DOI:
10.1039/D3PY00559C
(Paper)
Polym. Chem., 2023,
14, 3749-3760
Investigating AIE behaviors of amphiphilic AIEgen-based polymers through self-assembly architectures and hydrophobic core arrangements†
Received
19th May 2023
, Accepted 20th July 2023
First published on 21st July 2023
Abstract
Nanoscale polymersomes showing a prominent aggregation-induced emission (AIE) effect have enormous potential in biomedical applications. In this study, three types of amphiphilic polymers were synthesized using reversible addition–fragmentation chain transfer self-condensing vinyl polymerization. These polymers had linear, branched, and star-like structures, and were composed of hydrophobic and hydrophilic units, namely (2-(4-vinylphenyl)ethene-1,1,2-triyl)tribenzene (TPEE) and N,N-dimethylacrylamide (DMA), respectively. We found that when the ratio of TPEE and DMA was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (n/n), the star-like polymers showed significantly stronger emission than linear and branched polymers, due to tighter aggregation of TPEE in the polymersomes derived from intermolecular self-assembly of the star-like polymers. However, when the hydrophilic chain length was shortened, the AIE behavior of the star-like polymers was weaker than that of the linear polymers, possibly due to greater steric hindrance of TPEE during the transformation of micelles to vesicles within the linear polymers. We also employed the star-like polymer-derived nanoparticles for living cell imaging with two types of cells (A549 and HepG2). This study offers considerable insights into designing an AIE polymer structure for better emission behaviors.
:1 (n/n), the star-like polymers showed significantly stronger emission than linear and branched polymers, due to tighter aggregation of TPEE in the polymersomes derived from intermolecular self-assembly of the star-like polymers. However, when the hydrophilic chain length was shortened, the AIE behavior of the star-like polymers was weaker than that of the linear polymers, possibly due to greater steric hindrance of TPEE during the transformation of micelles to vesicles within the linear polymers. We also employed the star-like polymer-derived nanoparticles for living cell imaging with two types of cells (A549 and HepG2). This study offers considerable insights into designing an AIE polymer structure for better emission behaviors.
Introduction
Emergent fluorescent macromolecules have gained a great deal of attention in a wide range of application fields including cell imaging,1–3 chemical sensing,4,5 drug delivery and tracing,6,7 and photothermal therapy.8 Unexpected chromophore-free luminescent polymers are one of the booming topics due to the multi-color emissions under different excitation levels. Several polymers have been reported to have exceptional emissive properties, such as poly(amido amine),9 polyester1,10 polysiloxane,7 poly(amino ester),11 poly(ether amide), etc.12 The presence of a hyperbranched backbone in the polymer assemblies leads to an intriguing luminescence phenomenon attributed to the clustering of atoms through inter- and/or intrachain interactions within the polymer assemblies. This unique emission behavior, also referred to as cluster-triggered emission (CTE), is a result of the distinctive structural characteristics provided by the hyperbranched backbone.13–15 Benefiting from unveiling the origination of unconventional fluorescence, many strategies were explored to synthesize novel polymer backbones with versatile adaptabilities6,7 or to investigate the effects of polymer structures on fluorescence behaviors.16,17 While developing polymers with unexpected luminescence, AIE-active macromolecules have become dominant owing to their advantageous emission characteristics in the aggregated state, especially combined with self-assembly serving as the core-forming block. Aggregated hydrophobic blocks of self-assemblies effectively restrict the intramolecular motion of AIE luminous agents (AIEgen),18–20 which facilitates the integration of the inherent advantages of both to achieve broader applications. For instance, AIEgen-containing amphiphilic block polymers have been utilized as a facile platform to monitor the polymerization process,21 study self-assembly mechanism,22,23 clarify topology effects on chemical sensing,4 visualize the molecular weight change of supramolecular polymers,24 deliver the gene and trigger release with light irradiation.25 To date, vast efforts have been made to exploit novel application scenarios, yet few works focus on how amphiphilic polymer scaffolds and AIEgen arrays inside the core affect AIE behaviors, in which relationships between secondary structures and fluorescence features of polypeptides terminated with AIEgens were most studied.26–28 For example, Cai et al. proved that the rigid helical conformation of sulfono-γ-AA peptide effectively restricted the rotation of tetraphenyl ethylene (TPE) conjugated onto the peptide backbone, resulting in fluorescence even when not in the aggregated state. However, destabilization of the α helical structure, while replacing alanine residues with lysine residues, led to aggregation-dependent emission.28 In addition, Li et al. investigated that the position of TPE in micellar hydrophilic blocks had great influence on photoluminescence. Micelles, whose TPE units distributed randomly in hydrophilic blocks, displayed stronger emission compared to micelles where TPE units were incorporated into the outer terminal of hydrophilic blocks.29 Also, Zhang's research indicated that the greater hydrophilicity of poly(methacrylic acid) at a pH of 10.0–13.3 increased the space between the TPE inside star copolymers, leading to a sharp fluorescence decrease.30
Inspired from the hyperbranched architecture of unconventional fluorescent polymers,7,16,17 we conceive that the incorporation of AIEgen into a hyperbranched scaffold would significantly constrain its movement by an intertwist between multi-arms, resulting in exceptional AIE effects even at a unimolecular level. Herein, we incorporated TPEE as a hydrophobic moiety, as well as AIEgen, and DMA as a hydrophilic moiety to construct linear, branched and star-like amphiphilic polymers. The aim was to investigate the differences in AIE properties caused by self-assembly architectures and the arrangements of AIEgen inside the polymers. To further observe the transformations of the AIE intensity and self-assembly, the ratio of DMA and TPEE was reduced in both linear and star-like polymers. The results indicate that the AIE effect is the strongest in star-like polymers as well as polymersomes derived from them, particularly when the ratio of DMA to TPEE is 3:1 (n/n). In comparison, micelles from linear polymers and polymersomes from branched polymers exhibit weaker AIE effects. With the decreasing ratio of DMA and TPEE to 2:1 (n/n), vesicles from linear polymers show a stronger AIE effect compared to micelles from star-like polymers. Therefore, the AIE effect could be further intensified by precisely adjusting the polymer structures and proportion of hydrophilic to hydrophobic moieties.
Experimental section
Synthesis of polymers
Synthesis of poly(TPEE) (L).
The chain transfer agent 2-(((butylthio)carbonothioyl)thio)propanoic acid (RAFT, 11.9 mg, 0.05 mmol), initiator AIBN (1.64 mg, 0.01 mmol), and TPEE (179 mg, 0.5 mmol) were dissolved in 500 μl of THF and then the mixture was introduced into a 10 ml Schlenk tube. After freeze–pump–thaw cycles three times to deoxidize thoroughly, the mixture was heated to 80 °C for 20 h under the protection of nitrogen. After the reaction, the polymerized solution was added dropwise into methanol and centrifuged to collect the precipitate. The above purification procedure was repeated three times and then dried at reduced pressure. A yellow powdered solid was obtained as a final product.
Synthesis of poly(TPEE)-b-poly(DMA) (L-b-PDMA-1/L-b-PDMA-2).
Macro-RAFT L (32 mg), DMA (22/11 μl, 0.2/0.1 mmol), and initiator AIBN (0.328 mg, 0.002 mmol) were dissolved in 300 μl of THF and then the mixture was introduced into a 10 ml Schlenk tube. The polymerization process was the same as that of L. Purification was done in ether and the remaining procedures referred to that for L. A pale yellow powder was obtained as the final product.
Synthesis of poly(RAFT-MA)-co-poly(TPEE) (Hr).
A chain transfer agent as well as a branching agent 2-((2-(((butylthio)carbonothioyl)thio)propanoyl)oxy)ethyl methacrylate (RAFT-MA, 17.5 mg, 0.05 mmol), initiator AIBN (1.64 mg, 0.01 mmol), and TPEE (179 mg, 0.5 mmol) were dissolved in 500 μl of THF and then the mixture was injected into a 10 ml Schlenk tube. The polymerization and purification processes were the same as that for L.
Synthesis of poly(RAFT-MA)-co-poly(TPEE)-b-poly(DMA) (Hr-b-PDMA-1).
Macro-RAFT Hr (32 mg), DMA (22 μl, 0.2 mmol), and initiator AIBN (0.328 mg, 0.002 mmol) were dissolved in 300 μl of THF and then the mixture was injected into a 10 ml Schlenk tube. The polymerization and purification processes were the same as those for L-b-PDMA.
Synthesis of poly(RAFT-MA)-b-poly(TPEE) (Hb).
A chain transfer agent as well as a branching agent RAFT-MA (17.5 mg, 0.05 mmol) and initiator AIBN (1.64 mg, 0.01 mmol) were dissolved in 200 μl of THF and then the mixture was injected into a 10 ml Schlenk tube and deoxidized with freeze–pump–thaw cycles three times. Polymerization proceeded at 80 °C for 6 h under a nitrogen atmosphere. Then TPEE (179 mg, 0.5 mmol) was dissolved in 300 μl of THF and added to the above poly(RAFT-MA) solution for subsequent blocking. Purification was the same as that for L.
Synthesis of poly(RAFT-MA)-b-poly(TPEE)-b-poly(DMA) (Hb-b-PDMA-1/Hb-b-PDMA-2).
Macro-RAFT Hb (32 mg), DMA (22/11 μl, 0.2/0.1 mmol), and initiator AIBN (0.328 mg, 0.002 mmol) were dissolved in 300 μl of THF and then the mixture was injected into a 10 ml Schlenk tube. The polymerization and purification processes were the same as that for L-b-PDMA.
