
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of redox-responsive core–shell nanoparticles: insights into core-crosslinking efficiency†
Yannik
Olszowy
 ,
Janick
Wesselmann
,
Shenja Fabienne
Over
,
Florian
Pätzold
and
Ralf
Weberskirch
*
,
Janick
Wesselmann
,
Shenja Fabienne
Over
,
Florian
Pätzold
and
Ralf
Weberskirch
*
Department of Chemistry and Chemical Biology, Polymer Hybrid Systems, TU Dortmund University, Otto-Hahn Straße 6, 44227, Dortmund, Germany. E-mail: ralf.weberskirch@tu-dortmund.de
First published on 25th July 2023
Abstract
Core-crosslinked micelles have become an important class of materials for biomedical applications. However, there has been little work attempting to quantify the efficiency of the core crosslinking reaction, instead residual polymers are usually removed by dialysis. In this work we have prepared core crosslinked micelles based on poly(2-ethyl-2-oxazoline)-block-poly(n-butyl acrylate-co-D,L-homocysteine thiolactone acrylamide). Core crosslinking was examined by the addition of six different di- and triamines and nucleophilic ring-opening of the thiolactone ring. By using size exclusion chromatography (SEC) we were able to quantify the amount of crosslinked micelle and free block copolymer and were able to optimize the crosslinking conditions in terms of temperature, reaction time and crosslinker equivalents to obtain up to 80% core-crosslinked micelles. Subsequently, micelles that were crosslinked with cystamine were degraded in the presence of dithiothreitol (DTT) and resulted in degradation times of 1.5 h to 5 h and depended strongly on the composition of the hydrophobic core as shown by dynamic light scattering (DLS) and size exclusion chromatography (SEC). Cytotoxicity assays of the core-crosslinked micelles and block copolymer precursors were performed with COS7 cells and revealed high cell viabilities up to 0.1 mg mL−1.
Introduction
In the last decade, polymeric micelles have been extensively investigated for drug delivery and diagnostic applications.1–3 Despite their advantages in terms of ease of preparation, large compositional variability, and scalability, micelles exhibit low stability and dissociate in very dilute medium below the critical micelle formation concentration which may lead to premature drug release.4 One way to increase the stability of micelles is to crosslink them, either through the shell or the core.5 Although shell-crosslinked particles have been shown to be stable at high dilution6 while maintaining good encapsulation efficiencies, core-crosslinked particles have received significantly more attention because intermolecular crosslinking between particles can be largely avoided and crosslinking can be performed at higher polymer and micelle concentrations.5 Among the various methods to stabilize polymer micelles in the core, disulfide bonds (S–S) play an important role due to the redox active binding, which can be cleaved under reductive conditions to release a potential drug load.7–10 In recent years, different methods have been developed to prepare nanoparticles with a disulfide-based crosslinker. One of these methods is based on preparing copolymers with disulfide functionalities in the side chain and achieving crosslinking by adding catalytic amounts of dithiothreitol (DTT) to enable a disulfide exchange reaction.9–14 A second method is based on introducing the disulfide functionality into the nanoparticle by a multifunctional crosslinker.15–18 It is noteworthy, however, that the cross-linking efficiency of the reaction is hardly studied in these cases. Instead, characterization of nanoparticles is typically performed by transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements which only provide qualitative evidence of particle formation. This also applies to many other crosslinking reactions of micelles, regardless of whether they involve micelle core or shell crosslinking.19–23 Only a few papers really tried to look deeper into the crosslinking efficiency, like the work of Schacher et al. using 1H HR MAS NMR spectroscopy.24 However, this method requires well-resolved NMR signals and thus depends very much on the block copolymer and crosslinking chemistry. In addition, this NMR method appears not to be suitable for strongly crosslinked polymer networks as the analysis of hydrogels with different crosslinking densities recently showed.25 Therefore, the solution to the problem, especially for medical applications, is that nanoparticles crosslinked by core or shell are purified by dialysis for several days after synthesis. To separate the nanoparticle from non-reacted precursor polymer before they are employed for further in vitro and in vivo experiments. This approach makes it both impossible to optimize the crosslinking reaction and subsequent nanoparticle formation. Moreover, residual free polymer may lead to premature drug release26 or can be responsible for the formation of an unwanted plasma protein corona.27Therefore, the goal of our research was to synthesize biodegradable nanoparticles based on amphiphilic block copolymers and to use size exclusion chromatography (SEC) to analyze the core crosslinking reaction. Poly(2-ethyl-2-oxazoline) was chosen as the hydrophilic block for its potential as a promising replacement polymer for polyethylene glycol (PEG).28,29 The hydrophobic block was composed of D,L-homocysteine thiolactone acrylamide and n-butyl acrylate. The high reactivity and selectivity of the thiolactone units in homo- and copolymers has been demonstrated in the past, for example in the fabrication of glycopolymer based nanoparticle,30 functional polymer beads,31 polymeric ionic liquids32 or enzyme functionalized polymeric films.33
The synthesis was carried out by a living, cationic, ring-opening polymerization of 2-ethyl-2-oxazoline to form the poly(2-ethyl-2-oxazoline) block followed by reversible addition–fragmentation chain-transfer (RAFT) polymerization of the second, hydrophobic block. Subsequent core crosslinking was performed by nucleophilic, ring-opening of the thiolactone ring with six different di- and triamine crosslinker. The efficacy of the core crosslinking reaction was analyzed by SEC measurements of the nanoparticle mixture that provide quantitative information of the mass fraction of nanoparticles versus free polymer precursor and thus allows to optimize the crosslinking reaction conditions. Furthermore, the degradation of nanoparticles crosslinked with cystamine by reductive cleavage of the disulfide bridge were monitored by dynamic light scattering (DLS) and SEC analysis. The fabrication and analysis scheme for the nanoparticles is depicted in Scheme 1.
 | ||
| Scheme 1 Summary of the block copolymer synthesis, micelle formation, micellar core-crosslinking, degradation and characterization by means of DLS, TEM and SEC. | ||
Experimental section
Chemicals
All chemicals were purchased from Sigma Aldrich, TCI, Alfa Aesar, Carbolution or Acros Organics and were used without further purification. Benzyl bromide was distilled under vacuum at 60 °C and stored under inert conditions over molecular sieves. 2-Ethyl-2-oxazoline was distilled under inert conditions at 130 °C with calcium hydride and stored over molecular sieves. Dry solvents were taken from a solvent drying system MB-SPS 800 from Braun under nitrogen atmosphere.NMR spectroscopy
The 1H-NMR and 13C-NMR spectra were recorded using Bruker DRX-400 (400 MHz), DRX-500 (500 MHz), or DRX-600 (600 MHz) FT NMR instruments. The chemical shift δ was measured in ppm and the coupling constants J were expressed in Hz.SEC measurements
For the SEC measurements an L-5000 LC controller and a 655A-11 liquid chromatograph from Merck Hitachi were used. The polymers measured were dissolved in N,N-dimethyl formamide (HPLC grade + 5 g L−1 LiBr) so that the mass concentration was 3 mg mL−1. Samples were filtered with a 0.22 μm PFTE syringe prefilter before measurement. One precolumn from PSS GRAM and two columns (PSS GRAM analytical 1000 Å and PSS GRAM analytical 30 Å) were used for the measurements. The column temperature was 35 °C and the flow rate was 1 mL min−1. Signal detection was performed using an RI Detector Smartline 2300 from Knauer. A poly(methyl methacrylate) and poly(styrene) calibration kit from PSS was used to calibrate the system. To ensure complete elution of the nanoparticles from the analytical columns, recovery experiments with and without the columns were performed as triplets and the recovery rate was determined via comparison of the different detector signals. Two different methods were used for the quantitative evaluation of nanoparticle formation. First, the integrals of the nanoparticle (INP) and the polymer (IP) in the nanoparticle sample were directly compared.
Second, the residual polymer signal of the nanoparticle sample (IP) was compared with the integral of the polymer before crosslinking (Iref.).

