
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of mechanically tough poly(dimethyl siloxane) through the incorporation of acetylated cyclodextrin-based topologically movable cross-links†
Daichi
Yoshida‡
 a,
Junsu
Park‡
ac,
Naoki
Yamashita
a,
Ryohei
Ikura
ac,
Nobu
Kato
f,
Masanao
Kamei
f,
Kentaro
Ogura
f,
Minoru
Igarashi
f,
Hideo
Nakagawa
e and
Yoshinori
Takashima
*abcd
a,
Junsu
Park‡
ac,
Naoki
Yamashita
a,
Ryohei
Ikura
ac,
Nobu
Kato
f,
Masanao
Kamei
f,
Kentaro
Ogura
f,
Minoru
Igarashi
f,
Hideo
Nakagawa
e and
Yoshinori
Takashima
*abcd
aDepartment of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan. E-mail: takasima@chem.sci.osaka-u.ac.jp
bInstitute for Advanced Co-Creation Studies, Osaka University, Yamadaoka, Suita, Osaka 565-0871, Japan
cForefront Research Center, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan
dInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Japan
eShin-Etsu Chemical Co., Ltd., 4-1, Marunouchi 1 chome, Chiyoda-ku, Tokyo 100-0005, Japan
fShin-Estu Chemical Co., Ltd., Silicone-Electronics Materials Research Center, 1-10, Hitomi, Matsuida-machi, Annaka-shi, Gunma 379-0224, Japan
First published on 16th June 2023
Abstract
Poly(dimethyl siloxane) (PDMS) has been widely utilized in various fields of research. However, the weak mechanical properties of PDMS have limited the widespread application of this material in industry. Herein, we incorporated a movable cross-link as a topological cross-link into linear thiol-modified PDMS chains to prepare PDMS elastomers. Triacetylated γ-cyclodextrin (TAcγCD) was modified on PDMS chains through thiol–ene click chemistry. Both the Young's modulus and the toughness of the obtained PDMS elastomers with an appropriate modification ratio (Young's modulus, 31.1 MPa; toughness 30 MJ m−3) were approximately one hundred times higher than those of chemically cross-linked PDMS elastomers (Young's modulus, 0.3 MPa; toughness, 0.47 MJ m−3). The PDMS elastomers have advantages in terms of energy dissipation mechanisms as well as mechanical properties. The movable cross-links contributed to larger mechanical hysteresis areas and faster relaxation behavior. Structural studies involving differential scanning calorimetry and X-ray scattering measurements revealed that phase separation occurred by the addition of cyclodextrin, which changed the glass transition temperatures. Moreover, the nanometer-scale phase separation structure was attributed to good mechanical properties. We expect that these topological cross-links in PDMS elastomers will expand the material design strategies for noncarbon-based elastomers.
1. Introduction
Biocompatible, transparent, and soft materials are desirable in a wide range of fields, such as industrial, medical, and academic fields. Poly(dimethyl siloxane) (PDMS) possesses the aforementioned features and thus been applied as a matrix material in various wearable sensors,1–9 wearable generators,10–14 and cellular culture matrices.15–21 However, the weak mechanical properties of PDMS have limited its industrial applications.22 Scientists have revealed that the energy dissipation mechanism is a key factor in improving the mechanical properties of carbon-based polymeric materials.As effective energy dissipation, sacrificial networks,23–27 chain length distributions,28 sliding motion of cross-links based on rotaxane structures,29–40 appropriate relaxation behaviors,41–44 and reversible dynamic bonds such as hydrogen bonds,45–47 π–π interactions,48 host–guest complexation,49–51 coordination bonds,52 and ionic bonds53,54 have been reported in carbon-based materials. Among the numerous strategies described above, the incorporation of reversible dynamic bonds55–58 and topological cross-links59 have mainly improved the mechanical properties of PDMS materials.
Topological cross-links are interesting, as cross-links themselves can move along polymeric chains. Herein, we report the preparation of mechanically tough PDMS elastomers by incorporating cooperatively movable topological cross-links consisting of PDMS chains bearing triacetylated γ-cyclodextrin (TAcγCD) and PDMS chains passing through TAcγCD. Instead of using conventional reactions to incorporate cyclodextrin onto PDMS using a Karstedt catalyst,60 we simply modified TAcγCD onto the PDMS main chain by metal-free thiol–ene click chemistry. These cooperatively movable cross-links have effectively improved the mechanical toughness of carbon-based polymeric materials. As polymeric chains and TAcγCD are covalently connected, TAcγCD moves along the passing PDMS and vice versa. In addition, movable cross-links have been used to successfully mix generally immiscible polymers, such as polystyrene mixed with poly(ethyl acrylate)37 and poly(dimethyl acrylamide) mixed with poly(ethyl acrylate-co-butyl acrylate).43 We studied these tough PDMS elastomers in terms of mechanical properties and structures using tensile tests, cyclic tensile tests, stress relaxation tests, differential scanning calorimetry (DSC), and ultrasmall- and small-angle X-ray scattering (USAXS and SAXS) measurements. We found that the topological movable cross-links enhanced the mechanical toughness of the PDMS elastomers.
2. Results and discussion
2.1. Preparation of CD-modified PDMS on their side chains
We designed thiol-modified PDMS (PDMS-SH) for further modification reactions through a thiol–ene reaction. To prepare PDMS-SH, we first polymerized (3-mercaptopropyl) methyl-dimethoxysilane (MMDMS) using HCl to obtain poly(MMDMS) (PMMS) (Scheme S1†). 1H nuclear magnetic resonance (NMR) measurements showed that PMMS was prepared, as the molar ratio between the methyl group and thiol group was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S1†). 29Si NMR measurements showed that the obtained PMMS was a hexamer (two end units and four repeating units, Fig. S2†). The PMMS was mixed with octamethylcyclotetrasiloxane and hexamethyldisiloxane in the presence of trifluoromethanesulfonic acid to produce PDMS-SH (Scheme S2†). According to the 1H and 29Si NMR spectra of PDMS-SH, the molar ratios between the dimethyl siloxane repeating unit and the thiol-modified repeating unit were 9:1 and 10:1, respectively (Fig. S3 and S4†). We chose a ratio of 9:1 on the basis of the 1H NMR spectrum.
:1 (Fig. S1†). 29Si NMR measurements showed that the obtained PMMS was a hexamer (two end units and four repeating units, Fig. S2†). The PMMS was mixed with octamethylcyclotetrasiloxane and hexamethyldisiloxane in the presence of trifluoromethanesulfonic acid to produce PDMS-SH (Scheme S2†). According to the 1H and 29Si NMR spectra of PDMS-SH, the molar ratios between the dimethyl siloxane repeating unit and the thiol-modified repeating unit were 9:1 and 10:1, respectively (Fig. S3 and S4†). We chose a ratio of 9:1 on the basis of the 1H NMR spectrum.
