
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic star copolymers obtained by the arm first approach for gene transfection†
Fannie
Burgevin
a,
Alexia
Hapeshi
a,
Ji-Inn
Song
a,
Marta
Omedes-Pujol
b,
Annette
Christie
b,
Christopher
Lindsay
b and
Sébastien
Perrier
 *acd
*acd
aChemistry Department, University of Warwick, Library Road, CV4 7AL, Coventry, UK. E-mail: s.perrier@warwick.ac.uk
bFormulation Technology Group, Syngenta, Jealotts Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
cWarwick Medical School, University of Warwick, Coventry CV4 7AL, UK
dMonash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), 399 Royal Parade, Parkville, Victoria 3152, Australia
First published on 3rd July 2023
Abstract
Cationic polymers can be used as vectors to transport and efficiently protect nucleic acids. In this work we describe the synthesis of dense star-like polymers of 2-dimethylaminoethyl acrylate (DMAEA) and 2-dimethylaminoethyl methacrylate (DMAEMA) and the hydrolysis of the DMAEA units side chains for the complexation and release of nucleic acids. The successful chain extension of p(DMAEA80-stat-DMAEMA20) with acrylamide monomers allowed the preparation of stars by the arm-first approach. Soluble stars with a high number of well-defined arms (Narm ∼55–100) were obtained with the introduction of non-cationic N-acrylmorpholine (NAM) prior to the crosslinking step. The influence of the architecture on the hydrolysis of the DMAEA units side chains was studied, with only small differences observed compared to the corresponding arm. All stars were able to complex a large (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 basepairs) plasmid DNA encoding for green fluorescent protein (GFP), and transfect HEK293T cells, with the larger, more charged star structure leading to higher transfection efficiency. Although the transfection efficiency is lower than that of the gold standard polyethylenimine (PEI), the stars much lower toxicity, at concentrations as high as 1 mg mL−1, make them viable transfection agents.
000 basepairs) plasmid DNA encoding for green fluorescent protein (GFP), and transfect HEK293T cells, with the larger, more charged star structure leading to higher transfection efficiency. Although the transfection efficiency is lower than that of the gold standard polyethylenimine (PEI), the stars much lower toxicity, at concentrations as high as 1 mg mL−1, make them viable transfection agents.
Introduction
Cationic and especially amine-based polymers have attracted great interest for their ability to interact with negatively charged nucleic acids and to form a complex that is often called polyplex.1 Delivery of DNA or RNA has a wide range of applications, from treating genetic disorders or diseases, such as cancer,2 to pest control.3 Since RNA and DNA are subject to rapid degradation by nucleases and hydrolysis, they need to be protected by delivery vectors to ensure efficient delivery.4,5 The interaction with cationic polymers give good protection and have good delivery efficiency, with the positive charge enhancing endocytosis-mediated cellular uptake.6–8 Polymeric vectors have significant advantages over viral vectors in that they have low immunogenicity1,9,10 and tuneable physical and chemical properties based on modular compositions, functions, chain length and architectures, as well as simple production with good reproducibility.11 The gold standard for gene delivery in therapeutics is polyethylenimine (PEI), due to its low cost and commercial availability.12 Although PEI is an efficient delivery vector, its applications are currently limited due to high cationic charge density causing high cytotoxicity.13,14This high toxicity has led to the use of alternative polymers to PEI, including chitosan,15 poly(2-dimethylamino ethyl methacrylate) (PDMAEMA), poly-L-lysine (PLL)16 and poly(N,N-dimethylaminopropyl acrylamide) (PDMAPAAm).17 In particular, PDMAEMA polymers have received great interest in the literature,18–20 due to their reduced toxicity compared to PEI and comparable transfection efficiency.21,22 However, these polymers do not have a release mechanism for their nucleotides payload, and thus have limited efficiency. A route to payload release is to use cationic moieties that can be cleaved in physiological media,23–29 a feature found in poly(acrylate)s such as poly(2-dimethylaminoethyl acrylate) (pDMAEA), where the charged pendant moiety can be hydrolysed from the polymer backbone.28–32 pDMAEA has been shown to bind effectively to RNA and DNA, and the release is triggered via hydrolysis of its positively charged side groups complexed to the nucleotides payload.28,29,32–34 In addition, the hydrolysis leads to negatively charged polymers (a process termed charge-shifting) which enable the release of the negatively charged RNA or DNA via electrostatic repulsion.25,26,34 However, in order to control the release process, the polyplex has to be stable over a period of time to enable transfection, followed by degradation/hydrolysis inside the cell to release the nucleotides. A downside of this approach is the fast hydrolysis of pDMAEA, which occur in time period varying from 1 to 10 hours.28 Our group recently showed that by copoluymerising DMAEA and DMAEMA, which is non-hydrolysable the hydrolysis of the resulting copolymer, and therefore release of the genetic material, can be controlled and delayed.25
Polymer architecture and molecular weight have shown to have an impact on gene delivery efficiency.35–37 High molecular weight polymers tend to show better DNA/RNA binding, cellular uptake and transfection efficiency whereas lower molecular weight polymers show less cytotoxicity.36 Dense structure such as branched star-shaped polymers have shown promising properties, attributed to the spherical shape of the resulting polyplex and the better and stronger condensation of DNA.20,21,25,26,33,38,39 Such structures can be synthesised efficiently via the arm first approach: linear polymeric arms are first synthesised using a controlled polymerisation technique such as reversible addition–fragmentation transfer (RAFT), and then a bifunctional crosslinker is introduced to covalently interconnect the end of the arms to form star-shaped structure.19,40–42 The synthesis of well defined structures have been demonstrated in the literature.43,44 Although the literature shows a few examples of DMAEMA-based star polymers,19,20,45,46 there are far fewer examples of polymer architectures based on the acrylate version DMAEA. For instance, RAFT has been employed to make linear chains with good control over a range of molecular weight from 3 000 g mol−1 to up to 8 600 g mol−1 with dispersity ranging from 1.17 to 1.26 at higher molecular weight.23,28 More recently Ros et al. reported the polymerisation of DMAEA using RAFT in a mixture of dioxane and water at pH 3–4 resulting in a higher molecular weight polymer (31 200 g mol−1, Đ = 1.25).47 Liao et al. synthesised 4-arm stars of DMAEA using tetrafunctional RAFT agent and used these materials to complex and delivery siRNA.33 Cook et al. synthesised branched copolymers of DMAEA and DMAEMA, obtaining polymer with molecular weight ranging from 17000 to 27000 g mol−1 but the uncontrolled nature of the reaction led to structures with very broad dispersity (Đ = 3.1–8.2).25 Similarly, Rolph et al. reported the synthesis of DMAEA core-crosslinked stars by chain extending linear homopolymers poly(ethylene glycol monomethyl ether acrylate) (pEGA) or poly(2-hydroxyethyl acrylate) with DMAEA and a divinyl crosslinker (Đ ∼2.2–2.9).48 Nitroxide-mediated polymerisation (NMP) has also been used to obtain linear chain with molecular weight up to 13000 g mol−1 and dispersities around 1.5.49 Finally, Whitfield et al. used Cu(0)-mediated reversible deactivation radical polymerisation to make stars using multifunctional initiator with good control (Đ ∼1.1), by keeping conversion low (around 30%) in order to circumvent termination and side reactions.50 Interestingly, block copolymers of pDMAEA have only been reported using Cu(0)-mediated RDRP,50 with block of DMAEA preferencially incorporated as the final block. For instance, Zhao et al. chain extended PDMS to obtain amphiphilic block copolymers that self-assemble in aqueous solutions.51 Tran et al. made copolymers with a first block of poly(dimethyl acrylate) and a second block of statistically copolymerised DMAEA, N-isopropylacrylamide and butyl acrylate to form thermoresponsive micelles nanoparticles.52 There is however no example of the chain extension of pDMAEA for the production of star copolymers.
In this paper, we investigate the synthesis of dense star-shaped copolymers with arms composed of 80% DMAEA and 20% DMAEMA and their use for nucleic acid complexation. The star are obtained by the arm-first approach using RAFT polymerisation, where linear polymeric chains are first formed, then interconnected covalently by introduction of a crosslinker.40–42 Chain extension of p(DMAEA-stat-DMAEMA) was first investigated, showing acrylamides are the most suitable to obtain a good chain extension. The dense stars were characterised by 1H NMR and DMF-GPC with triple detection and their hydrolysis was compared to linear copolymer equivalents. Finally, their potential for complexing and delivering a large (10000 basepairs) plasmid DNA (pDNA) encoding green fluorescent protein was assessed.