DLS analysis of particle size
A Zeta PALS from Brookhaven Instruments Cooperation was used for the DLS measurements. The measurements were analyzed using the BIC Particle Solutions software. Polymer solutions with a concentration of 0.5 mmol L−1 and nanoparticle solutions with a concentration of 1 mg mL−1 were used for sample measurements.TEM analysis
A Philips CM200 instrument with the Gatan Orius SC200 camera was used to acquire the TEM images. The samples were measured in water with a concentration of 0.01 mg mL−1. Uranyl acetate was used for contrasting. During sample preparation, 20 μL of sample was placed on a copper grid. After one minute, the drop was dried with a filter paper.Synthesis of 1,2-(butylthiocarbonothioylthio)propanoic acid T134
A solution of 1-butanethiol (2.51 mL, 23.29 mmol, 1.00 eq.) and triethylamine (3.68 mL, 26.78 mmol, 1.15 eq.) in dry dichloromethane (50 mL) was cooled to 0 °C and after 30 min carbon disulfide (1.55 mL, 25.61 mmol, 1.10 eq.) was added under ice bath cooling. After heating to rt over a period of 30 min, 2-bromopropionic acid (2.30 mL, 25.61 mmol, 1.10 eq.) was added and the reaction was stirred for another 2 h at rt. Now the solvent was removed and the residue was taken up in cyclohexane (100 mL). Then the organic phase was washed three times each with 10% HCl and deionized water and dried over MgSO4. After removing the solvent, the product was purified by column chromatography (SiO2, cyclohexane/ethyl acetate, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 3:1 → 2:1). The product was obtained as a yellow solid (2.66 g, 11.20 mmol, 48%). 1H-NMR (CDCl3): δ 0.95 (t, J = 7.34 Hz, 3 H, CH2CH2CH3), 1.45 (sext, J = 7.41 Hz, 2 H, CH2CH2CH3), 1.64 (d, J = 7.34 Hz, 3 H, CHCH3), 1.70 (m, 2 H, CH2CH2CH3), 3.38 (t, J = 7.52 Hz, 2 H, SCH2CH2), 4.88 (q, J = 7.34 Hz, 1 H, HOOCCH(CH3)S) ppm. 13C-NMR (CDCl3): δ 13.55, 16.56, 22.02, 29.84, 37.09, 47.41, 177.08 ppm.
:1 → 3:1 → 2:1). The product was obtained as a yellow solid (2.66 g, 11.20 mmol, 48%). 1H-NMR (CDCl3): δ 0.95 (t, J = 7.34 Hz, 3 H, CH2CH2CH3), 1.45 (sext, J = 7.41 Hz, 2 H, CH2CH2CH3), 1.64 (d, J = 7.34 Hz, 3 H, CHCH3), 1.70 (m, 2 H, CH2CH2CH3), 3.38 (t, J = 7.52 Hz, 2 H, SCH2CH2), 4.88 (q, J = 7.34 Hz, 1 H, HOOCCH(CH3)S) ppm. 13C-NMR (CDCl3): δ 13.55, 16.56, 22.02, 29.84, 37.09, 47.41, 177.08 ppm.
Synthesis of thiolactone acrylamide M135
The D,L-homocysteine hydrochloride (7.00 g, 45.57 mmol, 1.00 eq.) was placed in a 1/1 mixture of 1,4-dioxane and deionized water (100 mL) along with NaHCO3 (19.14 g, 227.83 mmol, 5.00 eq.). After cooling the reaction solution to 0 °C, acryloyl chloride (7.36 mL, 91.13 mmol, 2.00 eq.) was added dropwise and the reaction solution was then stirred overnight at rt. Next, the resulting solid was filtered off and 100 mL brine was added to the filtrate. The aqueous phase was extracted three times with ethyl acetate and the combined organic phases were dried over MgSO4. After removal of the solvent, the product was obtained as a white solid after recrystallization from dichloromethane (6.44 g, 37.61 mmol, 83%). 1H-NMR (CDCl3): δ 1.99 (dq, J = 6.97, 12.59 Hz, 1 H, SCH2CH2), 2.95 (quin, J = 5.75 Hz, 1 H, SCH2CH2), 3.27 (ddd, J = 1.10, 6.85, 11.25 Hz, 1 H, SCH2CH2), 3.39 (dt, J = 5.14, 11.37 Hz, 1 H, SCH2CH2), 4.62 (quin, J = 6.36 Hz, 1 H, NHCHCO), 5.70 (dd, J = 1.47, 10.27 Hz, 1 H, CH2CHCO), 6.17 (dd, J = 10.15, 17.00 Hz, 1 H, CH2CHCO), 6.31 (dd, J = 1.47, 17.00 Hz, 1 H, CH2CHCO) ppm. 13C-NMR (CDCl3): δ 27.59, 31.84, 59.42, 127.60, 129.92, 165.82, 205.62 ppm.Deprotection of cystamine dihydrochloride
To a suspension of cystamine dihydrochloride (200.00 mg, 0.90 mmol, 1.00 eq.) in 20 mL dichloromethane, an aqueous 1 M sodium hydroxide solution (78.15 mg, 1.95 mmol, 2.20 eq.) was added dropwise. After the end of the addition, stirring was performed for 30 min at rt. Now 20 mL of water was added and the phases were separated. The aqueous phase was extracted three times with dichloromethane and then the combined organic phases were dried over MgSO4. After removal of the solvent, the product was obtained as a slightly yellowish oil (108.25 mg, 0.71 mmol, 80%). 1H-NMR (CDCl3): δ 1.36 (br s, 4 H, CH2NH2), 2.72 (t, J = 6.2 Hz, 4 H, CH2NH2), 2.97 (t, J = 6.2 Hz, 4 H, SCH2CH2) ppm. 13C-NMR (CDCl3): δ 40.59, 42.57 ppm.Synthesis of the RAFT functionalized homopolymers PP1–PP3
2-Ethyl-2-oxazoline (50.00 eq., PP1: 2.00 mL, 19.77 mmol; PP2: 3.00 mL, 29.76 mmol) was dissolved in dry acetonitrile (1 mL per 3 mmol) and benzyl bromide (1.00 eq., PP1: 46.97 μL, 0.39 mmol; PP2: 70.59 μL, 0.59 mmol) was added as initiator at rt. After stirring for 3 h at 120 °C, the polymerization was terminated by adding the RAFT reagent T1 (2.00 eq., PP1: 188.52 mg, 0.79 mmol; PP2: 283.36 mg, 1.19 mmol) and a triethylamine (3.00 eq., PP1: 164.44 μL, 1.19 mmol; PP2: 247.17 μL, 1.78 mmol) and stirring for 72 h at 50 °C. Then the solvent was removed and the residue was taken up in chloroform and washed twice with 5 wt% NaHSO4 solution, dried, and most of the solvent was removed. The polymer was precipitated at least three times from cold diethyl ether. The product was obtained as a yellow solid after drying at vacuum.PP1 (yield = 1.34 g, 0.23 mmol, 66%). 1H-NMR (CDCl3): δ 0.91 (m, 3 H, CH2CH2CH3) 1.09 (s, 167 H, COCH2CH3), 1.41 (m, 2 H, CH2CH2CH3), 1.55 (m, 3 H, CHCH3), 1.65 (m, 2 H, CH2CH2CH3), 2.28–2.37 (m, 113 H, COCH2CH3), 3.43 (s, 228 H, NCH2CH2), 4.24 (s, 2 H, NCH2CH2), 4.49 (m, 2 H, C6H5CH2), 4.78 (s, 1 H, OOCCH(CH3)S) ppm.
PP2 (yield = 2.70 g, 0.50 mmol, 86%). 1H-NMR (CDCl3): δ 0.91 (m, 3 H, CH2CH2CH3) 1.09 (s, 150 H, COCH2CH3), 1.40 (m, 2 H, CH2CH2CH3), 1.55 (m, 3 H, CHCH3), 1.66 (m, 2 H, CH2CH2CH3), 2.27–2.37 (m, 102 H, COCH2CH3), 3.43 (s, 205 H, NCH2CH2), 4.24 (s, 2 H, NCH2CH2), 4.49 (m, 2 H, C6H5CH2), 4.77 (s, 1 H, OOCCH(CH3)S) ppm.