TAcγCD-modified PDMS was prepared through a thiol–ene reaction between triacetylated 6-acrylamido methylether-γ-cyclodextrin (TAcγCDAAmMe) and PDMS-SH in the presence of 1-pentene (Pen) to protect residual thiol groups (PDMS-TAcγCD-Pen(x), where x refers to the mol% of TAcγCD among all repeating units, as shown in Fig. 1a and Scheme S3.† In addition, we carried out PDMS modification with various x values from 1 to 3 (1, 1.5, 1.8, 2, 2.3, 2.5, and 3) to confirm the effect of TAcγCD (Table S1†). The 1H NMR measurements of PDMS-TAcγCD-Pen(x) showed that the modification ratios similarly corresponded to the feeding ratios with the representative samples (x = 1, 2, and 3) (Fig. S5–7†). The PDMS-TAcγCD-Pen(x) samples were found to have x values of 0.8, 1.46, and 2.54 when the intent was to prepare samples with x = 1, 2, and 3, respectively, implying that the modification reaction proceeded almost quantitatively (Table S2†). The 13C NMR spectrum also confirmed that the desired polymers were prepared (Fig. S8†). Subsequently, we prepared a negative control consisting of chemical cross-links (CCPDMS), as shown in Fig. 1b, Scheme S4, and Fig. S9.† The obtained PDMS-TAcγCD-Pen(x) and CCPDMS films were colorless and transparent, as shown in Fig. 1c and d. Moreover, we also confirmed the chemical structures of the obtained polymers by Fourier transform infrared spectroscopy (Fig. S10†).
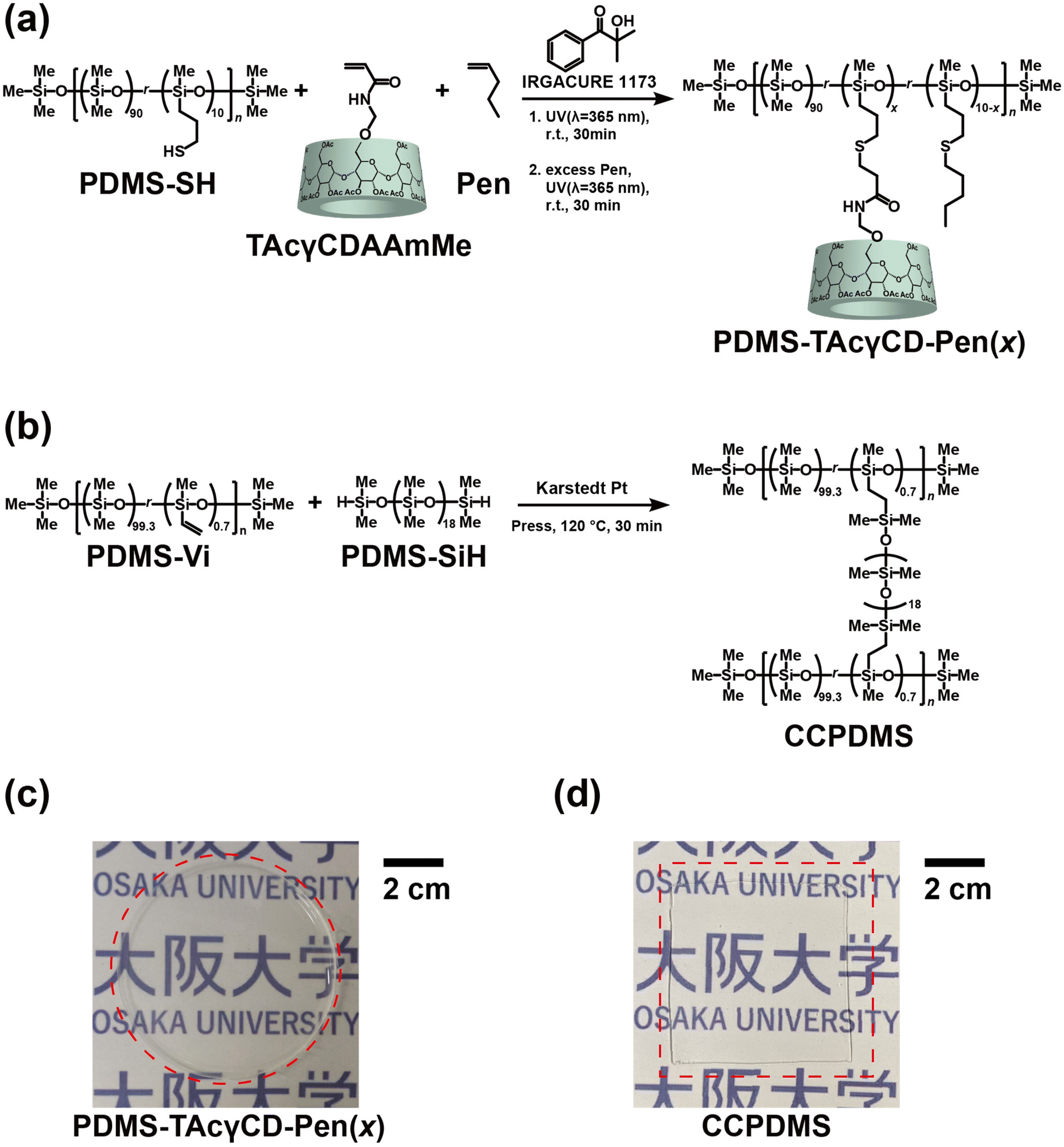 | ||
| Fig. 1 Preparation scheme of (a) PDMS-TAcγCD-Pen(x), where x refers to mol% TAcγCD in the repeating units, and (b) CCPDMS. Photographs of (c) PDMS-TAcγCD-Pen(x) and (d) CCPDMS. | ||
2.2. Mechanical properties of the CD-modified PDMS
The mechanical properties of PDMS-TAcγCD-Pen(x) were evaluated through tensile tests. The stress–strain curves of PDMS-TAcγCD-Pen(x) with various x = 1, 1.5, 1.8, 2, 2.3, 2.5 and 3 were plotted, as shown in Fig. 2a. When x = 1, the stress–strain curve was triangle-shaped. The initial slopes of the stress–strain curves, which defined the Young's modulus, increased gradually with increasing x. The positive correlation between the Young's modulus and x implied that cross-links existed in