DOI:
10.1039/D3PY00278K
(Paper)
Polym. Chem., 2023,
14, 2829-2837
Alternating cationic copolymerizations of vinyl ethers and sequence-programmed cyclic trimer consisting of one vinyl ether and two aldehydes for ABCC-type periodic terpolymers†
Received
16th March 2023
, Accepted 22nd May 2023
First published on 22nd May 2023
Abstract
Alternating cationic copolymerizations of 2-chloroethyl vinyl ether (CEVE; unit A) and cyclic trimers prepared from the cyclotrimerization of one vinyl monomer (unit B) and two aldehyde molecules (unit C) were investigated to obtain polymers with ABCC-type monomer sequences. The kinds of vinyl monomers and aldehyde molecules incorporated into the cyclic trimers affected the crossover frequencies during the copolymerizations with CEVE. In particular, a cyclic trimer consisting of isobutyl VE (IBVE) and 2-nonenal (NNE) was highly effective for alternating copolymerizations with CEVE at appropriate monomer concentrations. The obtained alternating copolymers potentially had three different sequences of ABCC-, ACCB-, and ACBC-types due to the different addition and ring-opening modes of the cyclic trimer. Structural analysis of the products obtained by methanolysis of the alternating copolymers revealed that ABCC-type periodic sequences were mainly obtained (83% under optimized conditions).
Introduction
In the polymerization of two or more monomers, the difference in monomer sequences is an important structural parameter of the polymers.1,2 The ultimate goal of synthetic polymer chemistry is to obtain polymers with defined monomer sequences comparable with those of natural macromolecules such as proteins and DNA.3 The polymerization protocols used to control monomer sequences are roughly divided into the following three categories: the iterative method, direct polymerization, and the sequence-programmed monomer strategy.4 Each method has inherent disadvantages, such as time or yield losses and limitations of applicable monomers. In the sequence-programmed monomer strategy, reactive oligomers containing periodic monomer sequences are synthesized and subsequently polymerized to yield polymers with periodic sequences.5–14 Although the preparations of such monomers generally require multiple steps consisting of iterative reactions, isolations, and purifications, a high-molecular-weight (high-MW) polymer with the desired monomer sequences can be obtained. In addition, copolymerization of sequence-programmed monomers and other monomers widens the accessibility of monomer sequences.15–17
In the copolymerization of the same types of monomers, such as vinyl monomers, using chain-growth polymerization, homopropagation and crossover reactions occur statistically based on the reactivity ratios of comonomers, which results in sequence-uncontrolled polymers in most cases. On the other hand, copolymerizations of different types of monomers potentially achieve highly selective propagation reactions due to the reaction selectivity of propagating species derived from different types of monomers. For example, concurrent vinyl-addition and ring-opening copolymerizations of vinyl and cyclic monomers are often difficult, while several studies have overcome the inertness and realized the crossover reactions between vinyl and cyclic monomers.18–23 Such reaction selectivity in the copolymerization of different types of monomers can be useful for the control of monomer sequences, particularly when one or more comonomers do not exhibit homopolymerizability.
The structures of active chain ends derived from vinyl monomers and cyclic monomers are highly important for successful copolymerization by frequent crossover reactions. During cationic copolymerization, oxonium ions, which are active species of oxygen-containing cyclic monomers, do not react with vinyl monomers. Therefore, carbocation generation by ring-opening reactions of oxonium ions is indispensable for copolymerizing vinyl monomers and oxygen-containing cyclic monomers by crossover reactions.24 For example, ring-opening reactions of cyclic acetals generate alkoxy-adjacent carbocations that are structurally similar to vinyl ether (VE)-derived carbocations. The cationic copolymerization processes of vinyl monomers and cyclic acetals were reported several decades ago.22,23 Our group has conducted systematic studies of the effects of the ring members and substituents of cyclic acetals on copolymerization behaviors.25 Additionally, the one-pot synthesis of ABC-type periodic terpolymers has been achieved by an AB-type sequence-programmed cyclic acetal synthesis and subsequent alternating cationic copolymerizations with VEs (unit C).26
In this study, a novel system for sequence-regulated polymer synthesis was developed based on sequence-programmed monomer synthesis and alternating cationic copolymerizations (Scheme 1). We focused on cyclic trimers consisting of one vinyl monomer and two aldehydes, which were generated by the side reactions during alternating cationic copolymerizations of vinyl monomers and conjugated aldehydes.27,28 Cyclic trimers are regarded as cyclic acetal monomers with structures that are similar to that of 2-methyl-1,3-dioxane (MDOX). Thus, cyclic trimers potentially undergo cationic copolymerization with vinyl monomers. According to a systematic investigation of monomer structures, alternating cationic copolymerizations were shown to proceed when 2-nonenal (NNE)-containing cyclic trimers were used under optimized conditions. The alternating copolymer potentially has three different monomer sequence types—ABCC,29,30 ACCB, and ACBC—due to the three possible ring-opening modes. The monomer sequences of the obtained polymers could be determined by structurally analyzing the acid methanolysis products. The results indicated that the ABCC- and ACBC-type periodic sequences were obtained via the ring-opening reactions of cyclic trimers. The ratios of the two types of sequences were affected by the polymerization conditions. Specifically, the ABCC-type periodic sequences were mainly generated by copolymerizing 2-chloroethyl VE (CEVE) and a cyclic trimer consisting of isobutyl VE (IBVE) and NNE without added bases (ABCC-type ratio = 83%).
 |
| | Scheme 1 Synthesis of the ABCC-type periodic terpolymer via alternating cationic copolymerization of VEs and cyclic trimers. | |
Experimental section
See the ESI† for Materials, Polymerization procedure, Acid methanolysis of product copolymers, and Characterization subsections.