Characterization
To prepare nanoparticle suspension, polymers dissolved in 100 μl of THF were added into 900 μl of water with high-speed stirring for 15–30 min and then the mixture was bubbled with Ar for removing THF. The samples for transmission electron microscopy (TEM) were prepared by dropping 10 μl of nanoparticle suspension on copper mesh coated with a carbon film and stewed for 30–45 min for the precipitation of nanoparticles. The redundant suspension was removed with dust-free paper and then the samples were dried for 48 h naturally without staining. Bright field images were acquired with an FEI Tecnai G2 spirit microscope operating at 120 kV and 1.48 μA. Dynamic light scattering (DLS) was conducted using a Malvern Zetasizer Nano ZS instrument in pure water. Samples were tested in 10 cm plastic cuvettes with a 173° scattering angle. Fluorescence spectra were recorded on an FL-970 spectrometer with the excitation wavelength at 360 nm and emission wavelength at 370–700 nm. The relative fluorescence quantum yields (ΦF) of nanoparticles were calculated referring to quinine sulfate as the standard with ΦF = 0.54 in 0.1 M H2SO4 solution at a 360 nm excitation. UV-vis spectra were recorded using a UV-8000 spectrometer.
Density functional theory (DFT) calculations
Geometry optimizations were performed with Gaussian 16 software by using the B3LYP functional that mixes the exact Hartree–Fock (HF) exchange with the expression for the three-exchange functional: Lee, Yang, and Parr with the use of the 31D basis set. GaussView 6.0 and GaussSum 3.0 programs were used for the visualization of our data.
Results and discussion
Synthesis and characterization of polymers
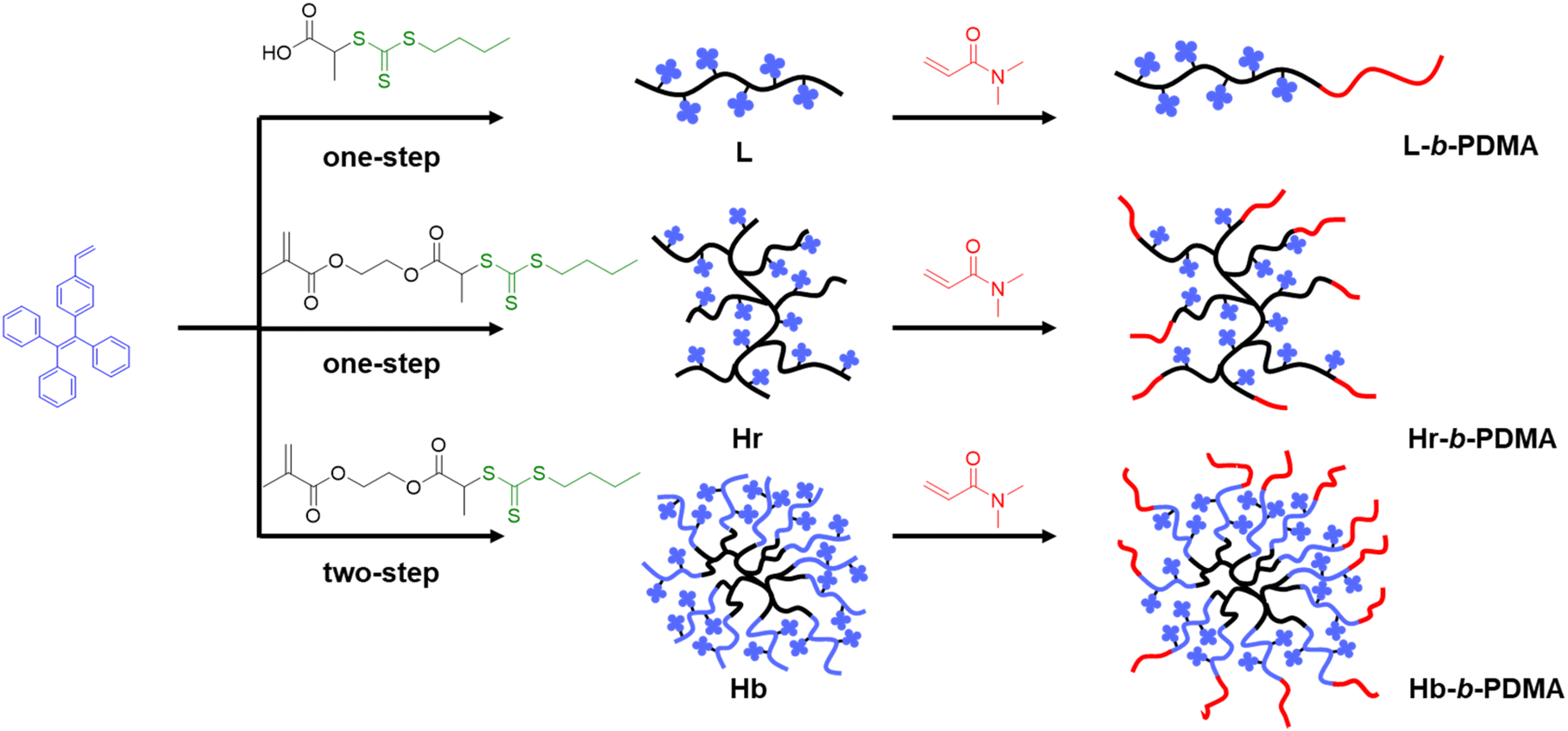
We first synthesized three hydrophobic macro-RAFT L, Hr and Hb. Fig. 1 displays a diagram of the synthesized polymers, while Fig. S2† depicts the concrete routes. 1H NMR spectra and GPC trace are illustrated in Fig. S4–S9 and Tables S1–S3.† It is worth noting that the polymerization of Hb was not controllable, resulting in two types of polymers with enormous molecular weight differences in this study (Fig. S7 and Table S1†). The heavier type was close to L and Hr, yet the smaller one was far less than that of L and Hr. The broad distribution of Hb and Hb-b-PDMA can be attributed to the uneven distribution of trithiocarbonate in the branched poly(RAFT-MA) cores, which eventuated diverse amounts of TPEE blocks subsequently, and accordingly caused broad distribution of Hb, as well as Hb-b-PDMA. Moreover, in our initial GPC analysis using THF as the solvent, the hydrophilic PDMA and amphiphilic copolymers showed broad and asymmetrical peaks. This suggests poor resolution, likely due to the inadequate solvation of the polymers and potential interactions with the column matrix. However, after reevaluating our GPC measurements using DMF as the solvent, we observed more symmetric peaks, particularly for the Hb and Hb-b-PDMA (Fig. S10†). This improved peak resolution indicates a better separation of the polymers and a reduced interaction with the stationary phase of the column. These results underscore the superior suitability of DMF as an eluent for the GPC analysis of PDMA polymers.
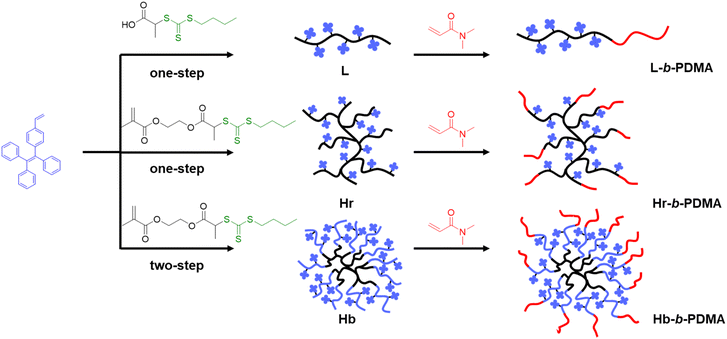 |
| | Fig. 1 Synthesis of linear (L), branched (Hr) and star-like (Hb) polymers. | |
AIE contrast of L, Hr and Hb
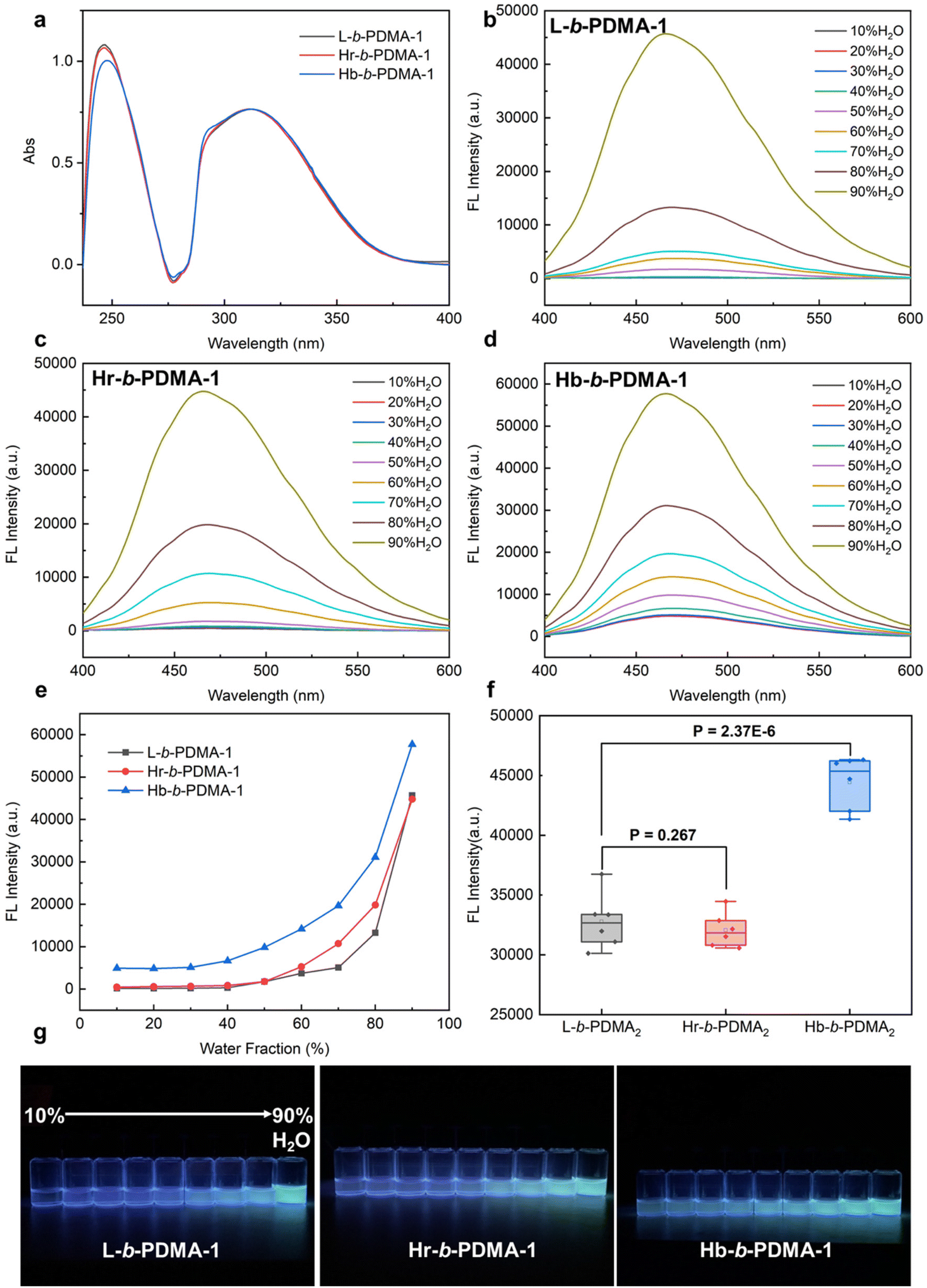