PP3 (yield = 2.69 g, 0.59 mmol, 82%). 1H-NMR (CDCl3): δ 0.92 (t, 3 H, J = 7.5 Hz, SCH2CH2CH2CH3), 1.11 (m, 163 H, NCOCH2CH3), 1.42 (m, 2 H, SCH2CH2CH2CH3), 1.57 (m, 3 H, OCOCH2CH3), 1.67 (m, 2 H, SCH2CH2CH2CH3), 2.17–2.54 (br, 109H, NCOCH2CH3), 3.17–3.66 (br(t), 237 H, J = 7.2 Hz, 54 x NCH2CH2, 1 x NCH2CH2, 1 x SCH2CH2CH2CH3), 4.11–4.34 (br, 2 H, NCH2CH2), 4.48–4.66 (br, 2 H, PhCH2N), 4.79 (m, 1 H, OCOCH).).
Synthesis of the M1 containing amphiphilic block copolymers P2.1–4
The macroinitiator (1.00 eq.) and the crosslinking monomer M1 (n eq.) were dissolved in 1,4-dioxane (0.5 mL mmol−1M1) and degassed three times by the freeze–pump–thaw method. AIBN (0.2 eq.) was then added and the reaction was stirred for 24 h at 90 °C. Now the polymer was purified by precipitation from cold diethyl ether. The product was obtained as a yellowish solid after drying at vacuum.P2.1 (0.15 g, 0.025 mmol, 89%). 1H-NMR (CDCl3): δ 0.92 (s, 3 H, CH2CH2CH3), 1.12 (s, 150 H, COCH2CH3), 1.40 (s, 4 H, CH2CH2CH3), 1.65 (s, 6 H, CH2CH2CH3), 2.30–2.39 (m, 110 H, COCH2CH3), 3.45 (s, 208 H, NCH2CH2), 4.12 (s, 3 H, NCH2CH2), 4.51–4.96 (m, 7 H, C6H5CH2, NHCHCO) ppm.
P2.2 (0.18 g, 0.026 mmol, 95%). 1H-NMR (CDCl3): δ 0.91 (s, 3 H, CH2CH2CH3), 1.11 (s, 150 H, COCH2CH3), 1.40–1.64 (m, 20 H, CH2CH2CH3), 2.29–2.38 (m, 118 H, COCH2CH3), 3.43 (s, 223 H, NCH2CH2), 4.12 (s, 3 H, NCH2CH2), 4.51–4.94 (m, 11 H, C6H5CH2, NHCHCO) ppm.
P2.3 (0.20 g, 0.026 mmol, 92%). 1H-NMR (CDCl3): δ 0.93 (s, 3 H, CH2CH2CH3), 1.12 (s, 150 H, COCH2CH3), 1.42–1.66 (m, 32 H, CH2CH2CH3), 2.30–2.40 (m, 135 H, COCH2CH3), 3.45 (s, 230 H, NCH2CH2), 4.12 (s, 3 H, NCH2CH2), 4.52–4.95 (m, 16 H, C6H5CH2, NHCHCO) ppm.
P2.4 (0.25 g, 0.029 mmol, 99%). 1H-NMR (CDCl3): δ 0.90 (s, 3 H, CH2CH2CH3), 1.09 (s, 150 H, COCH2CH3), 1.40–1.63 (m, 43 H, CH2CH2CH3), 2.28–2.37 (m, 142 H, COCH2CH3), 3.43 (s, 229 H, NCH2CH2), 4.12 (s, 3 H, NCH2CH), 4.59–4.91 (m, 21 H, C6H5CH2, NHCHCO) ppm.
Synthesis of the M1 and n-butyl acrylate containing amphiphilic block copolymers P1/P2.5–12
The macro-RAFT agent (1.00 eq.) and the hydrophobic monomers n-butyl acrylate (m eq.) and/or M1 (n eq.) were dissolved in 1,4-dioxane (0.5 mL mmol−1M1) and degassed three times by the freeze–pump–thaw method. AIBN (0.2 eq.) was then added and the reaction was stirred for 24 h at 90 °C. The polymer was purified by precipitation from cold diethyl ether and obtained as a yellowish solid after drying at high vacuum.P2.5 (0.13 g, 0.019 mmol, 70%). 1H-NMR (CDCl3): δ 0.92 (s, 15 H, CH2CH2CH3), 1.11 (s, 155 H, COCH2CH3), 1.35 (s, 14 H, CH2CH2CH3), 1.58 (s, 20 H, CH2CH2CH3), 2.29–2.39 (m, 122 H, COCH2CH3), 3.45 (s, 210 H, NCH2CH2), 4.02 (s, 12 H, OCH2CH2), 4.54–4.97 (m, 7 H, C6H5CH2, NHCHCO) ppm.
P2.6 (0.16 g, 0.022 mmol, 77%). 1H-NMR (CDCl3): δ 0.87 (s, 16 H, CH2CH2CH3), 1.06 (s, 155 H, COCH2CH3), 1.29 (s, 17 H, CH2CH2CH3), 1.53 (s, 27 H, CH2CH2CH3), 2.24–2.34 (m, 124 H, COCH2CH3), 3.39 (s, 219 H, NCH2CH2), 3.97 (s, 12 H, OCH2CH2), 4.51–4.90 (m, 11 H, C6H5CH2, NHCHCO) ppm.
P2.7 (0.20 g, 0.024 mmol, 85%). 1H-NMR (CDCl3): δ 0.93 (s, 17 H, CH2CH2CH3), 1.12 (s, 155 H, COCH2CH3), 1.35 (s, 18 H, CH2CH2CH3), 1.59 (s, 30 H, CH2CH2CH3), 2.30–2.40 (m, 135 H, COCH2CH3), 3.45 (s, 231 H, NCH2CH2), 4.03 (s, 14 H, OCH2CH2), 4.56–4.95 (m, 16 H, C6H5CH2, NHCHCO) ppm.
P2.8 (0.17 g, 0.024 mmol, 84%). 1H-NMR (CDCl3): δ 0.92 (s, 26 H, CH2CH2CH3), 1.12 (s, 155 H, COCH2CH3), 1.36 (s, 19 H, CH2CH2CH3), 1.59 (s, 26 H, CH2CH2CH3), 2.29–2.39 (m, 118 H, COCH2CH3), 3.45 (s, 220 H, NCH2CH2), 4.03 (s, 19 H, OCH2CH2), 4.57–4.96 (m, 6 H, C6H5CH2, NHCHCO) ppm.
P2.9 (0.20 g, 0.024 mmol, 88%). 1H-NMR (CDCl3): δ 0.92 (s, 29 H, CH2CH2CH3), 1.11 (s, 155 H, COCH2CH3), 1.35 (s, 27 H, CH2CH2CH3), 1.58 (s, 30 H, CH2CH2CH3), 2.29–2.39 (m, 133 H, COCH2CH3), 3.44 (s, 234 H, NCH2CH2), 4.02 (s, 24 H, OCH2CH2), 4.57–4.96 (m, 12 H, C6H5CH2, NHCHCO) ppm.
P2.10 (0.21 g, 0.023 mmol, 82%). 1H-NMR (CDCl3): δ 0.92 (s, 26 H, CH2CH2CH3), 1.10 (s, 155 H, COCH2CH3), 1.34 (s, 25 H, CH2CH2CH3), 1.57 (s, 47 H, CH2CH2CH3), 2.29–2.39 (m, 141 H, COCH2CH3), 3.44 (s, 236 H, NCH2CH2), 4.01 (s, 22 H, OCH2CH2), 4.54–4.96 (m, 16 H, C6H5CH2, NHCHCO) ppm.