Sequence-programmed cyclic trimer synthesis
The following is a description of the typical procedure. A glass tube equipped with a three-way stopcock was dried using a heat gun (Ishizaki; PJ-206A; blow temperature of ∼450 °C) under dry nitrogen. Toluene, 1,4-dioxane (1,4-DO), IBVE, and benzaldehyde (BzA) were added successively into the tube using dry syringes. The reaction was started by the addition of a prechilled EtAlCl2 solution (200 mM) in toluene/n-hexane. After 24 h, the reaction was terminated with aqueous ammonia. The quenched mixture was diluted with dichloromethane and washed with water. The volatile materials were then removed under reduced pressure at 50 °C to yield the polymer. When the obtained product contained a significant amount of impurities, the crude product was purified using silica gel column chromatography [n-hexane/ethyl acetate (EA)]. During the cyclotrimerizations of VEs and NNE, the second portion of VEs was added to the reaction mixture at a predetermined time to increase NNE conversion. For example, the cyclotrimerizations of isopropyl VE (IPVE) and NNE were conducted via the following steps. Toluene (17.4 mL), tetrahydrofuran (THF; 2.4 mL) as an added base, IPVE (1.7 mL), and NNE (4.1 mL) were placed successively into a glass tube using dry syringes. The reaction was initiated by adding a prechilled EtAlCl2 solution in n-hexane/toluene (500 mM; 3.0 mL) at 0 °C. Then, 1.7 mL of IPVE was added at a predetermined interval (10 min). The reaction was terminated by adding water with a small amount of aqueous ammonia (approximately 10 mL; 0.3 wt%). The quenched reaction mixture was diluted with dichloromethane and washed with water. The organic layer was evaporated under reduced pressure to remove the remaining volatile materials. The crude product was purified using silica gel column chromatography (n-hexane/EA = 22/1 v/v; final yield = 44%).
Results and discussion
Selective cyclotrimerizations of various vinyl monomers and aldehydes
Cyclotrimerizations of various vinyl monomers and aldehydes were conducted to obtain cyclic trimers bearing various structures (Scheme 2). The electronic and steric environments around the carbocations generated by ring-opening reactions of cyclic trimers were tunable by the types of vinyl monomers and aldehydes incorporated into cyclic trimers (Table 1). First, a cyclic trimer consisting of IBVE and BzA was produced using EtAlCl2 as a Lewis acid catalyst with 1,4-DO at −78 °C (entry 1 in Table 1, and see Fig. S1–S3† for the 1H NMR, 13C NMR, and ESI-MS spectra). A cyclic trimer was generated quantitatively, as in the case of the previous study on the alternating cationic copolymerizations of IBVE and BzA.27 NNE, which is a linear conjugated aldehyde, underwent relatively selective cyclotrimerizations with VEs (yields of cyclic trimers = 33–53%; entries 2–4 in Table 1) to yield a cyclic trimer; a 3,4-dihydro-2H-pyran derivative and a VE homopolymer were obtained as side products (Fig. S4† for the 1H NMR spectrum of the product obtained in the cyclotrimerizations of IBVE and NNE). The cyclic trimer was isolated using silica gel column chromatography [see Fig. S5–S7 (IBVE), S8–S10 (CEVE), and S11–S13 (IPVE)† for the 1H NMR, 13C NMR, and ESI-MS spectra of the cyclic trimers separated by silica gel chromatography]. The reactions of styrene (St) derivatives, such as p-methylstyrene (pMeSt) and 1,1-diphenylethylene (DPE) (entries 5 and 6 in Table 1), were also examined. The cyclotrimerizations of St derivatives and BzA were promoted by Bi(OTf)3 or GaCl3, unlike the highly selective cyclotrimerizations of VEs and BzA by EtAlCl2 as in the previous reports on the cyclotrimerization of pMeSt or DPE and aldehyde.31,32 For pMeSt, the concentration of BzA was set to approximately a fifth of that of pMeSt to promote cyclotrimerization and suppress pMeSt homopolymerization (entry 5 in Table 1 and Fig. S14† for the 1H NMR spectrum of the cyclic trimer separated by silica gel column chromatography). In contrast, DPE was demonstrated to be efficient for selective cyclotrimerization (yield of cyclic trimer ≥ 99%) (entry 6 in Table 1 and Fig. S15†).
 |
| | Scheme 2 Cyclotrimerization of various vinyl monomers and conjugated aldehydes. | |
Table 1 Cyclotrimerization of various vinyl monomers and aldehydesa
| Entry |
Vinyl monomer |
(M) |
Ald |
(M) |
Catalyst |
(mM) |
Time |
Conv.b (%) |
Yield of cyclic trimerb (%) |
| Vinyl monomer |
Ald |
|
[THF] = 0 (entries 1 and 6), 1.0 (entries 2–4), or 0.58 M (entry 5), [1,4-DO] = 1.0 (entry 1) or 0 M (entries 2–6), in toluene (entry 1) or dichloromethane (entries 2–6) at −78 (entries 1 and 6) or 0 °C (entries 2–5).
