
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymers from sugars and isothiocyanates: ring-opening copolymerization of a D-xylose anhydrosugar oxetane†
Ella F.
Clark
 ac,
Gabriele
Kociok-Köhn
b,
Matthew G.
Davidson
ac and
Antoine
Buchard
*ac
ac,
Gabriele
Kociok-Köhn
b,
Matthew G.
Davidson
ac and
Antoine
Buchard
*ac
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
bMaterials and Chemical Characterisation Facility (MC2), University of Bath, UK
cUniversity of Bath Institute for Sustainability, Bath, UK
First published on 22nd May 2023
Abstract
A D-xylose 3,5-anyhydrosugar (D-Ox) has been applied in the ring-opening copolymerisation (ROCOP) with isothiocyanates to form alternating AB-type copolymers with imidothiocarbonate linkages (Mn ≥ 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1). Catalysed by an Al(III)-based aminotrisphenolate complex, ROCOP proceeds with high selectivity with four aromatic isothiocyanates, including di-isothiocyanates used for crosslinking, to form thermally robust polymers (Td,5% >228 °C) with a range of high glass-transition temperatures (76–134 °C). Kinetic studies show a reaction order of two with respect to the binary catalytic system, and a first order with respect to D-Ox. When using an Al(III) porphyrin complex as catalyst, the reactivity can be tailored to form exclusively a non-polymerisable cyclic thionocarbamate byproduct. The topology, thermal and physical properties of the polymer could be altered by adding a difunctional isothiocyanate crosslinker. The synthesis of di and triblock copolymers was possible by using difunctional (macro)initiators and by exploiting the living character of the ROCOP process, which allowed chain-extension by the ring-opening polymerisation (ROP) of lactide. The polymers readily degrade under acidic and basic conditions. Photodegradation without additives is also possible with an 85% decrease in molar mass in one week. The potential of the imidothiocarbonate linkages for metal capture has been investigated, and good affinity for Cu2+ ions is seen.
900 g mol−1). Catalysed by an Al(III)-based aminotrisphenolate complex, ROCOP proceeds with high selectivity with four aromatic isothiocyanates, including di-isothiocyanates used for crosslinking, to form thermally robust polymers (Td,5% >228 °C) with a range of high glass-transition temperatures (76–134 °C). Kinetic studies show a reaction order of two with respect to the binary catalytic system, and a first order with respect to D-Ox. When using an Al(III) porphyrin complex as catalyst, the reactivity can be tailored to form exclusively a non-polymerisable cyclic thionocarbamate byproduct. The topology, thermal and physical properties of the polymer could be altered by adding a difunctional isothiocyanate crosslinker. The synthesis of di and triblock copolymers was possible by using difunctional (macro)initiators and by exploiting the living character of the ROCOP process, which allowed chain-extension by the ring-opening polymerisation (ROP) of lactide. The polymers readily degrade under acidic and basic conditions. Photodegradation without additives is also possible with an 85% decrease in molar mass in one week. The potential of the imidothiocarbonate linkages for metal capture has been investigated, and good affinity for Cu2+ ions is seen.
Introduction
Sugar derived polymers possess huge potential to combat concerns surrounding petroleum derived plastics.1–5 Not only are they bio-derived, but they can also impart useful properties such as high thermal stabilities and increased functionality.6 The addition of sulfur into a polymer chain can also improve the physical and thermal properties of the polymer, such as higher degradation temperatures (Td) and melting temperature (Tm).7,8 Sulfur has also been found to introduce interesting properties to a polymer such as the enhancement of electrical and optical characteristics and high affinity to metals.9,10 The photodegradation of sulfur-containing polymers has also been reported.11–15Ring-opening copolymerisation (ROCOP) can be applied to a diverse array of monomers, incorporating properties from both monomers to selectively produce highly functional polymers.16,17 Incorporation of sulfur into polymers has been recently reported via the ROCOP of isothiocyanates (ITCs) with epoxides, catalysed by Lewis pairs or lithium alkoxide salts, forming exclusively monoimidothiocarbonate linkages.18–21 Similarly, the ROCOP of ITCs with episulfides, catalysed by bis(triphenylphosphine)iminium chloride (PPNCl), has been shown to selectively form poly(diimidothiocarbonates).22,23 Recently, LiOBn was applied to sequence selective ring-opening terpolymerisation in which poly(monoimidothiocarbonate) and poly(diimidothiocarbonate) blocks were synthesised with high selectivity.15,21,24 Although highly active ROCOP of epoxides and ITCs has been achieved with numerous organocatalysts (triethylborane and excess phosphazene base, phosphazenium benzoxide) and organolithium catalysts (t-BuOLi and LiOBn), transition metal catalysis is yet to be reported. Furthermore, ROCOP of oxetanes is usually thermodynamically more challenging than with epoxides due to the comparatively low ring strain. To the best of our knowledge, ROCOP between an oxetane and ITCs has not been reported.
Our group, among others, is interested in D-xylose as a promising sustainable feedstock, which can be derived into 3,5-anhydro-1,2-O-isopropylidene-α-D-xylofuranose, D-Ox, an oxetane monomer containing a xylofuranose core. Both the cationic and anionic homopolymerisation of D-Ox has been reported, with polymers showing excellent thermal properties.25,26
The ROCOP of D-Ox with heteroallenes, namely, CO2 and CS2, as well as cyclic anhydrides has been reported.27–29 These studies found 1,2-cyclohexanediamino-N,N-bis(3,5-di-t-butylsalicyilidene)-chromium(III), CrSalen and PPNCl to be an efficient system to the catalysis the ROCOP of D-Ox, however this system has not been applied to ITCs and has been shown to be a poor catalyst for the ROCOP of isocyanates.30 Recently, Pang and co-workers reported the ROCOP of p-tosyl isocyanate using a Mn(III)–Cl salen complex, which allowed for controlled microstructure variation.31 Herein we have explored the metal catalysed ROCOP of D-Ox and ICTs to combine the useful properties of the sugar-based monomers with that of four aromatic ITCs, including di-isothiocyanates used for crosslinking.