P2.11 (1.03 g, 0.131 mmol, 99%). 1H-NMR (CDCl3): δ 0.92 (m, 3 H, CH2CH2CH3), 0.96–1.18 (s, 165 H, COCH2CH3), 1.36–1.87 (br, 31 H, CH2CH, CH2CH2CH3), 2.16–2.49 (br, 130 H, NCOCH2CH3, CH2CH), 2.50–2.78 (br, 17 H, HNCHCH2), 3.12–3.63 (br(t), 249 H, NCH2CH2, NCH2CH2, HNCHCH2CH2S, SCH2CH2CH2CH3), 4.00–4.29 (br, 4 H, NCH2CH2O), 4.46–5.10 (br, 14 H, C6H5CH2, HNCHCO) ppm.
P2.12 (0.95 g, 0.121 mmol, 89%). 1H-NMR (CDCl3): δ 0.92 (m, 29 H, COOCH2CH2CH2CH3, SCH2CH2CH2CH3), 0.98–1.17 (br, 165 H, NCOCH2CH3), 1.35 (s, 22 H, COOCH2CH2CH2CH3, SCH2CH2CH2CH3), 1.58 (s, 30 H, COOCH2CH2CH2CH3, SCH2CH2CH2CH3), 2.11–2.51 (br, 132 H, NCOCH2CH3, CH2CH), 2.53–2.88 (br(s), 10 H, HNCHCH2CH2S), 3.24 (m, 12 H, HNCHCH2CH2S), 3.28–3.54 (br(t), 227 H, NCH2CH2, NCH2CH2, SCH2CH2CH2CH3), 4.02 (s, 17 H, NCH2CH2O, COOCH2CH2CH2CH3), 4.40–5.10 (br, 8 H, C6H5CH2, HNCHCO) ppm.
Synthesis of nanoparticles
For a typical procedure the block copolymer (40.00 mg, 1.00 eq. based on the M1 units per polymer chain) and amine crosslinker (V1–V6) (n eq.) were dissolved in deionized water to give a 1 mM polymer solution. After treatment in an ultrasonic bath for five minutes, the reaction solution was stirred for a time t at a temperature T. Afterwards the solution was lyophilized and taken up in chloroform. The product was obtained by precipitation in cold diethyl ether and drying at vacuum as a white to yellowish solid.DLS degradation experiments
For the nanoparticle degradation experiments, 1 mg of each particle was dissolved in 5 mL methanol containing 1% v/v triethylamine. Now, the sample was degassed with argon for 30 min and then dithiothreitol was added if necessary to give a 10 mM or 10 μM solution. The sample was then transferred to a cuvette and measured for 12 h with a total of 90 individual measurements using DLS.SEC degradation experiments
To detect the degradation of the nanoparticles by SEC, 4 mg of the particles were dissolved in 20 mL of PBS-buffer (10 mM) and degassed for 30 minutes with argon. Dithiothreitol was added to the solution if necessary, to give a 10 mM solution. After stirring for 18 h at 37 °C, the sample was lyophilized. The residue was dissolved in little chloroform and the insoluble solid was centrifuged off. The degraded nanoparticle was obtained by precipitating the supernatant from cold diethyl ether. The solid obtained was dissolved in 0.5 mL N,N-dimethyl formamide (5 g L−1 LiBr) and analyzed by SEC.Cell viability
To verify the biocompatibility of the nanoparticles, viability tests were performed using Cell Counting Kit 8 (CCK-8). For this purpose, COS-7 cells (an African green monkey kidney fibroblast-like cell line obtained from ATCC, approximately 10000 cells per well) were cultured in a 96-well plate in 90 μL each of a medium consisting of DMEM, 10% FBS, sodium pyruvate and penicillin–streptavidin, and 10 μL of different concentrations of nanoparticles or polymer solutions (in PBS buffer) for 24 h at 37 °C and 5% CO2. Subsequently, 10 μL of CCK-8 solution were added to the wells and incubated for an additional 4 h. After mixing the solutions for 1 min, the absorbance samples were analyzed at a wavelength of 450 nm using a microplate reader Synergy2 from BioTeK and analyzed using Gen5 software. All samples were prepared and measured in triplets and referenced to samples without any polymer or nanoparticle.
Results & discussion
Synthesis of the block copolymer precursor
The amphiphilic precursor block copolymers were synthesized in a two-step reaction. In the first step, two hydrophilic poly(2-ethyl-2-oxazoline) (PEtOx) prepolymers PP1/2 were prepared by the ring-opening cationic polymerization (CROP) of 2-ethyl-2-oxazoline with benzyl bromide as initiator.36 After 3 h polymerization at 120 °C, the reaction was terminated with the carboxylic acid functional CTA T1 (Scheme 1).37,38 The degree of polymerization was 56 for PP1 and 51 for PP2 as determined by 1H-NMR spectroscopy (Table 1). Analysis by 1H-NMR revealed a quantitative termination reaction. The hydrophobic polymer block was composed of D,L-homocysteine thiolactone acrylamide M1 and eventually n-butyl acrylate as a co-monomer to increase the hydrophobicity. M1 was prepared by reacting acryloyl chloride with D,L-homocysteine hydrochloride and served in the final block copolymers as potential crosslinking site (Scheme 2). | ||
| Scheme 2 Synthesis of the amphiphilic block copolymer precursors, their micelle formation and subsequent core-crosslinking exemplary with cystamine. | ||
| Polymer | DPx/y/za (theo.) |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n
(g mol−1)
n
(g mol−1) |
n
(g mol−1) |
Đ | d h (H2O) (nm) (PDI) |
|---|---|---|---|---|---|
| a Determined via1H-NMR analysis relative to the EtOx signal. b Determined by SEC, measured in DMF + 5 g L−1 LiBr (PMMA standard) (c = 3 mg mL−1). c Determined by DLS measurements in deionized water (c = 0.1–0.5 mmol L−1). d Poorly soluble in H2O. | |||||
| PP1 | 56 (50) | 5880 | 4700 | 1.17 | — |
| PP2 | 51 (50) | 5390 | 4870 | 1.21 | — |
| PP3 | 55 (50) | 5780 | 6570 | 1.06 | — |
| P1 | 56/10/10 (56/10/10) | 8870 | 6220 | 1.24 | 19 ± 2 (0.158 ± 0.016) |
| P2.1 | 51/0/5 (51/0/5) | 6250 | 5470 | 1.32 | 10 ± 2 (0.387 ± 0.033) |
| P2.2 | 51/0/10 (51/0/10) | 7100 | 5920 | 1.36 | 16 ± 2 (0.249 ± 0.028) |
| P2.3 | 51/0/14 (51/0/15) | 7790 | 6520 | 1.42 | 27 ± 3 (0.215 ± 0.016) |
| P2.4 | 51/0/19 (51/0/20) | 8640 | 7090 | 1.44 | 45 ± 10d (0.279 ± 0.009) |
| P2.5 | 51/5/5 (51/5/5) | 6890 | 6130 | 1.35 | 18 ± 2 (0.222 ± 0.027) |
| P2.6 | 51/5/10 (51/5/10) | 7740 | 6380 | 1.42 | 21 ± 2 (0.199 ± 0.012) |
| P2.7 | 51/6/15 (51/5/15) | 8730 | 6860 | 1.44 | 27 ± 4d (0.285 ± 0.017) |
| P2.8 | 51/9/5 (51/10/5) | 7400 | 6060 | 1.39 | 18 ± 2 (0.162 ± 0.018) |
| P2.9 | 51/11/10 (51/10/10) | 8500 | 6590 | 1.43 | 32 ± 5 (0.178 ± 0.008) |
| P2.10 | 51/10/14 (51/10/15) | 9070 | 7160 | 1.45 | 42 ± 11d (0.301 ± 0.046) |
| P2.11 | 55/0/12 (55/0/15) | 7840 | 9160 | 1.09 | 19 ± 1 (0.156 ± 0.023) |
| P2.12 | 55/8/6 (55/10/10) | 7830 | 9450 | 1.09 | 19 ± 2 (0.191 ± 0.030) |
The second block was synthesized by RAFT technique. The macro-RAFT polymers PP1 and PP2 (Fig. S10/S11†) with their RAFT end group were used for the polymerization of n-butyl acrylate and M1. For polymer P1 a ratio of 1:10:10 of macro-RAFT agent PP1, M1 and n-butyl acrylate was used. The final composition was determined via1H-NMR (Fig. 1) and fits the theoretical values. Signal d was used to reference the polymer and signals j (M1) and f (n-butyl acrylate) were used to calculate the polymer composition.
 | ||
| Fig. 1 1H-NMR spectrum of block copolymer P1 (400 MHz in CDCl3). | ||
Polymer P1 was further analyzed by SEC and DLS (Table 1) and was used to optimize the reaction conditions of the crosslinking reaction with the biodegradable diamine cystamine.