Initially, the diversity of AIE was explored among L, Hr and Hb. To eliminate the influence of TPEE content in the three polymers, the distinctions in TPEE were ascertained at equivalent mass concentrations using UV-vis spectroscopy. As shown in Fig. 2a, the concentration of TPEE in Hb was slightly higher, about 1.07 times that in L and Hr. Therefore, the concentration was 0.1 mg ml−1 for both L and Hr and 0.093 mg ml−1 for Hb for the subsequent AIE tests. Fluorescence spectra and images of L, Hr and Hb under a UV lamp are shown in Fig. 2b–f. It is evident that the fluorescence intensity of Hb was much stronger than that of L and Hr in mixed solvents as the water fraction increased. At a low water content (10–30%, v/v), L and Hr displayed negligible fluorescence, yet Hb still exhibited weak illumination. Dispersed single molecular polymers existed mostly in a vast majority of good solvents (THF), in which the vibration and rotation of benzene rings depended on the intramolecular arrangements of TPEE. The spatial proximity of TPEE chains stretched from hyperbranched poly(RAFT-MA) cores made them impede each other so that Hb retained the relatively brighter fluorescence. As the proportion of water increased, the primary factor contributing to the AIE effect was intermolecular aggregation. For Hb, the multidirectional dense distributions of TPEE arms and interchain block in single molecules were synergistically beneficial for impeding TPEE motion, when compared with incomplete packing through an unordered twist between linear chains of L. It could be noticed that the Hr, containing a branched scaffold, exhibited an even weaker AIE effect. We inferred that the copolymerization of TPEE and RAFT-MA resulted in an inferior degree of branching and less dense distribution of TPEE in the branched frame, which was adverse to the intramolecular TPEE aggregation and twist between TPEE arms like Hb.
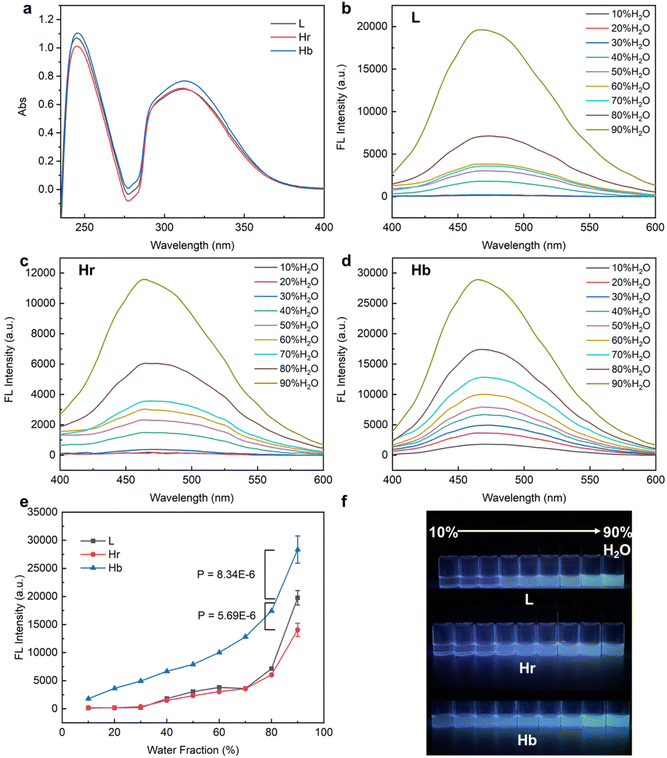 |
| | Fig. 2 AIE characteristics of L, Hr and Hb. (a) UV-vis spectra of L, Hr and Hb at the same mass concentration of 0.02 mg ml−1. Fluorescence spectra of L (b), Hr (c) and Hb (d) in a 10%–90% (v/v) H2O/THF mixture with 360 nm excitation and their conclusion (e). (f) fluorescence images of L, Hr and Hb in a 10%–90% (v/v) H2O/THF mixture under a UV lamp (365 nm). | |
AIE contrast of L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1
To further compare the AIE difference resulting from the self-assembly, L, Hr and Hb were used as macro-RAFTs to prepare block copolymers with hydrophilic DMA chains. The ratio of TPEE and DMA was determined as 1:3 (n/n) from 1H NMR spectra (Fig. S5†). Similarly, the concentrations of TPEE in the polymers, L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1, were similar at the same mass concentration (0.05 mg ml−1) as determined from UV-vis outcomes (Fig. 3a). The fluorescence of the block copolymers in 10%–90% (v/v) H2O/THF was measured and is shown in Fig. 3b–e and g. It is noticeable that the three amphiphilic polymers were much brighter (1.84–2.25 folds) than hydrophobic L, Hr and Hb under high water ratio (70–90%, v/v) conditions, even after a decrease in TPEE levels at the same mass concentration. This increase in fluorescence could be attributed to self-assembly induced boost. Horizontal statistical analysis indicated that the AIE effect of Hb-b-PDMA-1 gave an edge significantly (Fig. 3f, P < 0.001). This result can be attributed to the self-assembly modes. On the one hand, a unimer assembly of Hb-b-PDMA-1 formed mono-micelles where internal TPEE units were effectively restricted for maintaining fluorescence at low water content, distinguishing from the water-dependent interchain assembly of L-b-PDMA-1. Also, the increased water content could trigger the intermolecular assembly of Hb-b-PDMA-1, also known as a secondary assembly, to form a multi-molecule assembly with greater motional obstacle. The dual-assembly mode of Hb-b-PDMA-1 jointly contributes to the AIE effect. However, the AIE effect of Hr-b-PDMA-1 was similar to that of L-b-PDMA-1 even when it was expected to exhibit a similar dual-assembly mode. As mentioned above, the low branching degree of Hr-b-PDMA-1 resulted in a sparse distribution of TPEE and fewer TPEE arms, which were further apart spatially. As a result, the motions of the TPEE arms in the mono-micelles were not sufficiently blocked, and the flexibility of the TPEE chains was similar to that of dispersed L-b-PDMA-1.
