
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclopolymerization: a versatile approach toward multicyclic polystyrene and polystyrene-containing multicyclic copolymers†
Yamato
Ebii
a,
Yoshinobu
Mato
b,
Feng
Li
 b,
Kenji
Tajima
b,
Takuya
Yamamoto
b,
Takuya
Isono
*b and
Toshifumi
Satoh
*b
b,
Kenji
Tajima
b,
Takuya
Yamamoto
b,
Takuya
Isono
*b and
Toshifumi
Satoh
*b
aGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo 060-8628, Japan
bDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Sapporo 060-8628, Japan. E-mail: isono.t@eng.hokudai.ac.jp; satoh@eng.hokudai.ac.jp
First published on 5th June 2023
Abstract
Macromolecules with cyclic topologies have attracted significant attention because of their unique structures. Herein, we report the systematic synthesis of multicyclic polystyrene (mc-PS) via cyclopolymerization of α,ω-dinorbornenyl end-functionalized PS macromonomers mediated by the Grubbs third-generation catalyst (G3) under diluted conditions. By varying the initial macromonomer-to-G3 ratio, the number of cyclic units in the obtained mc-PS was controlled up to 239, which, compared to previously reported values, is more than 10 times higher. The molecular weight (Mn) of each cyclic unit was controlled between 1640 and 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 using macromonomers with different Mn. The use of mc-PS with different numbers of cyclic units revealed that upon increasing the number of cyclic units, the hydrodynamic volume and intrinsic viscosity per ring decreased and then reached constant values. Regarding mc-PS, the dependence of Mn on the glass transition temperature (Tg) was much stronger compared to that in monocyclic PS. Moreover, the Tg in the limit of infinite Mn was approximately 5 °C higher for mc-PS compared to that of linear PS and monocyclic PS, confirming the unique topological effect of the multicyclic structure. In addition, the cyclopolymerization of a block copolymer-type macromonomer consisting of PS and poly(rac-lactide) (PLA) or the statistical cyclocopolymerization of PS and PLA macromonomers afforded PS-containing multicyclic copolymers with various architectures. The developed cyclopolymerization strategy is more straightforward than previously known multicyclic PS and PS-containing multicyclic polymer syntheses and will provide a new avenue for fundamental studies and material applications.
100 using macromonomers with different Mn. The use of mc-PS with different numbers of cyclic units revealed that upon increasing the number of cyclic units, the hydrodynamic volume and intrinsic viscosity per ring decreased and then reached constant values. Regarding mc-PS, the dependence of Mn on the glass transition temperature (Tg) was much stronger compared to that in monocyclic PS. Moreover, the Tg in the limit of infinite Mn was approximately 5 °C higher for mc-PS compared to that of linear PS and monocyclic PS, confirming the unique topological effect of the multicyclic structure. In addition, the cyclopolymerization of a block copolymer-type macromonomer consisting of PS and poly(rac-lactide) (PLA) or the statistical cyclocopolymerization of PS and PLA macromonomers afforded PS-containing multicyclic copolymers with various architectures. The developed cyclopolymerization strategy is more straightforward than previously known multicyclic PS and PS-containing multicyclic polymer syntheses and will provide a new avenue for fundamental studies and material applications.
Introduction
Macromolecules with cyclic architectures have attracted significant attention because of their interesting properties stemming from the absence of polymer chain ends. For example, cyclic polymers have higher glass transition temperatures, higher densities, smaller hydrodynamic volumes and lower viscosities than their linear counterparts.1–6 Moreover, their cyclic polymer architecture is highly valuable for functional material applications, such as decreasing domain spacing for block copolymers,7 enabling superlubricity at interfaces8 and improving thermal stability of micelles.9 Therefore, interest in the synthesis, physical properties and functions of multicyclic polymers composed of two or more cyclic units has been increasing.Multicyclic polymers are typically classified into three categories based on their topology: fused-type, bridged-type and spiro-type (Fig. 1a). For fused- and spiro-type cyclic polymers, differences in the cyclic topology type were found to significantly affect their physical properties, such as hydrodynamic volume, melting temperature and critical micelle concentration.10–12 Several synthetic strategies have been proposed for the construction of different types of multicyclic polymers.10–20 For example, the coupling of monocyclic polymers through click reactions has been employed to produce fused- and spiro-type multicyclic polymers.10,13–15 An efficient coupling reaction, electrostatic self-assembly and covalent fixation, has also been applied for the synthesis of various multicyclic polymers, such as polytetrahydrofuran (PTHF).16–18 More recently, an intramolecular consecutive cyclization approach, in which a linear or star-shaped precursor intramolecularly cyclizes in a chain reaction manner, has been proposed as an efficient route to fused- and spiro-type multicyclic polymers.11,12 The majority of reported multicyclic polymers are composed of polyethers and polyesters, most likely because of their synthetic accessibility. Multicyclic polymers composed of general-purpose polymers, such as polystyrene (PS), with well-known fundamental characteristics, have been understudied despite their practical importance in plastics and elastomers.
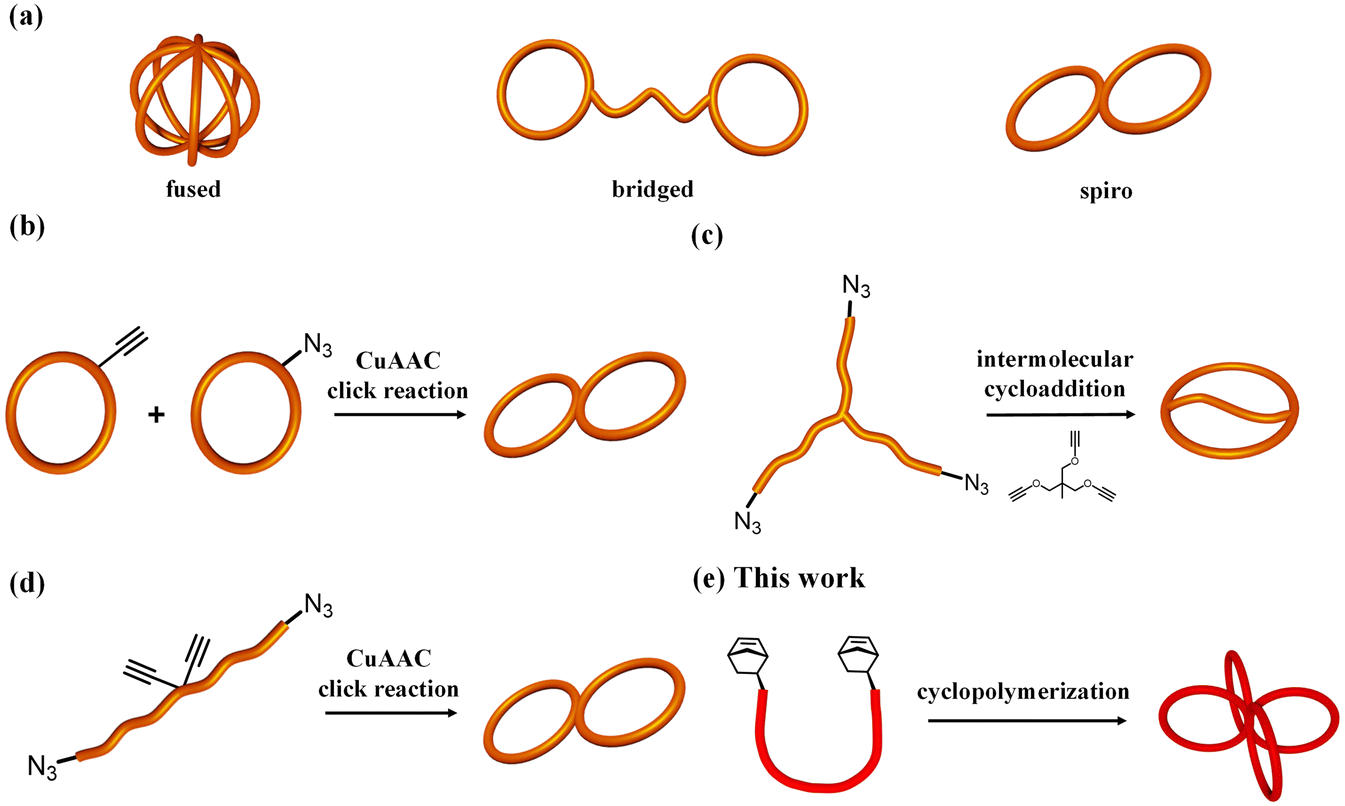 | ||
| Fig. 1 (a) Examples of diverse multicyclic topologies. Synthetic strategies for multicyclic PS: (b) intermolecular coupling of monocyclic PS (strategy (i)); (c) intermolecular cyclization of linear/star PS with a multifunctional linker (strategy (ii)); (d) intramolecular cyclization of linear/star PS precursors bearing functional groups at predetermined positions (strategy (iii)). (e) The cyclopolymerization of α,ω-dinorbornenyl end-functionalized PS described in this work. | ||
To date, several synthetic pathways for multicyclic PS (mc-PS) have been developed: (i) intermolecular coupling of monocyclic constituents,21–25 (ii) intermolecular cyclization of linear/star PS with a multifunctionalized linker,26–28 and (iii) intramolecular cyclization of linear/star PS precursors bearing functional groups at predetermined positions,29,30 as shown in Fig. 1b–d. Monteiro et al. reported a Cu-catalyzed azido/alkyne cycloaddition (CuAAC) click reaction of monocyclic units (i.e., strategy (i)) to produce 8-shaped and trefoil-shaped mc-PS.21 The same group also reported a stepwise CuAAC polymerization of monocyclic PS bearing azido and ethynyl groups to produce mc-PS with a higher number of ring units, up to 20.24 However, these approaches require multistep synthesis for the preparation of functionalized monocyclic PS, offering low yields. Moreover, because strategies shown in Fig. 1c and d require a custom-made PS precursor to construct each topology, the application of these strategies in the synthesis of mc-PS with more cyclic units is difficult. In summary, the current synthetic methods for mc-PS present issues in terms of difficulty in precursor preparation, inefficiency in ring formation and synthetic complexity due to multistep reactions, which make systematic mc-PS synthesis unrealistic. Therefore, establishing a synthetic approach to systematically produce mc-PS and elucidate the relationship between structure and properties and its industrial application is significant.