Results and discussion
Preliminary experiments
D-OX was prepared from D-xylose in a three-step procedure according to literature and distilled over CaH2 to give a 77% yield.29 The copolymerization of D-Ox with 3,5-bis(trifluoromethyl)phenyl isothiocyanate (ITC1), was first investigated with CrSalen and PPNCl (Fig. 1A) (Table S1,† entry 1). This catalyst system has been previously found to be effective in the ROCOP of D-Ox with cyclic anhydrides, CO2 and CS2, although, to the best of our knowledge, has yet to be reported for the ROCOP of isocyanates and isothiocyanates with epoxides.29 Polymerizations were initially trialed at [D-Ox]0:[ITC]0:[CrSalen]0:[PPNCl]0 loadings of 200:200:1:1, at 100 °C, and [D-Ox]0 = 1.52 mol L−1 in 1,2-dichlorobenzene (DCB) based on previous literature (Table S1,† entry 1). The appearance of a new broad multiplet at 5.88 ppm in the crude 1H NMR spectrum along with the concomitant decrease of starting material (22% conversion of D-Ox after 48 hours), indicated the formation of a ring opened product, which was later confirmed to be the anomeric protons of the polymeric imidothiocarbonate.29CrSalen was also applied for the first time to the ROCOP of cyclohexene oxide (CHO) and ITC1; the crude 1H NMR spectrum showed full conversion of CHO to the polyimidothiocarbonate in agreement with reported spectra (Table S1,† entry 2).19
 | ||
| Fig. 1 (A) Scheme for the copolymerization of D-Ox and ITC1. (B) Suggested routes towards formation of CX and poly(D-Ox-alt-ITCX). (C) 1H NMR spectra (500 MHz, CDCl3) of isolated poly(D-Ox-alt-ITC1), D-Ox and isolated C1. (D) Crystal structure of C1. | ||
Product characterisation
The crude 1H NMR spectrum showed novel major and minor multiplets in the anomeric region at 5.88 and 6.12 ppm respectively, with diffusion ordered spectroscopy (DOSY) NMR spectroscopy showing diffusion coefficients of 1.21 × 10−10 and 8.01 × 10−10 m2 s−1 respectively, confirming the formation of two main species (Fig. 1C and S13†). The major product was precipitated selectively from cold methanol and size-exclusion-chromatography (SEC; against narrow polystyrene standards) analysis of the isolated product confirmed the polymeric nature of the product (Mn,SEC = 7000 g mol−1. 1H NMR spectroscopy showed an equal ratio of D-Ox and ITC1 incorporated in the polymer chain, suggesting the expected alternating copolymer species, and confirmed by MALDI ToF spectrometry (Fig. S1 and S11† respectively).Selective imidothiocarbonate formation was confirmed by the presence of only one resonance at 158 ppm in the 13C NMR spectrum and a strong band at 1632 cm−1 in the Fourier-transform infrared (FTIR) spectrum (Fig. S2 and S6†).19 The single imine resonance also indicated consistent regioselective opening of D-Ox by the propagating sulfur anion. The alternative thionocarbamate linkage was not observed within the limit of NMR and IR spectroscopy in agreement with the work of Song and co-workers.191H–13C heteronuclear multiple bond correlation (HMBC) NMR spectroscopy showed correlation between the imine resonance and the xylofuranose component in exclusively the 3 and 5 positions indicating selective opening of D-Ox across the oxetane moiety (Fig. S4†).
The side product (C1) was isolated by precipitation from hexane from a solution of side product and monomer mix, with single crystals of C1 isolated from hexane and chloroform in a 5:1 ratio. In contrast to the polymer, a single resonance in the 13C NMR spectrum at 186 ppm confirmed complete formation of the cyclic thionocarbamate (Fig. S17†). This was supported by X-ray diffraction data of single crystals of C1 (Fig. 1D).
Catalyst screening
Aluminium and iron amino-tris(phenolate) (AlTris and FeTris) complexes were applied to the ROCOP of D-Ox with ITC1 due to their reported success in the ROCOP of epoxides and cyclic anhydrides where one of the monomers is sterically encumbered.32–34 Both complexes were found to greatly increase the rate of copolymerisation with D-Ox. A conversion of 84% of D-Ox could be achieved with AlTris in 6 hours with SEC showing molar masses (Mn,SEC) up to 21300 g mol−1 and a moderate dispersity (ĐM) of 1.63 (Table 1, entry 1). The crude 1H NMR spectrum showed that only 2% of converted D-Ox formed C1, the rest being polymer. With FeTris, 85% conversion of D-Ox was achieved after 6 hours, 6% being C1, which likely resulted in lower molar mass polymers (Mn,SEC = 15900 g mol−1, ĐM = 1.62, Table S1,† entry 7).
| Entry | Catalysta | Temp (° C) |
D-Ox:AlTris |
Solvent | [D-Ox]0 (mol L−1) | Time (h) | Conv.b (%) | Polymer select.c % |
M
n,theod |
Mn,SECe (ĐM) |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Reactions carried out at [D-Ox]0 = 1.52 mol L−1 with a 200:200:1:1 ratio of D-Ox:ITC1:AlTris:PPNCl unless otherwise stated.
b Conversion of D-Ox determined by 1H NMR spectroscopy by relative integration of anomeric protons in D-Ox (CDCl3, δ = 6.26 ppm (d, J = 3.7 Hz)) and poly(D-Ox-alt-ITC1) (CDCl3, δ = 5.81 ppm (m)).
c Percentage of converted D-Ox to poly(D-Ox-alt-ITC1).
d Calculated as Mr(Cl) + ((Mr(D-Ox) + Mr(ITC1)) × [D-Ox]0/[PPNCl]0 × % conv./100).
e Calculated by SEC relative to polystyrene standards in THF eluent, ĐM = Mw/Mn.
f Reactions carried out without PPNCl.
|
||||||||||
| 1 | AlTris | 100 | 200:1 |
DCB | 1.52 | 6 | 84 | 98 | 72700 |
21300 (1.63) |
| 2 | AlTris | 100 | 200:1 |
None | 3.03 | 48 | 95 | 92 | 77100 |
20000 (1.64) |
| 3 | AlTris | 80 | 200:1 |
DCB | 1.52 | 24 | 97 | 95 | 81500 |
22300 (1.40) |
| 4 | AlTris | 80 | 200:1 |
Toluene | 1.52 | 24 | >99 | 97 | 86000 |
22000 (1.62) |
| 5 | AlTris | 80 | 200:1 |
Acetonitrile | 1.52 | 24 | 95 | 96 | 80700 |
28000 (1.50) |
| 6 | AlTris | 60 | 200:1 |
Toluene | 1.52 | 6 | 21 | 95 | 17800 |
12700 (1.17) |
| 7 | AlTris | 60 | 200:1 |
Toluene | 1.52 | 24 | 60 | >99 | 53200 |
24300 (1.41) |
| 8 | AlTris | 80 | 200:1 |
Toluene | 1.00 | 6 | 27 | 96 | 24000 |
14000 (1.22) |
| 9 | AlTris | 80 | 200:1 |
Toluene | 1.00 | 24 | 74 | 99 | 64700 |
28200 (1.44) |
| 10 | AlTris | 80 | 200:1 |
Toluene | 1.52 | 24 | <1 | <1 | <1000 | <1000 |
| 11 | None | 80 | 200:1 |
Toluene | 1.52 | 24 | <1 | <1 | <1000 | <1000 |
| 12 | None | 80 | 200:1 |
Toluene | 1.52 | 24 | <1 | <1 | <1000 | <1000 |
| 13 | AlTris | 80 | 300:1 |
Toluene | 1.52 | 48 | 92 | 89 | 109000 |
28700 (1.62) |
| 14 | AlTris | 80 | 400:1 |
Toluene | 1.52 | 48 | 83 | 98 | 143600 |
34900 (1.71) |
| 15 | AlTris | 80 | 500:1 |
Toluene | 1.52 | 48 | 23 | 96 | 48800 |
20500 (1.30) |
When altering the ligand structure from an amino tris(phenolate) ligand to a porphyrin ligand, the catalyst selectivity switched towards formation of C1. Under identical conditions an aluminium porphyrin (AlPorph) complex was tested with PPNCl. After 6 hours, 56% conversion of D-Ox was observed; 7% of the D-Ox had converted to polymer whereas 49% had converted to C1 (Table S1,† entry 9). After 48 hours, 80% of overall species were side products (Table S1,† entry 8). Polymers isolated from methanol showed low molar masses (Mn,SEC = 4000 g mol−1).