Twelve other block copolymers P2.1–12 with varying monomer compositions were produced with the aim of getting further insights how crosslinking reaction is affected by the block copolymer composition as well as the crosslinker. The polymers were characterized with SEC and the results show a narrow molar mass distribution and good dispersities of 1.09 to 1.4 which is typical for a controlled radical polymerization (Table 1 and Fig. S2†). The DLS analysis indicate that the size of the polymeric micelles correlates with the length of the hydrophobic block. Thus, the micelles increase in size (P2.1 = 10 ± 2 nm; P2.4 = 45 ± 10 nm) when the hydrophobic block is extended (P2.1 = 5 units; P2.4 = 19 units). The same trend is visible for the polymers P2.5–P2.7 and P2.8–P2.10. The solubility of the polymers decreases with increasing hydrophobicity. The most hydrophobic polymers (P2.4, P2.7 and P2.10) are poorly soluble in water. P2.11/P2.12 were used for to study the effect of different crosslinker length and functionality.
Nanoparticle formation and quantitative analysis by SEC
An important goal of this work was to investigate the efficiency of the micellar core-crosslinking reaction as a function of various reaction parameters. As mentioned in the introduction, in most examples of nanoparticle synthesis, the success of core or shell crosslinking is only qualitatively demonstrated via DLS or TEM measurements.18–23 However, detailed studies on how efficient the reaction is under predefined conditions are lacking. This makes it difficult to optimize the reaction conditions and minimize the amount of free polymer. In this work we investigated thiolactone as a possible crosslinker in more detail as it is known to react very rapidly and quantitatively with nucleophiles such as primary amines under ring-opening.39,40As a crosslinker we used the biodegradable diamine cystamine (Scheme 1) that has already been used in related nanoparticle synthesis studies.17 To obtain well-defined spherical nanoparticles, the block copolymers were dissolved in H2O and the resulting micellar solution was afterwards crosslinked in the micellar core to form the final nanoparticles. The resulting disulfide linkages are biodegradable under reductive conditions and make nanoparticle formation reversible. To investigate the effectiveness of the crosslinking reaction we used SEC analysis to characterize the resulting nanoparticles. To ensure that the SEC results can be used quantitatively, we performed a recovery experiment. For this experiment, a polymer (P1) and a nanoparticle (NP1.1) sample were measured with and without the analytical column to ensure complete elution of the substrates. These measurements were performed as triplets and the calculated recovery rates were 90 ± 3% for P1 and 92 ± 4% for NP1.1 (Table S3†) suggesting that the SEC method can be used to quantitatively determine the composition of the nanoparticle and precursor polymer mixtures after the crosslinking reaction. Since the RI detector is a mass detector, integration of the signal areas is directly proportional to the mass of a component. In the case of a mixture of nanoparticles and precursor polymers two well separated signals appear in the SEC elugram and can be used to determine the mass proportion of each component. In addition, we used DLS and TEM measurements to analyze particle size and morphology.
P1 with ten crosslinkable thiolactone functionalities per polymer chain was used to investigate the influence of crosslinker equivalents, the temperature and the reaction time on the nanoparticle formation efficiency. We used cystamine (V3) as crosslinker, which leads to degradable nanoparticles due to the reductively cleavable disulfide group. The first experiments were carried out at 40 °C for 24 h and the amount of crosslinker was varied from 0.125 to 5.0 eq. (Fig. 2A). In theory, 0.5 eq. of the crosslinker based on the crosslinking functionalities should be sufficient to ensure effective crosslinking. From 0.125 to 1.0 eq. the amount of nanoparticles increased from about 20 wt% to around 80 wt%. As can be seen from Fig. 2, we calculated always two values for the crosslinking efficiency. The first value (Fig. 2A–C, black bar) was derived by the direct comparison of the two signals from the respective SEC elugrams (Fig. 2 on the right). The second value (blue bare) was obtained by comparing the integrals of nanoparticle mixture, i.e. nanoparticle and precursor block copolymer, with an external reference of the same polymeric precursor at the same concentration as. If the values of the two methods of evaluation are nearly the same as can be seen for the usage of 0.125 to 1.0 eq. cystamine in Fig. 2A no material is getting lost during sample preparation and filtration with a 0.22 μm PTFE filter or by other interaction with the SEC columns. However, when the crosslinking efficiency values of the two evaluation methods deviate from each other as can be seen for the usage of 2 eq. and 5 eq. cystamine as crosslinker (Fig. 2A and Table S1,†NP1.9 and NP1.10) larger particles with poor solubility are formed during the crosslinking process. These larger particles are then removed by filtration with the 0.22 μm PTFE filter when preparing the sample for the SEC analysis and thus do not appear anymore in the SEC elugram. But it is precisely the occurrence of insoluble particles that can be captured by the external reference method, which is what makes SEC analysis so useful.
 | ||
| Fig. 2 Left: Influence of (A) eq. of crosslinker, (B) reaction temperature and (C) reaction time on the conversion of polymer P1 to the crosslinked nanoparticles determined via SEC (black: direct comparison of elugram areas; blue: external comparison with a defined polymeric reference at the same sample concentration); right: corresponding SEC elugrams of the different nanoparticle mixtures (see Table S1†). | ||
The DLS measurements support the results of the SEC analysis. After the crosslinking reaction all nanoparticles show narrow distributed spherical particles of around 20 nm in H2O which fits the micellar size of P1. In methanol, as a non-selective solvent for the amphiphilic polymers, only covalently crosslinked nanoparticles can be detected (Table S1†). The sizes also fit to the micellar size of P1, suggesting that the particles are densely crosslinked and are not able to swell in organic solvents. TEM measurements of NP1.1 show spherical particles with a diameter of around 25 nm, which is in good agreement with the DLS measurements in methanol (Fig. S1†). For further experiments 0.5 eq. of diamine were used as the proportion of nanoparticles formed is very high and the formation of insoluble products through presumably intermolecular crosslinking reactions can be prevented. The impact of the reaction temperature (Fig. 2B) shows around 80% crosslinking efficiency at room temperature and 40 °C, while increasing the temperature to 60 °C or 80 °C leads again to insoluble nanoparticles. The SEC elugram for 60 °C shows a shorter retention time of the first signal and therefore suggests an increase in molar mass for this nanoparticle. This increase is also visible in the DLS measurements (Table S1†). The particle size increases from 17 ± 2 nm to 129 ± 20 nm. Therefore, a crosslinking temperature of 40 °C was chosen as the optimal reaction temperature. The investigation on the reaction time (Fig. 2C) indicates that the reaction is completed after 20 h.
