strong contrast in s-SNOM phase data, allowing for further verification of the location of the carbon dot particle. We note that often large areas of the hydrogel surface had to be scanned before any carbon dot particles partially above the surface were identified, and that no carbon dot particles were found either entirely or mostly above the surface of the hydrogel. It is therefore assumed that the CDs embedded within the 3D-VAT prints do not congregate on the surface of the material but instead are distributed throughout the matrix.
3D printing of hydrogels in aqueous plotting media
As mentioned above, 3D printing is developing rapidly in many fields. One of them is the use of 3D printing in medicine to create hydrogel materials with versatile applications. As proven in research of Tomal et al.51 it is possible to print polymer hydrogels by photopolymerization. Therefore, this paper attempts to use synthesized carbon materials as eco-friendly components, as efficient initiating systems for printing polymer hydrogels. Thus, a system of acrylic monomer HEA and water in a mass ratio of 1:1 was prepared together with the initiating system IOD/CDs (2/0.2 wt%) to produce polymer hydrogels with a certain structure under the influence of a laser light source @405 nm (spot diameter 50 μm) with an intensity of about 100 mW cm−2 (DLW technique). Pictures of the obtained hydrogel printout are shown in Fig. 17. The experiment resulted in a hydrogel printout with a thickness of ∼4 mm and good optical resolution. In addition, we proved that the application of the newly synthesized nanoparticles in hydrogel production was successful. As can be seen in the figure, the hydrogel with doped dots fluoresces green, this is synonymous with the most bathochromic shift of the emission spectrum of CA-NH3-CDs dots (as mentioned earlier in this article) (Fig. 13).
 | ||
| Fig. 13 3D hydrogel printout made by the free-radical photopolymerization of a HEA/water (1/1 wt%) mixture in the presence of a two-component photoinitiating system based on CA-NH3-CDs (0.2 wt%) and IOD (2 wt%). | ||
In addition, an experiment was conducted using ethylenediamine-doped dots synthesized in the laboratory. Two identical printouts were made. One contained CDs after the full purification procedure described in section (purification of CDs). The second printout was based on unpurified CDs – containing a fluorophore formed during their synthesis. The purpose of the experiment was to check the effect of the fluorophore present in the CDs on the 3D printing process. Another goal was also to check the resolution of the resulting printouts. SpeedCure 938 initiator (2 wt%), as well as MDEA amine (2 wt%) were used to obtain 3D hydrogel materials with purified and non-purified CDs. These components were dissolved in HEA acrylate monomer with the addition of water (1/1 w/w). As can be seen in Fig. 14. the first printout is darker and has better, final optical resolution. In the case of the printout with untreated dots, there was a bleaching the material during printing (originally the composition for printing was dark brown). Another difference observed was the intensity of the light used for printing. In the first case, 4.08 mW cm−2 light of higher intensity was used. In the second case, the conditions were milder. The light intensity was lower and was 2.96 mW cm−2. Analyzing the aforementioned observations and the printing parameters that had to be chosen to successfully print the designed materials, it can be concluded that the presence of the fluorophore causes: (1) increases the rate of polymerization, which prompted a decrease in the curing time of a single layer and the intensity of the light source, (2) worsens the optical resolution of the print. The above conclusions show the importance of proper carbon dot purification in the context of studying their efficiency in photochemical processes. The presence of a fluorophore in the product, which is a by-product of their synthesis, affects the performance of the CDs and falsifies the results often shown in the literature.
 | ||
| Fig. 14 3D hydrogel printout made by the free-radical photopolymerization of a HEA/water (1/1 wt%) mixture in the presence of a two-component photoinitiating system based on (1) purity CA-ED2-CDs (0.2 wt%), IOD (2 wt%) and MDEA (2 wt%) and (2) CA-ED2-CDs before purification (with impurities and fluorophores). Printing parameters: (a) layer thickness – 100 μm, time to cure 1st layer – 90 s, time to cure layer – 30 s; (b) layer thickness – 100 μm, time to cure 1st layer – 90 s, time to cure layer – 30. | ||
Other than the mechanism we mentioned earlier, in which CDs can provide excellent results in 3D printing, we also wanted to test the performance according to the mechanism described in the article.63 Our goal was to test the composition with another amine. This was n-phenylglycine, which absorbs light better than the amine MDEA. So we decided to test nitrogen-doped CDs for printing in the presence of the n-phenylglycine (NPG) and monomers such as poly(ethylene glycol) diacrylate (PEGDA 585), 1,6 hexanediol diacrylate (HDDA), the reaction medium was also water, in which the CDs were dissolved. The results obtained are shown in Fig. 15. Obtained material was characterized by high optical resolution and thickness of 2 mm. For comparison, the same resin, but without of CDs were used for 3D printing (Fig. S28†). In addition, it is worth noting that n-phenylglycine alone is capable of initiating photopolymerization processes, as described in the article.64 Taking this into account, it is safe to say that printing with n-phenylglycine does not give clear information about the effect of other additives in the mixture of composition.