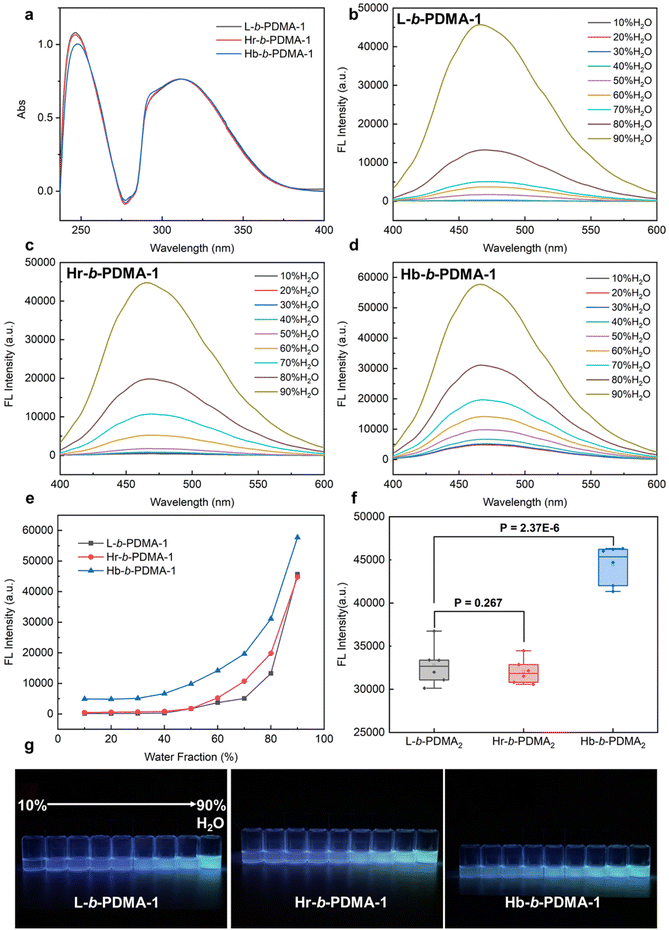 |
| | Fig. 3 AIE characteristics of L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1. (a) UV-vis spectra of L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1 at the same mass concentration of 0.05 mg ml−1. Fluorescence spectra of L-b-PDMA-1 (b), Hr-b-PDMA-1 (c) and Hb-b-PDMA-1 (d) in a 10%–90% (v/v) H2O/THF mixture with 360 nm excitation and their conclusion (e). (f) Fluorescence intensity difference statistics in a 90% H2O and 10% THF mixture. (g) Fluorescence images of L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1 under a UV lamp (365 nm). | |
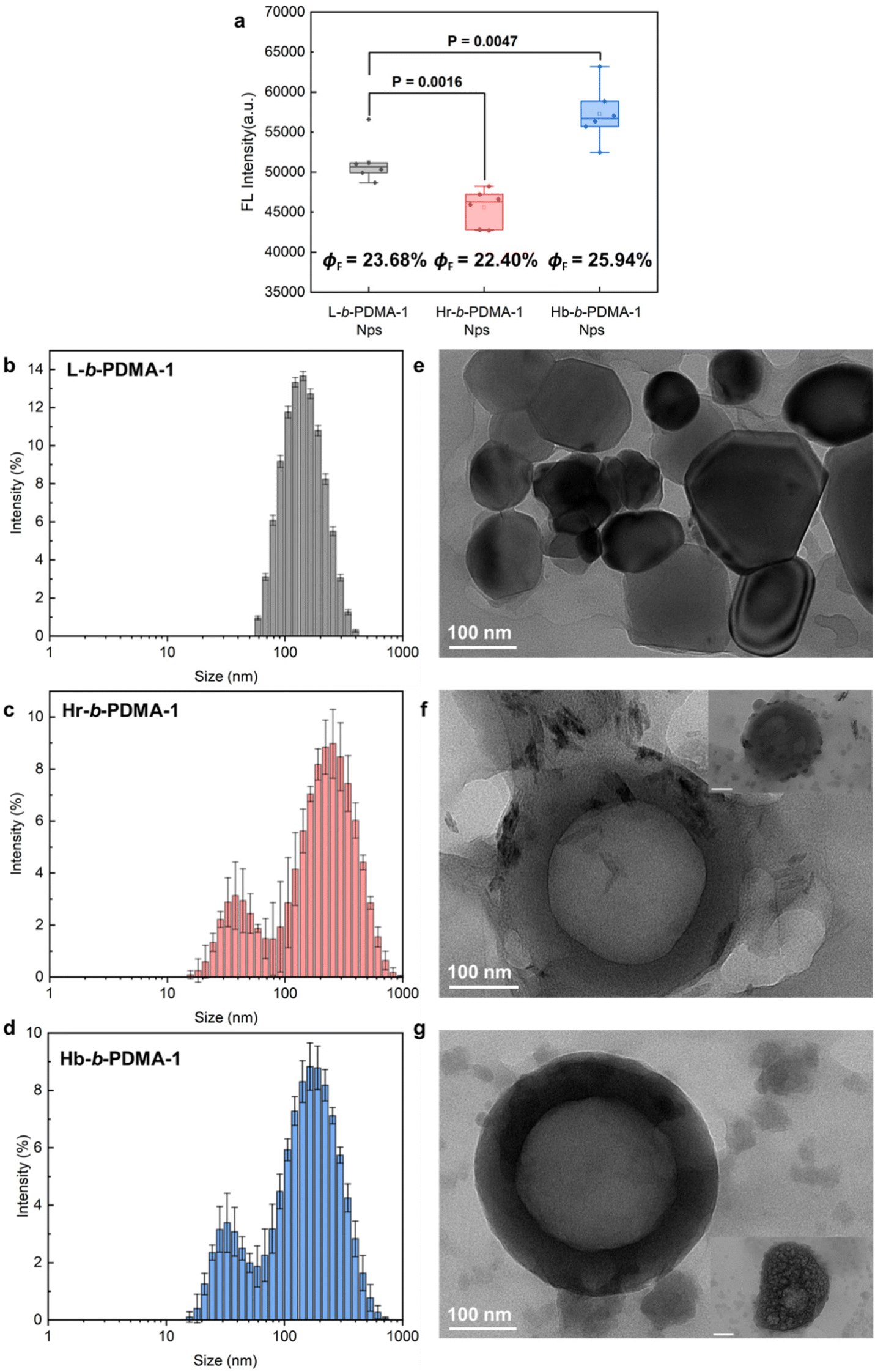
After the study of the influence of partial self-assembly on the AIE effect under low water ratio conditions, we compared the fluorescence differences of nanoparticles (Nps) prepared from L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1 to explore the impact of self-assembly on AIE properties. In Fig. 4a, Hb-b-PDMA-1 Nps display significantly stronger emission (P < 0.001) and higher ΦF than the others at the same concentration (0.1 mg ml−1) with the differences in TPEE amounts being eliminated (Fig. 3a); thus self-assembly structures played a critical role. Nps were further characterized through dynamic light scattering (DLS) and transmission electron microscopy (TEM). As presented in Fig. 4b–g, the Nps made from linear L-b-PDMA-1 showed narrow size distribution with a diameter around 150 nm (Fig. 4b), being consistent with TEM results (Fig. 4e). The Hr-b-PDMA-1 and Hb-b-PDMA-1 Nps, which were formed by a branched framework containing amphiphilic polymers, displayed identical patterns, in which the size distribution of twin peaks was observed as a result of small and larger Nps from intramolecular and intermolecular assembly, respectively. The above phenomenon was also referred to as secondary aggregation.31 The diameter of Hr-b-PDMA-1 NPs (40/220 nm) was slightly larger than that of Hb-b-PDMA-1 NPs (30/200 nm). It is speculated that if intramolecular or intermolecular packing from Hr-b-PDMA-1 is looser, it results in gaps being more spacious in the hydrophobic core. This may be used to explain why the emission of Hr-b-PDMA-1 Nps was inferior to that of L-b-PDMA-1 Nps (Fig. 4a).
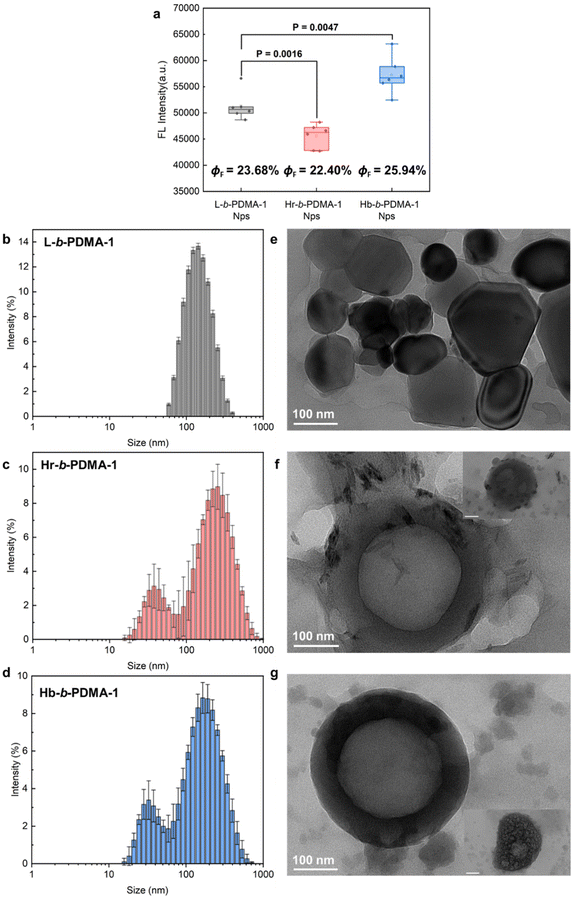 |
| | Fig. 4 Fluorescence comparisons and characterization of Nps. (a) Statistics of fluorescence intensity differences between L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1 Nps. Size distributions and TEM morphologies of Nps prepared from L-b-PDMA-1 (b and e), Hr-b-PDMA-1 (c and f) and Hb-b-PDMA-1 (d and g), respectively. ΦF: florescence quantum yields. Scale bar = 100 nm in the insets of f and g. | |
Regarding the morphologies of Nps, the L-b-PDMA-1 Nps appeared as shriveled and collapsed spheres (Fig. 4e), which were probably caused by the absence of an aqueous environment during testing. Hr-b-PDMA-1 and Hb-b-PDMA-1 intermolecularly assembled to polymersomes about 200 nm in size, and fuzzy blobs were regarded as mono-micelles (Fig. 4f and g). The potential formation procedure of polymersomes was also captured (insets in Fig. 4f and g). Based on the direct observations of Np morphologies, it was supposed that L-b-PDMA-1 could self-assemble to micelles, Hr-b-PDMA-1 and Hb-b-PDMA-1 leaned towards vesicles.
This observation was in line with a previous study by Wang et al.,32 who disclosed a self-assembly process of amphiphilic hyperbranched copolymers (HCMs) to vesicles through dissipative particle dynamics simulation. First, A–B or A–B–A shape mono-micelles emerged from microphase separation where A and B represented hydrophilic arms and a hydrophobic core, respectively. Then, distinctively, HCMs with a small hydrophilic fraction tended to undergo A–B type separation and two A–B shape mono-micelles would pack together in a head-to-tail manner to form a bilayer membrane. In contrast, A–B–A shape mono-micelles derived from large hydrophilic fraction HCMs stacked one by one to form a monolayer membrane. Finally, the membrane wrapped and fused to form vesicles. The dynamic process of polymersome formation could be interpreted based on the above simulation and a diagram is shown in Fig. 5. The highly branching core of Hb-b-PDMA-1 was able to densely pack the TPEE arms, resulting in efficient π–π stacking efficiency between mono-micelles in the hydrophilic interlayer of polymersomes. This led to a more compact arrangement of TPEE arms. In contrast, the sparse TPEE distribution and fewer TPEE arms in Hr-b-PDMA-1 hindered π–π stacking in polymersomes, resulting in looser packing and expanded gaps between mono-micelles. The core inside micelles prepared from L-b-PDMA-1 was spacious enough to allow for the free movement of internal TPEE blocks, leading to weaker accumulations of TPEE blocks compared to the Hb-b-PDMA-1 polymersomes.