In addition to multicyclic homopolymers, the interest in the synthesis of multicyclic copolymers is increasing. Hawker et al. reported that a monocyclic polymer consisting of PS-block-polyethylene exhibited a microphase-separated structure with a smaller domain-spacing than that in its linear counterpart.7 Borsali et al. revealed drastic differences in the morphologies of micelles made from linear and monocyclic PS-block-polyisoprene.31–34 Yamamoto et al. reported that a self-assembled micelle from cyclic poly(butyl acrylate)-block-poly(ethylene oxide) displayed significantly improved thermal stability compared to that of its linear counterpart.9 Even though research focused on monocyclic copolymers is promising for industrial applications, research on multicyclic copolymers is lacking. This is attributable to the complexity of current multicyclic polymer synthetic approaches, which impedes their application to the synthesis of even more complex multicyclic copolymers. Thus, the development of a versatile mc-PS synthetic approach that can be easily applied to the synthesis of PS-containing multicyclic copolymers is of great interest.
Herein, we report a versatile synthetic approach for mc-PS and PS-containing multicyclic copolymers based on cyclopolymerization. The synthesis of multicyclic polymers by the cyclopolymerization of α,ω-dinorbornenyl end-functionalized linear macromonomers via ring-opening metathesis polymerization (ROMP) using the Grubbs third generation catalyst (G3) has been previously reported by our group.35 When applying highly diluted conditions, the intramolecular cyclization process surpasses the intermolecular propagation, realizing multicyclic polymer formation in a chain growth manner virtually without intermolecular crosslinking. Although the systematic synthesis of multicyclic polymers with polyester and polyether main chains, such as polylactide, poly(ε-caprolactone), poly(ethylene glycol) and poly(tetrahydrofuran), has been achieved, the application of the cyclopolymerization approach for the synthesis of multicyclic polymers consisting of more common polymer main chains, such as polystyrene, remains unexplored. Herein, we demonstrate the effectiveness of the cyclopolymerization approach for the systematic synthesis of mc-PS with varying numbers and sizes of cyclic units (Fig. 1e and Scheme 1a). Several mc-PSs were synthesized and the relationships between the structure and the viscosity, hydrodynamic volume and glass transition temperature were comprehensively examined. Furthermore, the applicability of the developed approach for the synthesis of PS-containing multicyclic copolymers with various architectures was proven (Scheme 1b and c).
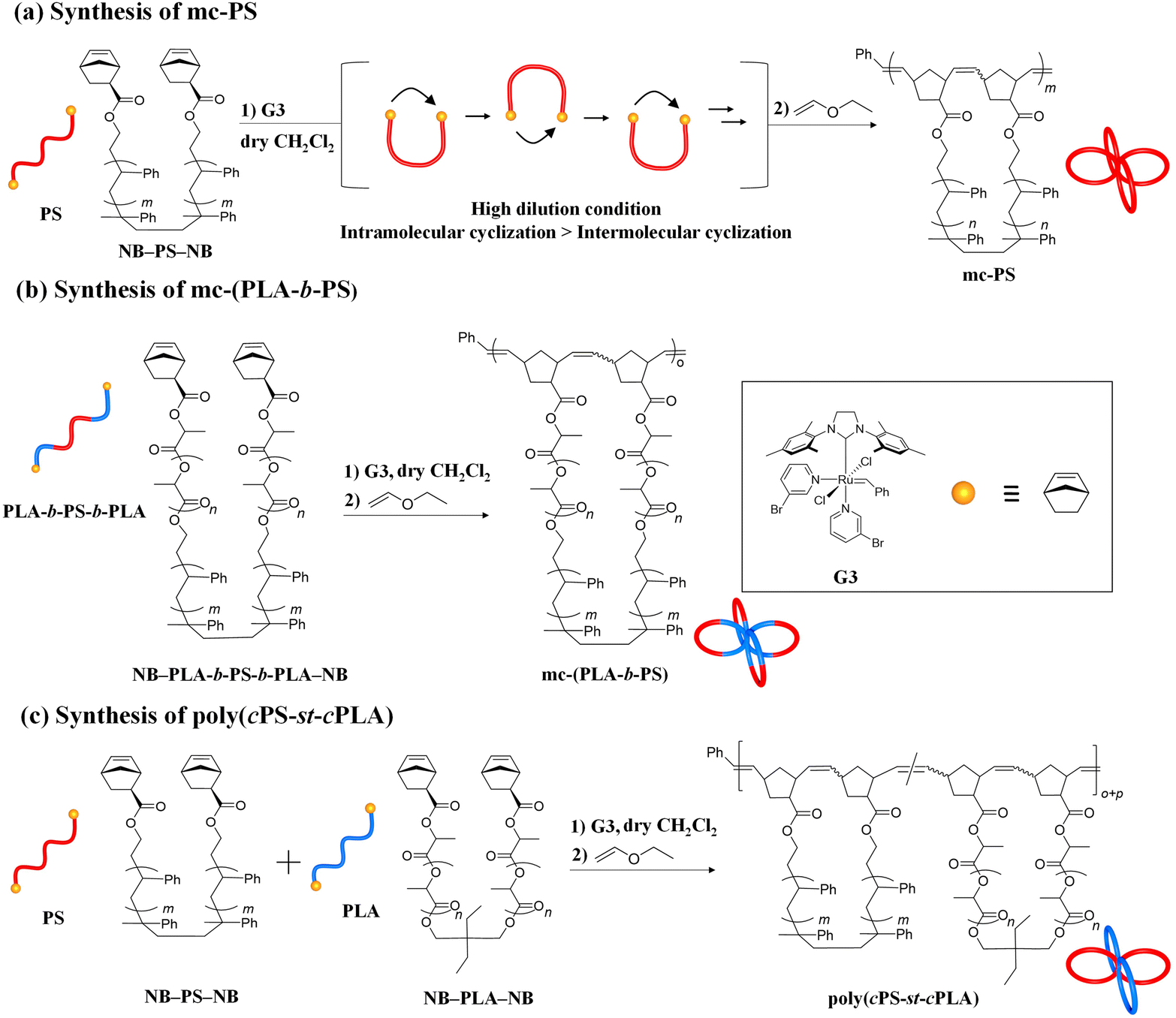 | ||
| Scheme 1 Synthesis of (a) mc-PS, (b) mc-(PLA-b-PS), and (c) poly(cPLA-st-cPS). | ||
Experimental
A typical procedure for the ROMP is as follows: in a three-necked flask, a stock solution of G3 (2.54 mL, 20.3 μmol as an 8.0 mol L−1 solution in dry CH2Cl2, 1.0 eq.) was quickly added to a stirred mixture of α,ω-dinorbornenyl-functionalized linear polystyrene (NB–PS1k–NB; Mn,NMR = 1640, 100 mg, 61.0 μmol, 3.0 eq.) in dry CH2Cl2 (122 mL) under an argon atmosphere. After 10 min, an excess amount of ethyl vinyl ether was added to the reaction mixture to terminate the ROMP. The crude product was purified by preparative GPC to remove the catalyst residue (see Fig. S1 in the ESI†), affording mc-PS1k as a brown solid (82.0 mg, 82.0%).Results and discussion
Synthesis of PS macromonomers
NB–PS–NB was synthesized via a condensation reaction between commercially available PS diol (HO–PS–OH) and exo-5-norbornenecarboxylic acid according to Scheme S2† and was used as the macromonomer (MM) in the cyclopolymerization approach. To synthesize mc-PS with cyclic units of various sizes, five different NB–PSx–NB macromonomers were prepared (x = 1k, 6k, 12k, 13k and 52k) with molecular weights ranging from 1k to 52k (Table 1). The quantitative esterification in each NB–PS–NB synthesis was confirmed using 1H NMR spectroscopy (Fig. S2–S6†).| Sample (MM) |