Determined by 1H NMR analysis of quenched reaction mixtures using pyridine or THF as the internal standard.
|
| 1 |
IBVE |
0.48 |
BzA |
0.98 |
EtAlCl2 |
20 |
24 h |
87 |
>99 |
88 |
| 2 |
IBVE |
0.50 |
NNE |
0.99 |
EtAlCl2 |
50 |
4 h |
>99 |
76 |
53 |
| 3 |
CEVE |
0.50 |
NNE |
1.0 |
EtAlCl2 |
50 |
24 h |
>99 |
73 |
42 |
| 4 |
IPVE |
0.52 |
NNE |
0.99 |
EtAlCl2 |
50 |
10 min |
>99 |
62 |
33 |
| 5 |
pMeSt |
0.34 |
BzA |
1.8 |
Bi(OTf)3 |
5.0 |
48 h |
>99 |
15 |
50 |
| 6 |
DPE |
0.50 |
BzA |
0.98 |
GaCl3 |
5.0 |
168 h |
92 |
92 |
>99 |
Cationic copolymerizations of CEVE and various cyclic trimers
The cyclic trimer consisting of IBVE and BzA was employed in a cationic copolymerization with CEVE using an initiating system suited for controlled alternating copolymerizations of various VEs and conjugated aldehydes (Scheme 3A).27 Both CEVE and the cyclic trimer were consumed in a reaction using GaCl3 as a Lewis acid catalyst in conjunction with ethanesulfonic acid (EtSO3H) with 1,4-DO in toluene at −78 °C. 1,4-DO was used as a weak Lewis base for the suppression of the side reactions as in our previous studies on the cationic copolymerizations of VEs and aldehydes.27,28 A product of Mn = 1.1 × 103 was obtained under these conditions (entry 1 in Table 2). The initiation reaction most likely occurred via the addition reaction of EtSO3H with the cyclic trimer or VE. Another possible initiation reaction potentially occurs via ring opening triggered by the interaction between GaCl3 and the cyclic trimer.261H NMR analysis of the polymer revealed that the cyclic trimer was introduced into the main chain by crossover reactions. However, the average numbers of CEVE/cyclic trimer units per block were approximately 25/1.0 (these values are based on the assumption that CEVE homopolymer chains are not generated), suggesting that CEVE homopropagation reactions preferentially occurred during copolymerization. To suppress CEVE homopropagation reactions, the initial concentration of the cyclic trimer was set to twice that of CEVE. The 1H NMR analysis of the obtained polymer indicated that the amount of the incorporated cyclic trimer increased (Fig. S16†). However, the result of the acid methanolysis of the obtained polymer indicated unsuccessful copolymerization. The peak top of the MW distribution (MWD) curve did not shift to the lower-MW region after methanolysis (Fig. 1A). If the copolymerization is successful, acid-labile acetal structures will be generated in the main chain of a copolymer. The results revealed that CEVE homopolymer chains, which did not have acid-degradable units, were predominantly produced during copolymerization. Furthermore, the copolymerizations of CEVE and the cyclic trimers consisting of BzA and St derivatives were examined with the IBEA/TiCl4/SnCl4 initiating system, which was suitable for the controlled cationic copolymerization of VEs and the cyclic acetals.33 However, the crossover reactions did not occur during the polymerizations, likely because of the steric hindrance around the cyclic acetal structures, and instead, CEVE homopolymers were obtained (entries 3 and 4 in Table 2). These results indicated that cyclic trimers consisting of BzA and vinyl monomers would not be suitable for copolymerization with CEVE.
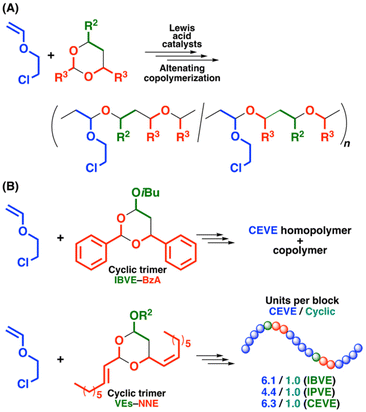 |
| | Scheme 3 Cationic copolymerization of CEVE with (A) various cyclic trimers and (B) cyclic trimers consisting of VEs and BzA or NNE. | |
 |
| | Fig. 1 MWD curves of the polymers obtained by the cationic copolymerization of CEVE and various cyclic trimers (black line; after purification by preparative GPC; entries 2 and 5 in Table 2 and entries 6 and 8 in Table 3) and their acid methanolysis products (purple line). CEVE and various cyclic trimers prepared from (A) BzA or (B) NNE. | |
Table 2 Cationic copolymerization of CEVE and various cyclic trimersa
| Entry |
CEVE (M) |
Cyclic trimer |
(M) |
Time |
Catalyst |
Additives |
Toluene/dichloromethane |
Conv.b (%) |
M
n × 10−3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
M
w/Mnc |
Units per blockd |
| CEVE |
Cyclic trimer |
CEVE |
Cyclic trimer |
| |
|
|
[Cationogen]0 = 4.0 mM (EtSO3H for entries 1, 2, and 5 or IBEA for entries 3 and 4), at −78 °C.
Determined by the 1H NMR analysis of quenched reaction mixtures (entries 1, 2, 4, and 5) or gas chromatography and the 1H NMR analysis of the product polymer (entry 3).
Determined by gel permeation chromatography (GPC; polystyrene standards).
Estimated by the 1H NMR analysis of the obtained products.
The MWD curve of the acid methanolysis products indicated that CEVE homopolymers were predominantly produced (Fig. 1A).