With this optimized set of reaction conditions, the impact of polymer architecture was examined. For this purpose, a total of ten block copolymers (P2.1–P2.10) with varying amounts of the crosslinking monomer M1 and n-butyl acrylate were synthesized (Table 1). The crosslinking results with cystamine indicate that at least ten repetition units of M1 should be present to ensure an efficient crosslinking. The addition of five or ten units of n-butyl acrylate increases the turnover for five repetition units of M1 (NP2.1, NP2.5 and NP2.8). For higher amounts of M1, this effect is not visible. Interestingly, the polymers with the largest hydrophobic block (NP2.4, NP2.7 and NP2.10) yield nanoparticles with larger molar masses and broader distributions. Moreover, these particles are poorly soluble in H2O and are thus not suitable for medical applications. The results, however, clearly demonstrate that the composition of the hydrophobic block has a huge effect on the crosslinking efficiency and final nanoparticle formation (Fig. 3 and Table S2†).
 | ||
| Fig. 3 Top: Influence of the amount of crosslinking monomer M1 and hydrophobic monomer n-butyl acrylate as well as their ratio on the nanoparticle formation determined via SEC (black: direct comparison of elugram areas; blue: comparison with polymeric reference); bottom: corresponding SEC elugrams of the nanoparticles; core-crosslinking conditions: 0.5 eq. cystamine relative to the thiolactone repeating units, 40 °C and 24 h. | ||
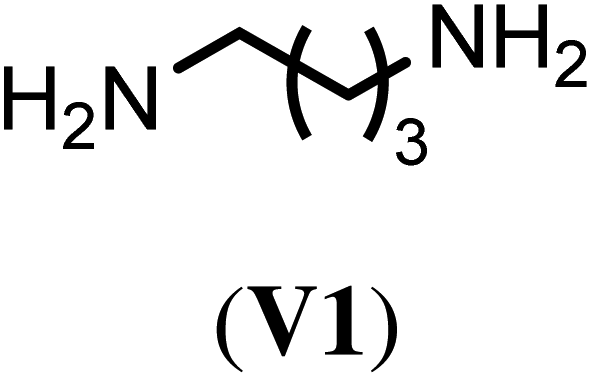
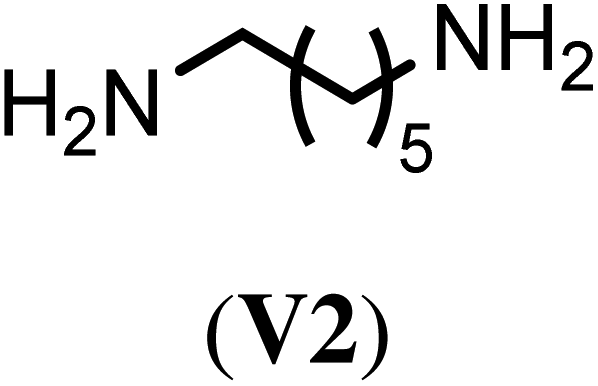
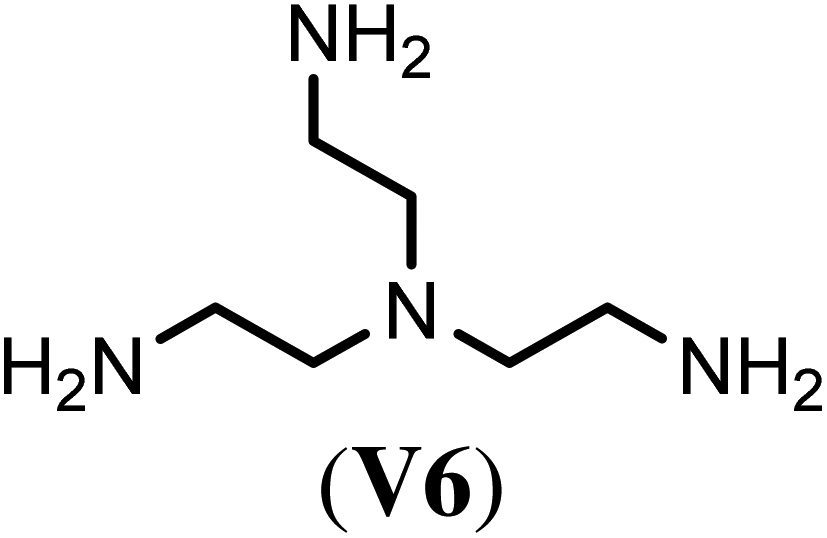
With the optimized reaction conditions, we tested then a set of different bi- and trifunctional amino crosslinker and how crosslinker length and functionality affects crosslinking efficiency. As a precursor polymer we used block copolymer P2.11 that contains a poly(D,L-homocysteine thiolactone acrylamide) in the hydrophobic block and P2.12 with a mixture of n-butyl acrylate/thiolactone monomers (Table 1). As a crosslinker, we studied four aliphatic diamines with four to eight atoms in length (V1–V4), 4,4′-diaminodiphenyl methane (V5) as a an aromatic diamine and tris(2-aminoethyl)amine (V6) as an aliphatic triamine. Nanoparticle formation was again carried out with the optimized reaction conditions of 24 h reaction time at 40 °C and 0.5 eq. of the diamino crosslinker relative to the thiolactone units. Only in the case of the trifunctional amine V6 0.33 eq. and 0.50 eq. of crosslinker were utilized. We prepared for each crosslinker/precursor block copolymer pair nanoparticles and analyzed them by DLS in H2O as a selective solvent and MeOH as a non-selective solvent to get first insights into the core-crosslinking reaction and nanoparticle formation. As can be seen from Tables S3 and S4,† all crosslinkers formed nanoparticles with exception of the aromatic diamine V5 where no particles could be detected by DLS measurements in MeOH. Best crosslinking efficiencies were obtained for the aliphatic crosslinker with a length of 6 to 8 atoms with up to 90% (Table 2, NP3.3, NP4.3) while shorter crosslinker with 4 C-atoms (Table 2, NP3.1, NP4.1) resulted in crosslinking efficiencies of 77% and 66% The aromatic diamine V5, which was used as a solid (Table 2, NP3.5) but also in a dissolved form (Table 2, NP3.6) did not result in any crosslinking due to its low solubility and nucleophilicity. Crosslinking efficiencies did not increase when using trifunctional amines with efficiencies of 70% to 87%. Moreover, there was no clear trend visible if the hydrophobic block was solely composed of thiolactone units (P2.11, Table 2) or a copolymer with n-butyl acrylate (P2.12, Table 2). In summary, the best crosslinking efficiencies were found for crosslinkers of a length of 6 to 8 C-atoms (V2–V4).
| Crosslinker | Nano-particlea | Crosslinking efficiencyb (%) | Nano-particlec | Crosslinking efficiencyb (%) |
|---|---|---|---|---|
| All crosslinkers were used in 0.5 eq. unless otherwise noted.a Nanoparticles derived from precursor polymer P2.11.b Determined by SEC (DMF + 5 mg mL−1 LiBr) with linear PMMA standard, rounded to the last digit.c Nanoparticles derived from precursor polymer P2.12.d The crosslinker 4,4′-diaminodiphenylmethane (V5) was partially dissolved in toluene.e Use of 0.33 eq. of the crosslinker tris(2-aminoethyl)amine (V6).f No signal for nanoparticles could be detected by SEC. | ||||
|
NP3.1 | 77 | NP4.1 | 66 |
|
NP3.2 | 87 | NP4.2 | 74 |

|
NP3.3 | 90 | NP4.3 | 90 |

|
NP3.4 | 82 | NP4.4 | 86 |

|
NP3.5 | 4 | NP4.5 | —f |
| NP3.6 | 0 | NP4.6 | —f | |
|
NP3.7 | 81 | NP4.7 | 88 |
| NP3.8 | 70 | NP4.8 | 87 | |
A careful look at the SEC data suggests that another reason could limit the crosslinking efficiency to about 90%. As can be seen in Fig. 4 for the example of NP4.3 and NP.4.4, the non-crosslinked polymer in the nanoparticle mixture does not show the same retention time as the precursor polymer P2.12 but is always shifted to longer retention times, i.e. smaller molar masses. A comparison with the macroRAFT PP3 shows an identical retention time, indicating that the non-crosslinked polymer could be homopolymer. These differences in retention times are found for all polymers in the nanoparticle mixture compared to the starting block copolymer (see also Fig. S34 and S35†).