M
n,NMRa |
M
n,SECb |
Đ |
M
n,MALSc |
|---|---|---|---|---|
| a Determined by 1H NMR. b Determined by size exclusion chromatography (SEC) equipped with a refractive index detector using PS standards. c Determined using triple-detection SEC equipped with multi-angle light scattering, viscosity and refractive index detectors (SEC-MALS-Visco) in chloroform. | ||||
| NB–PS1k–NB | 1640 | 1220 | 1.55 | — |
| NB–PS6k–NB | 6000 | 5800 | 1.09 | 6470 |
| NB–PS12k–NB | 11600 |
11500 |
1.07 | 12300 |
| NB–PS13k–NB | 12300 |
12500 |
1.22 | 13400 |
| NB–PS52k–NB | 55600 |
54400 |
1.06 | 52100 |
Synthesis of mc-PS via cyclopolymerization
The synthesis of mc-PS was conducted using the MM with the smallest molecular weight (1k), that is, NB–PS1k–NB. Because the G3-initiated ROMP is known to proceed in a living manner, the initial MM-to-G3 ratio determines the average number of cyclic units of the resulting mc-PS. A [NB–PS1k–NB]0/[G3]0 ratio of 3/1 was applied to obtain the target mc-PS with three cyclic units (run 1 in Table 2). Under highly diluted conditions ([NB–PS1k–NB]0 = 0.500 mM) in CH2Cl2, the cyclopolymerization proceeded without gelation, affording a soluble product in 82.0% yield. The SEC elution peak of the product clearly shifted to the high molecular weight region compared to that of NB–PS1k–NB, while maintaining the unimodal shape (Mn,SEC = 3870, Đ = 1.28, Fig. 2a). This result indicates that the controlled cyclopolymerization proceeds by suppressing uncontrolled intermolecular reactions. The 1H NMR analysis suggested that the ROMP, the elementary reaction of the cyclopolymerization, proceeded quantitatively, as evidenced by the complete disappearance of the signals due to the norbornenyl group (a, b: 6.18–6.00; c, d: 3.14–2.91 ppm) and the emergence of broad signals due to the polynorbornene backbone (Fig. 2b). Furthermore, the absolute Mn value determined by SEC-MALS-Visco (Mn,MALS) was 5850 and the number of cyclic units was calculated to be 3.6, which is close to the theoretical value. Thus, the cyclic units can be controlled by the living manner of the G3-initiated ROMP. Finally, MALDI-TOF MS was employed to reveal further structural details (Fig. 3). The spectra showed a series of repeated peaks with approximately 104 Da unit intervals corresponding to repeating PS. The peaks observed in the range of m/z = 2000–4000 Da were assigned to the desired mc-PS containing two cyclic units, whereas those observed in the range of m/z = 4000–6000 Da were assigned to the desired mc-PS containing three cyclic units. For example, the peaks at m/z = 2800.88 and 4407.60 (denoted by ○ and Δ, respectively) closely matched the calculated masses for the dimer containing 14 styrene repeating units ([M + Ag]+ = 2800.52) and the trimer containing 24 styrene repeating units ([M + Ag]+ = 4407.49), respectively. No other peaks were present apart from those of the desired mc-PS. This implies that mc-PS was produced through alternating intramolecular cyclization and intermolecular growth reactions. Overall, these structural characterization results confirmed that mc-PS was synthesized via cyclopolymerization as intended. To systematically characterize mc-PS, we attempted to synthesize mc-PS with more cyclic units by varying the ratio of [MM]0/[G3]0. Under the established conditions of [MM]0/[G3]0 = 6/1 or 9/1, the desired mc-PSs with 7.4 and 10.4 cyclic units were obtained (runs 2 and 3 in Table 2). To date, the highest number of cyclic units recorded in multicyclic PS is approximately 20, as reported by Monteiro et al.24 To overcome this limitation, the cyclopolymerization was conducted at [MM]0/[G3]0 = 20/1, 50/1, 75/1 and 100/1 (runs 4–7 in Table 2). The SEC analysis revealed that the cyclopolymerization proceeded without obvious side reactions, even at an [MM]0/[G3]0 ratio of 100/1 (Fig. S7†). Owing to the living polymerization nature, the number of the resulting cyclic units increased upon increasing the [MM]0/[G3]0 ratio and reached the expected value (= [MM]0/[G3]0). The highest number of cyclic units was 239, which is more than ten times the number reported by Monteiro et al. Thus, the number of cyclic units could be easily controlled by varying the initial MM-to-G3 ratio, enabling the synthesis of mc-PS with a high number of cyclic units. This enabled us to study the effect of the number of cyclic units on the physical properties of PS.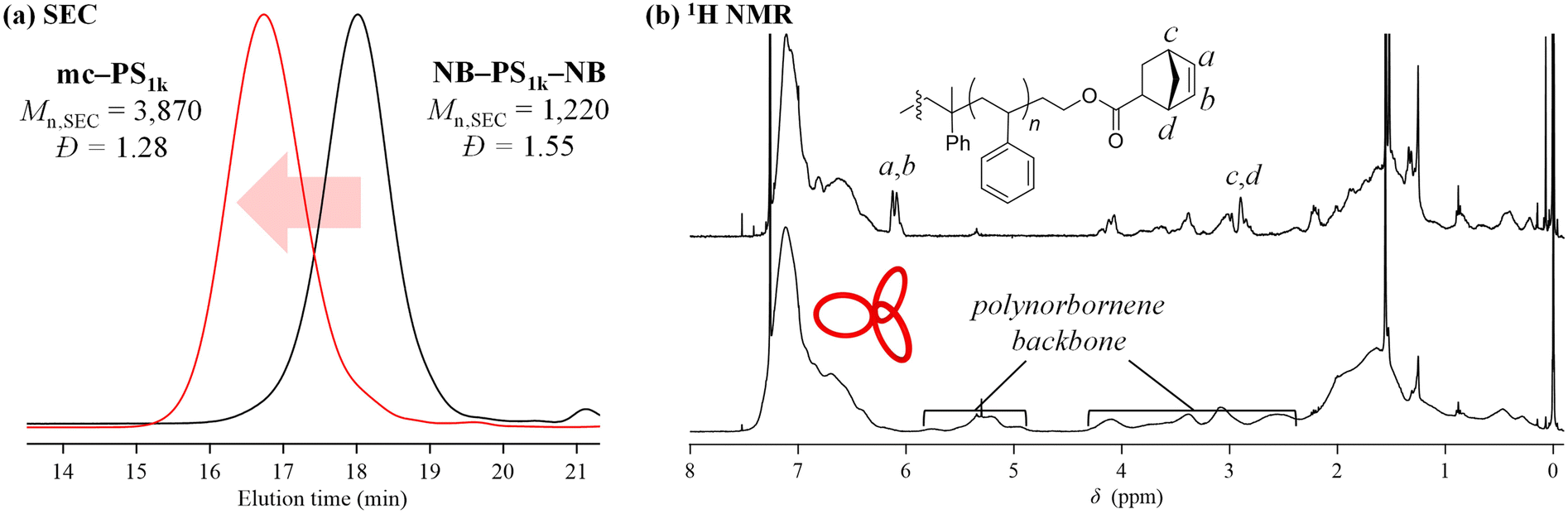 | ||