Not determined due to negligible crossover reactions.
|
| 1 |
0.50 |
IBVE–BzA |
0.50 |
70 h |
GaCl3 (4.0 mM) |
1,4-DO (1.0 M) |
10/0 |
37 |
11 |
1.1 |
1.48 |
(25)e |
(1.0)e |
| 2 |
0.40 |
IBVE–BzA |
0.80 |
24 h |
GaCl3 (10 mM) |
1,4-DO (1.0 M) |
10/0 |
72 |
7 |
2.1 |
1.51 |
(11)e |
(1.0)e |
| 3 |
0.20 |
pMeSt–BzA |
0.20 |
30 s |
TiCl4/SnCl4 (4.0 mM/20 mM) |
EA (20 mM) |
9/1 |
74 |
2 |
5.5 |
1.07 |
45 |
1.0 |
| 4 |
0.20 |
DPE–BzA |
0.20 |
5 min |
TiCl4/SnCl4 (4.0 mM/20 mM) |
EA (20 mM) |
9/1 |
>99 |
0 |
4.2 |
1.09 |
n.d.f |
n.d.f |
| 5 |
0.39 |
IBVE–NNE |
0.40 |
25 h |
GaCl3 (10 mM) |
1,4-DO (0.47 M) |
10/0 |
52 |
21 |
2.5 |
1.59 |
6.1 |
1.0 |
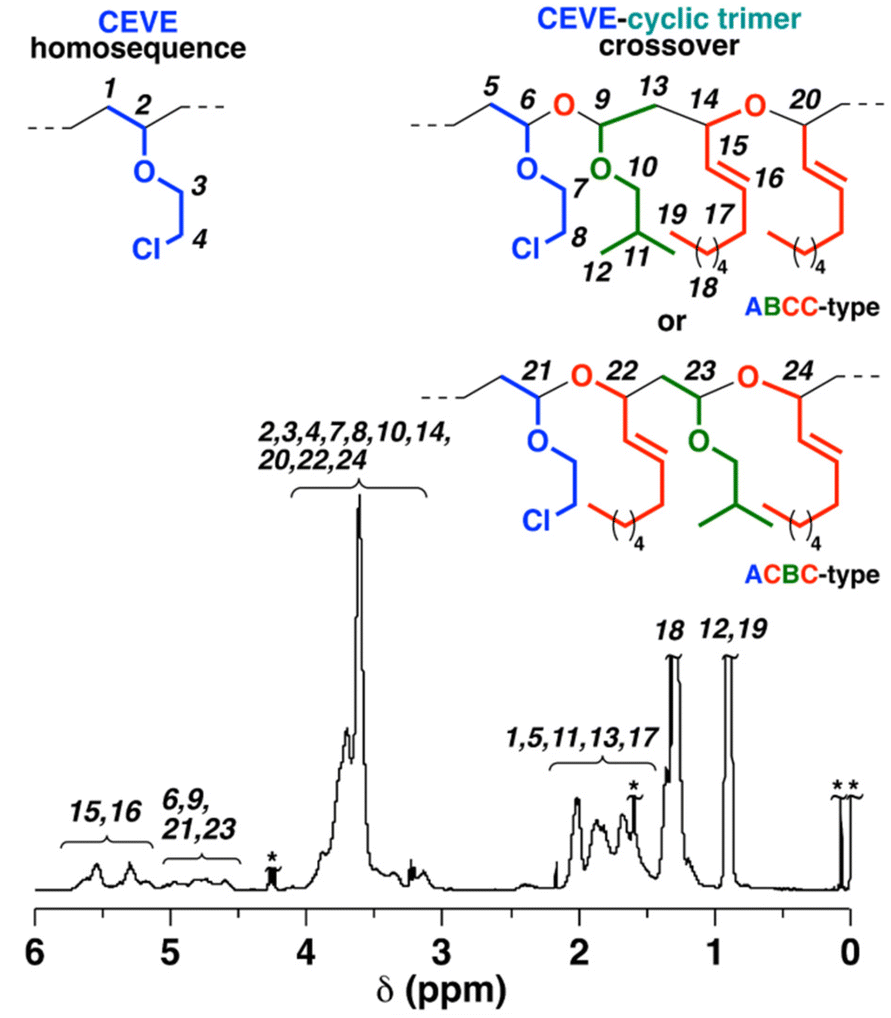
Unlike BzA-containing cyclic trimers, NNE-containing cyclic trimers were highly effective for copolymerization with CEVE (entry 5 in Table 2 and Scheme 3B). Indeed, in our previous study on alternating cationic copolymerizations of VEs and the cyclic trimers, the ring number and substituents of the cyclic trimers affected the crossover frequencies during the copolymerizations.33 The concurrent cationic vinyl-addition and ring-opening copolymerization processes of CEVE and the IBVE–NNE cyclic trimer were conducted under conditions similar to those used for the cationic copolymerizations of CEVE and the IBVE–BzA cyclic trimer, yielding a polymer of Mn = 2.5 × 103 (entry 5 in Table 2). The introduction of the cyclic trimer derived from crossover reactions was confirmed by the 1H NMR analysis of the obtained polymer (Fig. 2). The olefin structures derived from NNE were assigned to the peaks at 5.1–5.8 ppm (peaks 15 and 16). The peaks at 4.5–5.1 ppm (peaks 6, 9, 21, and 23) were attributed to the acetal structures generated by the crossover reaction from CEVE to the cyclic trimer. From the integral ratios of the olefin moiety and CEVE homosequences (peaks 2–4), the average numbers of CEVE and the cyclic trimer units per block were estimated to be 6.1 and 1.0, respectively, indicating that the cyclic trimer consisting of IBVE and NNE underwent frequent crossover reactions with CEVE, differing from the IBVE–BzA cyclic trimer. The absence of cyclic trimer homopropagation was assumed from the result that the homopolymerization of the cyclic trimer did not proceed under the same conditions as those for copolymerization (see Fig. S17† for the MWD curve of the product obtained in the cationic homopolymerization of the cyclic trimer consisting of IBVE and NNE).
 |
| | Fig. 2
1H NMR spectrum of the polymer obtained by the copolymerization of CEVE and the cyclic trimer consisting of IBVE and NNE [entry 5 in Table 2; after purification by preparative GPC; in CDCl3 at 30 °C; *water, grease, TMS, etc.; the structures derived from crossover reactions were determined using the 1H NMR and ESI-MS spectra of the methanolysis product of the alternating polymer of CEVE and the IBVE–NNE cyclic trimer (vide infra)]. | |
In addition, the MWD curve obviously shifted to the lower-MW region after acid methanolysis (Fig. 1B, left), indicating that acid-labile acetal moieties were incorporated into all the polymer chains by successful copolymerization. The Mn value of the acid methanolysis product was consistent with the average numbers of CEVE and the cyclic trimer units per block, as shown above.