PDMS-TAcγCD-Pen(x) and that TAcγCD played an important role in forming the cross-links. CD forms rotaxane structures under appropriate conditions. Based on the rotaxane structures, sliding cross-links29,30 and movable cross-links36,38,43 have been reported in carbon-based materials, suggesting probable movable cross-links involving TAcγCD in PDMS-TAcγCD-Pen(x).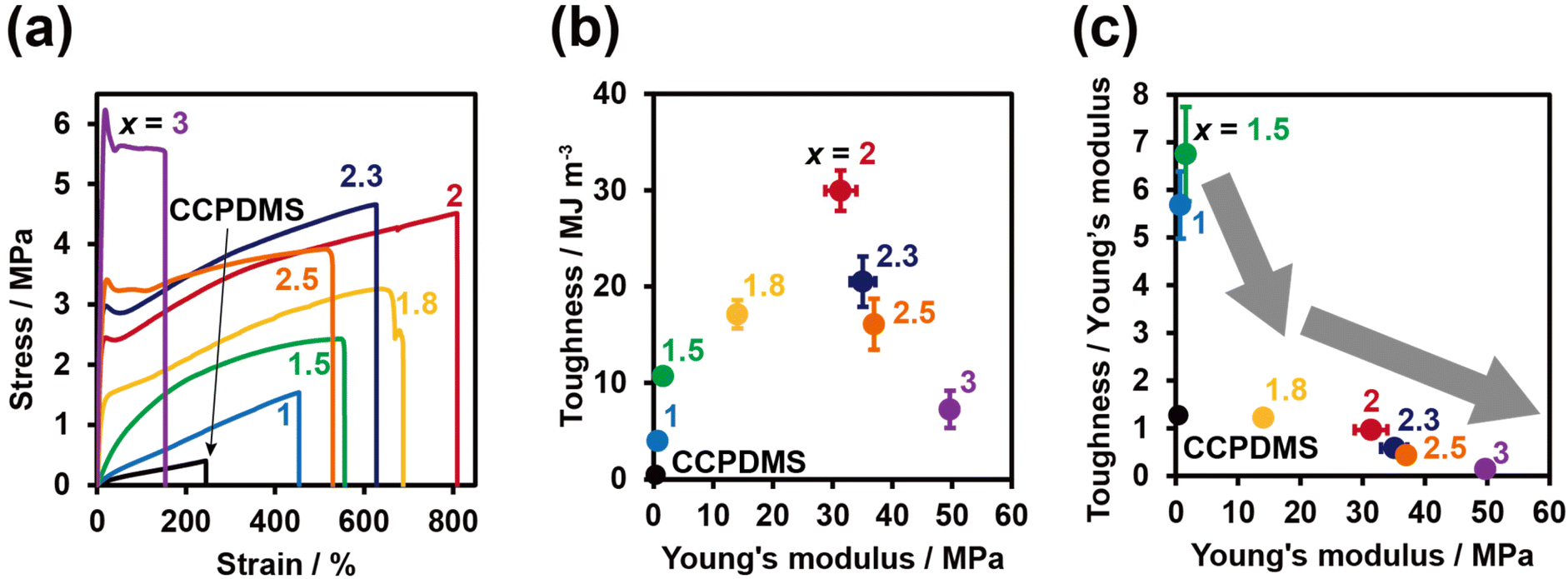 | ||
| Fig. 2 (a) Stress–strain curves, (b) two-dimensional toughness versus Young's modulus plots, and (c) toughness/Young's modulus versus Young's modulus plots of PDMS-TAcγCD-Pen(x) with various TAcγCD contents. | ||
The elongation at break, defined as the strain at a fracture point, gradually increased until x = 2. The addition of more than 2 mol% TAcγCD gradually decreased the elongation at break. This tendency was completely consistent with that previously reported for movable cross-links.36 An excessive amount of TAcγCD limited the movable ranges. Therefore, we postulated that TAcγCD formed movable cross-links in PDMS-TAcγCD-Pen(x).
We calculated the toughness from the area between the stress–strain curves and the strain axis. A two-dimensional plot of toughness versus the Young's modulus showed that the toughness was maximized when x = 2 (Fig. 2b). The similarity between this trend and that of the elongation at break indicates that the elongation break is the major factor influencing the toughness. The toughness of CCPDMS was extremely low compared with that of all PDMS-TAcγCD-Pen(x) samples. The movable cross-links contributed to the energy dissipation due to the sliding motion of TAcγCD along the PDMS main chain. Regarding the change in toughness itself, we plotted the mean toughness/mean Young's modulus (toughness/Young's modulus) versus the Young's modulus, as the Young's modulus usually influences the toughness (Fig. 2c). From x = 1.5 to x = 1.8, the toughness/Young's modulus showed a steep decrease, while the decrease became moderate when x ≥ 1.8. That is, a sufficient amount of TAcγCD contributed to maintaining the toughness with increasing Young's modulus. We postulated that the movable cross-links started working effectively when x ≥ 1.8.