 | ||
| Fig. 4 SEC chromatograms of degradable nanoparticle NP4.3 (left) and NP4.4 (right) crosslinked with cystamine (left) and 1,8-diaminooctane (right) measured in DMF (3 mg mL−1) + 5 g L−1 LiBr at a flow rate of 1 mL min−1 at 35 °C. | ||
Nanoparticle degradation experiments
For use in medicine as a drug delivery vehicle or in diagnostics biodegradability of the nanoparticles is of great importance.41–43 It is well known that disulfide bonds can be degraded under a reductive intracellular environment.11–18 To investigate the degradation of our nanoparticles, in a reductive intracellular environment a 10 mM solution of dithiotreitol (DTT) in PBS buffer was used as reducing agent.11,16 Nanoparticle degradation was again tracked using SEC and should be visible by the disappearance of the nanoparticle signal. To ensure complete degradation, the experiments were performed for 18 h at 36 °C. As a non-degradable reference particle we prepared another nanoparticle from P1 and used 1,6-hexyldiamine (HDA) as a non-degradable crosslinker (Table S1†). Fig. 5 shows the SEC elugrams of the degradable (NP1.1) and non-degradable (NP1.2) nanoparticle before and after degradation. The high molecular weight signal for NP1.1 completely disappears and indicates a complete degradation, while there were still two signals present in the elugram of NP1.2. | ||
| Fig. 5 SEC chromatograms of degradable nanoparticle NP1.1, crosslinked with cystamine (A) and non-degradable nanoparticle NP1.2, crosslinked with 1,6-hexyldiamine (B), before (black) and after crosslinking (blue) as well after degradation for 18 h in 10 mM PBS-buffer at 36 °C and 10 mM DTT (red) measured in DMF + 5 g L−1 LiBr. | ||
Surprisingly, the ratio of the two signals changed which provides evidence of a partial nanoparticle degradation of around 30% for NP1.2. A possible explanation could be that the thiols which are formed during the ring-opening reaction with the diamine can also be oxidized to disulfides as has been suggested by Du Prez et al.40 and thus would contribute to the crosslinking process. To test this hypothesis, we prepared a nanoparticle from P1 by reaction with 1-hexylamine for 24 h at 40 °C. Subsequent SEC analysis showed two overlapping signals that were not baseline separated. However, when applying the same integration limits as for the other nanoparticles the nanoparticle to polymer ratio was around 30 to 70 (Fig. S24A†). After treatment with 10 mM DTT for 24 h at 36 °C, only precursor polymer was again observed (Fig. S24B†) supporting the hypothesis of additional disulfide bond formation after ring-opening of the thiolactone units with amines. To gain better insight into the kinetics of nanoparticle degradation long-term DLS measurements were performed in the non-selective solvent methanol. The measurements were performed for 12 h at room temperature with different concentrations of the reducing agent DTT. In addition to measurements with 10 mM and 0 mM DTT, we also made measurements with 10 μM DTT because this concentration reflects the reductive environment of the extracellular environments like in blood vessels44 to verify that the nanoparticles are not degraded prematurely but only in the desired target tissue.
Fig. 6 shows the hydrodynamic diameter of the detected nanoparticles as well as the count rate. Without DTT, the count rate decreases to around 50% of the initial value independent of the used crosslinker while the diameter remains largely unchanged. At intracellular conditions (10 mM DTT) the count rate and diameter of NP1.1 rapidly decreases, indicating a degradation of the covalent crosslinks. After approximately 2 h, count rate (around 5% of initial value) and diameter remain at a constant level suggesting that the degradation has been completed at this point. For the non-degradable nanoparticle the curve progression of the high DTT concentration matches the measurement without DTT. Therefore, it can be assumed that the intracellular reductive conditions are not able to degrade the non-degradable nanoparticle. At the extracellular reductive conditions, the diameter and count rate for both nanoparticles are comparable to those measurements without DTT, assuming that even the degradable nanoparticle does not degrade under these conditions and therefore making it suitable for biomedical applications such as targeted drug delivery.
 | ||
| Fig. 6 Degradation experiments of NP1.1 (degradable) and NP1.2 (non-degradable) monitored via DLS. The samples were dissolved in MeOH with 1% v/v TEA and degassed with argon for 30 min. Afterwards DTT was added to the solution and the measurement was started. The hydrodynamic diameter is shown in black and the countrate in blue. | ||
In addition to the strength of the reductive environment, other parameters such as the block copolymer composition should also be investigated. Therefore, the degradation rate of the nanoparticles NP2.1–4 and NP2.6/9 was investigated to examine the influence of variable crosslinker concentrations and hydrophobicity of the nanoparticle core. The point of full degradation where diameter and count rate remain constant was set as point of the complete degradation and was determined by DLS measurements (Fig. S3†). The results indicate that the degradation rate decreases from around 1.5 to 5.5 h when increasing the crosslinking density (Table 3). Increasing the hydrophobicity of the nanoparticle core decreases the degradation time to some extend from 3 h to 2.0/2.2 h (Table 3). A possible reason for this observation could be that the additional n-butyl acrylate units also dilute the crosslinker density of the nanoparticle core. The resulting nanoparticles lead to a less densely crosslinked nanoparticle core, which possibly as a consequence also hinders the diffusion of DTT less than a densely crosslinked nanoparticle.
| NP | DPx/y/za (theo.) | Cross.b (%) | t deg. (h) |
|---|---|---|---|
| a Determined via1H-NMR end group analysis of polymeric precursors. b Determined by SEC measurements in DMF + 5 g L−1 LiBr. c Determined via degradation DLS measurements in MeOH (with 1% v/v TEA) with 10 mM DTT. | |||
| NP2.1 | 51/0/5 (51/0/5) | 49 | 1.5 |
| NP2.2 | 51/0/10 (51/0/10) | 78 | 3 |
| NP2.3 | 51/0/14 (51/0/15) | 84 | 5 |
| NP2.4 | 51/0/19 (51/0/20) | 86 | 5.5 |
| NP2.6 | 51/5/10 (51/5/10) | 80 | 2 |
| NP2.9 | 51/11/10 (51/10/10) | 82 | 2.2 |
Cell viability
To ensure that the nanoparticles as well as the polymeric precursors are nontoxic, they were tested in a cell viability assay. Different polymer and nanoparticle concentrations were incubated for 24 h with COS-7 cells and analyzed using CCK-8. Neither the polymer nor the nanoparticle show toxic effects, even at higher concentrations up to 1 mg mL−1 (Fig. 7). | ||
| Fig. 7 In vitro cell viability of COS-7 cells measured after incubation of different concentrations of polymer P1 (black) and nanoparticle NP1.2 (blue) for 24 h in comparison to samples without any polymer or nanoparticle present (n = 3). Determination by CCK-8. | ||
The cell viability for the polymer samples stays constant until 200 μg mL−1 and decreases to around 80% for the higher concentrations, while the viability for the nanoparticles remains constant for the entire concentration range studied.
Conclusions
In conclusion, we have prepared core-crosslinked micelles from amphiphilic precursor block copolymers. Core cross-linking was carried out by reacting di- or triamines with the thiolactone moieties of the hydrophobic micellar core. The reaction conditions and the influence of the block copolymer architecture on the crosslinking reaction were investigated in a systematic manner by using SEC analysis to quantify the mass fraction of free polymers and nanoparticles after the crosslinking reaction. Using block copolymer precursors with at least ten repetition units of the thiolactone containing monomer M1 and cross-linking conditions of 0.5 eq. of cystamine for 20 h at 40 °C resulted in approximately 80–90% crosslinking efficiency. Moreover, by investigation six different di- and triamines as potential crosslinker best crosslinking efficiencies of more than 80% were found for aliphatic diamines of 6 to 8 atoms in length. The subsequent nanoparticle degradation of cystamine core-crosslinked micelles with DTT was monitored using SEC and long term DLS measurements and the influence of the crosslinking density and hydrophobicity of the micellar core were investigated. Complete degradation was achieved after 1.5–5.5 h. Finally, the biocompatibility of a precursor block copolymer and a core-crosslinked micelle was verified by cell viability assay with COS-7 cells and neither the polymer nor the nanoparticle show significant cell toxicity up to 1 mg mL−1. The simplicity of the SEC analysis, independent of the chemistry of the polymeric micelles and the crosslinker used, make this method particularly interesting to optimize the crosslinking reaction and to draw conclusions about the amount of free polymer in the final nanoparticle.Conflicts of interest
The authors declare no competing financial interest.References
- N. Kamaly, Z. Y. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- M. Ghezzi, S. Pescina, C. Padula, P. Santi, E. Del Favero, L. Cantu and S. Nicoli, J. Controlled Release, 2021, 332, 312–336 CrossRef CAS PubMed.