Optimisation and control
Building from the high reactivity and selectivity of the AlTris complex, optimisation of the reaction was done to minimise the formation of side products. Polymerisation at 100 °C in the absence of solvent resulted in the solidification of the reaction mixture which limited monomer conversion (95% vs. >99% in solution; Table S2,† entry 3), although Mn,SEC of 20000 g mol−1 could still be achieved (Table 1, entry 2). The temperature was then reduced with the aim of decreasing the production of C1. At 80 °C in DCB, polymer selectivity was 95% (detected by 1H NMR spectroscopy) (Table 1, entry 3). Toluene and acetonitrile were then trialed to minimise the use of halogenated solvents and high polymerisation selectivity was maintained (Table 1, entries 4 and 5). Further reduction in temperature to 60 °C resulted in no detection of C1 by 1H NMR spectroscopy but much reduced conversions (Table 1, entries 6 and 7). Reducing the initial concentration of D-Ox to 1.0 mol L−1 at 80 °C resulted in reduced rates too, but with no significant improvement in selectivity (Table 1, entries 8 and 9). Control experiments showed that without the simultaneous presence of both AlTris and PPNCl no polymerisation was observed (Table 1, entries 10–12).
The D-Ox:AlTris ratios were then decreased to 300:1, 400:1 and 500:1, keeping [D-Ox]0 constant, with the aim of synthesising high molar mass polymers (Table 1, entries 13, 14 and 15 respectively). The rate was significantly affected by lowering the catalyst concentration and full conversion was not achieved after 48 hours at any low loading of catalyst. However, higher molar mass polymers were still obtained at ratios of 300:1 and 400:1 (Mn,SEC = 28700 and 34900 g mol−1 respectively), although at 500:1 the negative effect of impurities was too great to achieve high molar mass polymers.
To demonstrate the living character of the ROCOP process, a sequence addition experiment was performed. Reactions were initially carried out at 80 °C in toluene with 0.25 mmol of D-Ox and ITC1 (D-Ox:ITC1:AlTris:PPNCl loadings of 50:50:1:1) and monitored by 1H NMR spectroscopy and SEC. After 24 hours the conversion of D-Ox was >99% and an additional 1 mmol of D-Ox and ITC1 were added. A conversion of >99% was achieved after 24 hours. An increase in the molar mass was observed upon addition of the second batch of monomers (Mn,SEC before second monomer batch addition = 7600 g mol−1, ĐM = 1.47; Mn,SEC after second monomer batch addition = 13300 g mol−1, ĐM = 1.56, Fig. S14†). 1H DOSY NMR spectroscopy of the polymer showed a single diffusion coefficient (1.11 × 10−11 m2 s−1, Fig. S15†) for all resonances associated with poly(D-Ox-alt-ITC1), consistent with chain extension, albeit to a lesser degree than expected (likely from the introduction of additional chain transfer agent impurities (vide infra)).
Plots of conversion against Mn(SEC) and ĐM showed a linear increase in Mn(SEC) against D-Ox conversion (Fig. S12†). At intermediate conversions, SEC analysis showed bimodal distributions leading to higher dispersities, probably from concurrent initiation by chloride anions and by diprotic impurities in D-Ox, as typically observed in ROCOP. MALDI-ToF mass spectrometry corroborated this as three polymer series were detected (Fig. S11†) with repeat units of 443 g mol−1. The presence of residual protic impurities, acting as chain transfer agents, also meant that despite the living nature of the ROCOP, Mn,SEC values were consistently lower than theoretical ones (Mn,theo) (e.g., for Table S1†, entry 3 Mn,theo = 55000 g mol−1vs. Mn,SEC = 21000 g mol−1).
Optimisation towards formation of C1 and reactivity
Using the aluminium porphyrin complex AlPorph, the reaction selectivity could be altered to form 100% of C1, with no sign of polymer or monomer in the 1H crude NMR spectrum. This was achieved at 120 °C, at D-Ox:ITC1:AlPorph:PPNCl loadings of 100:100:1:1 in DCB. A suggested mechanism is shown in Fig. 1B. Compared to AlTris, the porphyrin ligand clearly influences the relative energy barrier between the propagation and back-biting steps. Selectivity towards thionocarbamate formation over the imidothiocarbonate during backbiting was attributed to greater stability of the nitrogen anion, which may also be enhanced by the porphyrin ligand system.
ROP of C1 was attempted with KOtBu and 18-crown-6-ether at 120 °C (D-Ox:ITC1:KOtBu:18-crown-6-ether loadings of 100:00:1:1) and with AlTris and PPNCl at 80 °C (D-Ox![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :ITC1:AlTris:PPNCl loadings of 100:100:1:1). After 24 hours under both conditions, no polymeric species was detected by SEC and the crude 1H NMR spectra showed no change to C1. The lack of reactivity of C1 under identical conditions to that of D-Ox/ITC1 ROCOP indicates the formation of C1 is irreversible and therefore not likely to be an intermediate in the polymerisation. Without closer reaction monitoring, it is however unclear at this stage if any propagation happens before back-biting or if back-biting occurs straight after initiation.
:ITC1:AlTris:PPNCl loadings of 100:100:1:1). After 24 hours under both conditions, no polymeric species was detected by SEC and the crude 1H NMR spectra showed no change to C1. The lack of reactivity of C1 under identical conditions to that of D-Ox/ITC1 ROCOP indicates the formation of C1 is irreversible and therefore not likely to be an intermediate in the polymerisation. Without closer reaction monitoring, it is however unclear at this stage if any propagation happens before back-biting or if back-biting occurs straight after initiation.
Kinetic studies
To gain a deeper understanding of reaction kinetics, [D-Ox] vs. time data were acquired using in situ FTIR spectroscopy by analysing the decrease in the absorption intensity at 1087 cm−1 corresponding to a νC–O vibration in D-Ox. The catalyst and cocatalyst were used in a 1:1 ratio and varied simultaneously. The reaction rate was evaluated for [AlTris–PPNCl] = 15 and 20 mmol L−1, using a 1.12 mol L−1 concentration of D-Ox in toluene at 80 °C, with triethylsilane as an internal standard for offline NMR analysis.
Visual kinetic analysis developed by Burés and co-workers revealed a second order dependence with respect to [AlTris–PPNCl] (Fig. 2A).35 It may be that the catalyst and co-catalyst both have an order of one and work in tandem: AlTris activating the monomer and PPNCl stabilising the anion. Alternatively, non-covalent dimeric aluminium species may be formed in situ as reported in the literature.36 A more in-depth kinetic analysis would be required to delineate the role of each component of this binary catalytic system.
 | ||
| Fig. 2 Kinetic data acquired by in situ FTIR spectroscopy, varying catalyst loading, [D-Ox] = 1.12 mol L−1 at 80 °C in toluene. (A) Visual kinetic analysis plot of normalised time against [D-Ox] at [cat] = [AlTris–PPNCl] = 20 and 15 mmol L−1. (B) Logarithmic plot of [D-Ox] vs. time from 0–75% conversion of D-Ox, [AlTris–PPNCl] = 20 mmol L−1. | ||
Semi-logarithmic plots of ln([D-Ox]) against time from 0–75% conversion showed linear fits indicative of a first-order dependence on D-Ox concentration. At a catalyst loading of 20 mmol L−1kobs was found to be 3.18 (±0.01) × 10−4 s−1 (Fig. 2B) These results suggests that the rate determining step is the ring-opening of the oxetane. The formation of C1 was not detectable by in situ FTIR spectroscopy or in the final crude 1H NMR spectrum, and a direct correlation between polymer production and D-Ox consumption was observed with no induction period.