2.3. Energy dissipation properties of the CD-modified PDMS
As the results of the tensile tests implied the formation of movable cross-links in PDMS-TAcγCD-Pen(x) and energy dissipation effects, we investigated their energy dissipation properties through stress-relaxation tests and cyclic tensile tests. The cyclic tensile tests were performed with various strains, as summarized in Table S3.† The stretched specimens were then returned directly to their original size. In addition, the specimens were gradually stretched more with increasing cycle numbers. The strain for the cyclic tensile tests was chosen based on the elongation at break of each sample. We calculated the hysteresis loss values defined as ratios between an area during the stretching process and an area during the returning process (Fig. S11†). The hysteresis loss values at the last three cycles were averaged, and the results were summarized in Fig. 3a. CCPDMS showed only 10% hysteresis loss, which was consistent with the behavior of conventional cross-linked materials. In contrast, the hysteresis loss of PDMS-TAcγCD-Pen(x) increased gradually with increasing x until x = 2, which was consistent with previously reported polymeric materials consisting of movable cross-links.36 Moreover, the hysteresis loss was maintained at more than 60% when x ≥ 2 despite the decreased strain.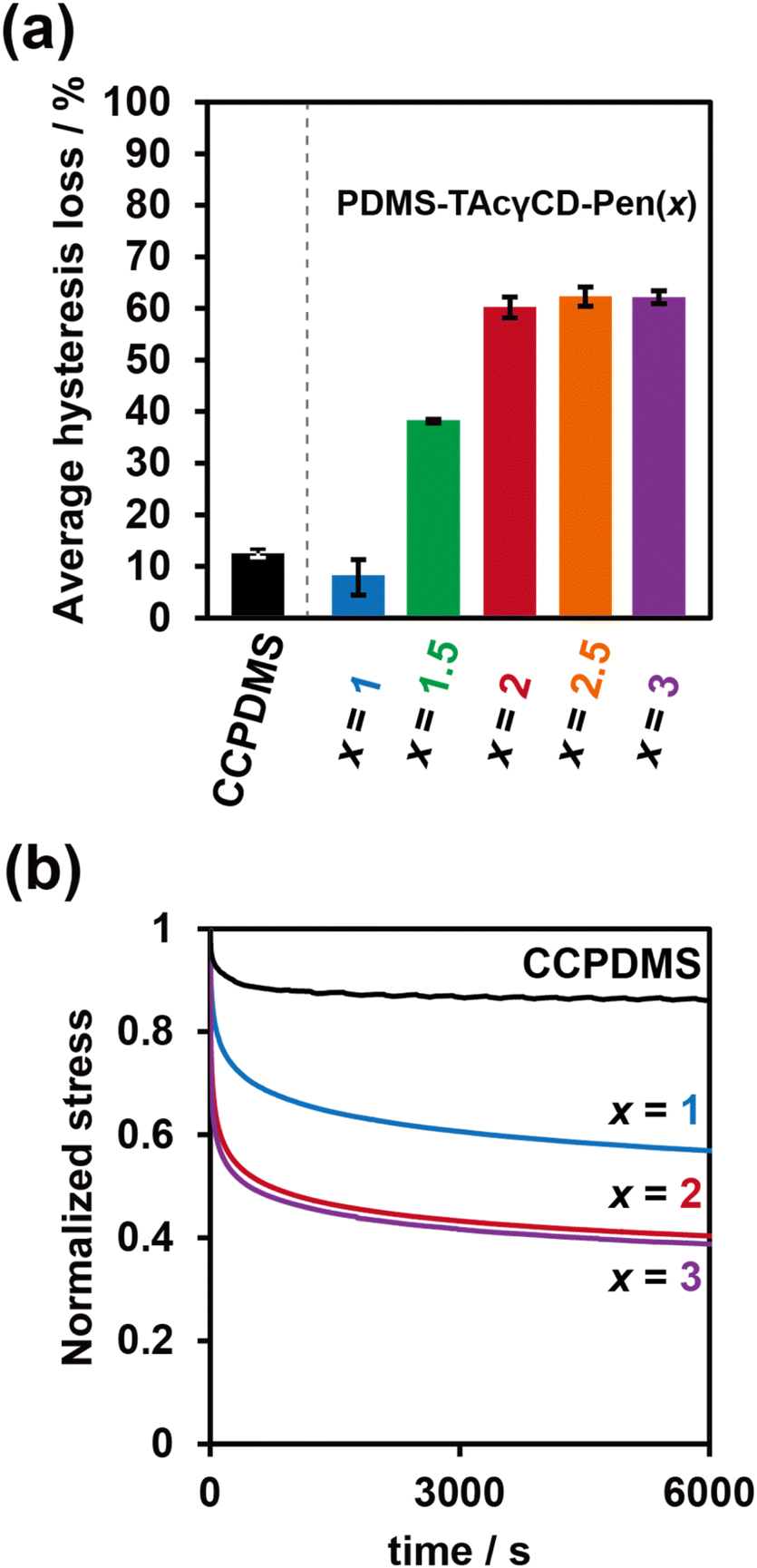 | ||
| Fig. 3 (a) Averaged hysteresis loss calculated from the last three cycles and (b) stress relaxation curves of CCPDMS and PDMS-TAcγCD-Pen(x) with various TAcγCD contents. For the stress relaxation curves, the stresses were normalized with initial stresses of each sample. | ||
Subsequently, stress relaxation tests were carried out with various strains, as summarized in Table S4.† Basically, the specimens were stretched until a preset strain and held for 6000 seconds. The stress relaxation behaviors were plotted with a normalized stress in Fig. 3b. For a quantitative evaluation of the stress relaxation behaviors, we tried to fit the stress relaxation curves based on Kohlrausch–Williams–Watts (KWW) models (1).
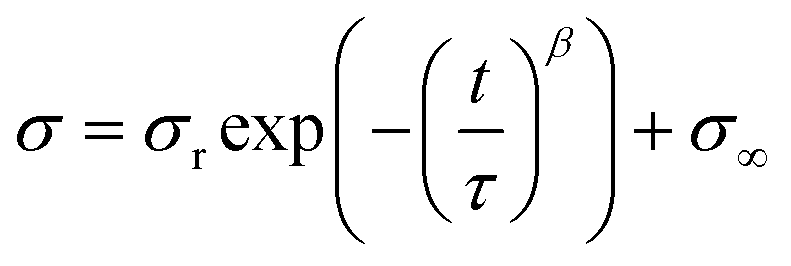 | (1) |
| PDMS materials | Relaxable components | Residual components | ||
|---|---|---|---|---|
| σ r /σoe | τ (second) | β | σ ∞ /σoe | |
| a Relaxable stress. b Relaxation time. c Stretching exponent. d Residual stress. e Initial stress. | ||||
| CCPDMS | 0.16 | 255 | 0.34 | 0.84 |
| PDMS-TAcγCD-Pen(1) | 0.40 | 161 | 0.37 | 0.62 |
| PDMS-TAcγCD-Pen(2) | 0.65 | 36 | 0.30 | 0.44 |
| PDMS-TAcγCD-Pen(3) | 0.58 | 35 | 0.29 | 0.43 |
Compared with CCPDMS, PDMS-TAcγCD-Pen(x) had a shorter relaxation time with increasing amounts of TAcγCD. TAcγCD was considered an important factor for stress relaxation. The relaxable stresses of PDMS-TAcγCD-Pen(x) were larger than those of CCPDMS. Moreover, the relaxable stress was maximized with x = 2, corresponding to the results of the tensile tests.
When we carried out the fitting for the entire 6000 seconds, the obtained fitting curves appeared to be fitted well at glance. However, the fitting curves were not matched as well as the curves fitted with the initial 600 seconds range (Fig. S13 and Table S5†). The R2 value of CCPDMS when fitted over 6000 seconds (0.990) was smaller than that obtained until 600 seconds (0.997). Since the presence of movable cross-links based on TAcγCD shortens the relaxation time,37,38 we discussed the relaxation behaviors focusing on the early stage by analyzing the fitting curves. The longer relaxation time of PDMS-TAcγCD-Pen(1) and PDMS-TAcγCD-Pen(3) compared with CCPDMS are logically unreasonable and support the suitability of the fitting within the initial 600 seconds. Although the fitting curve of CCPDMS matched well at longer time regimes (>600 seconds), those from PDMS-TAcγCD-Pen(x) did not match well and exhibited more relaxation at longer times. These results implied an additional relaxation mechanism that relaxes stress at longer times, in addition to the movable cross-links that relax stress at short time.