Cationic copolymerizations of CEVE and cyclic trimers of VEs and NNE under various conditions
To achieve ABCC-, ACCB-, or ACBC-type periodic sequences by alternating copolymerizations of CEVE and the cyclic trimer, appropriate reaction conditions were investigated; there was a particular focus on solvents, VE units incorporated into cyclic trimers, and monomer concentrations. Initially, the cationic copolymerizations of CEVE and the IBVE–NNE cyclic trimer were conducted in a mixture of toluene/n-hexane (6/4 v/v; entry 1 in Table 3 and Fig. 3). The 1H NMR spectrum of the obtained polymer indicated that the average number of CEVE and the cyclic trimer units per block were estimated to be 4.0 and 1.0; these results suggested that the crossover reactions occurred more frequently in the mixed solvent than in toluene alone (CEVE/cyclic trimer = 6.1/1.0; entry 5 in Table 2). The kinds of VEs incorporated into cyclic trimers affected copolymerization behaviors. The frequencies of crossover reactions increased when the cyclic trimer consisting of NNE and IPVE, a more reactive VE than IBVE, was used (entry 6 in Table 3). A polymer with shorter CEVE blocks (CEVE/cyclic trimer = 4.4/1.0; entry 6 in Table 3) than the IBVE–NNE counterpart (CEVE/cyclic trimer = 6.1/1.0; entry 5 in Table 2) was obtained. However, the use of the CEVE–NNE cyclic trimer resulted in copolymerization with less frequent crossover reactions (CEVE/cyclic trimer = 6.3/1.0; entry 8 in Table 3). These results indicated that the use of cyclic trimers containing more reactive VEs was suitable for crossover reactions with CEVE.
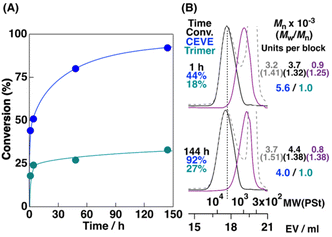 |
| | Fig. 3 (A) Time–conversion curves of the cationic copolymerization of CEVE and the cyclic trimer consisting of IBVE and NNE and (B) MWD curves of the obtained polymers (dashed line), portions after purification by preparative GPC (solid line), and their acid methanolysis products (purple line) (entry 1 in Table 3). | |
Table 3 Cationic copolymerization of CEVE and a cyclic trimer consisting of VEs and NNEa
| Entry |
CEVE (M) |
Cyclic trimer |
(M) |
Additives |
(mM) |
Toluene/hexane (v/v) |
Time |
Conv.b (%) |
M
n × 10−3c |
M
w/Mnc |
Units per blockd |
ABCC/ACBC ratioe |
| CEVE |
Cyclic trimer |
CEVE |
Cyclic trimer |
|
[EtSO3H]0 = 4.0 (entries 1–6 and 8) and 2.0 mM (entry 7); and [GaCl3]0 = 10 mM, at −78 °C.
Determined by 1H NMR analysis of quenched reaction mixtures.
Determined by GPC (polystyrene standards).
Estimated by the 1H NMR analysis of the obtained products after purification by preparative GPC. Numbers with two decimal places are shown when the values are very close to 1.00.
Determined by the 1H NMR analysis of the acid methanolysis products.
|
| 1 |
0.40 |
IBVE–NNE |
0.40 |
1,4-DO |
2.0 × 102 |
6/4 |
144 h |
92 |
27 |
3.7 |
1.51 |
4.0 |
1.0 |
— |
| 2 |
0.20 |
IBVE–NNE |
1.0 |
— |
6/4 |
1 min |
92 |
20 |
4.3 |
1.41 |
1.7 |
1.0 |
86/14 |
| 3 |
0.10 |
IBVE–NNE |
1.0 |
— |
6/4 |
10 min |
>99 |
23 |
4.2 |
1.52 |
1.07 |
1.0 |
83/17 |
| 4 |
0.10 |
IBVE–NNE |
1.0 |
— |
8/2 |
10 min |
>99 |
24 |
4.0 |
1.40 |
1.06 |
1.0 |
82/18 |
| 5 |
0.10 |
IBVE–NNE |
1.0 |
EA |
20 |
6/4 |
30 min |
>99 |
23 |
3.0 |
1.50 |
1.03 |
1.0 |
74/26 |
| 6 |
0.40 |
IPVE–NNE |
0.40 |
1,4-DO |
4.7 × 102 |
10/0 |
25 h |
43 |
8 |
1.6 |
1.76 |
4.4 |
1.0 |
— |
| 7 |
0.049 |
IPVE–NNE |
0.50 |
— |
9/1 |
5 min |
61 |
7 |
2.0 |
1.61 |
1.04 |
1.0 |
40/60 |
| 8 |
0.41 |
CEVE–NNE |
0.40 |
1,4-DO |
4.7 × 102 |
10/0 |
96 h |
60 |
20 |
3.5 |
1.63 |
6.3 |
1.0 |
— |
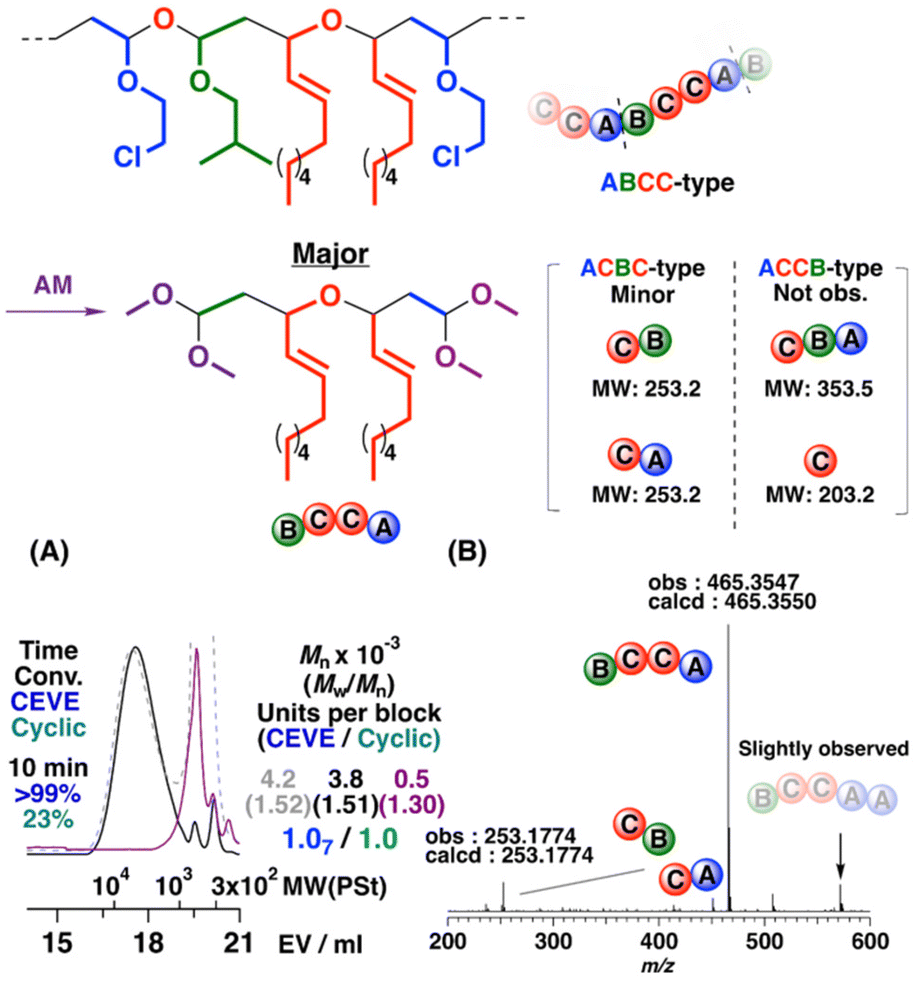