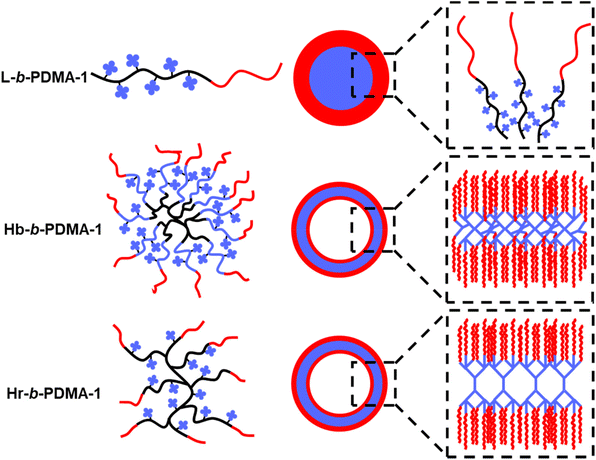 |
| | Fig. 5 Self-assembly diagram of L-b-PDMA-1, Hr-b-PDMA-1 and Hb-b-PDMA-1. | |
AIE contrast of L-b-PDMA-2 and Hb-b-PDMA-2
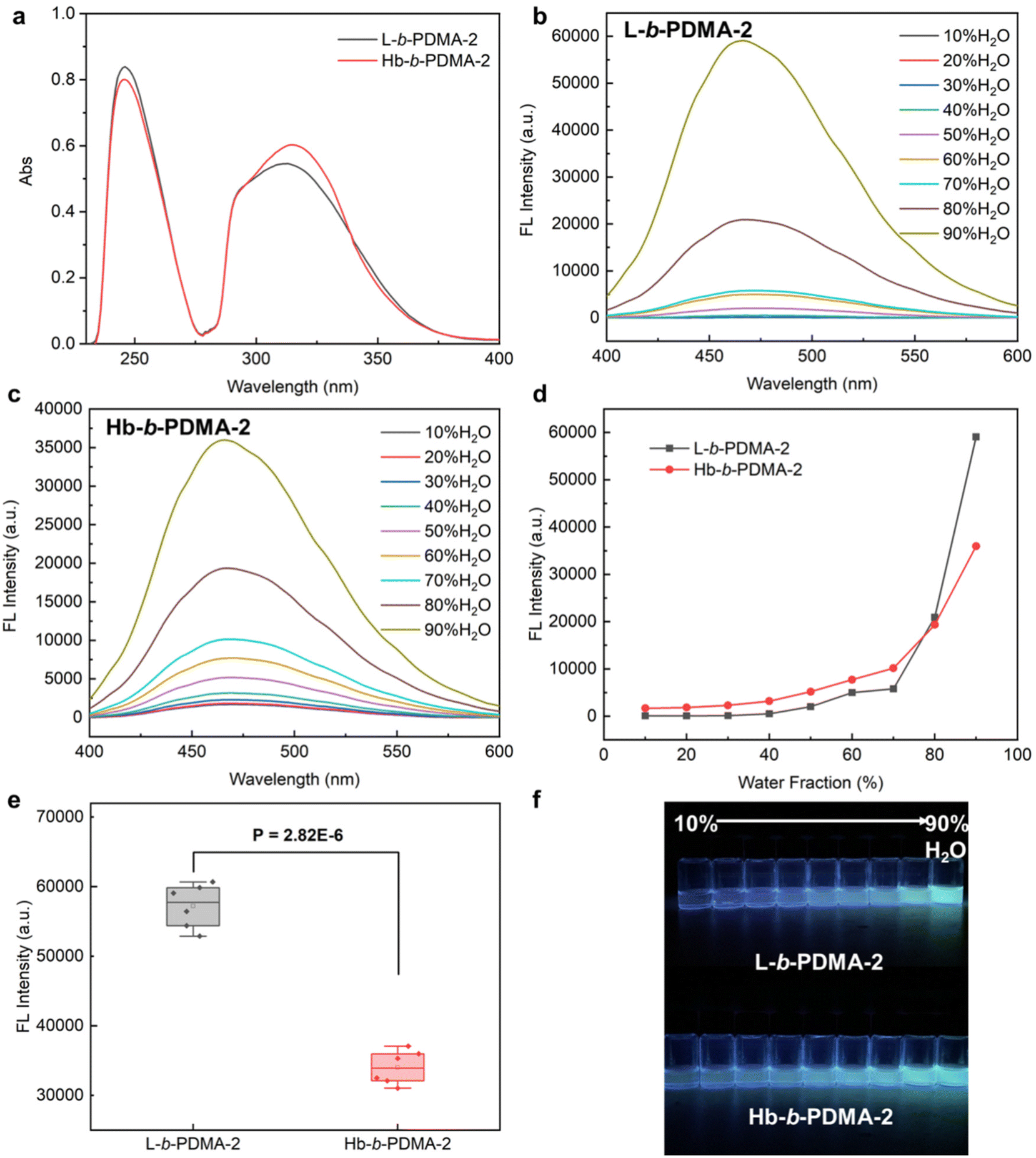
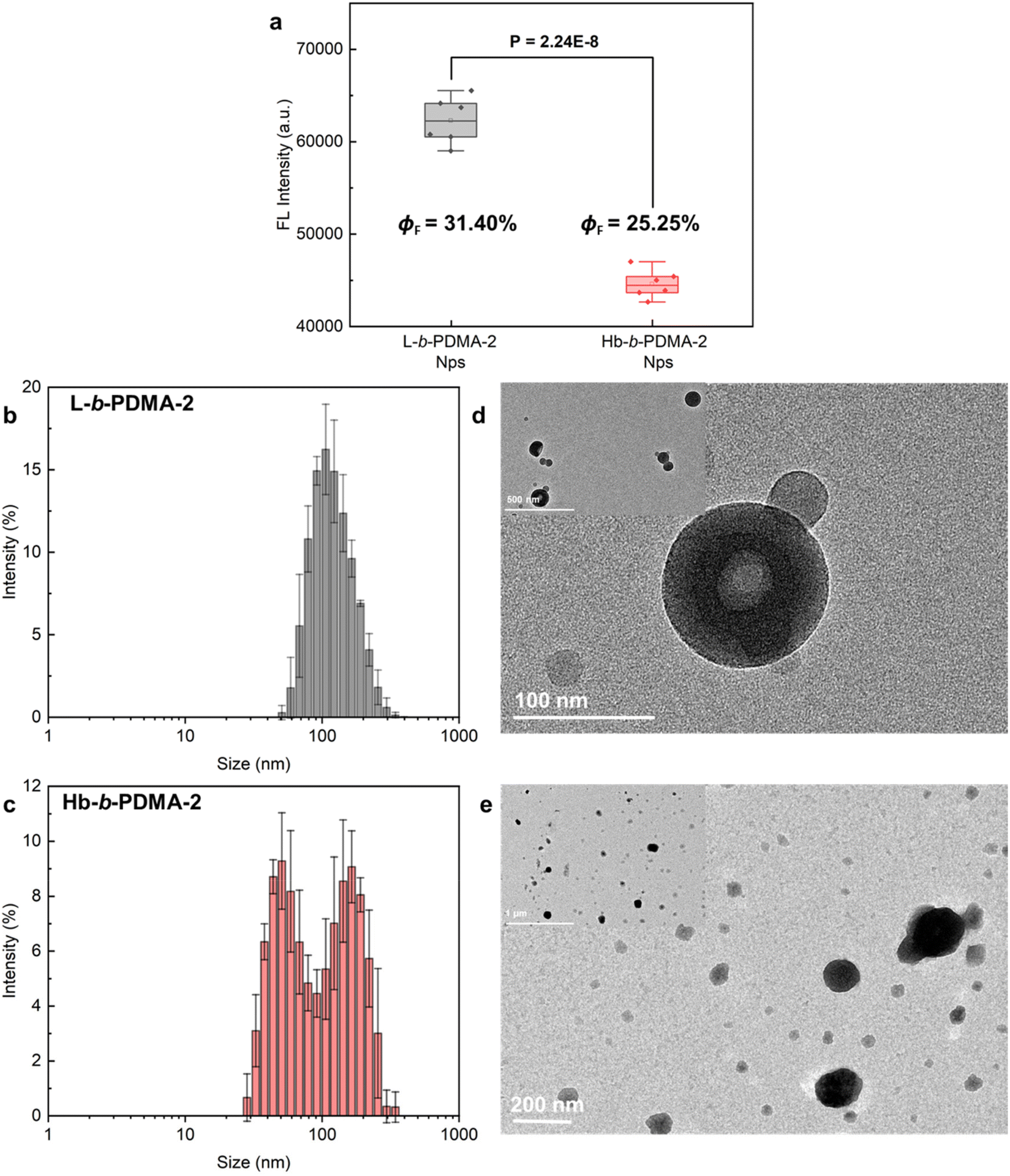
To explore the consistency of the aforementioned observations with shorter hydrophilic chains, L-b-PDMA-2 and Hb-b-PDMA-2 were synthesized with an increased ratio of TPEE to DMA of 1:2 (n/n) (Fig. S6†). UV-Vis spectra demonstrated that the TPEE content in L-b-PDMA-2 was approximately 0.9 times that in Hb-b-PDMA-2 (Fig. 6a). Therefore, 0.1 mg ml−1 L-b-PDMA-2 and 0.09 mg ml−1 Hb-b-PDMA-2 were used for further AIE comparisons. The AIE characteristics of the two polymers are shown in Fig. 6b–d. The emission changes of Hb-b-PDMA-2 with increasing water fraction were considered to be similar to those of Hb-b-PDMA-1 (Fig. 3c). Unexpectedly, L-b-PDMA-2 revealed striking reversion, in which the emission intensity increased sharply in an exponential manner with a 70% (v/v) water fraction and surpassed that of Hb-b-PDMA-2 by 1.7 times (Fig. 6d). Statistical t-test illustrated the significance of the above outcomes (P < 0.001, Fig. 6e).