2.4. Structural studies of CD-modified PDMS on a molecular scale
We first studied the internal structures of PDMS-TAcγCD-Pen(x) at the molecular scale using NMR and DSC. We carried out two-dimensional NOESY NMR measurements to confirm our hypothesis. The PDMS-SH underwent thiol–ene modification with Pen in the presence of TAcγCD (no vinyl group) to prepare PDMS-Pen/TAcγCD (Fig. 4a and Scheme S5†). The mixture showed significant cross-peaks between interior protons (H3 and H5) and methyl groups on the repeating units, suggesting rotaxane structures (Fig. 4b).59,61 This NOESY NMR spectrum indicated that movable cross-links formed in PDMS-TAcγCD-Pen(x) during the thiol–ene modification reaction. Consequently, the formation of movable cross-links was confirmed by mechanical tests and NMR spectroscopy.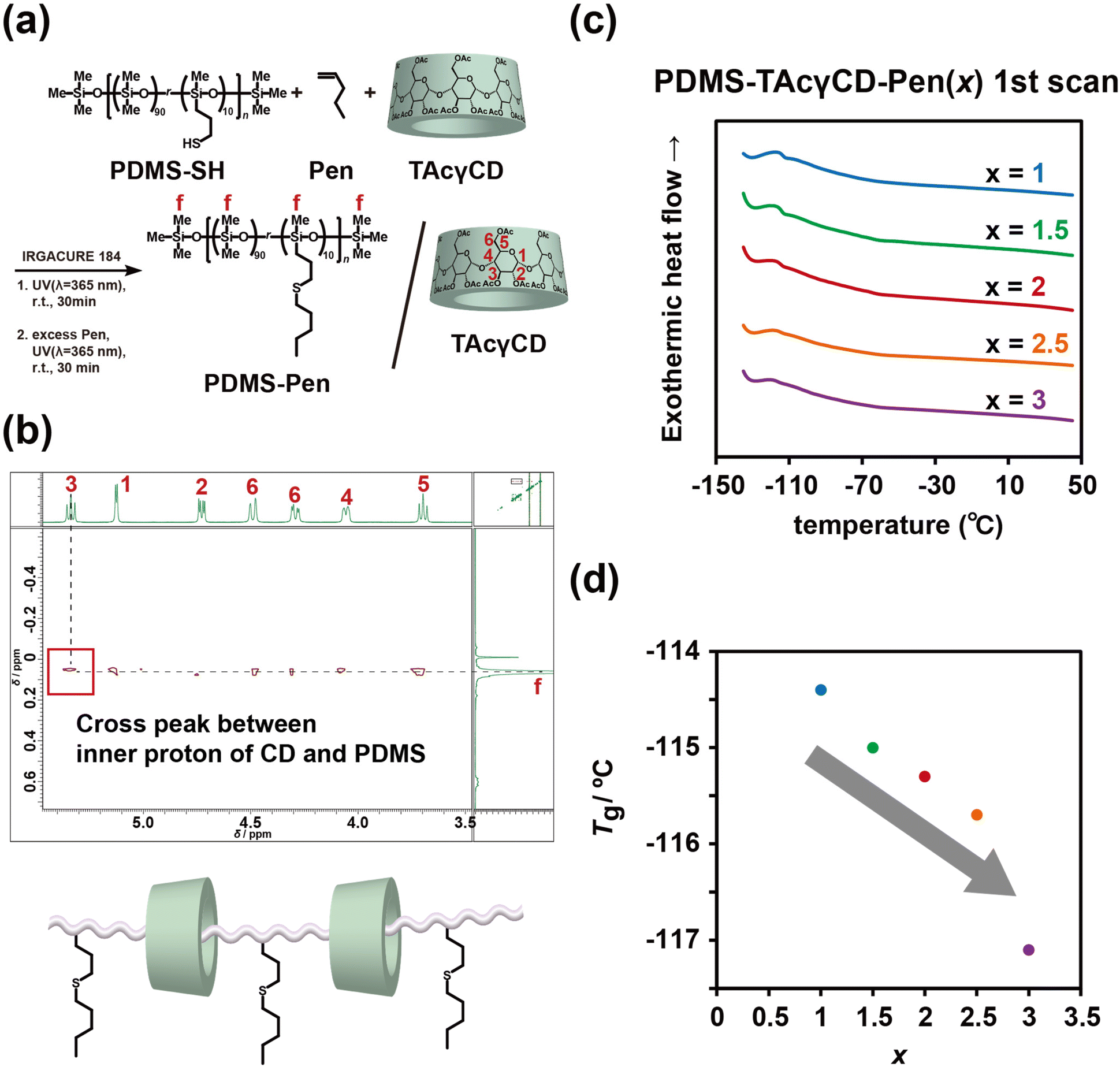 | ||
| Fig. 4 (a) Chemical structures of PDMS-Pen and TAcγCD for structural studies through NMR. (b) Two-dimensional NOESY NMR spectrum of a mixture of PDMS-Pen and TAcγCD and a proposed topologically bound structure based on the NOESY NMR spectrum. (c) DSC thermograms of PDMS-TAcγCD-Pen(x) obtained from the first heating scan. (d) Summary of the glass transition temperature with various amounts of TAcγCD. | ||
Then, we investigated the thermal properties of PDMS-TAcγCD-Pen(x). The samples were cooled to −140 °C and heated to 50 °C with a 10 °C min−1 scan rate (first scan, Fig. 4c). All of them showed glass transition temperatures (Tg) of approximately −120 °C. The relationship between the TAcγCD content and Tg were plotted in Fig. 4d. In general, more cross-links usually lead to a higher Tg due to the decrease in free volume. However, interestingly, more TAcγCD resulted in a lower Tg. These results can be explained in terms of free volume theory.62 The extremely low Tg of PDMS-TAcγCD-Pen(x) implied very large free volumes due to flexible bonds. Recently, it was revealed that only ∼22 mol% of the fed TAcγCD participated in forming movable cross-links in poly(ethyl acrylate) (PEA) elastomers.63 Furthermore, the majority of the movable cross-links were untrapped.