| Fig. 2 (a) SEC traces of NB–PS1k–NB and mc-PS1k obtained from run 1 in Table 2 (refractive index (RI) detection; eluent, THF; flow rate, 1.0 mL min−1). (b) 1H NMR spectra of NB–PS1k–NB (upper) and mc-PS1k (lower, run 1 in Table 2) in CDCl3 (400 MHz). | ||
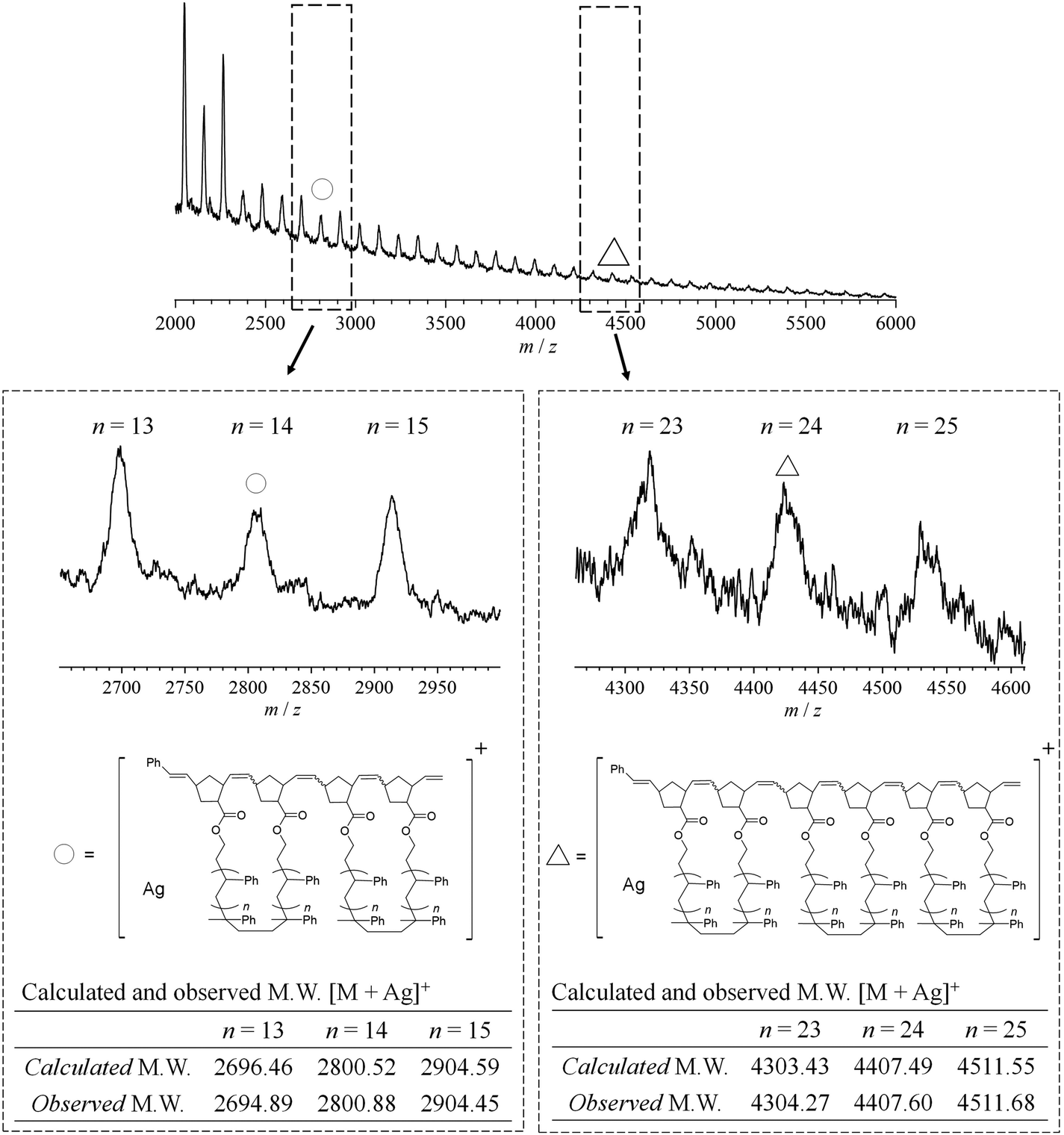 | ||
| Fig. 3 MALDI-TOF-MS analysis of mc-PS1k (run 1 in Table 2). | ||
| Run | MM | [MM]0/[G3]0 | [MM]0 (mM) | Time (min) |
M
n,MALSb |
Đ | Number of cyclic unitsd | R h (nm) | [η]b (mL g−1) | T g (°C) | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: temperature, room temperature; atmosphere, Ar; solvent, CH2Cl2. b Determined using SEC-MALS-Visco in chloroform. c Determined using SEC with THF and PS as the standard. d The average number of cyclic units in the obtained mc-PS was estimated as (Mn of mc-PS)/(Mn of NB–PS–NB). | |||||||||||
| 1 | NB–PS1k–NB (Mn,NMR = 1640) | 3/1 | 0.500 | 10 | 5850 | 1.28 | 3.6 | 1.8 | 6.4 | 99.8 | 82.0 |
| 2 | 6/1 | 0.500 | 10 | 12200 |
1.21 | 7.4 | 2.5 | 7.8 | 105.3 | 80.3 | |
| 3 | 9/1 | 0.500 | 10 | 17100 |
1.16 | 10.4 | 2.9 | 8.8 | 106.1 | 90.8 | |
| 4 | 20/1 | 0.500 | 15 | 72300 |
1.14 | 44.1 | 5.9 | 18.0 | 109.2 | 78.0 | |
| 5 | 50/1 | 0.500 | 30 | 82200 |
1.12 | 53.8 | 6.5 | 20.2 | 110.6 | 99.6 | |
| 6 | 75/1 | 1.000 | 60 | 180000 |
1.40 | 109.8 | 9.7 | 32.0 | 112.0 | 97.0 | |
| 7 | 100/1 | 1.000 | 80 | 392000 |
1.42 | 239.1 | 15.0 | 55.7 | 113.2 | 68.0 | |
| 8 | NB–PS6k–NB (Mn,MALS = 6470) | 3/1 | 0.125 | 150 | 22000 |
1.11 | 3.4 | 9.6 | 3.8 | 103.7 | 90.1 |
| 9 | 6/1 | 0.125 | 180 | 35500 |
1.12 | 5.5 | 10.2 | 4.5 | 111.2 | 72.2 | |
| 10 | 9/1 | 0.150 | 540 | 50600 |
1.07 | 7.8 | 11.6 | 5.0 | 111.4 | 72.3 | |
| 11 | NB–PS12k–NB (Mn,MALS = 12300) |
3/1 | 0.150 | 90 | 54400 |
1.44 | 4.4 | 5.5 | 16.1 | 105.2 | 98.0 |
| 12 | 6/1 | 0.150 | 540 | 109600 |
1.21 | 8.9 | 6.6 | 17.1 | 110.9 | 99.1 | |
| 13 | 9/1 | 0.150 | 540 | 155100 |
1.36 | 12.6 | 7.8 | 19.2 | 111.8 | 79.2 | |
| 14 | NB–PS52k–NB (Mn,MALS = 52100) |
3/1 | 0.100 | 330 | 497400 |
1.42 | 10.2 | 18.0 | 79.1 | 106.3 | 69.4 |
To elucidate the effect of ring size on the physical properties of PS, mc-PSs with larger ring sizes were synthesized by the cyclopolymerization of NB–PS6k–NB and NB–PS12k–NB (runs 8–13 in Table 2). Intermolecular cyclization would be less likely to occur if the molecular weight of the MM increased; therefore, the cyclopolymerization of NB–PS6k–NB and NB–PS12k–NB was conducted at lower substrate concentrations, at an [MM]0/[G3]0 ratio of 3/1 where [MM]0 = 0.125 or 0.150 mM (runs 8 and 11 in Table 2). Despite the increased molecular weight of the MM, the desired mc-PSs were successfully synthesized without gelation. The SEC elution peak of the products clearly shifted to a higher molecular weight region compared to that of the MM, and the 1H NMR data suggested that the ROMP proceeded quantitatively (Fig. S8–S11†). The synthesized mc-PS6k and mc-PS12k had 3.4 and 4.4 repeating units, respectively, which were close to the theoretical values. Moreover, mc-PSs with more cyclic units were successfully synthesized by applying [MM]0/[G3]0 = 6/1 and 9/1, while thorough optimization of the substrate concentration and reaction time was necessary (runs 9, 10, 12 and 13 in Table 2). However, we were unable to synthesize mc-PSx (x = 6k and 12k) composed of a large number of cyclic units (>20) because of the competitive deactivation of G3 during cyclopolymerization when applying a higher [MM]0/[G3]0 ratio. Nevertheless, we confirmed that the number of cyclic units could be regulated to some degree.