- J. Kaur, V. Mishra, S. K. Singh, M. Gulati, B. Kapoor, D. K. Chellappan, G. Gupta, H. Dureja, K. Anand, K. Dua, G. L. Khatik and K. Gowthamarajan, J. Controlled Release, 2021, 334, 64–95 CrossRef CAS PubMed.
- E. Blanco, H. F. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- S. Fukushima, K. Miyata, N. Nishiyama, N. Kanayama, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2005, 127, 2810–2811 CrossRef CAS PubMed.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- X. Y. Qiu, Y. Qu, B. B. Guo, H. Zheng, F. H. Meng and Z. Y. Zhong, J. Controlled Release, 2022, 341, 498–510 CrossRef CAS PubMed.
- X. F. Zhao, X. L. Gu, H. L. Sun, R. Cheng, Z. Y. Zhong and C. Deng, Biomacromolecules, 2020, 21, 104–113 CrossRef PubMed.
- J. Y. Tao, W. D. Fei, H. X. Tang, C. Q. Li, C. F. Mu, H. Y. Zheng, F. Z. Li and Z. H. Zhu, Mol. Pharmaceutics, 2019, 16, 786–797 CrossRef CAS PubMed.
- M. Li, L. Ling, Q. Xia and X. Li, RSC Adv., 2021, 11, 12757–12770 RSC.
- N. Chan, S. Y. An and J. K. Oh, Polym. Chem., 2014, 5, 1637–1649 RSC.
- M. Wagner, A. Krieger, M. Minameyer, B. Hämisch, K. Huber, T. Drewello and F. Gröhn, Macromolecules, 2021, 54, 2899–2911 CrossRef CAS.
- K. Leer, G. Cinar, J. L. Solomun, I. Martin, I. Nischang and A. Traeger, Nanoscale, 2021, 13, 19412–19429 RSC.
- L. Zhang, W. Liu, L. Lin, D. Chen and M. H. Stenzel, Biomacromolecules, 2008, 9, 3321–3331 CrossRef CAS PubMed.
- Z. Zhang, L. Yin, C. Tu, Z. Song, Y. Zhang, Y. Xu, R. Tong, Q. Zhou, J. Ren and J. Cheng, ACS Macro Lett., 2013, 2, 40–44 CrossRef CAS PubMed.
- A. P. Bapat, J. G. Ray, D. A. Savin and B. S. Sumerlin, Macromolecules, 2013, 46, 2188–2198 CrossRef CAS.
- X. Song, K. Yuan, H. Li, S. Xu and Y. Li, ACS Appl. Bio Mater., 2020, 3, 2455–2465 CrossRef CAS PubMed.
- N. N. Bayram, G. T. Ulu, M. Topuzogulları, Y. Baran and S. D. Isoglu, Macromol. Biosci., 2022, 22, 2100375 CrossRef CAS PubMed.
- P. Weber, M. Dzuricky, J. Min, I. Jenkins and A. Chilkoti, Biomacromolecules, 2021, 22, 4347–4356 CrossRef CAS PubMed.
- R. Jin, J. Sun, L. F. Zhou, X. L. Guo and A. N. Cao, Biomater. Sci., 2020, 8, 2507–2513 RSC.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11(10), 4985–4998 CrossRef PubMed.
- F. Fang, M. Li, J. F. Zhang and C. S. Lee, ACS Mater. Lett., 2020, 2, 531–549 CrossRef CAS.
- J. K. Elter, G. Sentis, P. Bellstedt, P. Biehl, M. Gottschaldt and F. H. Schacher, Polym. Chem., 2018, 9, 2247–2257 RSC.
- M. Sallouh, P. Degen, W. Hiller and R. Weberskirch, Polymer, 2015, 56, 141–146 CrossRef CAS.
- J. C. Li, L. Sun, Y. Liu, H. J. Yao, S. Jiang, P. Yunzhu, Y. J. Li and Y. G. Zhang, Nanomedicine, 2019, 15(1), 108–118 CrossRef CAS PubMed.
- I. Alberg, S. Kramer, C. Leps, S. Tenzer and R. Zentel, Macromol. Biosci., 2021, 21, 2000414 CrossRef CAS PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12(7), 986–998 CrossRef CAS PubMed.
- T. Lorson, M. M. Lubtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- Y. Chen, P. Espeel, S. Reinicke, F. E. Du Prez and M. H. Stenzel, Macromol. Rapid Commun., 2014, 35, 1128–1134 CrossRef CAS PubMed.
- D. Valverde, R. Porcar, R. Prinzi, S. V. Luis, B. Altava and E. García-Verdugo, Polym. Chem., 2022, 13, 4973–4979 RSC.
- S. Montolio, O. Zagorodko, R. Porcar, M. I. Burguete, S. V. Luis, H. Tenhu and E. García-Verdugo, Polym. Chem., 2017, 8, 4789–4797 RSC.
- R. Reinicker, H. C. Rees, P. Espeel, N. Vanparijs, C. Bisterfeld, M. Dick, R. R. Rosencrantz, G. Brezesinski, B. G. de Geest, F. E. Du Prez, J. Pietruszka and A. Böker, ACS Appl. Mater. Interfaces, 2017, 9, 8317–8326 CrossRef PubMed.
- A. Krieg, C. Weber, R. Hoogenboom, C. Remzi Becer and U. S. Schubert, ACS Macro Lett., 2012, 1(6), 776–779 CrossRef CAS PubMed.
- S. Reinicke, P. Espeel, M. M. Stamenovic and F. E. Du Prez, ACS Macro Lett., 2013, 2(6), 539–543 CrossRef CAS PubMed.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21(1), 151–208 CrossRef CAS.
- A. Krieg, C. Weber, R. Hoogenboom, C. R. Becer and U. S. Schubert, ACS Macro Lett., 2012, 1, 776–779 CrossRef CAS PubMed.
- M. N. Leiske, M. Lai, T. Amarasena, T. P. Davis, K. J. Thurecht, S. J. Kent and K. Kempe, Biomaterials, 2021, 274, 120843 CrossRef CAS PubMed.
- S. Reinicke, P. Espeel, M. M. Stamenović and F. E. Du Prez, ACS Macro Lett., 2013, 2, 539–543 CrossRef CAS PubMed.
- P. Espeel and F. E. Du Prez, Eur. Polym. J., 2015, 62, 247–272 CrossRef CAS.
- S. Su and P. S. Kang, Nanomaterials, 2020, 10, 656 CrossRef CAS PubMed.
- A. George, P. A. Shah and P. S. Shrivastav, Int. J. Pharm., 2019, 561, 244–264 CrossRef CAS PubMed.
- N. Shreyash, M. Sonker, S. Bajpai and S. K. Tiwary, ACS Appl. Bio Mater., 2021, 4, 2307–2334 CrossRef CAS PubMed.
- G. Wu, Y. Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 489–492 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data of the nanoparticle series NP1.X, NP2.X (Tables S1 and S2), SEC elugrams of the block copolymers P2.1–P2.12, 1H NMR of monomers, crosslinker, macroRAFT and block copolymers and 13C NMR of cystamine and the monomer M1 (Fig. S4 to S22), DLS data from the nanoparticles NP1.X series in MeOH, SEC data from macroRAFT PP3 and block copolymers P2.11 and P2.12 (Fig. S32) and nanoparticles derived from P2.11 and P2.12 with six different crosslinker (Fig. S33 and S34). See DOI: https://doi.org/10.1039/d3py00641g |
| This journal is © The Royal Society of Chemistry 2023 |