Experimental procedures
Materials
2-(Dimethylamino)ethyl acrylate (DMAEA; Sigma-Aldrich, 98%), 2-(dimethylamino)ethyl methacrylate (DMAEMA; Sigma-Aldrich, 98%), methyl acrylate (Sigma-Aldrich, 99%), methyl methacrylate (Sigma-Aldrich, 99%), acryloylmorpholine (NAM; Sigma-Aldrich, 97%), ethyleneglycol diacrylate (EGDA; Sigma-Aldrich, 90%), N,N′-methylenbis(acrylamide) (NAM, Sigma-Aldrich, 99%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, Alfa Aesar, 98%), (4-cyano pentanoic acid) ethyl trithiocarbonate (CPATC) was synthesised according to previously reported protocols.53 pDNA (pHR′ CMV GFP plasmid) was provided by Dr John James (University of Wawick) and isolated using a Qiagen Maxi Prep kit, following the established kit protocol. Agarose (Sigma-Aldrich), Tris-Borate-EDTA buffer (TBE), Gel loading dye purple (6×) (BioLabs), GelRed (Biotium), ethidium bromide (Sigma-Aldrich, 500 mg mL−1), sterile water molecular biology reagent (Sigma-Aldrich). Human embryonic kidney cells 293 (HEK293T, CRL-3216) (American type culture collection (ATCC)). Foetal bovine serum (FBS) (LabTech.com).
Gel permeation chromatography (GPC) was performed in DMF, an Agilent infinity II MDS instrument equipped with differential refractive index (DRI), viscometry (VS), dual angles light scattering (LS) and variable wavelength UV detectors was utilised. The system was equipped with 2× PLgel Mixed D columns (300 × 7.5 mm) and PLgel 5 μm guard column. The eluent was DMF with 5 mmol NH4BF4 additive. Samples were run at 1 mL min−1 at 50 °C. When using DRI detection for lower molecular weight polymers, poly(methyl methacrylate) standards (Agilent EasyVials) were used for calibration between 955000–550 g mol−1. Analyte samples were filtered through a nylon membrane with 0.22 μm pore size before injection. Respectively, experimental molar mass (Mn,GPC) and dispersity (Đ) values of synthesised polymers were determined by conventional calibration or triple detection using Agilent GPC/SEC software.
Proton nuclear magnetic resonance spectra (1H NMR) were recorded on a Bruker Advance 300 or 400 (300 or 400 MHz) at 27 °C using CDCl3 or D2O as solvents. Chemical shift values (δ) reported in ppm, and the residual proton signal of the solvent used as internal standard.
Atomic force microscopy (AFM)
AFM images were taken using an Asylum Research MFP-3D stand alone atomic force microscope. Samples were prepared by drop casting 5 μL of aqueous polymer solution at 0.025 mg mL−1 onto freshly cleaved mica, leaving to stand for 1 minute, tipping substrate and drying with filter paper, then under a stream of nitrogen.The hydrolysis study of the copolymers were performed in NMR tubes at 10 mg mL−1 in D2O. The reaction was followed by 1H NMR spectroscopy.
Agarose gel electrophoresis
Polyplexes were prepared at various ratio of nitrogen from the polymer to phosphorous from the nucleic acid (N/P ratios), by mixing the correct amount of polymer stock solution with 25 μL of a stock solution of pDNA at 0.1 mg mL−1 for a total solution of 50 μL. Polyplexes were vortexed and incubated for 30 min. Prior to loading, 25 μL of loading dye was added to each sample. Agarose gel was prepared by heating agarose (2 g) dissolved in 200 mL of 10% TBE buffer in the microwave until complete dissolution. The solution was cooled down for 20 minutes and 22 μL of GelRed was added. The mixture was poured into the gel caster and combed was inserted. The gel was left to set for 25 minutes at room temperature. The agarose gel electrophoresis were run in 10% TBE buffer. 15 μL of polyplexes were loaded into the agarose gel wells. The final gels were visualised under UV illumination.Ethidium bromide displacement assays
pDNA (15 μg mL−1) and ethidium bromide (1 μg mL−1) were incubated for 10 minutes at room temperature. And 50 μL of this solution was transferred to a 96 wellplate containing polymers at different concentrations corresponding to the different N/P ratios. After 20 minutes incubation fluorescence intensity (λEx = 525 nm, λEm = 605 nm) was measured using Biotek instruments Citation 3 cell imaging multi-mode reader. The maximum fluorescence was defined with controls containing only pDNA with ethidium bromide.Transfection
HEK293T cells were seeded in a 24 well plate at a density of 1 × 105 cells in 1 mL per well and left overnight. The culture medium was replaced by 300 μL Optimem® cell culture media (Thermo Fisher Scientific) without foetal bovine serum (FBS). Polyplexes were prepared by mixing pDNA solution and polymer solution. The solution was then incubated at room temperature for 60 minutes. After 60 minutes the media was replaced by fresh Optimem® plus polyplex solution (350 μL Optimem® and polyplex with final concentration of pDNA of 10 μg mL−1). The cells were incubated for 5 hours under 5% CO2 humidified atmosphere, then the wells were washed with warm medium. Media was replaced with fresh DMEM containing FBS. After 48 hours incubation (including 5 hours incubation with the polyplex), the cells were washed with PBS (0.5 mL). The cells were harvested using 150 μL of trypsin/EDTA. 300 μL of DMEM containing FBS was then added and the cell suspension was centrifuged. The cell pellet was resuspended in 100 μl PBS and 100 μL of 8% formaldehyde was added. The samples were left for 15 min at room temperature to fix cells and centrifuged. The cell pellets were washed with cold PBS (200 μL) twice. Cells were analysed using LSRIII flow cytometer (using a 488 nm laser with a 530/30 filter and a 561 nm laser with a 585/15 filter).Polymer cytotoxicity
HEK293T cells were seeded in a 96-well plate at 10000 cells per well and left to incubate for 24 hours at 37 °C in DMEM. Polymers were dissolved in serum free DMEM at 1.1 mg mL−1 and filtered through 0.22 μm filter. FBS was added and the concentration of polymer adjusted to 1 mg mL−1. The media was replaced by the media containing the polymer, serial dilution was used to incubate the cells with polymers of different concentrations and incubated for 18 hours at 37 °C. After dry exposure, cell viability was measured by using XTT assay. Cell viability was determined in triplicate in three independent sets of experiments and their standard deviation was calculated.
Methods
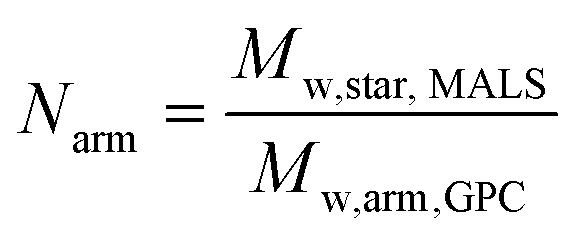 | (1) |
logM out of absolute molecular weight squared determined by light scattering against retention time with eqn (2) below: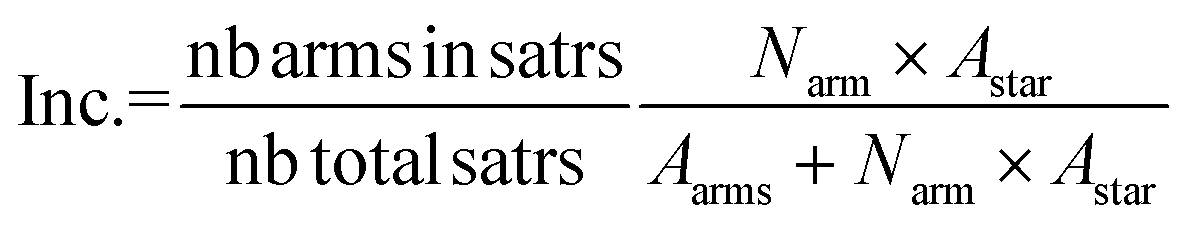 | (2) |
Synthesis
100 g mol−1, Đ = 1.02). 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.1 (–CO–O–CH2–CH2–), 2.5 ((–CO–O–CH2–CH2–), 2.3 (–C–(CH3)2 + –CH(CH2–)(CO–O–)), 2.0–1.2 (backbone).
300 g mol−1, Đ = 1.09). 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.1–3.2 (–CO–O–CH2–CH2– from DMAEA and DMAEMA + –O–CH2–CH2–N–), 2.2–2.8 ((–CO–O–CH2–CH2– from DMAEA and DMAEMA + –CH2–CH–CO– from NAM), 2.3–1.5 (–C–(CH3)2 + –CH(CH2–)(CO–O–) from DMAEA and DMAEMA + –CH2–CH– NAM backbone), 2.0–1.2 (backbone).
500 g mol−1, Đ = 1.11). 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.1–3.2 (–CO–O–CH2–CH2– from DMAEA and DMAEMA + –O–CH2–CH2–N–), 2.2–2.8 ((–CO–O–CH2–CH2– from DMAEA and DMAEMA + –CH2–CH–CO– from NAM), 2.3–1.5 (–C–(CH3)2 + –CH(CH2–)(CO–O–) from DMAEA and DMAEMA + –CH2–CH– NAM backbone), 2.0–1.2 (backbone).
900 g mol−1, Đ = 2.26). 1H NMR (400 MHz, CDCl3): δ (ppm) = 4.1–3.2 (–CO–O–CH2–CH2– from DMAEA and DMAEMA + –O–CH2–CH2–N–), 2.2–2.8 ((–CO–O–CH2–CH2– from DMAEA and DMAEMA + –CH2–CH–CO– from NAM), 2.3–1.5 (–C–(CH3)2 + –CH(CH2–)(CO–O–) from DMAEA and DMAEMA + –CH2–CH– NAM backbone + bisacrylamide), 2.0–1.2 (backbone).