The cyclopolymerization of a high-molecular-weight MM, NB–PS52k–NB, was also carried out for the synthesis of mc-PS with a ring size of 52k. Surprisingly, mc-PS52k with an average number of cyclic units of 10.2 was obtained without gelation (Fig. S12†). The successful cyclopolymerization of such a high-molecular-weight MM demonstrates the versatility of this method for the systematic synthesis of mc-PS.
An attractive aspect of the cyclopolymerization strategy is the possibility of synthesizing multicyclic polymers on a gram scale, which is in contrast with previous synthetic methodologies that require multiple steps and cumbersome processes. Commercial scale-up has been a substantial issue, even for monocyclic polymers;36 the applicability of the presented approach to gram-scale synthesis is therefore a critical factor for the practical applications of mc-PS. In this study, the cyclopolymerization of 1.5 g of NB–PS13k–NB with an [MM]0/[G3]0 ratio of 3/1 where [MM]0/[G3]0 = 0.150 was conducted successfully. The cyclopolymerization proceeded without gelation, affording 1.39 g of mc-PS13k with 3.8 cyclic units in a 92.7% yield. Again, the SEC elution peak of the product clearly shifted to the higher molecular weight region compared to that of the MM, and the 1H NMR data suggested that the ROMP proceeded quantitatively (Fig. S13 and S14†).
Solution properties of mc-PS
The investigation of the relationship between the number and size of the cyclic units of multicyclic polymers and their physical properties is highly attractive, and acquiring such information is beneficial for materials design based on multicyclic polymers. The hydrodynamic radius (Rh) and intrinsic viscosity ([η]) of the developed mc-PS1k with varied numbers of cyclic units were investigated using SEC-MALS-Visco in chloroform. The [η] and Rh values of mc-PS1k were in the range of 6.4–55.7 mL g−1 and 1.8–15.0 nm, respectively, as summarized in Table 2. To further analyze the solution conformation of mc-PS1k, the [η] and Rh values were normalized by dividing them with the number of cyclic units in the molecules (denoted as [η,ring] and Rh,ring). The results showed that [η,ring] and Rh,ring decreased with an increasing number of cyclic units up to approximately 50 and then reached almost constant values (Fig. 4a). This suggests that the conformation of each cyclic unit in solution gradually became more compact after the number of cyclic units reached a certain value. Molecular simulations and experimental studies have demonstrated that the conformation of a bottlebrush polymer changes from spherical to cylindrical as the backbone DP increases.37–40 Thus, conformational changes similar to those seen in bottlebrush polymers may occur in mc-PS1k upon increasing the number of cyclic units.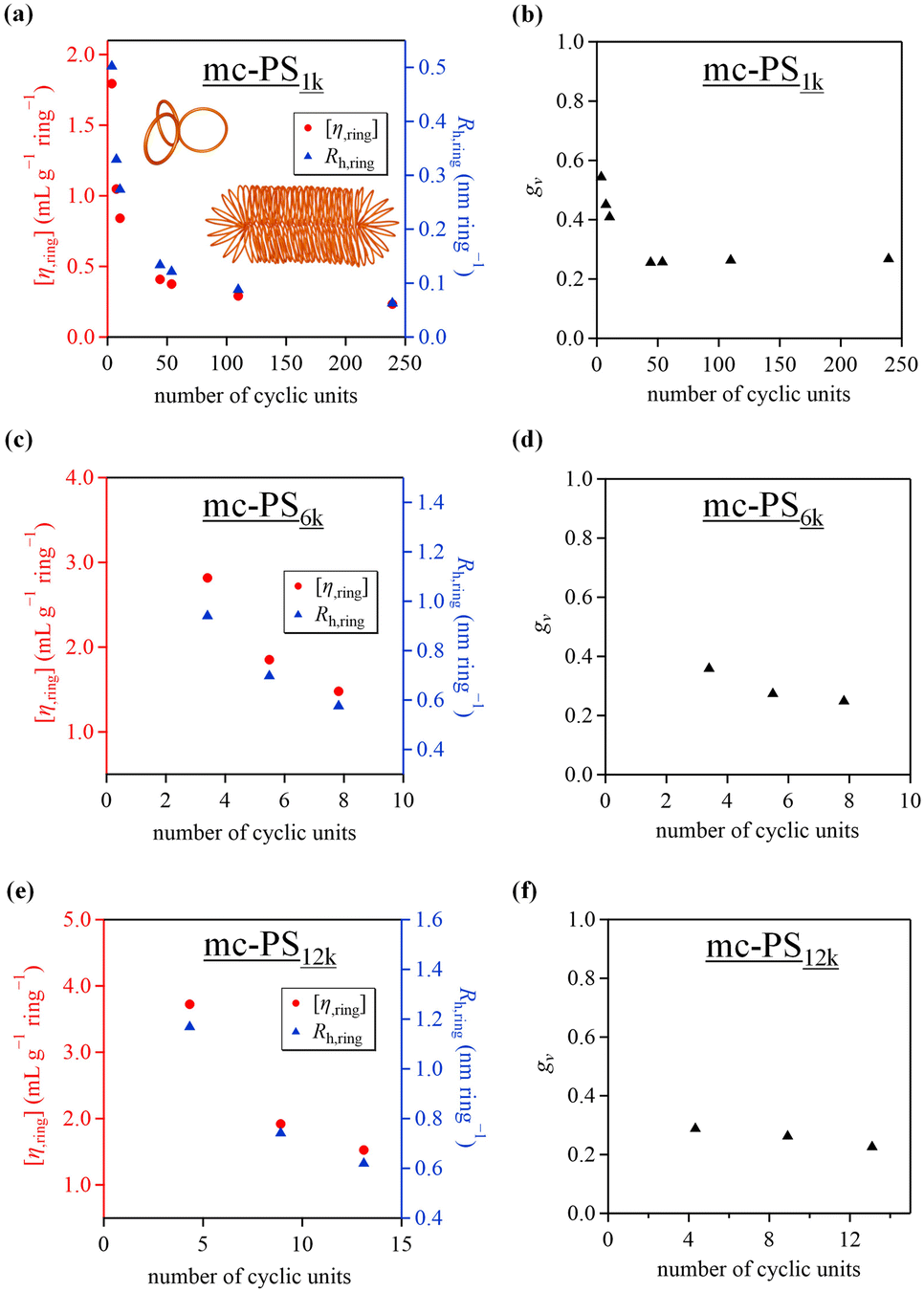 | ||
| Fig. 4 (a) Dependence of [η] (red) and Rh (blue) of mc-PS1k on the number of cyclic units (solvent, CHCl3). (b) Dependence of gv of mc-PS1k on the number of cyclic units. (c) Dependence of [η] (red) and Rh (blue) of mc-PS6k on the number of cyclic units (solvent, CHCl3). (d) Dependence of gv of mc-PS6k on the number of cyclic units. (e) Dependence of [η] (red) and Rh (blue) of mc-PS12k on the number of cyclic units (solvent, CHCl3). (f) Dependence of gv of mc-PS12k on the number of cyclic units. | ||
The degree of shrinkage of mc-PS1k in solution was investigated. The hydrodynamic volume contraction (gv) was determined from SEC-MALS-Visco using eqn (1).24
| gv = (Mn,app/Mn,abs)1.7 | (1) |
Solid state properties of mc-PS
Differential scanning calorimetry (DSC) was used to determine the Tg of mc-PS1k and linear PS in order to investigate the relationship between structure and Tg (Fig. S15 and S16†). Fig. 5a presents the Tg values of mc-PS1k and linear PS as a function of Mn. All mc-PS1k polymers showed a higher Tg than their linear counterparts, which can be attributed to their cyclic topology. For both linear and mc-PS1k, Tg increased with increasing Mn in the lower molecular weight region and finally converged to a constant value.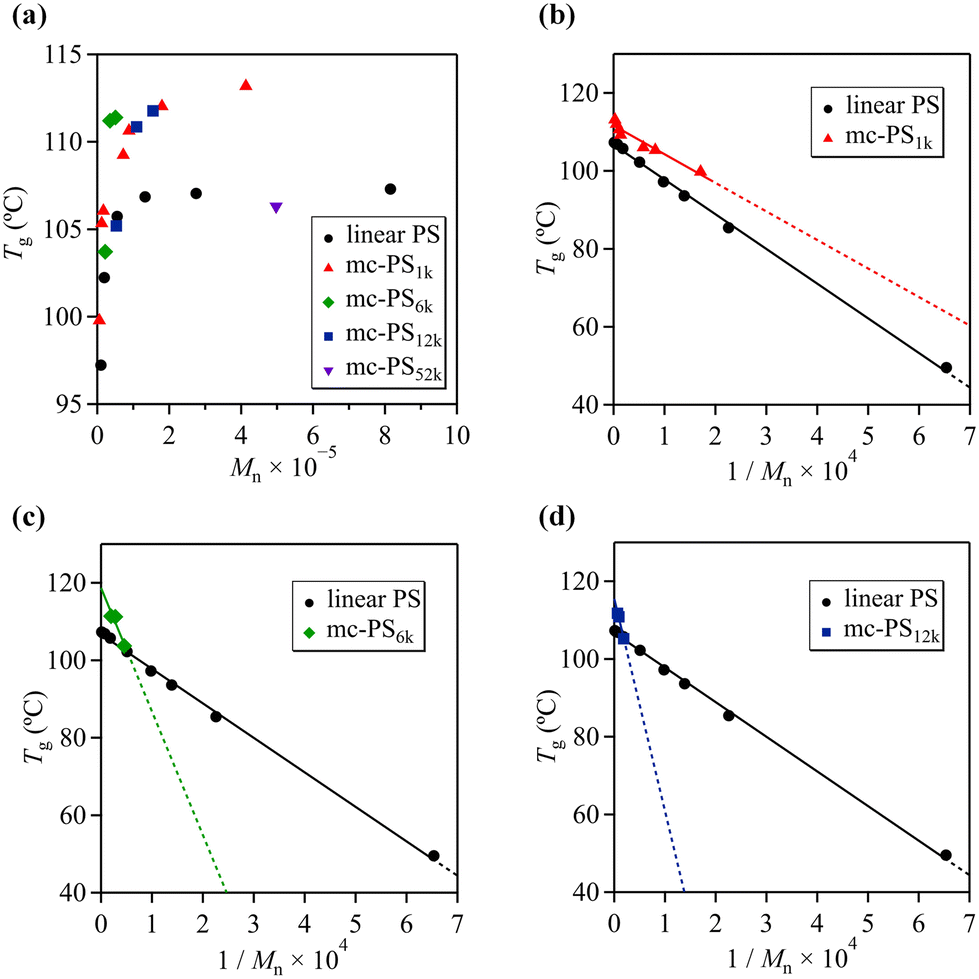 | ||
| Fig. 5 (a) Molecular weight dependence of glass transition temperature for mc-PS and linear PS. To fit the Flory–Fox equation, the glass transition temperature was plotted against the inverse of molecular weight of (b) mc-PS1k, (c) mc-PS6k and (d) mc-PS12k, plotted together with linear PS. | ||