Expanding the monomer scope
The procedure was then extended to a range of aliphatic and aromatic isocyanates with AlTris. ROCOP with aliphatic isocyanate, isopropyl isothiocyanate (ITC2) yielded no detectable polymeric species after 24 hours and only oligomeric species after 72 hours (Table 2, entries 1 and 2). 2-Chloroethyl isothiocyanate (ITC3), produced entirely the cyclic back-biting product, C3.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Comonomer (ITC ratio)a | Time (h) | Conv.b (%) | Polymer select.c (%) |
M
n,theod |
M
n,SECe |
Đ
Me |
T
gf (°C) |
T d,5% (°C) |
| Reactions carried out at [D-Ox]0 = 1.52 mol L−1 in toluene with a 200:200:1:1 ratio of D-Ox:ITC:AlTris:PPNCl at 80 °C unless otherwise stated, reaction quenched when stirring stopped.a Ratio of moles of specified ITC.b Conversion of D-Ox determined by 1H NMR spectroscopy by relative integration of anomeric protons in D-Ox (CDCl3, δ = 6.26 ppm (d, J = 3.7 Hz)) and poly(D-Ox-alt-ITCX) (CDCl3, δ = 6.03–5.81 ppm (m)).c Percentage of converted D-Ox to poly(D-Ox-alt-ITCX).d (Calculated as Mr(Cl) + ((Mr(D-Ox) + Mr(ITCX)) × [D-Ox]0/[PPNCl]0 × % conv./100).e Calculated by SEC relative to polystyrene standards in THF eluent, ĐM = Mw/Mn.f Values taken from second heating cycle. |
|||||||||
| 1 | ITC2 | 24 | 2 | >99 | 1100 | <1000 | — | — | — |
| 2 | ITC2 | 72 | 17 | 94 | 8800 | 1700 | 1.06 | — | — |
| 3 | ITC3 | 24 | 62 | 0 | <1000 | <1000 | — | — | — |
| 4 | ITC3 | 72 | 81 | 0 | <1000 | <1000 | — | — | — |
| 5 | ITC4 | 24 | 41 | 95 | 24000 |
6600 | 1.50 | 111 | 264 |
| 6 | ITC4 | 72 | 84 | 62 | 38100 |
9100 | 1.59 | 109 | 248 |
| 7 | ITC5 | 24 | 40 | 74 | 21900 |
4600 | 1.15 | 75 | 228 |
| 8 | ITC5 | 72 | 61 | 62 | 24400 |
4400 | 1.12 | 80 | 237 |
| 9 |
ITC6:ITC4 (2.5:97.5) |
72 | 50 | 90 | 30800 |
9200 | 2.10 | 105 | 274 |
| 10 |
ITC6:ITC4 (5:95) |
72 | 29 | 93 | 16600 |
29100 |
2.41 | 108 | 273 |
| 11 |
ITC6:ITC4 (7.5:92.5) |
72 | 61 | 84 | 31400 |
11300 |
3.91 | 116 | 261 |
| 12 |
ITC6:ITC4 (10:90) |
72 | 72 | 85 | 37600 |
— | — | 132 | 267 |
| 13 |
ITC6:ITC4 (20:80) |
72 | — | — | — | — | 134 | 175 | |
| 14 |
ITC6:ITC4 (50:50) |
72 | — | — | — | — | 129 | 208 | |
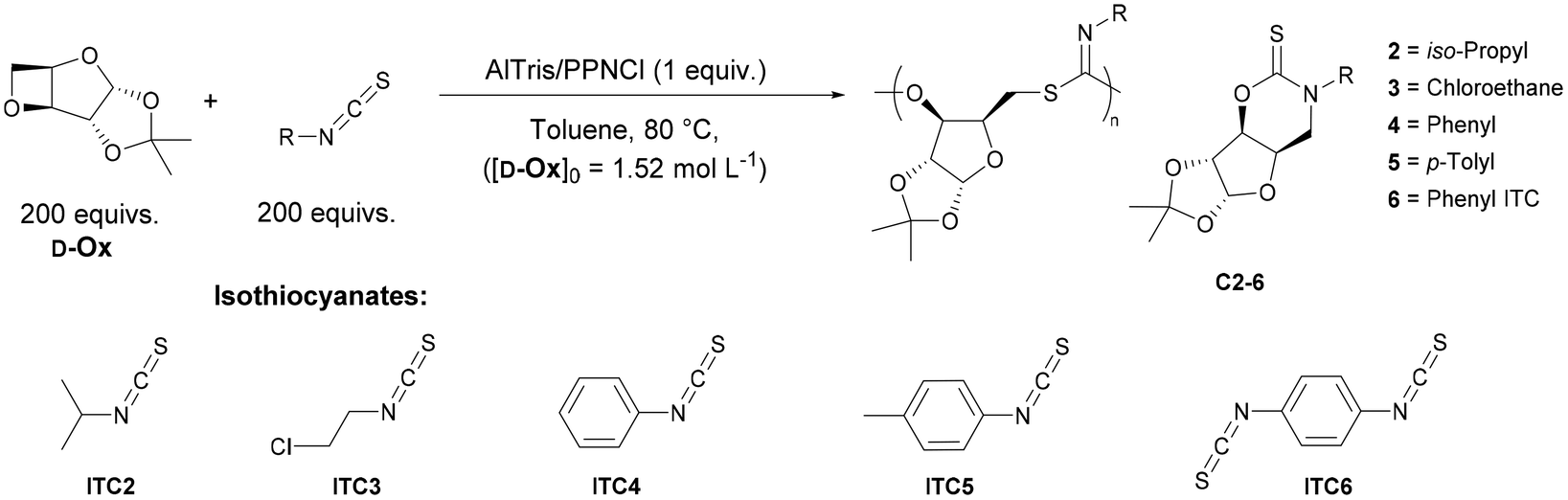
For all aromatic substrates tested, alternating AB copolymers were primarily synthesised. ROCOP with phenyl isothiocyanate (ITC4) and p-tolyl isothiocyanate (ITC5) proceeded with considerably lower conversions compared with ITC1 (conversion after 24 hours was 41 and 44% compared to >99% respectively), indicating the electron withdrawing groups significantly enhance the reactivity of the isothiocyanate towards ROCOP with D-Ox (Table 2, entries 5–8).
Poly(D-Ox-alt-ITC4) and poly(D-Ox-alt-ITC5) could be isolated with Mn,SEC up to 9100 and 4600 g mol−1 respectively, with any conversion of D-Ox after 24 hours mostly being converted to C4/5. This switch in catalyst regime to favour polymerisation with aromatic substrates versus aliphatic suggests the aromatic substituent has a part in directing catalysis towards ROCOP. We propose that aliphatic substituents increase the nucleophilicity of the nitrogen atom, favouring thionocarbamate formation which results in cyclisation (see suggested mechanism, Fig. 1B).