For a cyclic trimer, copolymerization at a higher initial concentration than that of a VE drastically promoted the crossover reactions. A polymer of Mn = 4.3 × 103 was obtained in the copolymerization at CEVE/IBVE–NNE cyclic trimer concentrations of 0.20 M/1.0 M (entry 2 in Table 3; dashed line in Fig. 4A). In the 1H NMR spectrum of the obtained polymer (Fig. 5 upper), the peak intensities of the structures derived from VE-to-cyclic trimer crossover reactions (peaks 2, 5, 17, and 19) were obviously larger than those obtained at equal concentrations of CEVE and the cyclic trimer (Fig. 2 and 5 upper). The integral ratio indicated that the average numbers of CEVE and the cyclic trimer units per block were estimated to be 1.7 and 1.0, respectively. The acid methanolysis of this polymer produced a compound of Mn = 0.5 × 103 (the purple line in Fig. 4A). The generation of the low-MW product after acid methanolysis indicated that acetal moieties were introduced into the main chain by frequent crossover reactions.
 |
| | Fig. 4 (A) MWD curves of the polymer obtained by the cationic copolymerization of CEVE and the cyclic trimer consisting of IBVE and NNE (dashed line), a portion after purification by preparative GPC (solid line), and its acid methanolysis product (purple line) and (B) ESI-MS spectrum of the acid methanolysis product (entry 2 in Table 3). | |
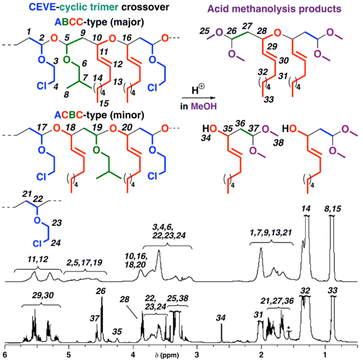 |
| | Fig. 5
1H NMR spectra of the polymer obtained by the cationic copolymerization of CEVE and the cyclic trimer consisting of IBVE and NNE and its acid methanolysis product (entry 2 in Table 3), after purification by preparative GPC, in CDCl3 at 30 °C; *water; structures of ABCC- and ACBC-type sequences derived from alternating propagation reactions are shown. Non-periodic structures, such as AABCC- and AAABCC-type sequences, containing CEVE homosequences also exist (labels 21–24 are used for CEVE homosequences). | |
Structural analysis of the methanolysis product obtained above revealed the ring-opening mode of the cyclic trimer. The possible reaction mechanisms of the copolymerization are summarized in Scheme 4. Adding a cyclic acetal to the VE-derived carbocation formed an oxonium ion. Two possible oxonium ions were generated by the reaction of the oxygen atom at the 1- or 3-position of the cyclic acetal. An oxonium ion was subsequently converted to a carbocation via a ring-opening reaction. The oxonium ion derived from the reaction of the oxygen atom at the 1-position had two acetal moieties; hence, two different carbocations were generated through paths (i) and (ii). The other possible oxonium ion afforded different carbocations through path (iii). Therefore, three different monomer sequences—ABCC-, ACCB-, and ACBC-type—were potentially generated through different addition and cleavage modes. Methanolysis products had structures corresponding to the constitutional repeating units of the original polymers as a result of the cleavage of the acetal moieties; this phenomenon was very useful in revealing the ring-opening mode. Indeed, the ESI-MS analysis of the methanolysis product (Fig. 4B) detected peaks assigned to structures composed of four monomer units (CEVE/IBVE/NNE = 1/1/2; a circle symbol with n = 0; two VE side chains were converted into methoxy moieties by transacetalization with methanol) or two monomer units (NNE/CEVE or IBVE = 1/1; a square symbol with n = 0; a VE side chain was converted into a methoxy moiety). The other peaks [CEVE/IBVE/NNE = 2 or 3/1/2 (circle symbols with n = 1 or 2) and NNE/CEVE = 1/2 (a square symbol with n = 1)] were derived from blocks with CEVE homosequences. The result suggested that the cyclic acetal underwent a ring-opening reaction by the modes shown by paths (i) and (iii) in Scheme 4. Possible products derived from the other mode (path (ii) in Scheme 4) were not observed in the ESI-MS analysis.