However, a difference was observed in the Mn at which the Tg reached a constant value, approximately 50 000 for linear PS and 200 000 for mc-PS1k.
To further investigate the dependence of the Tg on the molecular weight, we employed the Flory–Fox equation:41Tg = Tg,∞ − AMn−1, where Tg,∞ is Tg in the limit of infinite Mn and A is an empirical parameter related to the free volume. Fig. 5b shows the plots of Tg for linear PS and mc-PS against the inverse of Mn, with linear fitting corresponding to the Fox–Flory equation. By extrapolation of Tg to the infinite molecular weight, Tg,∞ was determined to be 106.7 and 111.6 °C for linear PS and mc-PS, respectively. This confirms the topological effect on Tg. Monteiro et al. have reported that multicyclic PS shows a higher Tg than linear PS because the linkages between each cyclic unit can be considered as irreversible knots, lowering the conformational entropy of the coil.24 In contrast to multicyclic PS, the Tg,∞ of monocyclic PS is almost identical to that of linear PS.42 The distinct difference in Tg between linear and monocyclic polymers in the low molecular weight region is attributed to the presence and absence of a terminal structure. Meanwhile, in the high molecular weight range, the terminal contribution of the linear polymer becomes negligible; thus, the Tg of linear and monocyclic polymers converges to the same value. Therefore, the increased Tg,∞ of mc-PS over those of linear and monocyclic PS is a unique characteristic of the multicyclic topology.
The A values for the linear PS and mc-PS1k were 8.9 × 104 and 7.3 × 104, respectively, indicating a strong Tg–Mn dependence. For linear PS, such a strong Tg–Mn dependence can be explained by the free volume around the terminals. In contrast, the Tg–Mn dependence of mc-PS is explained by other factors: the steric repulsion of the cyclic side chains of mc-PS increases upon increasing the norbornene backbone DP, that is, the number of cyclic units, which reduces the free volume of the cyclic chain. Such a scenario is also different from that of the Tg–Mn dependence of monocyclic polymers, where Tg shows a weak dependence on the molecular weight.43–45
Next, we investigated the Tg–Mn dependence of mc-PSs with larger numbers of cyclic units (Fig. S17†). As expected, mc-PS6k and mc-PS12k also showed an increased Tg compared to that of linear PS with a comparable molecular weight. The Tg,∞ values of mc-PS6k and mc-PS12k were 118.7 and 115.5 °C, respectively. These values were higher than that of linear PS (106.7 °C), which implied the topological effect on the Tg, similar to that seen in mc-PS1k (Fig. 5c and d). Notably, the Tg of mc-PS52k (Mn = 497400) was determined to be 106.3 °C, which was almost identical to that of its linear PS counterpart (Mn = 275000, Tg = 107.0 °C). According to a previous study, linear and monocyclic PS with a molecular weight of 42k showed almost the same Tg.46 In other words, for molecular weights higher than 42k, no terminal effect was observed on Tg. This suggests that when the molecular weight of the cyclic units is greater than a certain value, Tg is not affected, even when the number of cyclic units is increased.
Various mc-PSs with comparable cyclic units and different ring sizes were used for investigating the dependence of Tg on the ring size: mc-PS1k (number of cyclic units = 3.6, ring size = 1k, run 1 in Table 1), mc-PS6k (number of cyclic units = 3.4, ring size = 6k, run 8 in Table 1) and mc-PS12k (number of cyclic units = 4.4, ring size = 12k, run 11 in Table 1). The Tg,∞ and A parameter were determined to be 105.5 °C and 3.0 × 103, respectively, using the Flory–Fox equation (Fig. 6). The Tg,∞ of mc-PS with a number of cyclic units of approximately 3–4 was close to that of linear PS. Such a finding can be explained by the same reason as that for mc-PS52k showing a comparable Tg to that of linear PS. The estimated A parameter was close to the reported value for monocyclic PS (4.5 × 103), which is about an order of magnitude lower than that of linear PS (8.9 × 104).42 Such a difference confirmed the smaller free volume of mc-PS compared to that of its linear counterpart.