Phenyl diisothiocyanate (ITC6) was added to ROCOP of D-Ox with ITC4 (2.5–50% ITC6 in ITC4) to form crosslinked polymers. SEC analysis of the isolated polymers showed broad traces typical of crosslinked polymers. Due to signal overlapping in the aromatic region, 1H NMR analysis could not be used to quantify the incorporation of cross-linker. However, as the ITC6 loading was increased, new signals in the 13C NMR in the aromatic region at 126.6 and 122.5 ppm were observed confirming incorporation of ITC6 into the polymer chain (Fig. S28†). Increasing the crosslinking (ITC6 >10%), resulted in solidification of the reaction mixture, presumably limiting conversion although NMR and SEC analysis was not possible due to insolubility.
Thermal analysis
Thermal analysis of poly(D-Ox-alt-ITC1) with a range of molar masses between 11300 and 26200 g mol−1 was undertaken to understand the impact of molar mass on the properties.25 All polymers analysed were found to have glass transition temperatures (Tg) between 106 to 117 °C, with increased molar mass resulting in increased Tg (Fig. S10†). Temperatures of onset of degradation (Td,5%) were found to be between 228 to 269 °C. These are higher than polyimidothiocarbonates synthesised from epoxide ROCOP with ITCs reported by Feng and co-workers (max Td 220 °C).20 This is likely due to the furanose ring, which has been previously found to improve the thermal stability of polymers.29 Mass spectrometry detected species during the primary degradation step with m/z = 44 and 55, which were attributed to S–C and C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O ions, indicating that the thermal break down of the imidothiocarbonate linkages occurs first (Fig. S9†). A small, secondary degradation step at higher temperatures was observed for all TGA with mass spectrometry of the degradation products showing species with m/z = 81 and 96. This is likely a secondary degradation of the furanose core.
C–O ions, indicating that the thermal break down of the imidothiocarbonate linkages occurs first (Fig. S9†). A small, secondary degradation step at higher temperatures was observed for all TGA with mass spectrometry of the degradation products showing species with m/z = 81 and 96. This is likely a secondary degradation of the furanose core.
Poly(D-Ox-alt-ITC4) showed similar thermal properties; however poly(D-Ox-alt-ITC5) possessed a markedly lower Tg, likely due to a combination of low molar mass and reduced polarity. As expected, thermal analysis of the crosslinked polymers showed high Tgs, increasing with crosslinking density, as well as multiple thermal degradation steps indicative of varied chain linkages, with higher crosslinking densities resulting in greater residual char (Fig. S29†).25
Hydrolytic and UV degradation
An attractive feature of sugar derived polymers is the high content of polarised bonds resulting in multiple sites for the degradation of the polymer backbone. It was envisioned that the imidothiocarbonate linkages would be susceptible to hydrolysis under both acidic and basic conditions. Poly(D-Ox-alt-ITC1) was thus stirred in a 1 mol L−1 HCl(aq) THF mixture at 50 °C and a rapid decrease in polymer molar mass was observed (by 85% within 5 hours, Fig. 3). Identification of the degradation products by 1H and 13C NMR spectroscopy proved challenging (Fig. S30 and S31†). Further analysis of the degradation products after 3 hours by 19F NMR spectroscopy was also inconclusive, albeit a new signal appearing downfield to that of the polymer (−62.8 vs. −63.1 ppm, respectively) (Fig. S32†).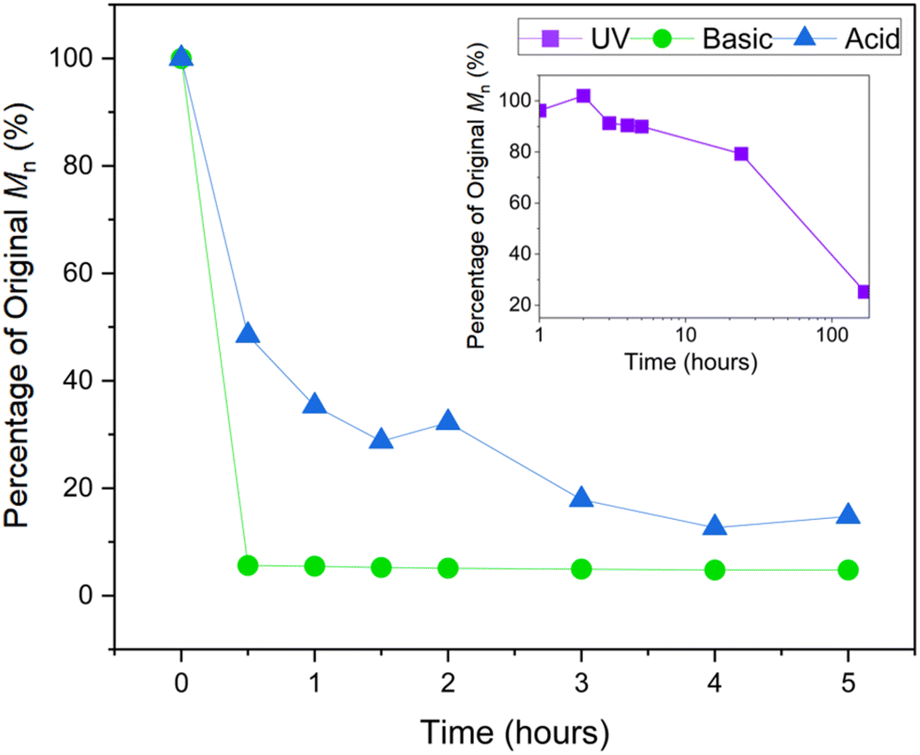 | ||
| Fig. 3 Degradation plots of poly(D-Ox-alt-ITC1) (Mn,SEC = 21800 g mol−1, ĐM = 1.33). Photodegradation conducted in THF exposed to 365 nm radiation. Basic and acidic degradation done in THF with aqueous NaOH and HCl respectively (1 mol L−1) at 50 °C. | ||
The polymer was also stirred in THF and NaOH(aq) at a 1 mol L−1 concentration at 50 °C: after 30 min no polymeric species was detected by SEC analysis (Fig. 3). Analysis of the degradation products by mass spectroscopy detected a major species of a molar mass of 444.07 g mol−1, the mass of the repeat unit and an additional proton, suggesting formation of a cyclic product (Scheme 1 and Fig. S38†). In the 13C NMR spectrum (Fig. S35†), the presence of an imine resonance at 159 ppm indicated the formation of a cyclic imidothiocarbonate, with the HMBC spectrum confirming coupling between the imine group and the xylofuranose moiety (Fig. S37†). A suggested mechanism for the basic degradation and back-biting towards the cyclic imidothiocarbonate product is shown on Scheme 1. Notably such back-biting and that seen during the ROCOP process proceeds from different chain-ends (alkoxide and carbamate, respectively), resulting in two different structural isomers.