The movable cross-link forming stage was the main difference between the previous PEA elastomers and PDMS-TAcγCD-Pen(x). In the case of PEA elastomers, TAcγCDAAmMe was dissolved in liquid ethyl acrylate and polymerized in a bulk state. In contrast, PDMS-TAcγCD-Pen(x) was prepared by modifying TAcγCDAAmMe onto PDMS-SH. As TAcγCD forms the rotaxane structure easily with the former approach,64 the majority of the cavity of fed TAcγCD for PDMS-TAcγCD-Pen(x) should be unoccupied. Moreover, the flexible chain-end of PDMS-TAcγCD-Pen(x) could interrupt further formation of the movable cross-links because chain-end recognition of TAcγCD to form the rotaxane structure was difficult. We postulated that the free volume in PDMS-TAcγCD-Pen(x) increased because the formation of entanglements was interrupted with the sterically bulky free TAcγCD. The increase in the free volume resulted in a lower Tg. For the second scan, we also carried out DSC under different conditions: all PDMS-TAcγCD-Pen(x) samples were first heated to 150 °C and then cooled to −140 °C. Then, they were heated to 150 °C again with the same scan rate (10 °C min−1) (Fig. S14†). The Tg measured by the second scans was always lower than that measured by the first scans. In addition, the decrease in Tg became moderate. Rapid cooling from 150 °C seemed to induce larger free volumes. In addition, the melting point of CCPDMS at −45 °C disappeared with TAcγCD modification. This result indicated that TAcγCD inhibited the crystallization of PDMS chains, supporting the larger free volumes with TAcγCD of PDMS-TAcγCD-Pen(x) compared with CCPDMS. The inhibited PDMS chain crystallization seemed to be observed in the additional relaxation in Fig. S12.†
2.5. Structural studies of PDMS-TAcγCD-Pen(x) in terms of phase separation
We studied the structures of PDMS-TAcγCD-Pen(x) using two-dimensional NOESY NMR spectroscopy and DSC measurements on a relatively small scale. We carried out USAXS and SAXS measurements on a larger scale (Fig. 5). We combined the USAXS profiles and SAXS profiles at the scattering vector q ∼ 10−1 nm−1 for comprehensive understanding over a wide range of scales. Considering the larger scale measured by USAXS measurements (q < 10−1 nm−1), PDMS-TAcγCD-Pen(x) had higher intensities than CCPDMS regardless of the amount of TAcγCD. However, the addition of more TAcγCD led to a decrease in intensities, implying more averaged internal structures. As PDMS and TAcγCD are organic–inorganic hybrid materials and acetylated saccharides, respectively, they generally prefer to separate when mixed. With a smaller amount of TAcγCD, the phase separation resulted in more heterogeneous internal structures because of localized TAcγCD. Subsequently, more TAcγCD caused more dispersed structures, which were described as lower scattering intensities.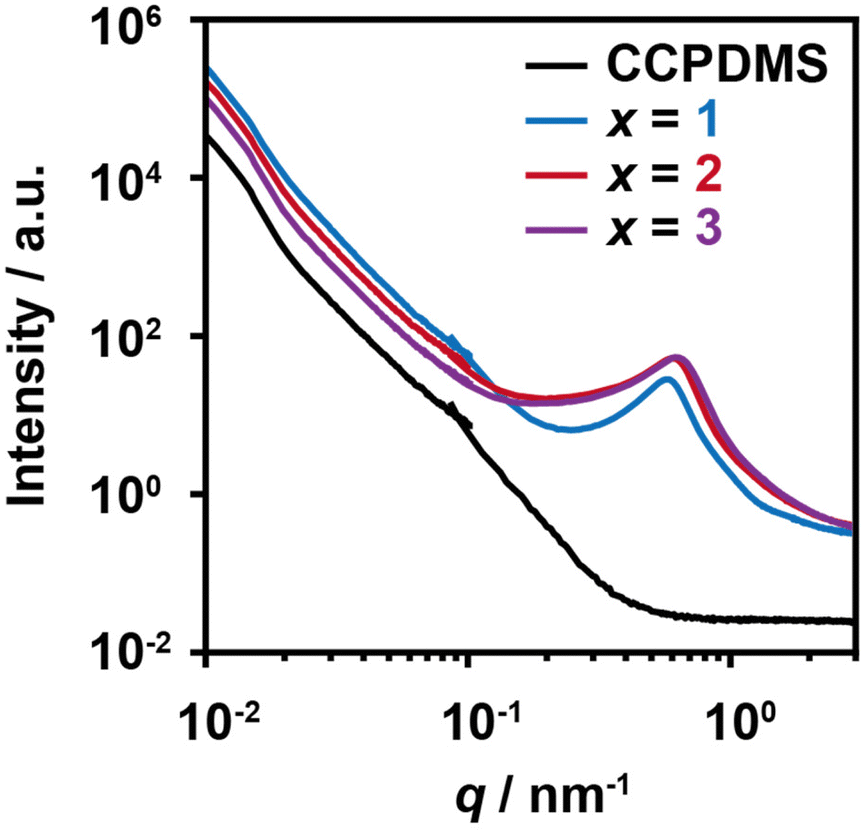 | ||
| Fig. 5 Combined X-ray scattering profiles of CCPDMS and PDMS-TAcγCD-Pen(x). The USAXS profiles and SAXS profiles covered q from 10−2 to 10−1 nm−1 and from 10−1 to 3 nm−1, respectively. | ||
Regarding the SAXS regime (q > 10−1 nm−1), PDMS-TAcγCD-Pen(x) showed significant peaks at approximately q = 0.6 nm−1, corresponding to a 10.5 nm domain spacing size. As the only difference between CCPDMS and PDMS-TAcγCD-Pen(x) was TAcγCD (Pen can be neglected because of its small size), we postulated that the peaks originated from the aggregation of TAcγCD. Interestingly, more TAcγCD shifted the peak to a larger q (smaller domain spacing sizes). More TAcγCD formed smaller and more dispersed structures, which was also supported by the USAXS measurements as a hierarchical structure. Namely, PDMS-TAcγCD-Pen(x) had a sea (PDMS matrix)-island (TAcγCD domains) phase separation structures. When we zoomed in the DSC thermograms, additional transitions were observed at approximately 120 °C (Fig. S15†). We postulated that these transitions correspond to the aggregation of TAcγCD confirmed in the SAXS regime.
In Fig. 3, we suggested a possibility of additional relaxation mechanism at longer relaxation time. The relaxation of aggregated TAcγCD seemed to be the additional relaxation mechanism. According to a recent report on the phase separated PDMS system,65 PDMS-TAcγCD-Pen(x) would contain a percolated phase separation structure and the percolation structure seemed to relax at the longer relaxation time. These phase separation structures of PDMS-TAcγCD-Pen(x) seemed to contribute to the improvement of high toughness.