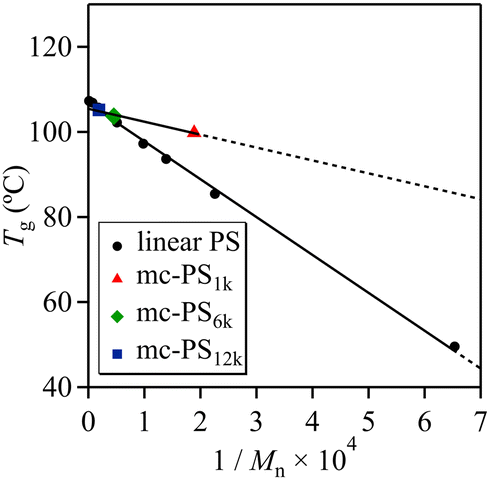 | ||
| Fig. 6 Plot of glass transition temperature vs. inverse of molecular weight for linear PS and mc-PS bearing comparable cyclic units: mc-PS1k (number of cyclic units = 3.6, ring size = 1k, run 1 in Table 1), mc-PS6k (number of cyclic units = 3.4, ring size = 6k, run 8 in Table 1) and mc-PS12k (number of cyclic units = 4.4, ring size = 12k, run 11 in Table 1). | ||
Application to PS-containing multicyclic copolymer synthesis
To demonstrate the versatility of the present cyclopolymerization approach, we synthesized PS-containing multicyclic copolymers with different architectures (Schemes 1b and c). Our aim was to enrich mc-PS with novel properties that cannot be obtained through its utilization alone, thus, expanding its possible applications. In this study, we focus on multicyclic copolymers composed of PS and poly(rac-lactide) (PLA) segments. Because the block copolymer consisting of PS and PLA is known to show a high Flory–Huggins interaction parameter, multicyclic copolymers possessing PS and PLA segments are expected to self-assemble into microphase-separated structures.47–50First, we investigated the cyclopolymerization of α,ω-norbornenyl end-functionalized PLA-block-PS-block-PLA triblock copolymer (NB–PLA-b-PS-b-PLA–NB; Mn,NMR = 15900, Mn,SEC = 24300, Đ = 1.04, see the ESI† for synthetic details) to synthesize multicyclic PLA-block-PS (mc-(PLA-b-PS)) according to Scheme 1b. Under established conditions, the cyclopolymerization of NB–PLA-b-PS-b-PLA–NB ([NB–PLA-b-PS-b-PLA–NB]0 = 0.15 mM, [MM]0/[G3]0 = 3/1; see Table S1†) yielded a soluble product. The SEC elution peak of the product clearly shifted to a higher molecular weight region (Mn,SEC = 55200, Đ = 1.82) compared to that of the MM (Fig. 7), and 1H NMR analysis suggested that the ROMP proceeded quantitatively (Fig. S18†). Although the Tg of the PS block could not be clearly observed, DSC analysis revealed that the Tg of the PLA segment of mc-(PLA-b-PS) was higher than that of the macromonomer (Fig. S19†), strongly suggesting the multicyclic architecture of the obtained mc-(PLA-b-PS). The number of cyclic units was determined from its Mn,MALS as 5.2. These results indicate that the cyclopolymerization strategy can be applied to the synthesis of PS-containing multicyclic copolymers. Furthermore, mc-(PLA-b-PS) copolymers possessing 6.3 and 8.8 cyclic units were successfully obtained by applying the [MM]0/[G3]0 = 6/1 and 9/1, respectively.
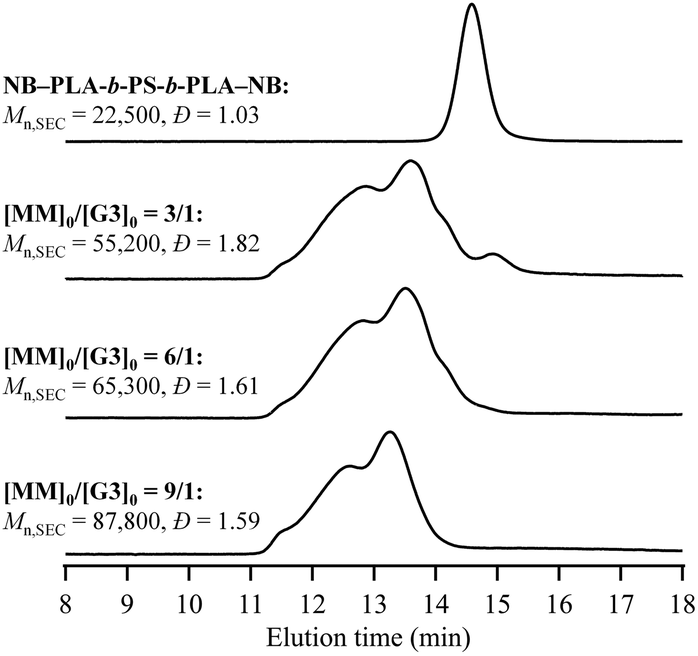 | ||
| Fig. 7 SEC traces of NB–PLA-b-PS-b-PLA–NB and mc-(PLA-b-PS) (refractive index (RI) detection; eluent, THF; flow rate, 1.0 mL min−1). | ||
A statistical copolymer of cyclic PS and cyclic PLA (poly(cPS-st-cPLA)) was synthesized, as shown in Scheme 1c. α,ω-Norbornenyl end-functionalized PLA (NB–PLA–NB) was synthesized following our previously reported method (Fig. S20 and S21†).35 The cyclocopolymerization of NB–PS6k–NB (Mn,NMR = 6000, Mn,SEC = 5800, Đ = 1.09) and NB–PLA6k–NB (Mn,NMR = 6700, Mn,SEC = 12100, Đ = 1.08) was conducted at [NB–PS6k–NB]0/[NB–PLA6k–NB]0/[G3]0 = 1/1/1 to afford a soluble product without gelation (Mn,SEC = 15300, Đ = 1.34, Table S2†). The SEC analysis of the product revealed that both macromonomers were quantitatively consumed (Fig. 8a), and 1H NMR analysis suggested the quantitative consumption of each norbornenyl group (Fig. S22†). Furthermore, the Mn,MALS of the products was determined to be 23800, from which the number of cyclic units was calculated to be 3.8, which is close to the theoretical value. The number of cyclic units and the ring size can also be controlled by changing the [NB–PS–NB]0/[NB–PLA–NB]0/[G3]0 ratio or using macromonomers with different Mn values (Fig. 8a and b). To provide evidence for the multicyclic architecture, we compared the Tg, Rh and [η] of poly(cPS-st-cPLA) (Mn,MALS = 40300, number of cyclic units = 6.5, run 20 in Table S2†) with those of the graft copolymer counterpart composed of linear PS and PLA side chains (poly(PS-st-PLA)) with comparable side-chain and main-chain lengths (Mn,MALS = 33800, number of graft units = 10.7; see synthetic details in Fig. S23–S25†). DSC analysis revealed that the Tg of each segment of poly(cPS-st-cPLA) was higher than that of poly(PS-st-PLA) (Fig. 8c). SEC-MALS-Visco measurements revealed the lower Rh and [η] compared to those of poly(PS-st-PLA) in the same molecular weight range (Fig. 8d and e). These results strongly suggest the multicyclic architecture of the obtained poly(cPS-st-cPLA). Overall, we demonstrated that multicyclic copolymers containing cyclic PS units with various architectures can be synthesized via the established cyclopolymerization approach. The microphase separation behavior of these multicyclic copolymers is currently being investigated by our group.
 | ||
| Fig. 8 SEC traces of (a) poly(cPS6k-st-cPLA6k) and (b) poly(cPS13k-st-cPLA13k), and their corresponding macromonomers (refractive index (RI) detection; eluent, THF; flow rate, 1.0 mL min−1). (c) DSC curves during the second heating process of poly(PS-st-PLA) (black) and poly(cPS-st-cPLA) (run 20 in Table S2,† green). (d) Rh conformation plots and (e) Mark–Hauwink–Sakurada plots for poly(PS-st-PLA) (black) and poly(cPS-st-cPLA) (run 20 in Table S2,† green). | ||
Conclusions
In this study, we synthesized multicyclic polystyrene (mc-PS) and PS-containing multicyclic copolymers via the cyclopolymerization of α,ω-dinorbornenyl end-functionalized macromonomers. Owing to the ease of macromonomer preparation (a one-step procedure using a commercially available reagent) and the high efficiency of the cyclopolymerization reaction, we systematically synthesized a series of mc-PSs, achieving the highest reported number of cyclic units (239) and molecular weight of cyclic units (52100). Compared to previously reported approaches that require multistep synthesis, the developed synthetic method is highly efficient and straightforward. Our synthetic approach allowed the systematic evaluation of the effects of structure on the viscosity, hydrodynamic volume and glass transition temperature, which are understudied. Furthermore, we successfully synthesized multicyclic copolymers composed of PS and poly(rac-lactide) with various architectures via the cyclopolymerization of block copolymer-type macromonomers or the cyclocopolymerization of two macromonomers, further proving the synthetic versatility of our approach. Given the industrial importance of PS and PS-containing copolymers, the insights revealed in this study will contribute to both fundamental studies and practical applications of multicyclic polymers.