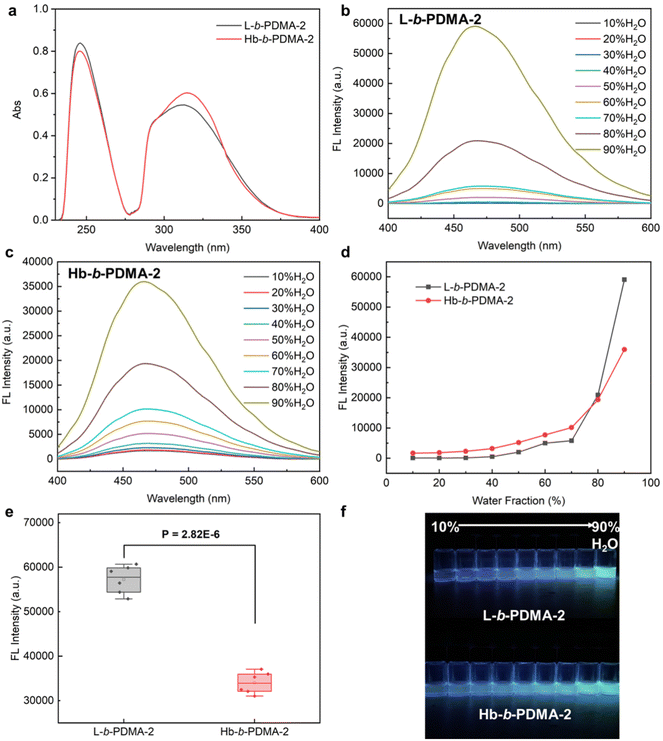 |
| | Fig. 6 AIE characteristics of L-b-PDMA-2 and Hb-b-PDMA-2. (a) UV-vis spectra of L-b-PDMA-2 and Hb-b-PDMA-2 at the same mass concentration of 0.02 mg ml−1. Fluorescence spectra of L-b-PDMA-2 (b) and Hb-b-PDMA-2 (c) in a 10%–90% (v/v) H2O/THF mixture with 360 nm excitation and their conclusion (d). (e) Fluorescence intensity difference statistics in a 90% H2O and 10% THF mixture. (f) Fluorescence images of L-b-PDMA-2 and Hb-b-PDMA-2 under a UV lamp (365 nm). | |
Shortening the hydrophilic fractions of the polymers resulted in a change in the self-assembly structures, leading to the observed reversion in AIE behaviour. To visualize the transformations, Nps produced from L-b-PDMA-2 and Hb-b-PDMA-2 were characterized. The fluorescence intensity of L-b-PDMA-2 Nps was significantly higher than that of Hb-b-PDMA-2 Nps (P < 0.001, Fig. 7a), as well as ΦF. The L-b-PDMA-2 Nps have a narrow distribution with diameters around 90 nm and presented polymersome structures (Fig. 7b and d), in agreement with previous reports that self-assembly structures transformed from spheres to worm-like rods and eventually to vesicles in pace with decreasing hydrophilic fractions.33–35 Identical dual peaks were observed in the distribution of Hb-b-PDMA-2 Nps where a proportion of smaller granules increased in contrast to Hb-b-PDMA-1 Nps (Fig. 7c). The presence of irregular sphere-like particles around 150 nm and amorphously smaller particles with widely scattered size from 50 to 100 nm was observed, and no polymersomes were observed. Dynamic simulations reported in the literature revealed that an increase in core hydrophobicity leads to the formation of conical mono-micelles, with an A–B shape, through microphase separation, which then further assemble to larger micelles.32,36
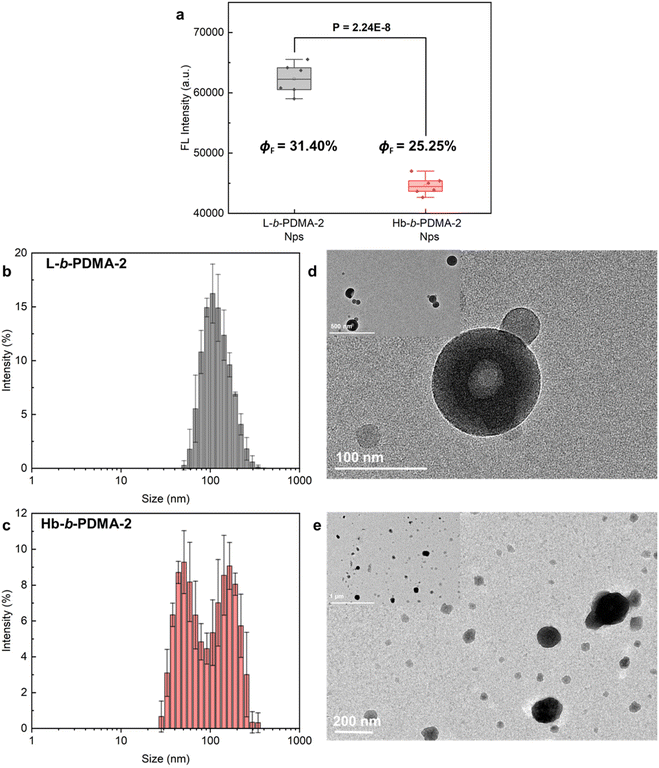 |
| | Fig. 7 Fluorescence comparisons and characterization of Nps. (a) Statistics of fluorescence intensity differences between L-b-PDMA-2 and Hb-b-PDMA-2 Nps. Size distributions and TEM morphologies of Nps prepared from L-b-PDMA-2 (b and d) and Hb-b-PDMA-2 (c and e), respectively. ΦF: florescence quantum yields. | |
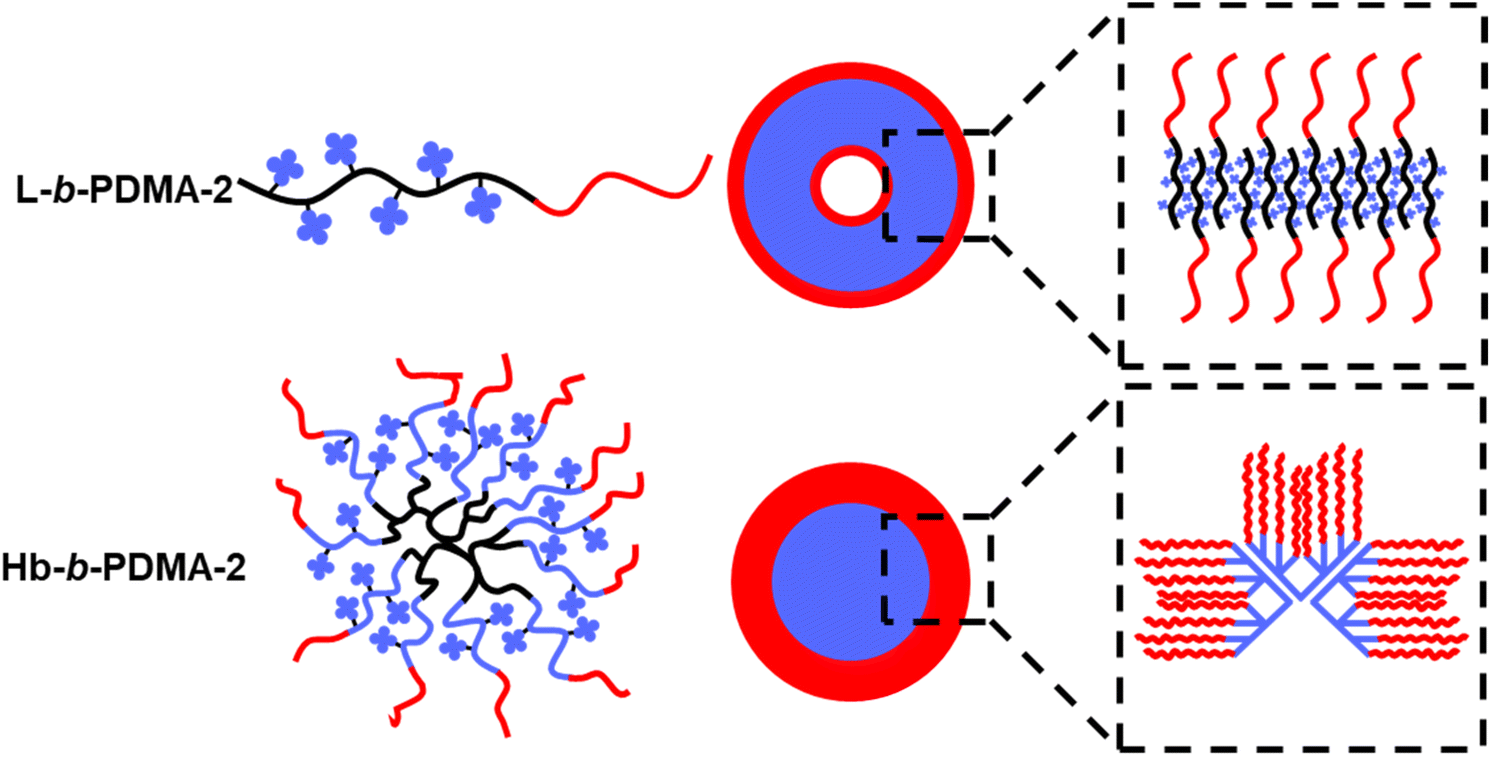
Based on the observed phenomena and reports, we present a self-assembly diagram of L-b-PDMA-2 and Hb-b-PDMA-2, as shown in Fig. 8. For L-b-PDMA-2, the reverse chains, which resembled the arrangement mode within a phospholipid bilayer, alternately aligned to wrap TPEE blocks inside and expose DMA blocks outside, constructing an membrane-like shape and ultimately fused to vesicles. Space impediments of TPEE blocks in polymersomes were more significant than those of micelles, resulting in sharp ascent of emission. In contrast, Hb-b-PDMA-2 had a heterogeneous assembly of A–B type mono-micelles, resulting in wide size distribution of secondarily assembled micelles and lower packing efficiency compared to the prior single polymersome containing intensive mono-micelles. This decentralized packing efficiency led to a weaker emission of Hb-b-PDMA-2 Nps.