 | ||
| Fig. 15 A printout made by photopolymerization of a HDDA/PEGDA/WATER (40/40/20 wt%) mixture in the presence of a two-component photoinitiating system based on CA-ED2-CDs (0.2 wt%) and N-phenyloglicyne (2 wt%). Printing parameters: layer thickness – 100 μm, time to cure 1st layer – 45 s, time to cure layer – 15 s. | ||
Conclusions
In the present study, three types of carbon dots were synthesized. Obtained materials were based on citric acid and were doped with nitrogen sources such as ammonia and ethylenediamine. The dots were obtained by the “bottom-up” method. In further steps, the spectroscopic properties of the obtained materials were examined and the high quality of the purification process was confirmed. Then efficient systems were developed to initiate the photopolymerization processes of different acrylate monomers, leading to polymeric hydrogel materials. The same compositions have been used in 3D printing processes, to obtain three-dimensional materials. The effectiveness of the developed carbon dot-based systems has been confirmed in both digital light processing (DLP) and direct laser writing (DIW) printing technologies. In addition, the significance of proper purification of the dots from the fluorophore, which affects the printing process and the final properties of the material, has been proven through 3D printing. It has been evidenced that when analyzing the performance of carbon dots in 3D printing processes, the presence of fluorophore falsifies the results, and proper purification of the dots allows their real potential to be explored. In addition, it has been proven that when analyzing in detail the ability of carbon dots to initiate photopolymerization processes in 3D printing, it is inappropriate to use N-phenylglycine as a co-initiator, as it has the ability to initiate the polymerization reaction and disturbs the analysis of the effect of carbon dots themselves on the printing process. We believe that the research presented in this manuscript will become the basis for further work on carbon dots in the context of the diverse use of photopolymerization processes and avoid errors affecting the misinterpretation of data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was funded by National Science Centre (NCN, Poland) from budget funds for science in the years 2022–2026, as a research project under the “OPUS” program (“Emerging strategy approaches for the design and functionalization of carbon dots as multifunctional, dynamic, green systems photoinitiators and photocatalysts involved in photopolymerisation processes”; project no: UMO-2021/41/B/ST5/04533).N. T. H. F would like to thank: Engineering and Physical Sciences Research Council (EPSRC) for funding (grant: EP/V012126/1 and EP/T517835/1). N. T. H. F. acknowledges the Sorby Centre for Electron Microscopy at the University of Sheffield for allowing electron microscopy and analysis to be performed. A. J. K. and A. I. T. acknowledge support by EPSRC grant EP/V007696/1.
References
- R. Wang, K. Q. Lu, Z. R. Tang and Y. J. Xu, Mater. Chem., 2017, 5, 3717–3734 RSC.
- H. Li, X. Yan, D. Kong, R. Jin, C. Sun, D. Du, Y. Lin and G. Lu, Nanoscale Horiz., 2020, 5, 218–234 RSC.
- T. C. Wareing, P. Gentile and A. N. Phan, ACS Nano, 2021, 15, 15471–15501 CrossRef CAS PubMed.
- K. Ghosal and A. Ghosh, Mater. Sci. Eng., C, 2019, 96, 887–903 CrossRef CAS PubMed.
- S. K. Debnath and R. Srivastava, Front. nanotechnol., 2021, 3, 644564 CrossRef.
- R. Knoblauch and C. D. Geddes, Materials, 2020, 13, 4004 CrossRef CAS PubMed.
- Z. Zhang, G. Yi, P. Li, X. Zhang, H. Fan, Y. Zhang, X. Wang and C. Zhang, Nanoscale, 2020, 12, 13899–13906 RSC.
- P. Kumar, S. Dua, R. Kaur, M. Kumar and G. Bhatt, RSC Adv., 2022, 12, 4714–4759 RSC.
- N. Gao, L. Huang, T. Li, J. Song, H. Hu, Y. Liu and S. Ramakrishna, J. Appl. Polym. Sci., 2020, 137 Search PubMed.
- W. Su, H. Wu, H. Xu, Y. Zhang, Y. Li, X. Li and L. Fan, Mater. Chem. Front., 2020, 4, 821–836 RSC.