Author contributions
Yamato Ebii: investigation, methodology, writing – original draft, visualisation. Yoshinobu Mato: investigation, methodology. Feng Li: investigation, supervision. Kenji Tajima: investigation, supervision. Takuya Yamamoto: investigation, supervision. Takuya Isono: investigation, supervision, writing – review and editing, funding acquisition. Toshifumi Satoh: conceptualization, writing – review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the MEXT Grant-in-Aid for Challenging Exploratory Research (19K22209), the JST CREST (JPMJCR19T4), the Photoexcitonic Project (Hokkaido University), the Creative Research Institute (CRIS, Hokkaido University), the Project of Junior Scientist Promotion at Hokkaido University, Eno Scientific Foundation, and the Tokyo Ohka Foundation for the Promotion of Science and Technology.References
- M. Schappacher and A. Deffieux, Macromolecules, 2001, 34, 5827–5832 CrossRef CAS.
- P. G. Santangelo, C. M. Roland, T. Chang, D. Cho and J. Roovers, Macromolecules, 2001, 34, 9002–9005 CrossRef CAS.
- S. J. Clarson and J. A. Semlyen, Polymer, 1986, 27, 1633–1636 CrossRef CAS.
- G. B. Mckenna, G. Hadziioannou, P. Lutz, G. Hild, C. Strazielle, C. Straupe, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 498–512 CrossRef CAS.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS PubMed.
- M. Gauthier-Jaques and P. Theato, ACS Macro Lett., 2020, 9, 700–705 CrossRef CAS PubMed.
- J. E. Poelma, K. Ono, D. Miyajima, T. Aida, K. Satoh and C. J. Hawker, ACS Nano, 2012, 6, 10845–10854 CrossRef CAS.
- G. Morgese, L. Trachsel, M. Romio, M. Divandari, S. N. Ramakrishna and E. M. Benetti, Angew. Chem., 2016, 128, 15812–15817 CrossRef.
- S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251–10253 CrossRef CAS PubMed.
- Y. Satoh, H. Matsuno, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Macromolecules, 2017, 50, 97–106 CrossRef CAS.
- Y. Mato, K. Honda, K. Tajima, T. Yamamoto, T. Isono and T. Satoh, Chem. Sci., 2019, 10, 440–446 RSC.
- Y. Mato, K. Honda, B. J. Ree, K. Tajima, T. Yamamoto, T. Deguchi, T. Isono and T. Satoh, Commun. Chem., 2020, 3, 97 CrossRef CAS PubMed.
- T. Isono, Y. Satoh, K. Miyachi, Y. Chen, S. I. Sato, K. Tajima, T. Satoh and T. Kakuchi, Macromolecules, 2014, 47, 2853–2863 CrossRef CAS.
- N. Kusuyama, Y. Daito, H. Kubota, Y. Kametani and M. Ouchi, Polym. Chem., 2021, 12, 2532–2541 RSC.
- Y. X. Zhang, Y. Wu, Y. L. Zhao, L. C. Zhang and K. Zhang, Macromolecules, 2021, 54, 6901–6910 CrossRef CAS.
- N. Sugai, H. Heguri, K. Ohta, Q. Meng, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 14790–14802 CrossRef CAS.
- N. Sugai, H. Heguri, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2011, 133, 19694–19697 CrossRef CAS PubMed.
- Y. Tezuka, Polym. J., 2012, 44, 1159–1169 CrossRef CAS.
- H. L. Zhang, W. Xu, C. Liu and C. Y. Hong, Polym. Chem., 2021, 12, 5357–5363 RSC.
- C. Ma, Y. Quan, X. Liao, R. Sun and M. Xie, Polym. Chem., 2023, 14, 2072–2079 RSC.
- D. E. Lonsdale and M. J. Monteiro, Chem. Commun., 2010, 46, 7945–7947 RSC.
- M. D. Hossain, J. C. Reid, D. Lu, Z. Jia, D. J. Searles and M. J. Monteiro, Biomacromolecules, 2018, 19, 616–625 CrossRef CAS PubMed.
- A. Pipertzis, M. D. Hossain, M. J. Monteiro and G. Floudas, Macromolecules, 2018, 51, 1488–1497 CrossRef CAS.
- M. Gavrilov, F. Amir, J. Kulis, M. D. Hossain, Z. Jia and M. J. Monteiro, ACS Macro Lett., 2017, 6, 1036–1041 CrossRef CAS PubMed.
- Z. C. Yan, M. D. Hossain, M. J. Monteiro and D. Vlassopoulos, Polymers, 2018, 10, 1–13 Search PubMed.
- J. Jeong, K. Kim, R. Lee, S. Lee, H. Kim, H. Jung, M. A. Kadir, Y. Jang, H. B. Jeon, K. Matyjaszewski, T. Chang and H. J. Paik, Macromolecules, 2014, 47, 3791–3796 CrossRef CAS.
- A. K. Mohanty, J. Ye, J. Ahn, T. Yun, T. Lee, K. S. Kim, H. B. Jeon, T. Chang and H. J. Paik, Macromolecules, 2018, 51, 5313–5322 CrossRef CAS.
- J. H. Jung, A. Kumar Mohanty, J. Ye, T. Lee, J. Ahn, Y. G. Lim, T. Chang and H. J. Paik, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 4020–4026 CrossRef CAS.
- G. Y. Shi and C. Y. Pan, Macromol. Rapid Commun., 2008, 29, 1672–1678 CrossRef CAS.
- X. Q. Xue, Y. J. Chen, Y. F. Li, K. Liang, W. Y. Huang, H. J. Yang, L. Jiang, Q. M. Jiang, F. L. Chen, T. Jiang, B. Z. Lin, B. B. Jiang and H. T. Pu, Chem. Commun., 2021, 57, 399–402 RSC.
- E. Minatti, R. Borsali, M. Schappacher, A. Deffieux, V. Soldi, T. Narayanan and J. L. Putaux, Macromol. Rapid Commun., 2002, 23, 978–982 CrossRef CAS.
- E. Minatti, P. Viville, R. Borsali, M. Schappacher, A. Deffieux and R. Lazzaroni, Macromolecules, 2003, 36, 4125–4133 CrossRef CAS.
- S. Lecommandoux and R. Borsali, Polym. Int., 2006, 55, 961–969 CrossRef.
- E. Di Cola, C. Lefebvre, A. Deffieux, T. Narayanan and R. Borsali, Soft Matter, 2009, 5, 1081–1090 RSC.
- T. Isono, T. Sasamori, K. Honda, Y. Mato, T. Yamamoto, K. Tajima and T. Satoh, Macromolecules, 2018, 51, 3855–3864 CrossRef CAS.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
- Z. Li, K. Zhang, J. Ma, C. Cheng and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5557–5563 CrossRef CAS PubMed.
- H. P. Hsu, W. Paul, S. Rathgeber and K. Binder, Macromolecules, 2010, 43, 1592–1601 CrossRef CAS.
- Z. Li, N. S. Lee and K. L. Wooley, J. Am. Chem. Soc., 2011, 133, 1228–1231 CrossRef CAS.
- S. L. Pesek, X. Li, B. Hammouda, K. Hong and R. Verduzco, Macromolecules, 2013, 46, 6998–7005 CrossRef CAS.
- T. G. Fox and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef CAS.
- L. Gao, J. Oh, Y. Tu, T. Chang and C. Y. Li, Polymer, 2019, 170, 198–203 CrossRef CAS.
- S. J. Clarson, J. A. Semlyen and K. Dodgson, Polymer, 1991, 32, 2823–2827 CrossRef CAS.
- L. Rique-Lurbet, M. Schappacher and A. Deffieux, Macromolecules, 1994, 27, 6318–6324 CrossRef CAS.
- H. Kammiyada, M. Ouchi and M. Sawamoto, Macromolecules, 2017, 50, 841–848 CrossRef CAS.
- Y. Gan, D. Dong and T. E. Hogen-Esch, Macromolecules, 1995, 28, 383–385 CrossRef CAS.
- A. S. Zalusky, R. Olayo-Valles, J. H. Wolf and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 12761–12773 CrossRef CAS PubMed.
- I. Keen, H. H. Cheng, A. Yu, K. S. Jack, T. R. Younkin, M. J. Leeson, A. K. Whittaker and I. Blakey, Macromolecules, 2014, 47, 276–283 CrossRef CAS.
- X. Li, Y. Liu, L. Wan, Z. Li, H. Suh, J. Ren, L. E. Ocola, W. Hu, S. Ji and P. F. Nealey, ACS Macro Lett., 2016, 5, 396–401 CrossRef CAS PubMed.
- K. Watanabe, S. Katsuhara, H. Mamiya, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Chem. Sci., 2019, 10, 3330–3339 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional data. See DOI: https://doi.org/10.1039/d3py00449j |
| This journal is © The Royal Society of Chemistry 2023 |