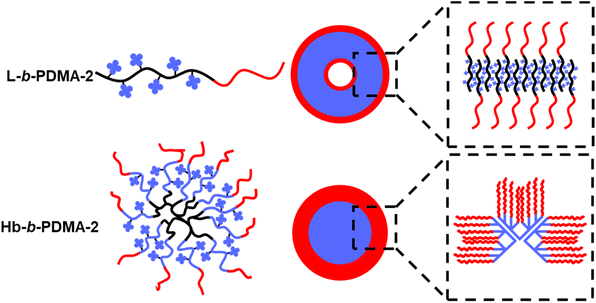 |
| | Fig. 8 Self-assembly diagram of L-b-PDMA-2 and Hb-b-PDMA-2. | |
DFT calculations
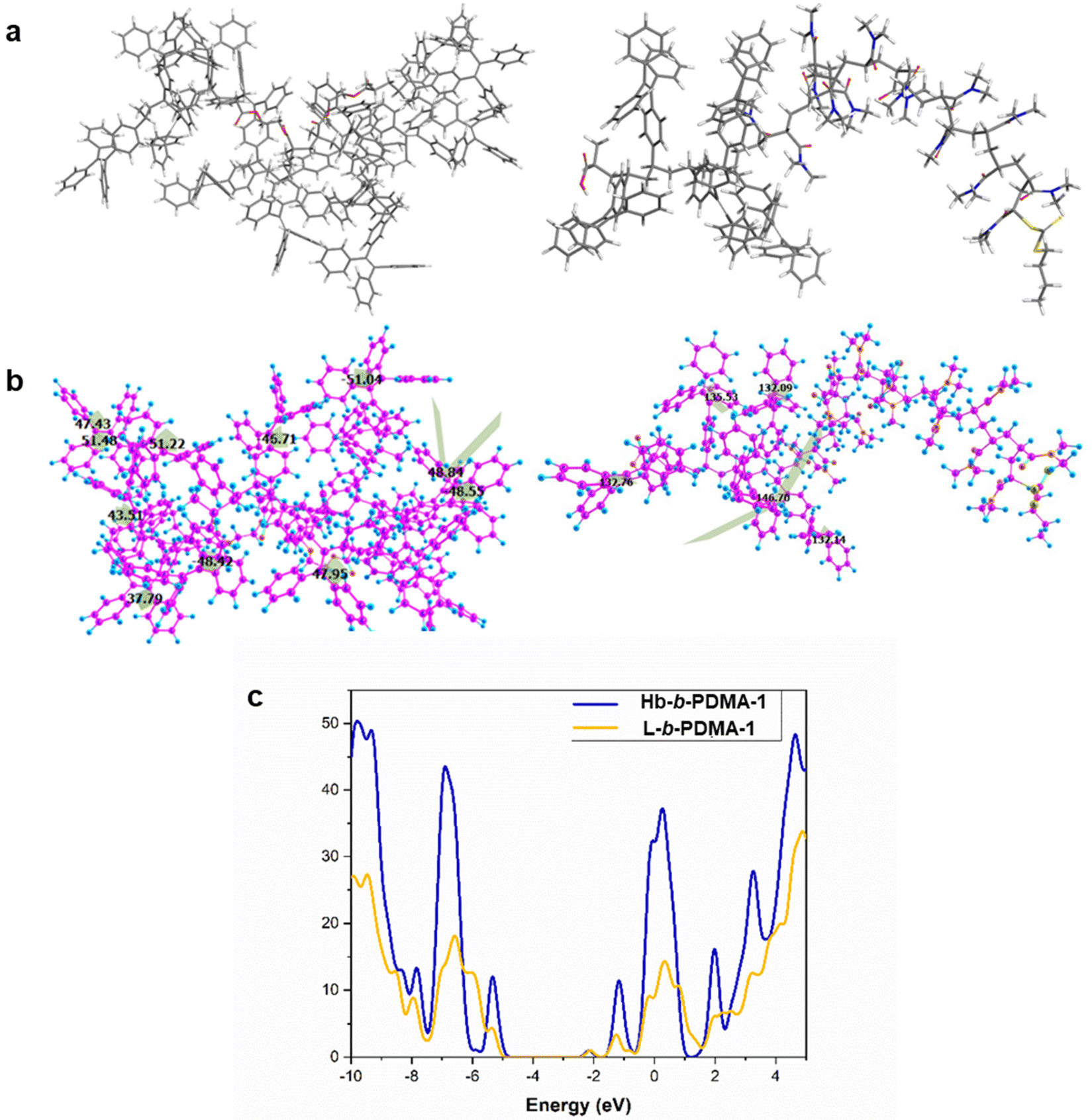
Furthermore, density functional theory (DFT) calculations were performed to unveil the excited state properties of both polymers, the star-like (Hb-b-PDMA-1) polymer and the linear one (L-b-PDMA-1). The optimized geometries of L-b-PDMA-1 and Hb-b-PDMA-1 at the ground state are illustrated in Fig. 9a and b. The calculated band gap of L-b-PDMA-1 is 3.16 eV with a HOMO of −5.26 eV and a LUMO of −2.10 eV, whereas Hb-b-PDMA-1 has a calculated band gap of 2.94 eV with a HOMO of −5.08 eV and a LUMO of −2.14 eV, as presented in Fig. 9c. The difference between the HOMO and LUMO for Hb-b-PDMA-1 is smaller than that of L-b-PDMA-1, which facilitates an intramolecular charge-transfer (ICT). The torsion angles of TPEE phenyl rings in the Hb-b-PDMA-1 optimized structure are in the range of 37° to 51°, promoting π–π stacking and enhancing the fluorescence efficiency in the solid state, unlike L-b-PDMA-1, which has higher dihedral angles up to 156° and a semi-planar structure. Moreover, the Hb-b-PDMA-1 polymer contains numerous TPEE phenyl groups, with many of them being out of the plane as shown in Fig. 9b, resulting in a non-planar structure that enhances the emission intensity. This property allows Hb-b-PDMA-1 to exhibit strong fluorescence in both the solid and solution states.
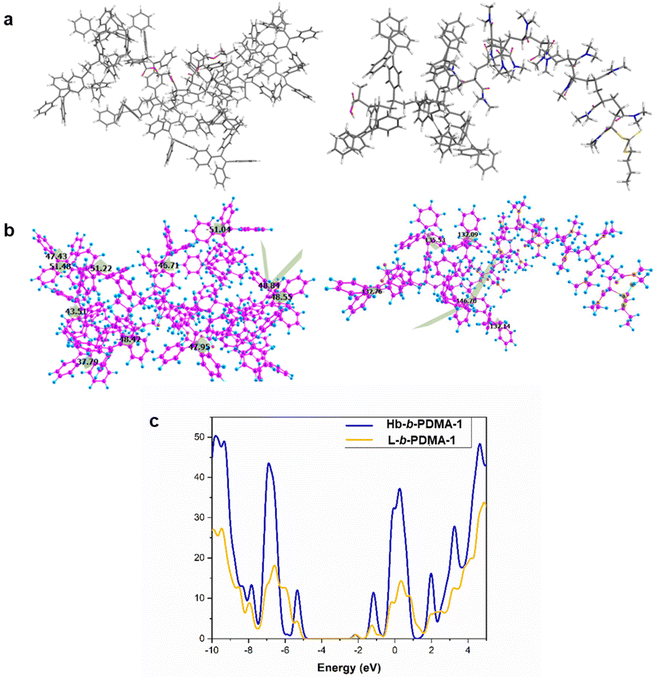 |
| | Fig. 9 DFT calculations of L-b-PDMA-1 and Hb-b-PDMA-1. (a and b) Optimized geometries and torsion angles for Hb-b-PDMA-1 (in the left) and L-b-PDMA-1 (in the right) with the Gaussian 16 package at the B3LYP/6-31G level. For Hb-b-PDMA-1, DMA tails were deleted and capped by an H atom. (c) DOS spectra of Hb-b-PDMA-1 (blue color) and L-b-PDMA-1 (yellow color). | |
Conclusions
In this study, we integrated TPEE as a hydrophobic and AIEgen moiety and DMA as a hydrophilic moiety to construct linear, branched and star-like polymers to investigate AIE difference induced by self-assembly architectures and internal arrangements of TPEE blocks. By maintaining a fixed TPEE and DMA ratio of 1:3 (n/n), we observed that the star-like polymer exhibited superior emission due to the dense arrangements in the polymersomes assembled through intermolecular packing. Linear and branched one glowed less brightly due to the broader space for TPEE block freedom in micelles and expanded gaps between mono-micelles caused by sparser TPEE arms in branching frames, respectively.
Furthermore, we increased the TPEE and DMA ratio to 1:2 (n/n) in the star-like and linear polymers for exploring transformations in emission strength and self-assembly architectures. Unexpectedly, the linear polymer showed significantly better AIE performance than the star-like polymer. The shorter hydrophilic chains in the linear structure facilitated the formation of polymersomes with highly compact conjugation between TPEE blocks. The star-like polymer, however, assembled heterogeneously to acquire particles with wide size distribution, showing that the mono-micelle constituting larger micelles differed in amount and consequently, decentralized intermolecular packing efficiency. The DFT calculations were conducted to prove that the TPEEs in star-like polymers were more crowded and the motion of the benzene rings was better limited. Finally, live cell imaging showed great potential in tracing the carrier of the star-like polymer. This study provides valuable insights into designing more efficient AIEgen-based polymers.
Abbreviations
| TPEE | (2-(4-Vinylphenyl)ethene-1,1,2-triyl)tribenzene |
| DMA |
N,N-Dimethyacrylamide |
| RAFT-MA | 2-((2-(((Butylthio)carbonothioyl)thio)propanoyl)oxy)ethyl methacrylate |
| AIE | Aggregation induced emission |
| L | Poly(TPEE) |
| Hr | Poly(RAFT-MA)-co-poly(TPEE) |
| Hb | Poly(RAFT-MA)-b-poly(TPEE) |
| L-b-PDMA | Poly(TPEE)-b-poly(DMA) |
| Hr-b-PDMA | Poly(RAFT-MA)-co-poly(TPEE)-b-poly(DMA) |
| Hb-b-PDMA | Poly(RAFT-MA)-b-poly(TPEE)-b-poly(DMA) |
| Nps | Nanoparticles. |
Author contributions
J. G. and Q. X. directed the project. L. W. and Z. L. conducted the polymer synthesis and AIE comparisons. L. W., L. L. and S. Z. conducted polymer and nanostructure characterization. L. W., S. L. and J. L. conducted the CCK8 assays and cell imaging. G. K. conducted the DFT calculations. All authors analyzed the data and discussed the results.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22071263), the Guangdong Province Zhujiang Talents Program (2019QN01Y127), and the Shenzhen Fundamental Research Program (JCYJ20200109110215774). The authors also would like to thank the Platform for Science and Technology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences for NMR test.
References
- C. P. Ma, Q. Q. Ling, S. D. Xu, H. N. Zhu, G. Zhang, X. Zhou, Z. G. Chi, S. W. Liu, Y. Zhang and J. R. Xu, Macromol. Biosci., 2014, 14, 235–243 CrossRef CAS PubMed.
- N. Kalva, S. Uthaman, E. H. Jang, R. Augustine, S. H. Jeon, K. M. Huh, I. K. Park and I. Kim, Dyes Pigm., 2021, 186, 108975 CrossRef CAS.
- X. L. Guan, L. Meng, Q. J. Jin, B. C. Lu, Y. B. Chen, Z. F. Li, L. Wang, S. J. Lai and Z. Q. Lei, Macromol. Mater. Eng., 2018, 303, 1700553 CrossRef.
- F. Ishiwari, M. Sakamoto, S. Matsumura and T. Fukushima, ACS Macro Lett., 2018, 7, 711–715 CrossRef CAS PubMed.
- P. Q. Nhien, W. L. Chou, T. T. K. Cuc, T. M. Khang, C. H. Wu, N. Thirumalaivasan, B. T. B. Hue, J. I. Wu, S. P. Wu and H. C. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 10959–10972 CrossRef CAS PubMed.
- Z. Z. Dong, Y. Z. Bi, H. R. Cui, Y. D. Wang, C. L. Wang, Y. Li, H. W. Jin and C. Q. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 23840–23847 CrossRef CAS PubMed.
- L. H. Bai, P. F. Yang, L. L. Guo, S. S. Liu and H. X. Yan, Biomacromolecules, 2022, 23, 1041–1051 CrossRef CAS PubMed.
- Y. N. Liu, H. Wang, S. L. Li, C. S. Chen, L. Xu, P. Huang, F. Liu, Y. Su, M. W. Qi, C. Y. Yu and Y. F. Zhou, Nat. Commun., 2020, 11, 1724 CrossRef CAS PubMed.
- W. I. Lee, Y. J. Bae and A. J. Bard, J. Am. Chem. Soc., 2004, 126, 8358–8359 CrossRef CAS PubMed.
- Y. Q. Du, H. X. Yan, W. Huang, F. Chai and S. Niu, ACS Sustainable Chem. Eng., 2017, 5, 6139–6147 CrossRef CAS.
- M. Sun, C. Y. Hong and C. Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581–20584 CrossRef CAS PubMed.
- A. Noel, Y. P. Borguet, J. E. Raymond and K. L. Wooley, Macromolecules, 2014, 47, 7109–7117 CrossRef CAS PubMed.
- E. Zhao, J. W. Y. Lam, L. M. Meng, Y. Hong, H. Q. Deng, G. X. Bai, X. H. Huang, J. H. Hao and B. Z. Tang, Macromolecules, 2015, 48, 64–71 CrossRef CAS.
- X. Y. Dou, Q. Zhou, X. H. Chen, Y. Q. Tan, X. He, P. Lu, K. Y. Sui, B. Z. Tang, Y. M. Zhang and W. Z. Yuan, Biomacromolecules, 2018, 19, 2014–2022 CrossRef CAS PubMed.
- S. X. Tang, T. J. Yang, Z. H. Zhao, T. W. Zhu, Q. Zhang, W. B. W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 RSC.
- L. H. Bai, H. X. Yan, T. Bai, L. L. Guo, T. L. Lu, Y. Zhao and C. J. Li, Biomacromolecules, 2020, 21, 3724–3735 CrossRef CAS PubMed.
- L. P. Fang, C. S. Huang, G. Shabir, J. L. Liang, Z. Y. Liu and H. F. Zhang, ACS Macro Lett., 2019, 8, 1605–1610 CrossRef CAS PubMed.
- N. L. C. Leung, N. Xie, W. Z. Yuan, Y. Liu, Q. Y. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
- G. F. Zhang, Z. Q. Chen, M. P. Aldred, Z. Hu, T. Chen, Z. L. Huang, X. G. Meng and M. Q. Zhu, Chem. Commun., 2014, 50, 12058–12060 RSC.
- Y. N. Zhang, H. J. Xu, X. D. Ma, Z. X. Shi, J. Yin and X. S. Jiang, Macromol. Rapid Commun., 2016, 37, 998–1004 CrossRef CAS PubMed.
- S. J. Liu, Y. H. Cheng, H. K. Zhang, Z. J. Qiu, R. T. K. Kwok, J. W. Y. Lam and B. Tang, Angew. Chem., Int. Ed., 2018, 57, 6274–6278 CrossRef CAS PubMed.
- N. Zhang, H. Chen, Y. Fan, L. Zhou, S. Trepout, J. Guo and M. H. Li, ACS Nano, 2018, 12, 4025–4035 CrossRef CAS PubMed.
- D. P. Zhang, Y. J. Fan, H. Chen, S. Trepout and M. H. Li, Angew. Chem., Int. Ed., 2019, 58, 10260–10265 CrossRef CAS PubMed.
- Q. Y. Li, H. W. Zhang, K. Lou, Y. B. Yang, X. F. Ji, J. T. Zhu and J. L. Sessler, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121746119 CrossRef CAS PubMed.
- Y. Y. Yuan, C. J. Zhang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 11419–11423 CrossRef CAS PubMed.
- S. F. Ji, M. Lin, Z. H. Li, L. L. Xu, X. H. Fu, G. S. Chen, Z. B. Li and J. Sun, Biomacromolecules, 2022, 23, 798–807 CrossRef CAS PubMed.
- L. Y. Lin, P. C. Huang, D. J. Yang, J. Y. Gao and J. L. Hong, Polym. Chem., 2016, 7, 153–163 RSC.
- Y. Shi, G. Q. Yin, Z. P. Yan, P. Sang, M. H. Wang, R. Brzozowski, P. Eswara, L. Wojtas, Y. X. Zheng, X. P. Li and J. F. Cai, J. Am. Chem. Soc., 2019, 141, 12697–12706 CrossRef CAS PubMed.
- S. Y. Wang, B. X. Jin, G. F. Chen, Y. J. Luo and X. Y. Li, Polym. Chem., 2020, 11, 4706–4713 RSC.
- Z. Zhang and N. Hadjichristidis, ACS Macro Lett., 2018, 7, 886–891 CrossRef CAS PubMed.
- Y. F. Zhou, W. Huang, J. Y. Liu, X. Y. Zhu and D. Y. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS PubMed.
- Y. L. Wang, B. Li, H. B. Jin, Y. F. Zhou, Z. Y. Lu and D. Y. Yan, Chem. – Asian J., 2014, 9, 2281–2288 CrossRef CAS PubMed.
- T. Uneyama and M. Doi, Macromolecules, 2005, 38, 5817–5825 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- Y. L. Wang, B. Li, Y. F. Zhou, Z. Y. Lu and D. Y. Yan, Soft Matter, 2013, 9, 3293–3304 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) of RAFT-MA. 1H NMR (400 MHz, CDCl3), GPC traces and molecular weights of L, Hr, Hb, L-b-PDMA-1/2, Hr-b-PDMA-1 and Hb-b-PDMA-1/2. Cytotoxicity of Hb-b-PDMA-1 to A549 and HepG2. See DOI: https://doi.org/10.1039/d3py00559c |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a