- X. Mu, M. Wu, B. Zhang, X. Liu, S. Xu, Y. Huang, X. Wang, D. Song, P. Ma and Y. Sun, Talanta, 2021, 221, 121463 CrossRef CAS PubMed.
- H. Huang, H. Ge, Z. Ren, Z. Huang, M. Xu and X. Wang, Front. Bioeng. Biotechnol., 2021, 9, 1–10 CrossRef CAS.
- J. Manioudakis, F. Victoria, C. A. Thompson, L. Brown, M. Movsum, R. Lucifero and R. Naccache, J. Mater. Chem. C, 2019, 7, 853–862 RSC.
- D. Li, W. Li, H. Zhang, X. Zhang, J. Zhuang, Y. Liu, C. Hu and B. Lei, ACS Appl. Mater. Interfaces, 2020, 12, 21009–21019 CrossRef CAS PubMed.
- J. Wang, R. Sheng Li, H. Zhi Zhang, N. Wang, Z. Zhang and C. Z. Huang, Biosens. Bioelectron., 2017, 97, 157–163 CrossRef CAS PubMed.
- H. J. Lee, J. Jana, Y. L. Thi Ngo, L. L. Wang, J. S. Chung and S. H. Hur, Mater. Res. Bull., 2019 DOI:10.1016/j.materresbull.2019.110564.
- L. Wang, Y. Wang, Y. Hu, G. Wang, S. Dong and J. Hao, J. Colloid Interface Sci., 2019, 539, 203–213 CrossRef CAS PubMed.
- X. Chen, X. Liu, P. Childs, N. Brandon and B. Wu, Adv. Mater. Technol., 2017 DOI:10.1002/admt.201700148.
- K. Qin, D. Zhang, Y. Ding, X. Zheng, Y. Xiang, J. Hua, Q. Zhang, X. Ji, B. Li and Y. Wei, Analyst, 2020, 145, 177–183 RSC.
- F. Yuan, Y. K. Wang, G. Sharma, Y. Dong, X. Zheng, P. Li, A. Johnston, G. Bappi, J. Z. Fan, H. Kung, B. Chen, M. I. Saidaminov, K. Singh, O. Voznyy, O. M. Bakr, Z. H. Lu and E. H. Sargent, Nat. Photonics, 2020, 14, 171–176 CrossRef CAS.
- C. Hu, M. Li, J. Qiu and Y. P. Sun, Chem. Soc. Rev., 2019, 48, 2315–2337 RSC.
- M. Bacon, S. J. Bradley and T. Nann, Part. Part. Syst. Charact., 2014, 31, 415–428 CrossRef CAS.
- P. K. Yadav, S. Chandra, V. Kumar, D. Kumar and S. H. Hasan, Catalysts, 2023, 13 Search PubMed.
- B. D. Mansuriya and Z. Altintas, 2018, 11, John Wiley & Sons.
- W. Kasprzyk, T. Świergosz, S. Bednarz, K. Walas, N. V. Bashmakova and D. Bogdał, Nanoscale, 2018, 10, 13889–13894 RSC.
- S. Fernandes, J. C. G. E. da Silva and L. P. da Silva, Materials, 2022, 15, 3446–3468 CrossRef CAS PubMed.
- S. Fernandes, J. C. G. Esteves da Silva and L. Pinto da Silva, NanoImpact, 2021, 23, 1–11 CrossRef PubMed.
- L. Cui, J. Wang and M. Sun, Rev. Phys., 2021, 6 Search PubMed.
- P. Fiedor and J. Ortyl, Materials, 2020, 13, 1–25 CrossRef PubMed.
- J. Ortyl, P. Fiedor, A. Chachaj-Brekiesz, M. Pilch, E. Hola and M. Galek, Sensors, 2019, 19, 1668–1680 CrossRef CAS PubMed.
- P. Fiedor, M. Pilch, P. Szymaszek, A. Chachaj-Brekiesz, M. Galek and J. Ortyl, Catalysts, 2020, 10, 284–304 CrossRef CAS.
- M. Topa, F. Petko, M. Galek and J. Ortyl, Sensors, 2020, 20, 3043–3050 CrossRef CAS PubMed.
- E. Hola, F. Morlet-Savary, J. Lalevée and J. Ortyl, Eur. Polym. J., 2023 Search PubMed.
- J. Ortyl, J. Wilamowski, P. Milart, M. Galek and R. Popielarz, Polym. Test., 2015, 48, 151–159 CrossRef CAS.
- J. Ortyl and R. Popielarz, J. Appl. Polym. Sci., 2013, 128, 1974–1978 CAS.
- E. Hola, A. Gruchała, R. Popielarz and J. Ortyl, Eur. Polym. J., 2021 Search PubMed.