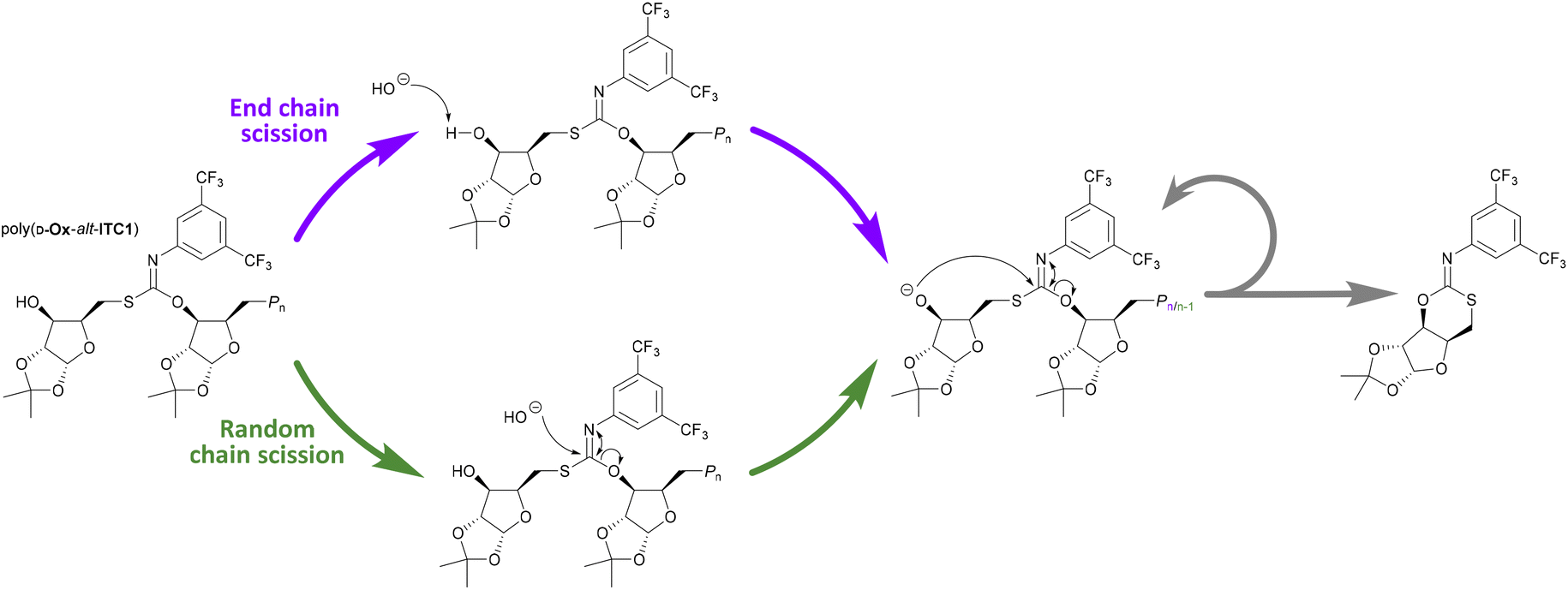 | ||
| Scheme 1 Two proposed pathways for basic degradation of poly(D-Ox-alt-ITC1). | ||
Our group, among others, has also recently reported photodegradation of sulfur containing polymers.12 Upon UV radiation at 365 nm for 5 hours without any additives in THF, the Mn of poly(D-Ox-alt-ITC1) decreased by 10%.12 After 24 hours and 1 week, the Mn had decreased by 20% and 85% respectively (Fig. 3). When stirred in the dark, the Mn of poly(D-Ox-alt-ITC1) did not decrease. Poly(D-Ox-alt-ITC1) also showed good thermal stability when stirred at 140 °C in tetrachloroethane, with no cyclic species detected by 1H NMR spectroscopy.
Polymer architecture variations
To synthesise more complex polymer architectures, initiating systems other than chloride anions (from PPNCl) were investigated under the identified standard ROCOP conditions (80 °C, [D-Ox]0 = 1.52 mol L−1 in toluene). Initial experiments with 1,8-diazobicyloco[5.4.0]undec-7-ene (DBU) and AlTris were undertaken. It was hypothesised that in the presence of a base, residual protic species in the monomer (specifically 1,2-O-isopropylidene-xylofuranose or IPXF) would form alkoxide species able in turn to initiate D-Ox ROCOP. At a AlTris:DBU:D-Ox:ITC1 ratio of 1:1:200:200, low dispersity poly(D-Ox-alt-ITC1) with moderate Mn was isolated (Mn = 6600 g mol−1ĐM = 1.13). Having established that initiation from protic impurities in the monomer was possible, an exogeneous alcohol initiator was next purposedly added.
Experiments with 1,4-butanediol (BD) as the initiator were undertaken at a BD:AlTris:DBU:D-Ox:ITC1 ratio of 1:2:2:40:40 (Fig. 4Ai). The crude 1H NMR spectrum showed 70% selectivity towards the polymer (Table S2,† entry 1). 1H NMR analysis of the isolated polymer revealed two new signals at 4.14 and 1.95 ppm which were attributed to BD incorporations into the polymer (Mn,SEC = 8400 g mol−1, ĐM = 1.20) (Fig. S40†). However, DOSY NMR spectroscopy showed two main diffusion coefficients for signals assigned to the polymer, likely due to initiation from both monomer protic impurities and BD (Fig. S41†). The initiating groups could not be assigned with MALDI ToF mass spectrometry although the repeating unit of 443 was observed (Fig. S42†).
 | ||
| Fig. 4 (A) (i) Schematic of D-Ox and ITC1 ROCOP initiated by BD in the presence of DBU, (ii) schematic of D-Ox and ITC1 ROCOP initiated by PEG under basic conditions, (iii) schematic of D-Ox and ITC1 ROCOP initiated by BD under basic conditions followed by chain extension by ROP of L-lactide, (iv) schematic of the synthesis of IMOT from PLA, D-Ox and ITC1. (B) (i) SEC traces of commercial PEG and an aliquot taken after ROCOP with D-Ox and ITC1 (in chloroform), (ii) SEC traces of an aliquots taken after ROCOP with D-Ox and ITC1 and chain extension by ROP of L-lactide. (C) The polymer structure of poly(LLA-b-(D-Ox-alt-ITC1)-b-LLA). | ||
The ratio of D-Ox:BD was then increased with the aim of making higher Mn polymers (Table S2,† entries 2–5). The molar mass increased with decreasing initiator loading up to 11000 g mol−1 at a BD:AlTris:DBU:D-Ox:ITC1 ratio of 1:2:2:200:200. Further decrease in initiator loading had no effect on molar masses, which was attributed to residual protic impurities outcompeting the BD as the predominating initiators.
To demonstrate the potential of the alcohol initiated ROCOP of D-Ox, initiation from a diol capped polymer was then attempted. Polyethylene glycol (PEG) was chosen as the bifunctional macroinitiator due to its simple structure and low Tg, a desirable property in the synthesis of ABA triblock copolymers for thermoplastic elastomers (Fig. 4Aii). ROCOP was carried out with commercial PEG with a molar mass of 6000 g mol−1 at a PEG:AlTris:DBU:D-Ox:ITC1 ratio of 1:2:2:100:100. Crude 1H NMR analysis showed a 91% conversion of D-Ox, with 85% selectivity towards poly(D-Ox-alt-ITC1). As anticipated, 1H NMR analysis of the isolated polymer showed it contained PEG, with a single resonance at 3.64 ppm (Fig. S43†). The crude SEC trace was bimodal which was attributed to the presence of two initiating species: the low molar mass species initiated from protic small molecule impurities (water or IPXF) and high molar mass species initiated by chain extension from PEG (Mn,SEC of PEG = 11000 g mol−1, ĐM = 1.14; Mn,SEC after chain extension = 19400 g mol−1, ĐM = 1.08) (Fig. 4Bi). This was confirmed with DOSY NMR which showed one species diffusing at 6.36 × 10−10 m2 s−1 (Fig. S44†). Chain extension of PEG with poly(D-Ox-alt-ITC1), lowered the Tm from 63 °C to 47 °C, determined by DSC, and only one Tg at −29 °C was observed, indicating no microphase separation between PEG and poly(D-Ox-alt-ITC1), likely due to short copolymer blocks (Fig. S45†).