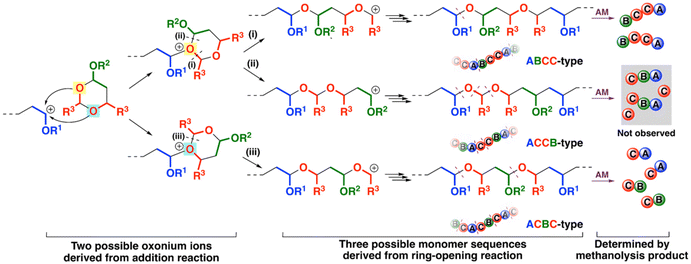 |
| | Scheme 4 The reaction pathway of the generation of the different sequence-regulated polymers (AM: acid methanolysis). | |
ABCC- and ACBC-type sequence generations were consistent with the 1H and 13C NMR spectra of the methanolysis product (Fig. 5 lower and Fig. S18†). For example, peaks at 3.8 ppm (peak 28) and 4.5 ppm (peak 26) of the 1H NMR spectrum were assigned to the two allyl methine protons adjacent to the oxygen atom and the two acetal methine protons, respectively, of the methanolysis product from the ABCC-type monomer sequences. In addition, the hydroxy, hydroxy-adjacent methine, and acetal peaks assigned to the product from ACBC-type monomer sequences emerged at 2.6, 4.25, and 4.6 ppm (peaks 34, 35, and 37). These assignments were confirmed by analysis of the 1H–1H correlation spectroscopy (COSY) NMR spectrum (Fig. S19†). The ABCC-type/ACBC-type ratio was calculated to be 86/14 by the integral ratios of two allyl methine protons adjacent to the oxygen atom and hydroxy-adjacent methine proton of each methanolysis product (peaks 28 and 35).
Alternating-like cationic copolymerizations were found to proceed when the concentration of the cyclic trimer was set to a tenth of that of CEVE due to the suppression of CEVE homopropagation (entries 3, 4, and 7 in Table 3). Moreover, the composition ratios of the ABCC- to ACBC-type monomer sequences were tuned by the polymerization conditions. For example, the ACBC-type monomer sequences were preferentially generated with EA; hence, the selectivity of generation of the ABCC-type monomer sequences decreased (ABCC-type ratio = 74%; entry 4 in Table 3). An alternating polymer with comparable amounts of the ACBC- and ABCC-type monomer sequences was obtained while copolymerizing CEVE and the IPVE–NNE cyclic trimer (entry 7 in Table 3 and see Fig. S20† for the 1H NMR spectra of the obtained polymer and its acid methanolysis product). In addition, the alternating cationic copolymerizations of CEVE and the IBVE–NNE cyclic trimer at −78 °C, which was the most suitable polymerization temperature (Table S1†), afforded a polymer with a relatively high ABCC-type ratio [ABCC-type ratio = 83%; entry 3 in Table 3; Mn = 4.2 × 103 (the dashed line in Fig. 6A)]. Although the average degree of polymerization of the obtained polymer was approximately 9, the MWD curve indicated that portions with MWs higher than 10 × 103, which corresponds to a degree of polymerization of approximately 21, were contained in the polymer. The average numbers of CEVE and the cyclic trimer units per block calculated by the 1H NMR spectrum of this polymer were 1.07 and 1.0, respectively, indicating the occurrence of alternating copolymerizations. The MALDI-TOF-MS spectrum of the obtained polymer (Fig. S21†) exhibited two series of peaks that had alternating intervals corresponding to the mass values of CEVE and the cyclic trimer. The unimodal MWD curve of the high-MW portion separated by preparative GPC shifted to the low-MW region after acid methanolysis (the purple line in Fig. 6A). In the ESI-MS spectrum of the acid methanolysis product (Fig. 6B), the major peak had an m/z value (465.3547) corresponding to the structure with four monomer units derived from the ABCC-type monomer sequences. Interestingly, the peaks derived from two or more CEVE units were extremely small; these results corroborated that ABCC-type periodic sequences were mainly generated.
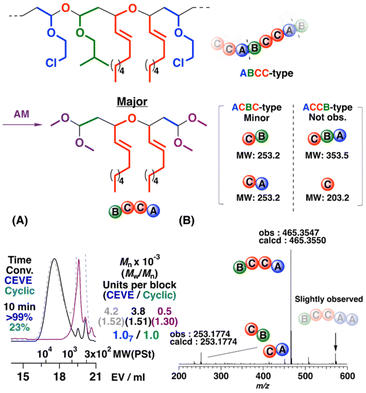 |
| | Fig. 6 (A) MWD curves of the polymer obtained by the cationic copolymerization of CEVE and the cyclic trimer consisting of IBVE and NNE (dashed line), a portion after purification by preparative GPC (solid line), and its acid methanolysis product (purple line) and (B) ESI-MS spectrum of the acid methanolysis product (entry 3 in Table 3). | |
Conclusion
The alternating copolymerizations of CEVE and the NNE-containing cyclic trimer were demonstrated to proceed under optimized conditions. Among the three possible sequences—ABCC-, ACCB-, and ACBC-type—in the polymer chain, the ABCC- and ACBC-type sequences were selectively generated, which was revealed by the structural analysis of the acid methanolysis product of the obtained polymer. Moreover, the ABCC-type periodic sequences were preferentially generated during the copolymerizations of CEVE and a cyclic trimer consisting of IBVE and NNE without added bases (ABCC-type ratio = 83%). By utilizing a cyclic trimer consisting of one VE and two aldehydes, we provided new insight into sequence-regulated polymerization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by JSPS KAKENHI Grants 18K05217 and 21J11360.
References
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- C. B. Anfinsen, Science, 1973, 181, 223–230 CrossRef CAS PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- I. Cho, Prog. Polym. Sci., 2000, 25, 1043–1087 CrossRef CAS.
- M. Ueda, Prog. Polym. Sci., 1999, 24, 699–730 CrossRef CAS.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 6 CrossRef PubMed.
- J. Zhang, M. E. Matta and M. A. Hillmyer, ACS Macro Lett., 2012, 1, 1383–1387 CrossRef CAS PubMed.
- H. Zhang, Z. Zhou, X. Cheng, B. Yu, Z. Luo, X. Li, M. A. Rahman and Y. Sha, Macromolecules, 2021, 54, 9174–9184 CrossRef CAS.
- O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 CrossRef CAS PubMed.
- Z.-L. Li, L. Li, X.-X. Deng, L.-J. Zhang, B.-T. Dong, F.-S. Du and Z.-C. Li, Macromolecules, 2012, 45, 4590–4598 CrossRef CAS.
- J. Li and J. He, ACS Macro Lett., 2015, 4, 372–376 CrossRef CAS PubMed.
- W. Xi, S. Pattanayak, C. Wang, B. Fairbanks, T. Gong, J. Wagner, C. J. Kloxin and C. N. Bowman, Angew. Chem., Int. Ed., 2015, 54, 14462–14467 CrossRef CAS PubMed.
- C.-H. Tsai, A. Fortney, Y. Qiu, R. R. Gil, D. Yaron, T. Kowalewski and K. J. T. Noonan, J. Am. Chem. Soc., 2016, 138, 6798–6804 CrossRef CAS PubMed.
- T. Yokozawa and T. Suzuki, Macromolecules, 1996, 29, 22–25 CrossRef CAS.
- Z. Zhang, Y.-Z. You, D.-C. Wu and C.-Y. Hong, Macromolecules, 2015, 48, 3414–3421 CrossRef CAS.
- N. Ousaka and T. Endo, Macromolecules, 2021, 54, 2059–2067 CrossRef CAS.
- T. Saegusa, Angew. Chem., Int. Ed. Engl., 1977, 16, 826–835 CrossRef.
- C. I. Simionescu, M. Grigoras, E. Bîcu and G. Onofrei, Polym. Bull., 1985, 14, 79–83 CrossRef CAS.
- B. L. Rivas, G. D. C. Pizarro and G. S. Canessa, Polym. Bull., 1988, 19, 123–128 CAS.
- H. Yang, J. Xu, S. Pispas and G. Zhang, Macromolecules, 2012, 45, 3312–3317 CrossRef CAS.
- M. Okada, Y. Yamashita and Y. Ishii, Makromol. Chem., 1966, 94, 181–193 CrossRef CAS.
- M. Okada and Y. Yamashita, Makromol. Chem., 1969, 126, 266–275 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2014, 47, 6635–6644 CrossRef CAS.
- K. Maruyama, A. Kanazawa and S. Aoshima, Polym. Chem., 2019, 10, 5304–5314 RSC.
- K. Maruyama, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 799–809 CrossRef CAS.
- Y. Ishido, R. Aburaki, S. Kanaoka and S. Aoshima, Macromolecules, 2010, 43, 3141–3144 CrossRef CAS.
- Y. Ishido, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4684–4693 CAS.
- C. D. Hurd, J. Chem. Educ., 1966, 43, 527–531 CrossRef CAS.
- S. M. Oon and D. G. Kubler, J. Org. Chem., 1982, 47, 1166–1171 CrossRef CAS.
- B. Sreedhar, V. Swapna, C. Sridhar, D. Saileela and A. Sunitha, Synth. Commun., 2005, 35, 1177–1182 CrossRef CAS.
- T. Naito, A. Kanazawa and S. Aoshima, Polym. Chem., 2019, 10, 1377–1385 RSC.
- K. Maruyama, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 4034–4045 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A part of the experimental section, polymerization data, NMR spectra, ad ESI-MS spectra. See DOI: https://doi.org/10.1039/d3py00278k |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Arihiro
Kanazawa
,
Arihiro
Kanazawa