- J. Ortyl, M. Galica, R. Popielarz and D. Bogdał, Pol. J. Chem. Technol., 2014, 16, 75–80 CrossRef CAS.
- W. Tomal, M. Pilch, A. Chachaj-Brekiesz, M. Galek, F. Morlet-Savary, B. Graff, C. Dietlin, J. Lalevée and J. Ortyl, Polym. Chem., 2020, 11, 4604–4621 RSC.
- A. Al Mousawi, C. Dietlin, B. Graff, F. Morlet-Savary, J. Toufaily, T. Hamieh, J. P. Fouassier, A. Chachaj-Brekiesz, J. Ortyl and J. Lalevée, Macromol. Chem. Phys., 2016, 217, 1955–1965 CrossRef.
- J. Ortyl, M. Galek, P. Milart and R. Popielarz, Polym. Test., 2012, 31, 466–473 CrossRef CAS.
- G. Temel and N. Arsu, J. Photochem. Photobiol., A, 2007, 191, 149–152 CrossRef CAS.
- W. Tomal, M. Pilch, A. Chachaj-Brekiesz, M. Galek, F. Morlet-Savary, B. Graff, C. Dietlin, J. Lalevée and J. Ortyl, Polym. Chem., 2020, 11, 4604–4621 RSC.
- E. Hola, M. Topa, A. Chachaj-Brekiesz, M. Pilch, P. Fiedor, M. Galek and J. Ortyl, RSC Adv., 2020, 10, 7509–7522 RSC.
- E. Hola, M. Pilch and J. Ortyl, Catalysts, 2020, 10, 1–28 CrossRef.
- J. Ortyl, K. Sawicz and R. Popielarz, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4522–4528 CrossRef CAS.
- E. Hola, M. Pilch, M. Galek and J. Ortyl, Polym. Chem., 2020, 11, 480–495 RSC.
- J. Ortyl, P. Fiedor, A. Chachaj-Brekiesz, M. Pilch, E. Hola and M. Galek, Sensors, 2019, 19, 1668 CrossRef CAS PubMed.
- M. Retailleau, A. Ibrahim, C. Croutxé-Barghorn, X. Allonas, C. Ley and D. Le Nouen, ACS Macro Lett., 2015, 4, 1327–1331 CrossRef CAS PubMed.
- Y. Liang, L. Chen, D. Zhuang, H. Liu, R. Fu, M. Zhang, D. Wu and K. Matyjaszewski, Chem. Sci., 2017, 8, 2101–2106 RSC.
- C. Kütahya, P. Wang, S. Li, S. Liu, J. Li, Z. Chen and B. Strehmel, Angew. Chem., 2020, 132, 3192–3197 CrossRef.
- W. Tomal, T. Świergosz, M. Pilch, W. Kasprzyk and J. Ortyl, Polym. Chem., 2021, 12, 3661–3676 RSC.
- J. A. Pan, J. C. Ondry and D. V. Talapin, Nano Lett., 2021, 21, 7609–7616 CrossRef CAS PubMed.
- Y. Wang, F. Yang, X. Li, F. Ru, P. Liu, L. Wang, W. Ji, J. Xia and X. Meng, Adv. Funct. Mater., 2019 DOI:10.1002/adfm.201904913.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS PubMed.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef CAS PubMed.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., 2015, 127, 14834–14846 CrossRef.
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Häusler, P. Kasák, W. Schnick and A. L. Rogach, Chem. Mater., 2017, 29, 10352–10361 CrossRef CAS.
- H. R. Harald Hantsche, Adv. Mater., 1993, 5, 778–1010 CrossRef.
- R. Dedryvère, M. Maccario, L. Croguennec, F. le Cras, C. Delmas and D. Gonbeau, Chem. Mater., 2008, 20, 7164–7170 CrossRef.
- J. Fang, Y. Wang, M. Kurashvili, S. Rieger, W. Kasprzyk, Q. Wang, J. K. Stolarczyk, J. Feldmann and T. Debnath, Angew. Chem., Int. Ed., 2023 DOI:10.1002/anie.202305817.
- W. Kasprzyk, T. Świergosz, P. P. Romańczyk, J. Feldmann and J. K. Stolarczyk, Nanoscale, 2022, 14, 14368–14384 RSC.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- X. Huang, M. Shi, H. Zhai, Y. Zhang, Y. Zhang and Y. Zhao, Polym. Chem., 2022, 14, 268–276 RSC.
- G. Ullrich, P. Burtscher, U. Salz, N. Moszner and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 115–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00726j |
| This journal is © The Royal Society of Chemistry 2023 |