Next, the synthesis of ABA-type triblock copolymers with poly L-lactic acid (PLLA) blocks was investigated. First, sequential addition of L-lactide to poly(D-Ox-alt-ITC1) was attempted (Fig. 4Aiiii). The poly(D-Ox-alt-ITC1) block was synthesised under standard conditions with BD and DBU as the initiating system (BD:AlTris:DBU:D-Ox:ITC1 loadings of 1:2:2:50:50) and monitored by 1H NMR spectroscopy and SEC. After 18 hours the conversion of D-Ox was >99% and 200 equivalents of L-lactide was then added. The reaction was quenched after 24 hours and crude 1H NMR spectroscopy showed 94% conversion of L-lactide to PLLA. DOSY NMR of the isolated polymer showed a single diffusion coefficient at 7.76 × 10−11 m2 s−1 (Fig. S48†) and confirmed formation of a true PLLA-poly(D-Ox-alt-ITC1) copolymer. An increase in the molar mass was also observed (Mn,SEC 4600 g mol−1 (ĐM = 1.21) before L-lactide addition; Mn,SEC 7500 g mol−1 (ĐM = 1.32) after LA ROP; Fig. 4Bii). 1H NMR analysis of the isolated block copolymer showed some broadening of the PLLA quartet at 5.16 ppm and the anomeric proton resonance at 5.88 ppm, which was attributed to chain scrambling occurring (Fig. S46†).
This was confirmed by the appearance of new signals between 175.21–169.36 ppm in the 13C NMR spectrum, confirming the formation of new carbonyl linkages (Fig. S47†). DSC analysis showed a single Tg (41 °C), lower than both PLA and poly(D-Ox-alt-ITC1) (Fig. S49†). This has been attributed to chain scrambling resulting in small blocks which inhibit polymer chain stacking.
Surprisingly, the synthesis of poly((D-Ox-alt-ITC1)-b-LLA-b-(D-Ox-alt-ITC1)) by addition of D-Ox and ITC1 to a telechelic PLLA macroinitiator, resulted in complete degradation of PLLA and no new polymeric species detected by SEC (Fig. 4Aiv). ((3,5-Bis(trifluoromethyl)phenyl)imino)-5-methyl-1,3-oxathiolan-4-one was isolated via sublimation and the structure was confirmed by NMR spectroscopy. 13C NMR analysis showed one carbonyl and imine peak at 172 and 154 ppm respectively and the HMBC spectrum showed correlation between the imine resonance and the methine at 5.07 ppm (Fig. 4Aiii and Figs. S51, S54†). It is worth noting that this oxathiolane does not incorporate any xylose moiety. A suggested mechanism, relying on ICT insertion, transesterification and back-biting steps, is shown in the ESI (Scheme S1†).
Metal coordination
The high functional group content of poly(D-Ox-alt-ITC1) prompted us to preliminarily assess its performance as metal ion absorbent. It was hypothesised that the imidothiocarbonate linkage could act as a coordinating group. Indeed, previous studies have found S and N containing polymers to be effective at removal of Hg2+ from aqueous solutions, with adsorption of Hg2+ often attributed to simultaneous binding to S and N.37–39 Similarly, polymeric Schiff bases have been found to form metal complexes, in particular with Cu2+.40–46Two metal ions were considered, Cu2+ and Co3+, which had been previously identified as toxic metals found in wastewater and which would allow colorimetric titration to monitor complexation.47 Initial tests with 0.1 mol L−1 [Co(NH3)6]Cl3 and Cu(OAc)2·H2O in water with poly(D-Ox-alt-ITC1) at 80 °C were carried out. Poly(D-Ox-alt-ITC1) remained insoluble in water at 80 °C with stirring. After 24 hours, each polymer was washed with water and centrifuged, until the supernatant was colourless, and dried in vacuo at 50 °C.
Polymers exposed to Cu2+ and Co3+ (polymer-Cu and polymer-Co respectively) were not soluble in common NMR solvents. After exposure to metal ions, polymer-Cu showed greater absorbance by FTIR spectroscopy at 1627 cm−1 and 1431 cm−1, corresponding to CN and S–C stretches respectively, and was therefore investigated further (Fig. S55†).41In situ IR spectroscopy (poly(D-Ox-alt-ITC1) stirred in a THF solution of Cu(OAc)2·H2O at 50 °C) showed these vibrations increased at the same rate and plateaued after 135 minutes suggesting Cu2+ interacts through the thioimidate moiety of the imidothiocarbonate linkage (Fig. S57†).
Further evidence of bidentate coordination was observed by Raman spectroscopy analysis of polymer-Cu. An increase in intensity and broadening of the vibrational modes at 1620 cm−1 and 1380 cm−1, corresponding to CN and S–C vibrations respectively was observed, indicative of multiple coordination modes (Fig. 5A). Moreover, vibrational modes at 416 and 749 cm−1 were observed in both the Raman spectra of the polymer-Cu and Cu(OAc)2·H2O, confirming Cu2+ adsorption to poly(D-Ox-alt-ITC1) and retention after washing.
 | ||
| Fig. 5 Metal coordination to polymers. (A) Raman spectra of Cu(OAc)2·H2O, polymer-Cu and poly(D-Ox-alt-ITC1). (B) UV-vis spectra of 0.33 and 0.066 mol L−1 aqueous Cu(OAc)2·H2O solutions before and after addition of polymer. | ||
Poly(D-Ox-alt-ITC1) was then stirred in the presence of different concentrations of Cu(OAc)2·H2O (0.33 and 0.066 mol L−1) and the UV-vis spectrum of the original solution was compared to that of the supernatant. A decrease in absorbance at 740 nm was observed for each of the supernatants (28% at 0.33 mol L−1 and 67% at 0.066 mol L−1) indicating a decrease in the concentration of the Cu(OAc)2·H2O in solution (Fig. 5B).
Thermogravimetric analysis was done to compare the percentage of residual char at 600 °C of the polymer-Cu and poly(D-Ox-alt-ITC1) (Fig. S56†). Decomposition of polymer-Cu resulted in a residual mass 5% greater than that of the poly(D-Ox-alt-ITC1), indicative of residual thermally stable Cu, likely copper oxides. As reported in literature, the thermal stability of poly(D-Ox-alt-ITC1) decreased upon coordination to Cu2+, with comparison of the thermal degradation profile indicating multiple degradation pathways in polymer-Cu.42 It is likely that Cu2+ catalyses the degradation of poly(D-Ox-alt-ITC1) at lower temperatures, however high thermal degradation temperatures are still maintained for polymer-Cu (Td,5% = 234 and 278 °C for polymer-Cu and poly(D-Ox-alt-ITC1 respectively).
Conclusions
The ROCOP of aromatic ITCs with an anhydro-functionalised xylofuranose derivative has been reported. Selectivity towards polymerisation and formation imidothiocarbonate linkages over formation of a cyclic thionocarbamate could be controlled through variation of the catalyst and reaction parameters. Kinetic studies showed a first order dependence of the reaction rate in oxetane monomer concentration, and a second order dependence on the concentration of binary catalyst/cocatalyst system (AlTris and PPNCl). All polyimidothiocarbonates showed high thermal stability (Td,5% >228 °C) and high Tg (76–132 °C). Crosslinking with difunctional ITCs lead to increased Tgs. Chemical and photo degradation has also been demonstrated under acidic, basic and UV light conditions. Block copolymers could be synthesised through variation of the initiator as well as through exploitation of the living nature of ROCOP. Finally, initial studies showed the polymer could be used as a metal ion adsorbent for Cu2+. Performances remain modest but call for further investigation into bio-derived and degradable polymers with high functional group content for metal coordination.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Royal Society (UF/160021 and URF\R\221027: fellowship to A. B., RGF\R1\180036 studentship to E. F. C.) for research funding.References
- G. L. Gregory, E. M. López-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- J. A. Galbis, M. D. G. García-Martín, M. V. De Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- R. Xiao and M. W. Grinstaff, Prog. Polym. Sci., 2017, 74, 78–116 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- J. Kawada, T. Lütke-Eversloh, A. Steinbüchel and R. H. Marchessault, Biomacromolecules, 2003, 4, 1698–1702 CrossRef CAS PubMed.
- V. B. Purohit, M. Pięta, J. Pietrasik and C. M. Plummer, Polym. Chem., 2022, 13, 4858–4878 RSC.
- E. Marianucci, C. Berti, F. Pilati, P. Manaresi, M. Guaita and O. Chiantore, Polymer, 1994, 35, 1564–1566 CrossRef CAS.
- R. K. Sadhir and K. F. Schoch, Chem. Mater., 1996, 8, 1281–1286 CrossRef CAS.
- B. A. Fultz, D. Beery, B. M. Coia, K. Hanson and J. G. Kennemur, Polym. Chem., 2020, 11, 5962–5968 RSC.
- C. Hardy, G. Kociok-Köhn and A. Buchard, Chem. Commun., 2022, 58, 5463–5466 RSC.
- Z. Florjańczyk, D. Raducha and A. Kozera-Szałkowska, Macromolecules, 1996, 29, 826–834 CrossRef.
- C. Hardy, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2023, 14, 623–632 RSC.
- S. Rupf, P. Pröhm and A. J. Plajer, Chem. Sci., 2022, 13, 6355–6365 RSC.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- T. Lai, P. Zhang, J. Zhao and G. Zhang, Macromolecules, 2021, 54, 11113–11125 CrossRef CAS.
- L. Song, M. Liu, D. You, W. Wei and H. Xiong, Macromolecules, 2021, 54, 10529–10536 CrossRef CAS.
- C. Chen, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 9474–9481 CrossRef CAS.
- D. Silbernagl, H. Sturm and A. J. Plajer, Polym. Chem., 2022, 13, 3981–3985 RSC.
- X.-F. Zhu, R. Xie, G.-W. Yang, X.-Y. Lu and G.-P. Wu, ACS Macro Lett., 2021, 10, 135–140 CrossRef CAS PubMed.
- X. F. Zhu, G. W. Yang, R. Xie and G. P. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115189 CAS.
- A. J. Plajer, ChemCatChem, 2022, 14, e202200867 CrossRef CAS.
- T. M. McGuire, J. Bowles, E. Deane, E. H. E. Farrar, M. N. Grayson and A. Buchard, Angew. Chem., Int. Ed., 2021, 60, 4524–4528 CrossRef CAS PubMed.
- T. Uryu, Y. Koyama and K. Matsuzaki, Makromol. Chem., 1984, 185, 2099–2107 CrossRef CAS.
- D. K. Tran, A. Z. Rashad, D. J. Darensbourg and K. L. Wooley, Polym. Chem., 2021, 12, 5271–5278 RSC.
- T. M. McGuire and A. Buchard, Polym. Chem., 2021, 12, 4253–4261 RSC.
- T. M. McGuire, E. F. Clark and A. Buchard, Macromolecules, 2021, 54, 5094–5105 CrossRef CAS.
- M. Jurrat, B. J. Pointer-Gleadhill, L. T. Ball, A. Chapman and L. Adriaenssens, J. Am. Chem. Soc., 2020, 142, 8136–8141 CrossRef CAS PubMed.
- C. Hu, X. Chen, M. Niu, Q. Zhang, R. Duan and X. Pang, Macromolecules, 2022, 55, 652–657 CrossRef CAS.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- L. Peña Carrodeguas, C. Martín and A. W. Kleij, Macromolecules, 2017, 50, 5337–5345 CrossRef.
- F. D. Monica and A. W. Kleij, ACS Sustainable Chem. Eng., 2021, 9, 2619–2625 CrossRef CAS.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- J. González-Fabra, F. Castro-Gómez, A. W. Kleij and C. Bo, ChemSusChem, 2017, 10, 1233–1240 CrossRef PubMed.
- F. Gorginpour, S. Moradinia, M. Daneshi and H. Zali-Boeini, Ind. Eng. Chem. Res., 2022, 61, 3694–3703 CrossRef CAS.
- S. Ravi, P. Puthiaraj, K. H. Row, D.-W. Park and W.-S. Ahn, Ind. Eng. Chem. Res., 2017, 56, 10174–10182 CrossRef CAS.
- S. Ravi and W.-S. Ahn, Microporous Mesoporous Mater., 2018, 271, 59–67 CrossRef CAS.
- L. Wang, J. Zhang, J. Sun, L. Zhu, H. Zhang, F. Liu, D. Zheng, X. Meng, X. Shi and F.-S. Xiao, ChemCatChem, 2013, 5, 1606–1613 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, K. Huang, Y. liu and J. Zhu, Macromolecules, 2020, 53, 2919–2931 CrossRef CAS.
- R. Antony, S. Theodore David Manickam, K. Saravanan, K. Karuppasamy and S. Balakumar, J. Mol. Struct., 2013, 1050, 53–60 CrossRef CAS.
- H. Shirakura, Y. Hijikata, J. Pirillo, T. Yoneda, Y. Manabe, M. Murugavel, Y. Ide and Y. Inokuma, Eur. J. Inorg. Chem., 2021, 2021, 1705–1708 CrossRef CAS.
- B. De Bruin, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2000, 39, 2936–2947 CrossRef CAS PubMed.
- G. Nasr, T. Macron, A. Gilles, Z. Mouline and M. Barboiu, Chem. Commun., 2012, 48, 6827–6829 RSC.
- F. García, J. Pelss, H. Zuilhof and M. M. J. Smulders, Chem. Commun., 2016, 52, 9059–9062 RSC.
- N. A. A. Qasem, R. H. Mohammed and D. U. Lawal, npj Clean Water, 2021, 4, 36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; catalyst screening data; spectroscopic and crystallographic data for C1; SEC traces, spectroscopic (NMR, MALDI-ToF MS) and thermal (TGA, DSC) data for the polymers; degradation and metal coordination data. CCDC 2253111. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3py00443k |
| This journal is © The Royal Society of Chemistry 2023 |