Results and discussion
Star synthesis
One of the key advantages of the arm-first approach is that it enables to build complex star architectures from already well-characterised linear chains, in this case copolymers of 80% DMAEA and 20% DMAEMA, by directly chain extending them with a suitable crosslinker to interconnect the arms.41,55–57 Several studies have shown the importance of finding the best parameters, such as the choice of crosslinker, in order to optimise the synthesis and obtain core crosslinked stars with narrower disersities.43,44 The arms were synthesised using (4-cyano pentanoic acid)yl ethyl trithiocarbonate (CPAETC) as chain transfer agent (CTA), as it has been shown to efficiently control the polymerisation of both acrylate and methacrylates25,53 with 4,4′-azobis(4-cyanovaleric acid) (ACVA) as initiator at 70 °C over 24 hours. The amount of initiator was kept low at a ratio of control transfer agent to initiator ([CTA]/[I]) of 20, in order to reduce the fraction of dead chains, which would lead to excess of linear chains side products in the final product.The synthesis of stars by the arm-first approach requires an efficient chain extension, to ensure arm crosslinking and to minimise the quantity of unattached linear chains.44 A range of difunctional crosslinking monomers are available, and the choice of monomer family can have a large impact on chain extension efficiency due to the RAFT fragmentation mechanism. An acrylate derivative was first considered to match the reactivity of DMAEA.25,48 A preliminary test was performed to verify that an acrylate functionality is suitable for the chain extension: a p(DMAEA-stat-DMAEMA) copolymer was chain extended with methyl acrylate (MA) ([M]/[mCTA] = 50) but showed poor reinitiation, and GPC analysis revealed a limited fraction of the first block shifted to higher molecular weight (Fig. 1). As the reinitiation with an acrylate was not efficient, an extension of the copolymer with an acrylamide derivative – N-acryloylmorpholine (NAM) was used as model monomer – was performed to assess mCTA reactivity. The reaction reached almost full NAM conversion, as shown by 1H NMR with almost total disappearance of the vinyl peaks at 6.5, 6.3 and 5.7 ppm. The GPC traces (Fig. 1) showed a clear shift of molecular weight distribution to higher molecular weights, suggesting effective chain extension, although a small amount of macroRAFT agent still did not reinitiate. From these results, acrylamide-based crosslinkers were identified as the most suitable choice for chain extension.
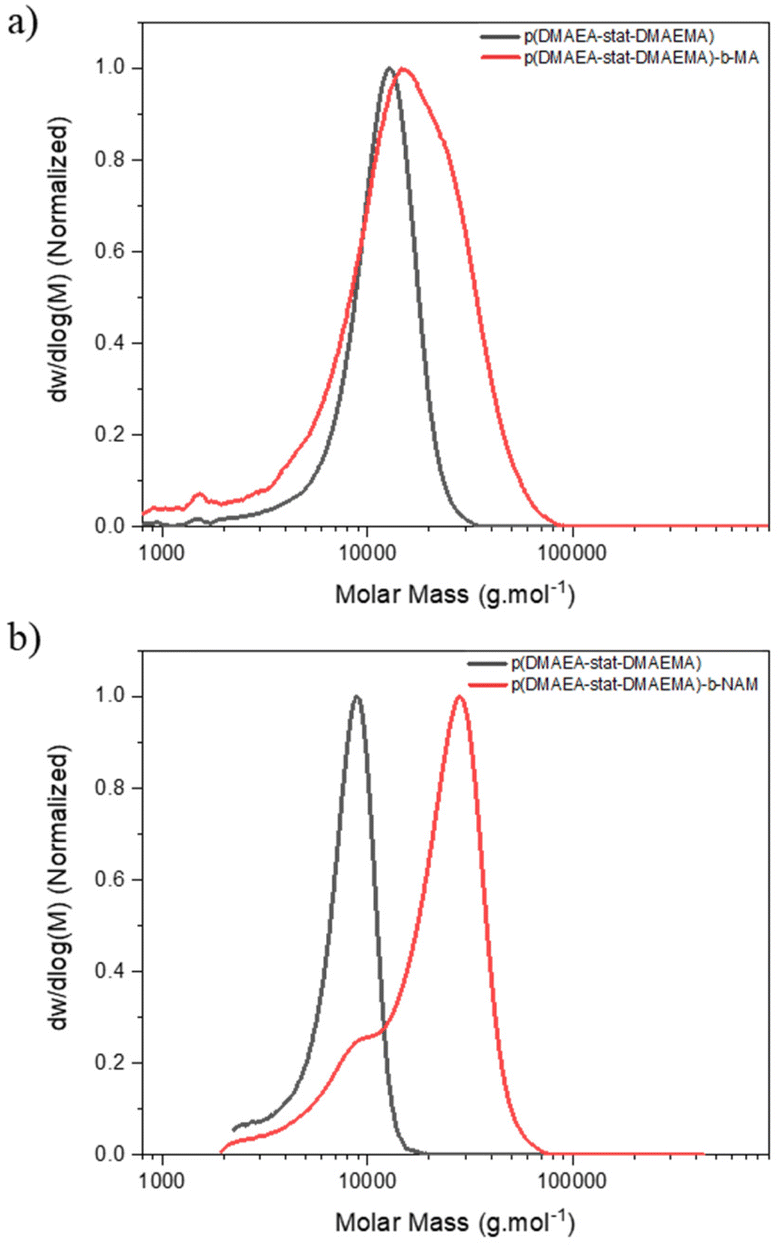 | ||
| Fig. 1 DMF-GPC chromatogram of chain extension of p(DMAEA-stat-DMAEMA) with (a) MA, (b) NAM. | ||
Methylenebis(acrylamide) was chosen as crosslinker (CL) to synthesise the stars by the arm-first approach. A purified linear p(DMAEA20-stat-DMAEMA5-b-NAM12) copolymer (Mn = 11100 g mol−1) previously synthesised was used as mCTA for chain extension. A block of NAM was added in a second step in order to obtain soluble stars after purification. Without the addition of NAM before crosslinking, the stars obtained after purification were not soluble, presumably due to the presence of charges. ACVA was used as initiator (I) at 70 °C over 3 hours (Scheme 1) and a [CL]/[mCTA] ratio of 3, a [CTA]/[I] ratio of 20 and a crosslinker concentration of 0.2 M were chosen for this reaction. The chain extension was followed by 1H NMR spectroscopy and GPC (Fig. 2a). 1H NMR showed that 95% of the double bonds of the crosslinker were consumed in 3 hours (Table 1), whilst GPC clearly provided evidence of the arms crosslinking into star copolymers, with the formation of a higher molecular weight species increasing in size over the course of the reaction. Star copolymers were obtained with a dispersity of 1.62 and molecular weight of 119900 g mol−1. Stars were then precipitated in a mixture of 70% cold diethyl ether and 30% dichloromethane, to selectively remove the unreacted arms, and dried (Fig. 2b). Similarly, stars with smaller arms (half the size) were synthesised (Star 2). Finally, in order to make the synthesis simpler by reducing the number of steps, NAM was copolymerised with DMAEA and DMAEMA in one-step to yield a statistical copolymer, rather than a block as previously described (Star 3, Table 2).
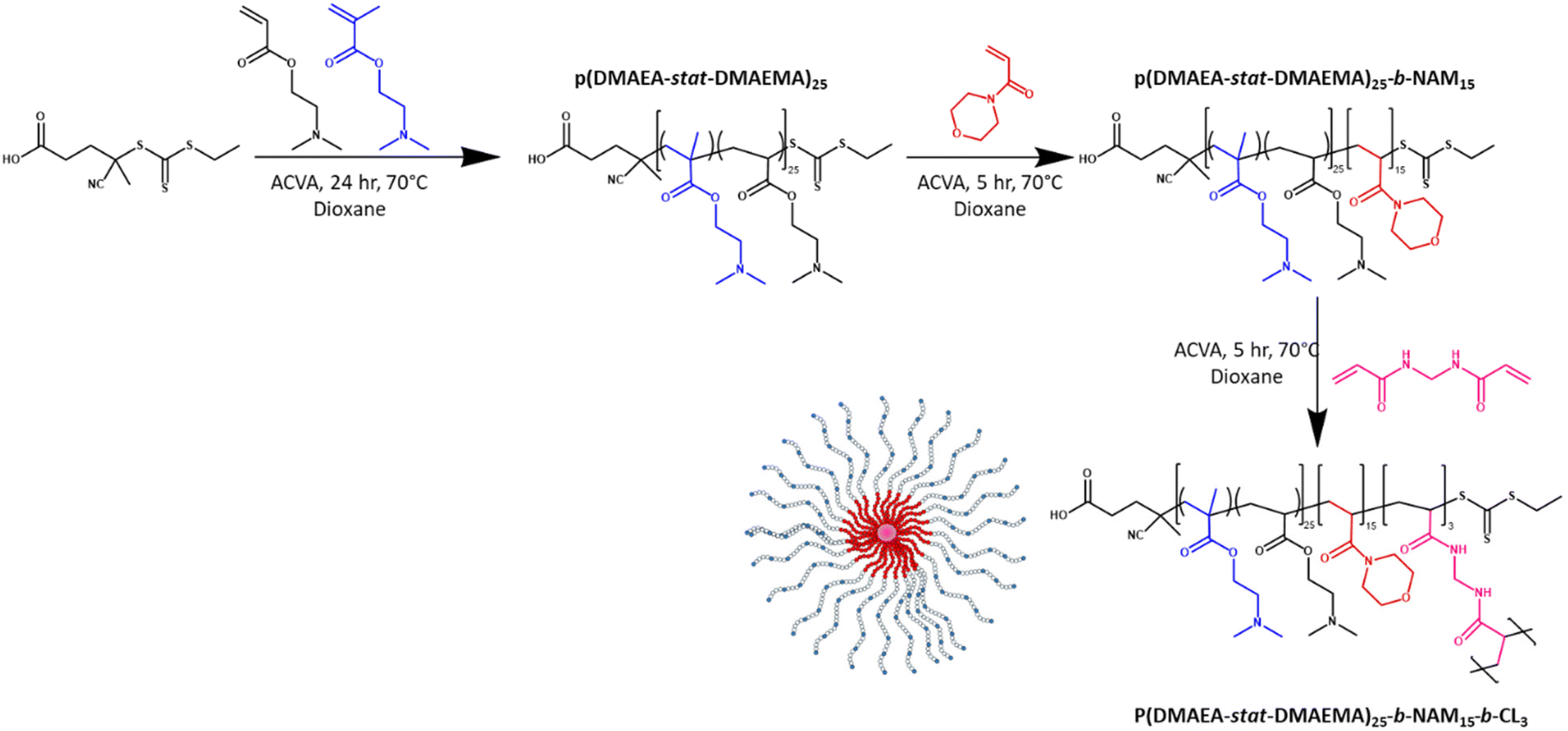 | ||
| Scheme 1 Synthesis of p(DMAEA-stat-DMAEMA)25-b-NAM15-b-CL3 star copolymer by the arm-first approach. | ||
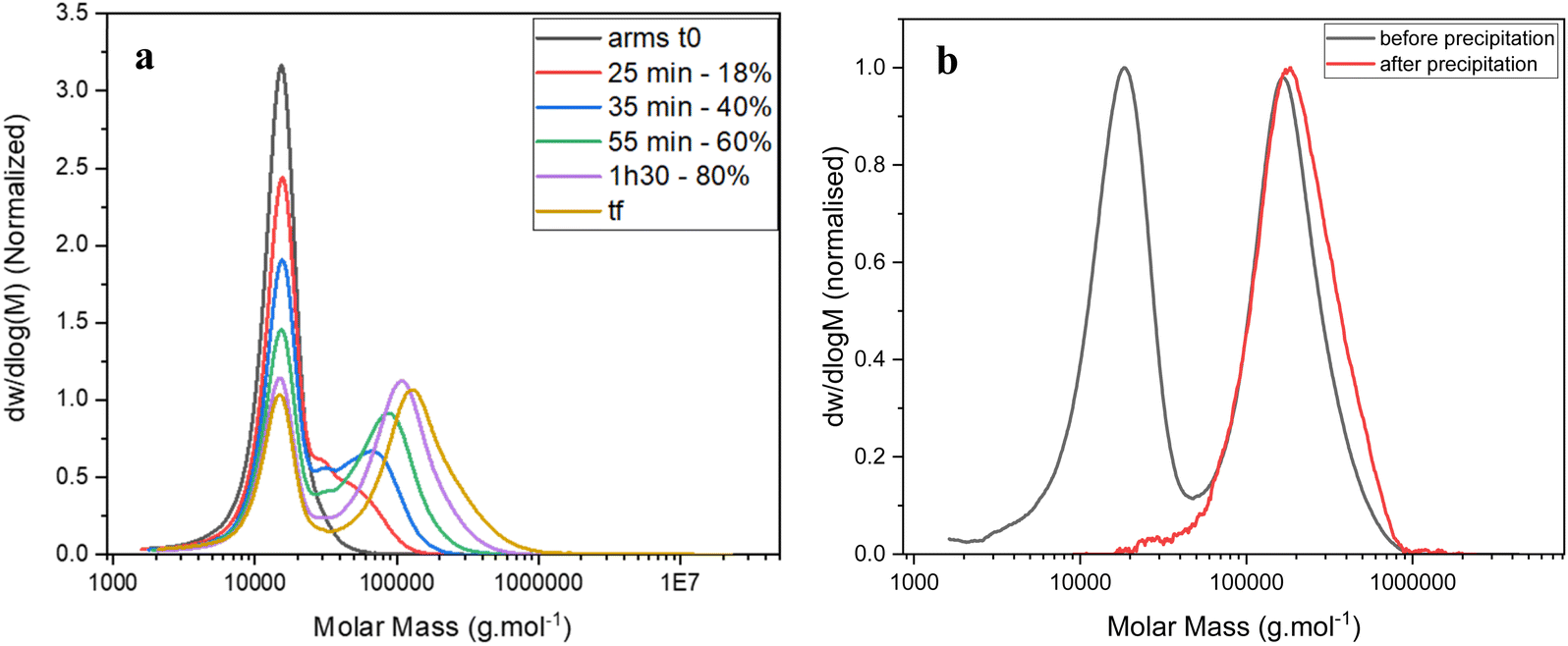 | ||
| Fig. 2 GPC traces of (a) the kinetic of star formation ([CL] = 0.2 M, [CL]/[mCTA] = 3, [mCTA]/[I] = 20) – samples are taken at 0 minute (t0), 25, 35 55 and 90 minutes and at the end of the reaction (tf – 180 minutes; 95% crosslinker conversion) – the percentage corresponds to the crosslinker conversion measured by 1H NMR; (b) star poymer at tf and after purification by precipitation, showing complete removal of unreacted arms. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| M n,th (g mol−1) | Conv.a (%) | M n, GPC (g mol−1) | M w, GPC (g mol−1) | Đ | A | R H (nm) | N arm | Arm incorp. (%) | |
| a Monomer (DMAEA/DMAEMA, NAM or crosslinker) conversions determined by 1H NMR. b Molar mass were determined by DMF-GPC, absolute molecular weight from light scattering detection, α = Kuhn–Mark–Houwink–Sakurada parameter from viscometry detector, Narm = number of arms per star, arm incorporation is calculated from RI detector. | |||||||||
| L (DMAEA-stat-DMAEMA)25 | 3500 | 91 | 11100 |
11300 |
1.02 | — | — | — | — |
| L (DMAEA-s-DMAEMA)25-b-NAM12 | 5500 | 95 | 15300 |
16600 |
1.09 | 0.41 | — | — | — |
| S (DMAEA-s-DMAEMA)25-b-NAM12 – Star 1 | — | 99 | 383900 |
868800 |
2.26 | 0.31 | 11 | 100 | 75 |
| L (DMAEA-stat-DMAEMA)12 | 1800 | 89 | 9100 | 9900 | 1.09 | — | — | — | — |
| L (DMAEA-s-DMAEMA)12-b-NAM7 | 2800 | 97 | 6200 | 6500 | 1.06 | 0.35 | — | — | — |
| S (DMAEA-s-DMAEMA)12-b-NAM7 – Star 2 | — | 99 | 41300 |
95800 |
2.32 | 0.24 | 5.5 | 65 | 83 |
| L (DMAEA-s-DMAEMA-s-NAM)40 | 5600 | 91 | 12500 |
13900 |
1.11 | 0.55 | — | — | — |
| S (DMAEA-s-DMAEMA-s-NAM)40 – Star 3 | — | 98 | 228800 |
380800 |
1.66 | 0.28 | 10 | 55 | 50 |
Star copolymer characterisation
The star copolymers were characterised by DMF-GPC and 1H NMR spectroscopy (Table 3). For the block-copolymers and the stars, triple detection was used to determine α and K values. The core crosslinked star copolymers made of the well-defined block copolymers (Đ < 1.1) showed higher dispersities (Đ = 2.2–2.35) as expected from such synthesis when the star core is obtained via crosslinking. As expected, Mark–Houwink plots yielded lower alpha values (α = 0.24–0.31) than their respective arms (α = 0.35–0.55). Analysis revealed that stars with longer arms have an average of 100 arms per star while the one with smaller arms have an average of 65 arms per star as calculated according to the eqn (1).44,54 For the stars with longer arms (Star 1), 75% of the linear chains were incorporated, against 83% for the stars with shorter arms (Star 2), presumably because of steric hindrance considerations, as the arms are shorter an there is a lower number of arm per star in Star 2. For the stars made with the statistically incorporated NAM, only 50% of the arms were incorporated. This can be explained by the less efficient reinitiation from DMAEA units compared to NAM. Hydrodynamic radius (RH) of the stars measured were in agreement as RH of the larger structures is double (10–11 nm) the one of the smaller one (5.5 nm).| Parameters | Star 1 | Star 2 | Star 3 |
|---|---|---|---|
a Error values represent the standard error associated with the fitted values.
b
N
arms represents the number of arms within each star and was held constant throughout the fitting procedure based on the number of arms determined via GPC analyses.
c
![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) g represents the number averaged radius of gyration of individual polymer chains within the star.
d
σ represents the standard deviation of the lognormal distribution in g.
g represents the number averaged radius of gyration of individual polymer chains within the star.
d
σ represents the standard deviation of the lognormal distribution in g.
|
|||
| I 0/cm−1 | 1.23 ± 0.01 | 1.13 ± 0.01 | 1.74 ± 0.02 |
| N arms | 100 | 65 | 55 |
|
g
/Å
|
77.3 ± 0.16 | 70.8 ± 0.21 | 97.9 ± 0.46 |
| σ | 0.22 ± 0.01 | 0.50 ± 0.01 | 0.59 ± 0.01 |
Stars were further characterised using SAXS and AFM. SAXS data were fitted to a polydisperse star polymer model (Fig. 3), which describes the scattering from linear Gaussian polymer chains crosslinked to a central core. The zero-angle intensity (I0) is proportional to the concentration and size of the individual stars. Other input parameters include the radius of gyration (Rg) of the individual polymer chains within the star, and the number of arms. Here, the number of arms was fixed according to the number of arms determined through GPC analysis. The Rg was fit to a lognormal distribution, where the number averaged Rg and the standard deviation were fitted parameters. The number average g was calculated for the three stars (Table 3), Star 1 had a g of 77 Å, while Star 3 with the same arm length had a g of 98 Å but with a higher standard deviation (0.59). Star 1 was better defined with a standard deviation of 0.22 (Fig. 3b). This was supported by AFM images (Fig. 4), more aggregation could be observed with Star 3 whereas better defined spheres could be observed for Star 1. Star 2 had a smaller g (71 Å) with a higher standard deviation of 0.50. This conclusion was confirmed by AFM, where small stars could be observed, forming large aggregates.
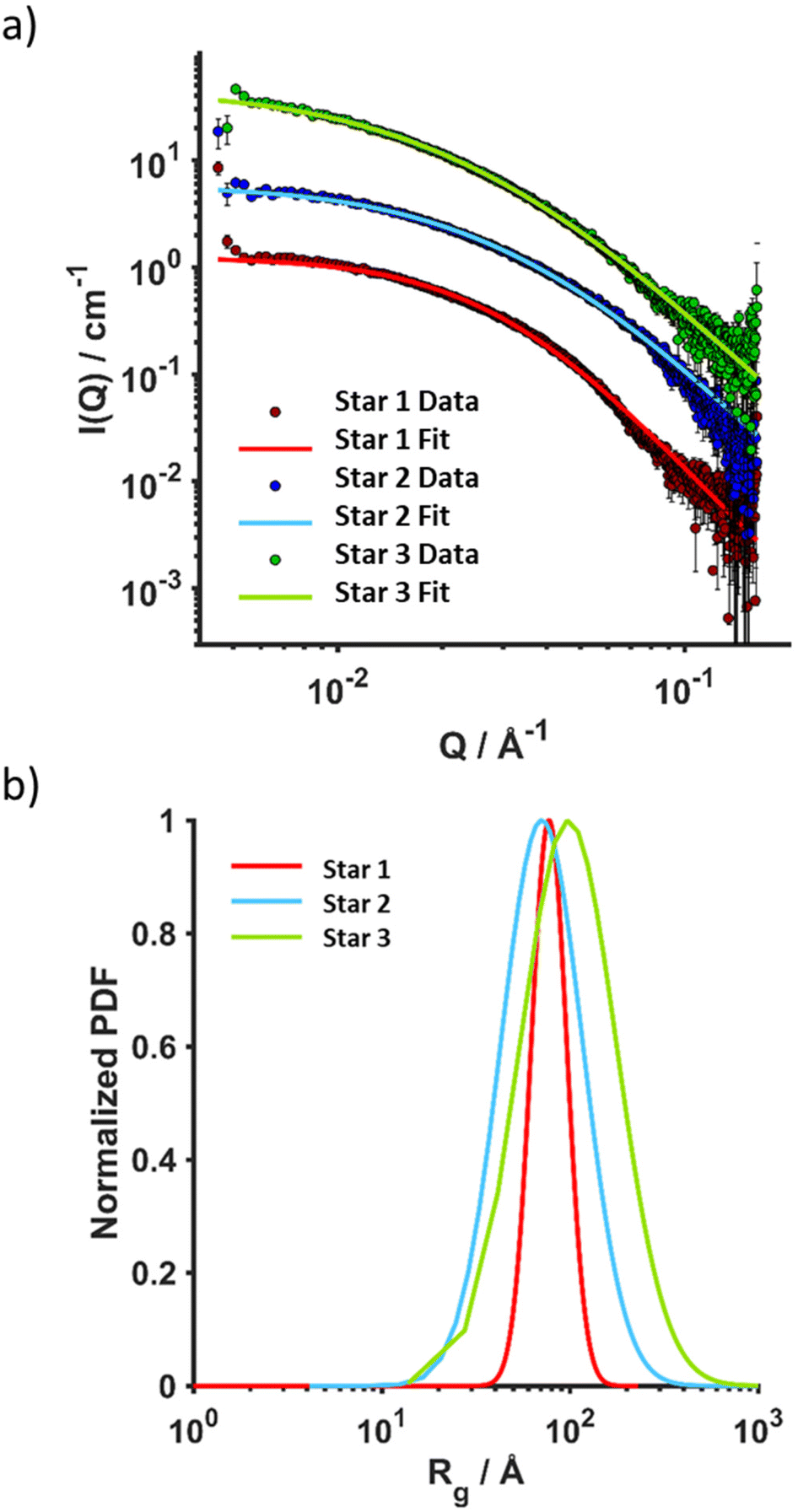 | ||
| Fig. 3 SAXS analysis in DMF (2 mg mL−1, 25 °C) of Star 1, Star 2 and Star 3. (a) Overlay of raw data and fittings (polydisperse star polymer model), (b) radius of gyration fit to a lognormal distribution. | ||
 | ||
| Fig. 4 AFM pictures of (a) Star 1, (b) Star 2 and (c) Star 3. The samples were prepared at 0.025 mg mL−1 onto freshly cleaved mica. | ||
Hydrolysis study
The effect on the hydrolysis of the DMAEA side chains of incorporating DMAEMA and NAM and of the size of the star architecture was investigated. The hydrolysis of the polymers were performed in NMR tubes in D2O at a concentration of 10 mg mL−1 and the reaction was followed by 1H NMR spectroscopy. Fig. 5a shows the spectra of star 1 over 13 days. When hydrolysis of the DMAEA side chain occurs, intensity of the peaks at 4.2 ppm, 2.7 ppm and 2.3 ppm decreased and sharp peaks appeared (3.8 ppm, 2.9 ppm and 2.6 ppm) due to small molecule by-product dimethyl aminoethanol (DMAE). The integrations of DMAE peaks in comparison to peak at 4.2 ppm were used to calculate the fraction of hydrolysis.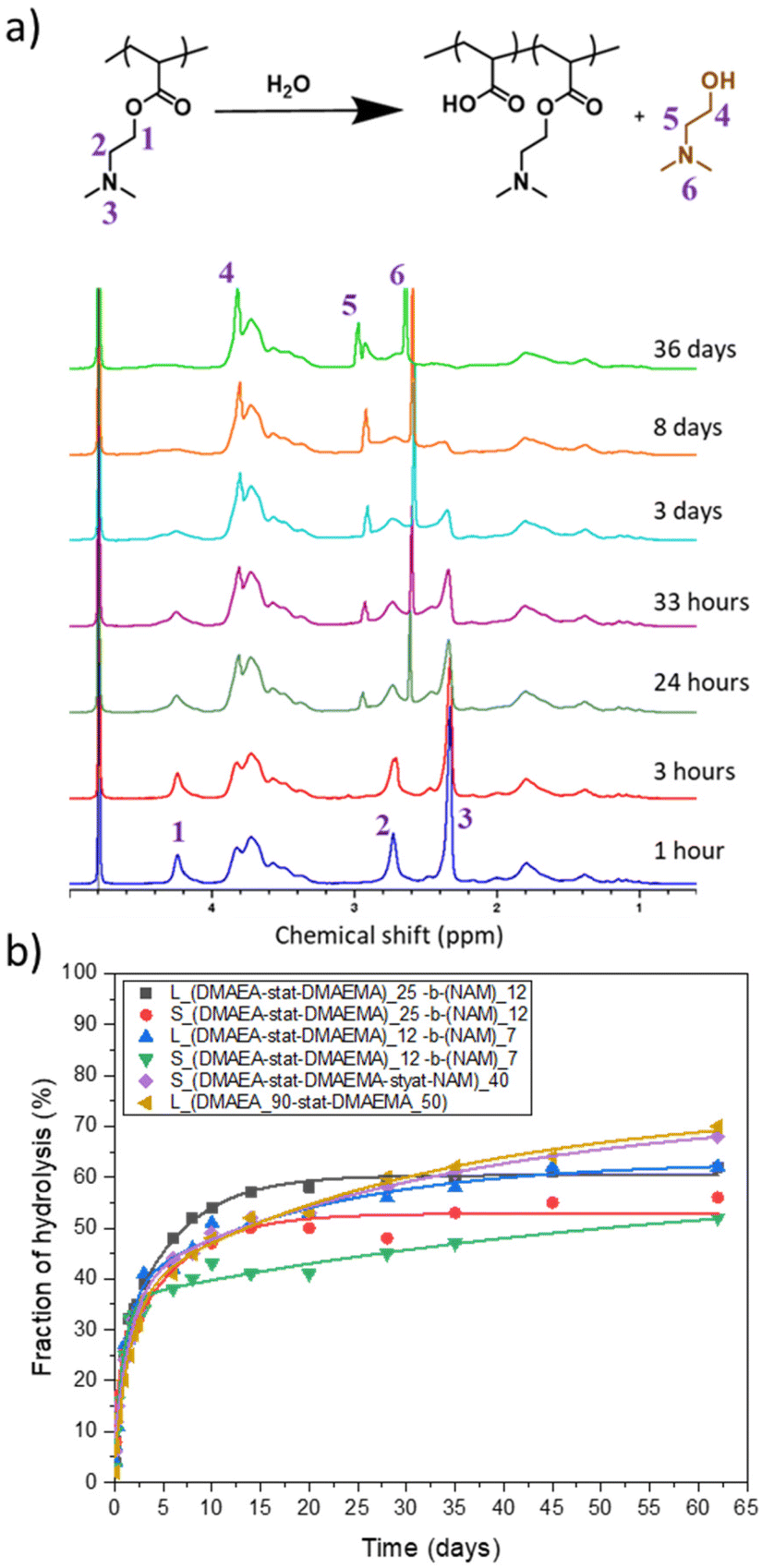 | ||
| Fig. 5 (a) 1H NMR spectrum illustrating hydrolysis of DMAEA side chains to release DMAE in D2O over 36 days for p(DMAEA-stat-DMAEAMA)25-b-NAM15 linear arms. (b) Hydrolysis kinetics of stars (S) and their corresponding arms (L) in D2O over 62 days determined by 1H NMR spectroscopy. | ||
The change from a linear to star structure was not expected to have significant influence on the hydrolysis, as indicated by previous research on branched architectures and stars with low number of arms.25,26,48 To confirm this, the hydrolysis of these structures was compared to the linear chains forming the star arms. Initially, all star copolymers hydrolysed quickly, exhibiting 30% hydrolysis in 2 days (Fig. 5b). Following this initial rapid phase, the rate of hydrolysis decreased, especially for the smaller star, Star 3, which continue to slowly hydrolyse from 33% at 2 days to 52% at 62 days. The star made of statistical DMAEA, DMAEMA and NAM arms reached 68% hydrolysis in 62 days. This might be due to the presence of the hydrophilic NAM units along the chains which improve accessibility of water molecules inside the dense structure. This was also observed by Gurnani et al., who noticed that increasing hydrophilicity by incorporating statistically hydroxyethyl acrylate (HEA) monomer led to faster hydrolysis of DMAEA.23 Compared to their arms, the hydrolysis of the stars was further slowed down and the final hydrolysis was lower, with hydrolysis reaching 56% and 52% for Star 1 and 2, respectively, whereas reaching 62% for both the arms. The accessibility to the reactive units and hydrophilicity of the polymer seems to influence the rate and extent of hydrolysis after the initial rapid period.
Polyplex formation
In order to assess the ability of the different star copolymers to complex nucleic acid, we explored polyplex formation with a 10000 basepairs plasmid DNA (pDNA) expressing green fluorescent protein (GFP). The polyplexes were characterised by agarose gel electrophoresis and ethidium bromide displacement assays. By varying the ratio of nitrogen from the polymer to phosphorus from the nucleic acid (N/P ratio), the amount of polymer necessary to fully complex the nucleic acid can be determined. Complexation was performed with pDNA at N/P ratios ranging from 0 to 10. The three star copolymers seemed to be able to fully complex pDNA from a N/P ratio of 2 (Fig. 6). Smears can be observed on the gels at N/P 1, suggesting most pDNA was complexed to the polymers but some nucleic acids were still free.
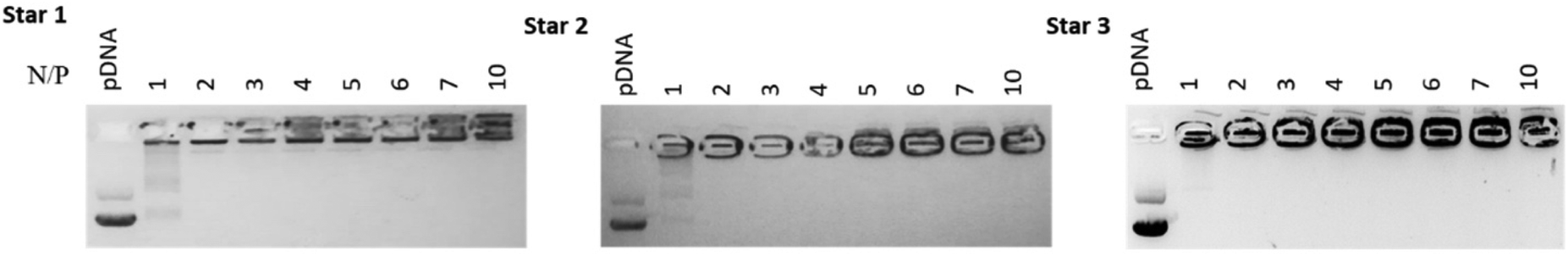 | ||
| Fig. 6 Agaraose gel electrophoresis pictures of complexation of pDNA with Star 1, 2 and 3 at N/P ratios of 0, 1, 2, 3, 4, 5, 6, 7 and 10. | ||
Ethidium bromide displacement assays were also performed to assess the strength of complexation between polymers and pDNA (Fig. 7). All compounds, including PEI, required much higher N/P ratio for displacing ethidium bromide. Displacement was at maximum at N/P ratio of 10 for all systems, with PEI displacing more pDNA (79% displacement) than the three stars. Star 1, which was designed as a larger star with dense cationic charge, seemed to bind more strongly to pDNA (over 40% displacement), than the smaller equivalent Star 2 (30% displacement) and Star 3, designed with a less dense cationic charge distribution, spaced out by the neutral NAM monomer (20% displacement). These results confirm the intuitive hypothesis that a more charged structure (Star 1) is more efficient at binding a nucleic acid, independently of its larger size.
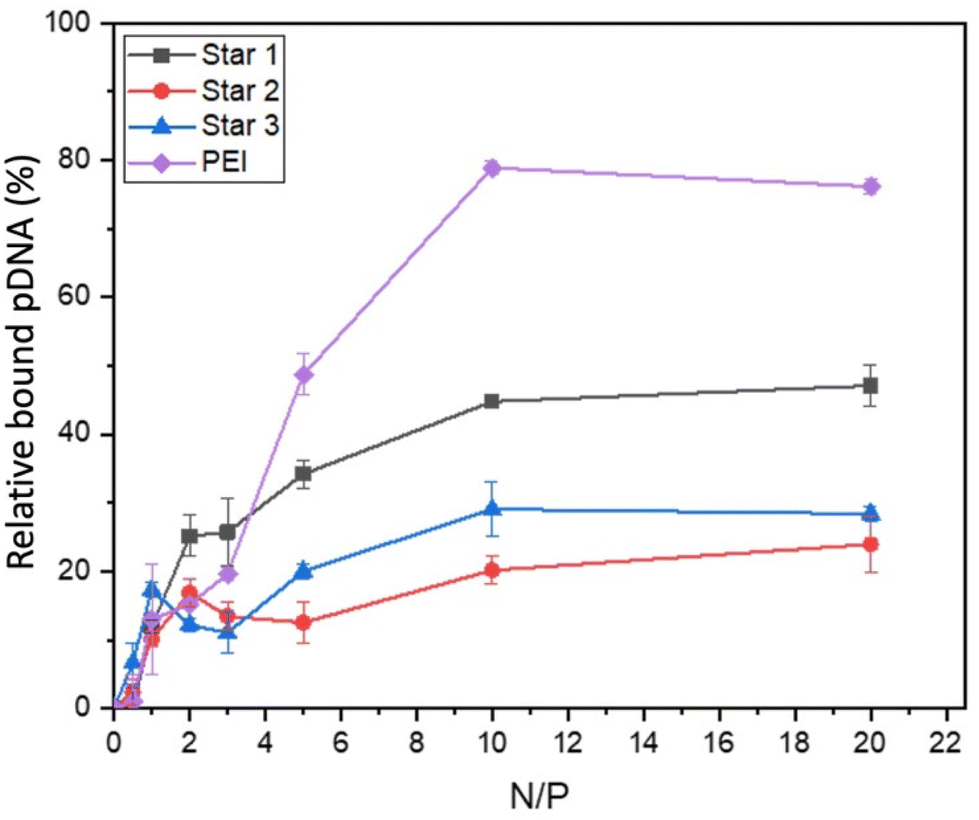 | ||
| Fig. 7 Ethidium bromide displacement of pDNA with Star 1, 2 and 3. | ||
Polymer cytotoxicity and transfection
Cytotoxicity of the polymers were first established using XTT assay on HEK293T cells (Fig. 8). HEK293T cells are a well known model to study cytotoxicity and transfection as they have a reliable growth, low maintenance and high transfectability.58 The XTT assay measures cellular metabolic activity, which is then used as an indicator for viability.59 Linear PEI was used as a reference and showed toxicity even at low concentrations (0.125 mg mL−1), as reported in literature.1 However, cells treated with either the star copolymers or the linear copolymer showed high viability of HEK293T cells, at concentrations as high as 1 mg mL−1. These results are surprising as typically, high molecular weight species have toxicity issues.14 These results are promising findings for further applications for these structures.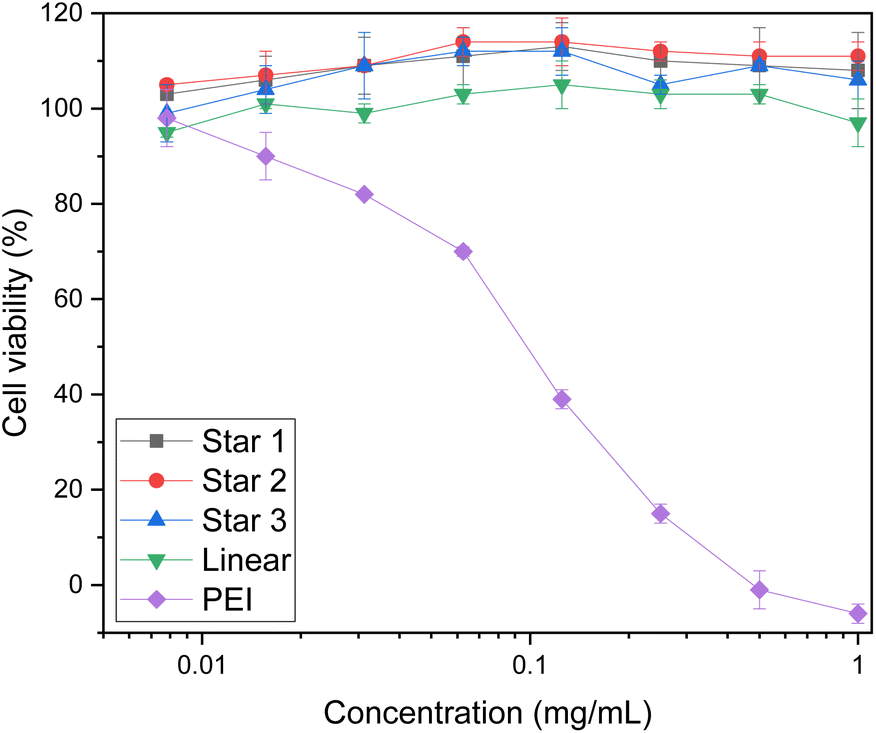 | ||
| Fig. 8 Toxicity of stars and linear copolymer of DMAEA and DMAEMA. Viability of HEK293T cells using XTT assay. | ||
Complexes of pDNA expressing GFP with Star 1, Star 2, Star 3 and p(DMAEA80-DMAEMA20) linear chain at N/P 20 were incubated with HEK293T cells. Cells were then incubated for 5 hours. After replacing the media, the cells were incubated for a further 48 hours to measure the transfection efficiency with these polymers compared to linear PEI (Fig. 9). Transfection efficiency is expressed as the percentage of cells expressing GFP, as measured by flow cytometry. Poor transfection rates were obtained for the stars compared to PEI, as a maximum of 7.6% transfection was measured for Star 1 and 5.1% for Star 2 and 1.6% for Star 3, while 31.8% was reached with linear PEI. However, the control linear copolymer of DMAEA and DMAEMA did not perform well either, as only 1.8% transfection was achieved. These results are in line with the ethidium bromide assays showing that the larger, more charged Star 1 was more efficient at binding pDNA.
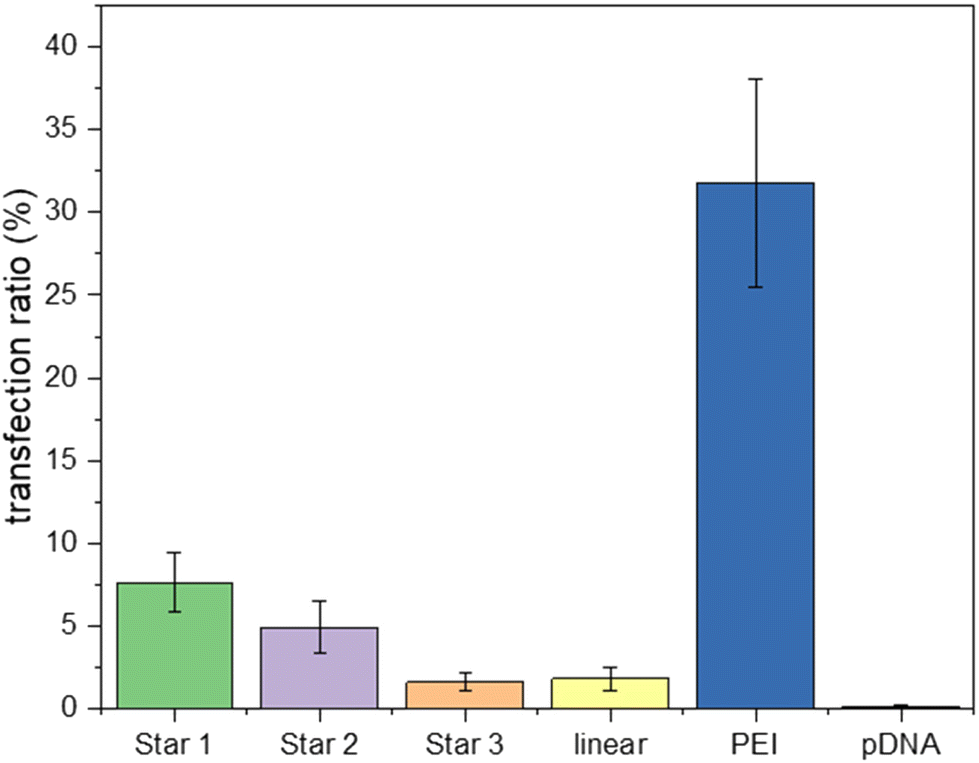 | ||
| Fig. 9 GFP pDNA transfection inHEK293T cell-line with polyplex (N/P 20) incubated for 48 hours growth. Samples were analysed by flow cytometry to determine fluorescence. | ||
Imaging using optical microscopy (see ESI†) showed large non-spherical aggregates (size of about 2 to 40 μm) corresponding to the polyplexes in these conditions and aggregation with the cells. In comparison, PEI-based polyplexes formed much smaller particles and caused less aggregation of the cells. Size is an important factor to obtain good transfection efficiency; usually particles under 500 nm result in efficient endocytosis mechanism.60,61 It is therefore possible that the size of the star-based polyplexes was responsible for the low transfection rates, with only small polyplexes being taken up by the cells. An alternative explanation for the poorer transfection efficiency when compared to that of PEI could be the lower cationic density of the pDMAEA based materials when compared to PEI. However, the higher cationic density of PEI is also source of very high cytotoxicity, as it disturbs the cell membrane, and limits the use of PEI for clinical applications. Therefore, although the star have lower transfection efficiency, their low toxicity still makes them viable materials as polyplexes.
Conclusions
Star copolymers were obtained via the chain extension of a poly(DMAEA) block with acrylamide monomers (NAM and bisacrylamide crosslinker). In order to obtain stable and soluble stars after purification and drying, a block of NAM was introduced before crosslinking to reduce the positive charge. This non-cationic part composed about 40% of the chain. Two stars of different size were synthesised with this non-cationic second block (arms: Mn ∼3500 g mol−1 and Mn ∼1800 g mol−1). A third star was synthesised with arms composed of statistical copolymer of NAM, DMAEA and DMAEMA (Mn ∼5600 g mol−1). All the architectures were obtained with a high number of arms (Narm ∼55–100) and good arm incorporation (50–83%). Better arm incorporation was observed with smaller arms and with arms extended from NAM and not DMAEA. Small differences in hydrolysis were observed after the initial period as the hydrolysed fraction of side chains were slightly higher for the arms compared to the stars. This can be explained by the dense and compact structure making the access for water more difficult for hydrolysis to occur. The three stars were able to complex a large (10000 basepairs) plasmid DNA expressing GFP, and were tested for transfection. The materials showed lower transfection efficiency than the gold standard PEI, presumably due to their lower positive charge density, but their much lower toxicity, as evidenced by XTT, still makes them a good alternative candidate for transfection applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Syngenta is gratefully acknowledged for the provision of a scholarship (F. B.). We thank the kind contribution of Ramon Garcia Maset for SEM, Andrew Kerr for AFM, Steve Huband for SAXS and Steve Hall for SAXS fitting.References
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- G. Lin, H. Zhang and L. Huang, Mol. Pharm., 2015, 12, 314–321 CrossRef CAS PubMed.
- S. Liu, M. Jaouannet, D. M. A. Dempsey, J. Imani, C. Coustau and K.-H. J. B. a. Kogel, Biotechnol. Adv., 2019, 107463 Search PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- J. Spit, A. Philips, N. Wynant, D. Santos, G. Plaetinck and K.-H. Kogel, Insect Biochem. Mol. Biol., 2017, 81, 103–116 CrossRef CAS PubMed.
- J. C. Kaczmarek, P. S. Kowalski and D. G. Anderson, Genome Med., 2017, 9, 60 CrossRef PubMed.
- N. P. Ingle, J. K. Hexum and T. M. Reineke, Biomacromolecules, 2020, 21, 1379–1392 CrossRef CAS PubMed.
- A. Elouahabi and J.-M. Ruysschaert, Mol. Ther., 2005, 11, 336–347 CrossRef CAS PubMed.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS PubMed.
- D. Bouard, N. Alazard-Dany and F. L. Cosset, Br. J. Pharmacol., 2009, 157, 153–165 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297 CrossRef CAS PubMed.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef CAS PubMed.
- M. Bauer, L. Tauhardt, H. M. L. Lambermont-Thijs, K. Kempe, R. Hoogenboom, U. S. Schubert and D. Fischer, Eur. J. Pharm. Biopharm., 2018, 133, 112–121 CrossRef CAS PubMed.
- S. P. Strand, S. Lelu, N. K. Reitan, C. de Lange Davies, P. Artursson and K. M. Vårum, Biomaterials, 2010, 31, 975–987 CrossRef CAS PubMed.
- S. B. Hartono, W. Gu, F. Kleitz, J. Liu, L. He, A. P. J. Middelberg, C. Yu, G. Q. Lu and S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS PubMed.
- R. Iwai, S. Kusakabe, Y. Nemoto and Y. Nakayama, Bioconjugate Chem., 2012, 23, 751–757 CrossRef CAS PubMed.
- J. K. Kiviaho, V. Linko, A. Ora, T. Tiainen, E. Järvihaavisto, J. Mikkilä, H. Tenhu, Nonappa and M. A. Kostiainen, Nanoscale, 2016, 8, 11674–11680 RSC.
- C. Boyer, J. Teo, P. Phillips, R. B. Erlich, S. Sagnella, G. Sharbeen, T. Dwarte, H. T. T. Duong, D. Goldstein, T. P. Davis, M. Kavallaris and J. McCarroll, Mol. Pharm., 2013, 10, 2435–2444 CrossRef CAS PubMed.
- T. J. Gibson, P. Smyth, M. Semsarilar, A. P. McCann, W. J. McDaid, M. C. Johnston, C. J. Scott and E. Themistou, Polym. Chem., 2020, 11, 344–357 RSC.
- S. Agarwal, Y. Zhang, S. Maji and A. Greiner, Mater. Today, 2012, 15, 388–393 CrossRef CAS.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- P. Gurnani, A. K. Blakney, R. Terracciano, J. E. Petch, A. J. Blok, C. R. Bouton, P. F. McKay, R. J. Shattock and C. Alexander, Biomacromolecules, 2020, 21, 3242–3253 CrossRef CAS PubMed.
- A. Cook, PhD thesis, University of Warwick, 2018.
- A. Cook, R. Peltier, M. Hartlieb, R. Whitfield, G. Moriceau, J. Burns, D. Haddleton and S. Perrier, Polym. Chem., 2018, 9, 4025–4035 RSC.
- R. Whitfield, A. Anastasaki, N. P. Truong, A. B. Cook, M. Omedes-Pujol, V. Loczenski Rose, T. A. Nguyen, J. A. Burns, S. b. Perrier and T. P. Davis, ACS Macro Lett., 2018, 7, 909–915 CrossRef CAS PubMed.
- N. P. Truong, W. Gu, I. Prasadam, Z. Jia, R. Crawford, Y. Xiao and M. J. Monteiro, Nat. Commun., 2013, 4, 1902 CrossRef PubMed.
- N. P. Truong, Z. Jia, M. Burges, N. A. McMillan and M. J. Monteiro, Biomacromolecules, 2011, 12, 1876–1882 CrossRef CAS PubMed.
- N. P. Truong, Z. Jia, M. Burgess, L. Payne, N. A. McMillan and M. J. Monteiro, Biomacromolecules, 2011, 12, 3540–3548 CrossRef CAS PubMed.
- M. McCool and E. Senogles, Eur. Polym. J., 1989, 25, 857–860 CrossRef CAS.
- L. Novo, L. Y. Rizzo, S. K. Golombek, G. R. Dakwar, B. Lou, K. Remaut, E. Mastrobattista, C. F. van Nostrum, W. Jahnen-Dechent, F. Kiessling, K. Braeckmans, T. Lammers and W. E. Hennink, J. Controlled Release, 2014, 195, 162–175 CrossRef CAS PubMed.
- H. T. Ho, M. L. Bohec, J. Frémaux, S. Piogé, N. Casse, L. Fontaine and S. Pascual, Macromol. Rapid Commun., 2017, 38, 1600641 CrossRef PubMed.
- X. Liao, N. D. Falcon, A. A. Mohammed, Y. Z. Paterson, A. G. Mayes, D. J. Guest and A. Saeed, ACS Omega, 2020, 5(3), 1496–1505 CrossRef CAS PubMed.
- T. A. Werfel, C. Swain, C. E. Nelson, K. V. Kilchrist, B. C. Evans, M. Miteva and C. L. Duvall, J. Biomed. Mater. Res., Part A, 2016, 104, 917–927 CrossRef CAS PubMed.
- J. Cai, Y. Yue, D. Rui, Y. Zhang, S. Liu and C. Wu, Macromolecules, 2011, 44, 2050–2057 CrossRef CAS.
- A. Aied, U. Greiser, A. Pandit and W. Wang, Drug Discovery Today, 2013, 18, 1090–1098 CrossRef CAS PubMed.
- A. C. Rinkenauer, S. Schubert, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2015, 3, 7477–7493 RSC.
- T. K. Georgiou, Polym. Int., 2014, 63, 1130–1133 CrossRef CAS.
- F. Dai, P. Sun, Y. Liu and W. Liu, Biomaterials, 2010, 31, 559–569 CrossRef CAS PubMed.
- D. P. Yang, M. N. N. L. Oo, G. R. Deen, Z. Li and X. J. Loh, Macromol. Rapid Commun., 2017, 38, 1700410 CrossRef PubMed.
- Q. Chen, X. Cao, Y. Xu and Z. An, Macromol. Rapid Commun., 2013, 34, 1507–1517 CrossRef CAS PubMed.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 3154–3160 CrossRef CAS.
- J. Ferreira, J. Syrett, M. Whittaker, D. Haddleton, T. P. Davis and C. Boyer, Polym. Chem., 2011, 2, 1671–1677 RSC.
- C. Bray, R. Peltier, H. Kim, A. Mastrangelo and S. Perrier, Polym. Chem., 2017, 8, 5513–5524 RSC.
- E. Vlassi, A. Papagiannopoulos and S. Pispas, Macromol. Chem. Phys., 2022, 223, 2200008 CrossRef CAS.
- K. Wang, H. Peng, K. J. Thurecht, S. Puttick and A. K. Whittaker, Polym. Chem., 2014, 5, 1760–1771 RSC.
- S. Ros, J. Wang, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2020, 53, 3514–3523 CrossRef CAS.
- M. S. Rolph, A. Pitto-Barry and R. K. O'Reilly, Polym. Chem., 2017, 8, 5060–5070 RSC.
- K. Bian and M. F. Cunningham, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 414–426 CrossRef CAS.
- R. Whitfield, A. Anastasaki, N. P. Truong, P. Wilson, K. Kempe, J. A. Burns, T. P. Davis and D. M. Haddleton, Macromolecules, 2016, 49, 8914–8924 CrossRef CAS.
- W. Zhao, P. Fonsny, P. FitzGerald, G. G. Warr and S. Perrier, Polym. Chem., 2013, 4, 2140–2150 RSC.
- N. T. D. Tran, Z. Jia, N. P. Truong, M. A. Cooper and M. J. Monteiro, Biomacromolecules, 2013, 14, 3463–3471 CrossRef CAS PubMed.
- S. C. Larnaudie, J. C. Brendel, K. A. Jolliffe and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1003–1011 CrossRef CAS.
- X. Cao, C. Zhang, S. Wu and Z. An, Polym. Chem., 2014, 5, 4277–4284 RSC.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- R. A. Patil, N. H. Aloorkar, A. S. Kulkarni and D. J. Ingale, Int. J. Pharm. Sci. Nanotechnol., 2012, 5(2), 1675–1684 Search PubMed.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Aust. J. Chem., 2006, 59, 719–727 CrossRef CAS.
- P. Thomas and T. G. Smart, J. Pharmacol. Toxicol. Methods, 2005, 51, 187–200 CrossRef CAS PubMed.
- B. Alonso, R. Cruces, A. Pérez, C. Sánchez-Carrillo and M. Guembe, J. Microbiol. Methods, 2017, 139, 135–137 CrossRef CAS PubMed.
- J. M. Morachis, E. A. Mahmoud and A. Almutairi, Pharmacol. Rev., 2012, 64, 505 CrossRef CAS PubMed.
- A. Gutjahr, C. Phelip, A.-L. Coolen, C. Monge, A.-S. Boisgard, S. Paul and B. Verrier, Vaccines, 2016, 4(4), 34 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00352c |
| This journal is © The Royal Society of Chemistry 2023 |