3. Conclusions
We successfully prepared mechanically tough PDMS elastomers by incorporating TAcγCD-originating movable cross-links based on the energy dissipation mechanism. Compared with the toughness of conventional chemically cross-linked PDMS elastomers (CCPDMS), that of PDMS elastomers with TAcγCD-originating movable cross-links (PDMS-TAcγCD-Pen(x)) was one hundred times larger with appropriate amounts of TAcγCD (x = 2). Moreover, we also studied their energy dissipation functions through cyclic tensile tests and stress relaxation tests. The movable cross-links effectively dissipated external energies to achieve high toughness. The internal structures of PDMS-TAcγCD-Pen(x) at different scales were investigated by NMR measurements, DSC measurements, USAXS, and SAXS measurements. According to these measurements, PDMS-TAcγCD-Pen(x) contained movable cross-links, and TAcγCD in the elastomers caused phase separation (aggregation of TAcγCD). Both movable cross-links and phase separation structures seemed to improve the mechanical properties.PDMS materials have been utilized in bio- and human-related applications due to their biocompatibility. A challenge limiting the wide application of PDMS materials is their weak mechanical properties. Our material designs involve cooperatively movable cross-links to achieve topologically cross-linked PDMS elastomers. To our knowledge, PDMS elastomers with topological cross-links have rarely been reported. We expect this report to be a pioneering work to expand the border lines of topologically cross-linked polymeric materials toward PDMS elastomers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST, the Core Research for Evolutional Science and Technology (CREST) program JPMJCR22L4 (Y. T.), COI-NEXT program JPMJPF2218, and the establishment of university fellowships toward the creation of science technology innovation, grant number JPMJFS2125 (D. Y.). The authors would like to thank Dr Keiichi Osaka (SPring-8, JASRI) and Dr Noboru Ohta for the synchrotron radiation scattering measurements. The synchrotron radiation experiments were performed at BL19B2 and BL40B2 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2021B1844 and 2021A1593, respectively). We also thank Dr Naoya Inazumi and the Analytical Instrumental Facility, Graduate School of Science, Osaka University, for supporting the NMR measurements and FT-IR spectroscopy.References
- N. Lu, C. Lu, S. Yang and J. Rogers, Adv. Funct. Mater., 2012, 22, 4044–4050 CrossRef CAS.
- M. Amjadi, A. Pichitpajongkit, S. Lee, S. Ryu and I. Park, ACS Nano, 2014, 8, 5154–5163 CrossRef CAS PubMed.
- X. Wang, Y. Gu, Z. Xiong, Z. Cui and T. Zhang, Adv. Mater., 2014, 26, 1336–1342 CrossRef CAS PubMed.
- J. Lee, S. Kim, J. Lee, D. Yang, B. C. Park, S. Ryu and I. Park, Nanoscale, 2014, 6, 11932–11939 RSC.
- Y. R. Jeong, H. Park, S. W. Jin, S. Y. Hong, S. S. Lee and J. S. Ha, Adv. Funct. Mater., 2015, 25, 4228–4236 CrossRef CAS.
- J. Lee, H. Kwon, J. Seo, S. Shin, J. H. Koo, C. Pang, S. Son, J. H. Kim, Y. H. Jang, D. E. Kim and T. Lee, Adv. Mater., 2015, 27, 2433–2439 CrossRef CAS PubMed.
- Q. Wang, M. Jian, C. Wang and Y. Zhang, Adv. Funct. Mater., 2017, 27, 1605657 CrossRef.
- Y. Gao, H. Ota, E. W. Schaler, K. Chen, A. Zhao, W. Gao, H. M. Fahad, Y. Leng, A. Zheng, F. Xiong, C. Zhang, L. C. Tai, P. Zhao, R. S. Fearing and A. Javey, Adv. Mater., 2017, 29, 1701985 CrossRef PubMed.
- W. Chen, L. X. Liu, H. B. Zhang and Z. Z. Yu, ACS Nano, 2021, 15, 7668–7681 CrossRef CAS PubMed.
- X. S. Zhang, M. Di Han, R. X. Wang, F. Y. Zhu, Z. H. Li, W. Wang and H. X. Zhang, Nano Lett., 2013, 13, 1168–1172 CrossRef CAS PubMed.
- J.-H. Lee, K. Y. Lee, M. K. Gupta, T. Y. Kim, D.-Y. Lee, J. Oh, C. Ryu, W. J. Yoo, C.-Y. Kang, S.-J. Yoon, J.-B. Yoo and S.-W. Kim, Adv. Mater., 2014, 26, 765–769 CrossRef CAS PubMed.
- W. Seung, M. K. Gupta, K. Y. Lee, K. S. Shin, J. H. Lee, T. Y. Kim, S. Kim, J. Lin, J. H. Kim and S. W. Kim, ACS Nano, 2015, 9, 3501–3509 CrossRef CAS PubMed.
- G. Zhao, Y. Zhang, N. Shi, Z. Liu, X. Zhang, M. Wu, C. Pan, H. Liu, L. Li and Z. L. Wang, Nano Energy, 2019, 59, 302–310 CrossRef CAS.
- T. G. Yun, M. Park, D. H. Kim, D. Kim, J. Y. Cheong, J. G. Bae, S. M. Han and I. D. Kim, ACS Nano, 2019, 13, 3141–3150 CrossRef CAS PubMed.
- E. Leclerc, Y. Sakai and T. Fujii, Biotechnol. Prog., 2004, 20, 750–755 CrossRef CAS PubMed.
- E. K. F. Yim, R. M. Reano, S. W. Pang, A. F. Yee, C. S. Chen and K. W. Leong, Biomaterials, 2005, 26, 5405–5413 CrossRef CAS PubMed.
- M. Murrell, R. Kamm and P. Matsudaira, Biophys. J., 2011, 101, 297–306 CrossRef CAS PubMed.
- E. Pedraza, M. M. Coronel, C. A. Fraker, C. Ricordi and C. L. Stabler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4245–4250 CrossRef CAS PubMed.
- B. Trappmann, J. E. Gautrot, J. T. Connelly, D. G. T. Strange, Y. Li, M. L. Oyen, M. A. C. Stuart, H. Boehm, B. Li, V. Vogel, J. P. Spatz, F. M. Watt and W. T. S. Huck, Nat. Mater., 2012, 11, 642–649 CrossRef CAS PubMed.
- S. Halldorsson, E. Lucumi, R. Gómez-Sjöberg and R. M. T. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- S. Xiao, J. R. Coppeta, H. B. Rogers, B. C. Isenberg, J. Zhu, S. A. Olalekan, K. E. McKinnon, D. Dokic, A. S. Rashedi, D. J. Haisenleder, S. S. Malpani, C. A. Arnold-Murray, K. Chen, M. Jiang, L. Bai, C. T. Nguyen, J. Zhang, M. M. Laronda, T. J. Hope, K. P. Maniar, M. E. Pavone, M. J. Avram, E. C. Sefton, S. Getsios, J. E. Burdette, J. J. Kim, J. T. Borenstein and T. K. Woodruff, Nat. Commun., 2017, 8, 1–13 CrossRef PubMed.
- R. M. Kappel, A. J. H. Klunder and G. J. M. Pruijn, Eur. J. Plast. Surg., 2014, 37, 123–128 CrossRef.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS PubMed.
- W. Huang, D. Restrepo, J. Y. Jung, F. Y. Su, Z. Liu, R. O. Ritchie, J. McKittrick, P. Zavattieri and D. Kisailus, Adv. Mater., 2019, 31, 1901561 CrossRef CAS PubMed.
- J. E. Mark, Acc. Chem. Res., 1994, 27, 271–278 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- C. Liu, N. Morimoto, L. Jiang, S. Kawahara, T. Noritomi, H. Yokoyama, K. Mayumi and K. Ito, Science, 2021, 372, 1078–1081 CrossRef CAS PubMed.
- D. Zhao, Z. Zhang, J. Zhao, K. Liu, Y. Liu, G. Li, X. Zhang, R. Bai, X. Yang and X. Yan, Angew. Chem., Int. Ed., 2021, 60, 16224–16229 CrossRef CAS PubMed.
- M. B. Yi, T. H. Lee, G. Y. Han, H. Kim, H. J. Kim, Y. Kim, H. S. Ryou and D. U. Jin, ACS Appl. Polym. Mater., 2021, 3, 2678–2686 CrossRef CAS.
- K. Mayumi and K. Ito, Polymer, 2010, 51, 959–967 CrossRef CAS.
- K. Mayumi, M. Tezuka, A. Bando and K. Ito, Soft Matter, 2012, 8, 8179–8183 RSC.
- K. Koyanagi, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2017, 50, 5695–5700 CrossRef CAS.
- R. Ikura, J. Park, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Macromolecules, 2019, 52, 6953–6962 CrossRef CAS.
- R. Ikura, S. Murayama, J. Park, Y. Ikemoto, M. Osaki, H. Yamaguchi, A. Harada, G. Matsuba and Y. Takashima, Mol. Syst. Des. Eng., 2022, 7, 733–745 RSC.
- C. Jin, J. Park, H. Shirakawa, M. Osaki, Y. Ikemoto, H. Yamaguchi, H. Takahashi, Y. Ohashi, A. Harada, G. Matsuba and Y. Takashima, Soft Matter, 2022, 18, 5027–5036 RSC.
- K. Iijima, D. Aoki, H. Otsuka and T. Takata, Polymer, 2017, 128, 392–396 CrossRef CAS.
- J. Sawada, D. Aoki, Y. Sun, K. Nakajima and T. Takata, ACS Appl. Polym. Mater., 2020, 2, 1061–1064 CrossRef CAS.
- S. Konishi, Y. Kashiwagi, G. Watanabe, M. Osaki, T. Katashima, O. Urakawa, T. Inoue, H. Yamaguchi, A. Harada and Y. Takashima, Polym. Chem., 2020, 11, 6811–6820 RSC.
- S. Konishi, J. Park, O. Urakawa, M. Osaki, H. Yamaguchi, A. Harada, T. Inoue, G. Matsuba and Y. Takashima, Soft Matter, 2022, 18, 7369–7379 RSC.
- Y. Kawai, J. Park, Y. Ishii, O. Urakawa, S. Murayama, R. Ikura, M. Osaki, Y. Ikemoto, H. Yamaguchi, A. Harada, T. Inoue, H. Washizu, G. Matsuba and Y. Takashima, NPG Asia Mater., 2022, 14, 1–11 CrossRef.
- L. Zhong, Y. Hao, J. Zhang, F. Wei, T. Li, M. Miao and D. Zhang, Macromolecules, 2022, 55, 595–607 CrossRef CAS.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 34, 2109063 CrossRef CAS PubMed.
- X. Huang, S. Nakagawa, H. Houjou and N. Yoshie, Macromolecules, 2021, 54, 4070–4080 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. MacKay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- J. Park, S. Murayama, M. Osaki, H. Yamaguchi, A. Harada, G. Matsuba and Y. Takashima, Adv. Mater., 2020, 32, 2002008 CrossRef CAS PubMed.
- Z. Huang, X. Chen, S. J. K. O'Neill, G. Wu, D. J. Whitaker, J. Li, J. A. McCune and O. A. Scherman, Nat. Mater., 2022, 21, 103–109 CrossRef CAS PubMed.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 8350–8369 CrossRef CAS PubMed.
- R. K. Bose, N. Hohlbein, S. J. Garcia, A. M. Schmidt and S. Van Der Zwaag, Phys. Chem. Chem. Phys., 2015, 17, 1697–1704 RSC.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- E. Filippidi, T. R. Cristiani, C. D. Eisenbach, J. H. Waite, J. N. Israelachvili, B. K. Ahn and M. T. Valentine, Science, 2017, 358, 502–505 CrossRef CAS PubMed.
- J. Kang, D. Son, G. J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. H. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef PubMed.
- J. C. Lai, X. Y. Jia, D. P. Wang, Y. B. Deng, P. Zheng, C. H. Li, J. L. Zuo and Z. Bao, Nat. Commun., 2019, 10, 1164 CrossRef PubMed.
- Y. Miwa, K. Taira, J. Kurachi, T. Udagawa and S. Kutsumizu, Nat. Commun., 2019, 10, 1828 CrossRef PubMed.
- D. Yoshida, J. Park, R. Ikura, N. Yamashita, H. Yamaguchi and Y. Takashima, Chem. Lett., 2023, 52, 93–96 CrossRef CAS.
- K. Kato, K. Inoue, M. Kidowaki and K. Ito, Macromolecules, 2009, 42, 7129–7136 CrossRef CAS.
- A. Noomen, S. Hbaieb, H. Parrot-Lopez, R. Kalfat, H. Fessi, N. Amdouni and Y. Chevalier, Mater. Sci. Eng., C, 2008, 28, 705–715 CrossRef CAS.
- H. Okumura, Y. Kawaguchi and A. Harada, Macromolecules, 2001, 34, 6338–6343 CrossRef CAS.
- R. P. White and J. E. G. Lipson, Macromolecules, 2016, 49, 3987–4007 CrossRef CAS.
- Y. Kashiwagi, O. Urakawa, S. Zhao, Y. Takashima, A. Harada and T. Inoue, Macromolecules, 2021, 54, 3321–3333 CrossRef CAS.
- R. Ikura, Y. Ikemoto, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Polymer, 2020, 196, 122465 CrossRef CAS.
- S. Ge, S. Samanta, B. Li, G. P. Carden, P. F. Cao and A. P. Sokolov, ACS Nano, 2022, 16, 4746–4755 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00282a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |