
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable glycopolymers for saRNA transfection†
Alessandra
Monaco
a,
Beatriz Dias
Barbieri
 b,
Gokhan
Yilmaz
a,
Robin J.
Shattock
*b and
C. Remzi
Becer
*a
b,
Gokhan
Yilmaz
a,
Robin J.
Shattock
*b and
C. Remzi
Becer
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: remzi.becer@warwick.ac.uk
bDepartment of Infectious Diseases, Section of Immunology of Infection, Imperial College London, Norfolk Place, London W21PG, UK. E-mail: r.shattock@imperial.ac.uk
First published on 12th May 2023
Abstract
Gene delivery is a powerful technique that is often exploited in the treatment of several diseases. Currently, most gene vectors are expensive virus-based systems, which can trigger immune responses. As a cheaper and safer alternative to these systems, biodegradable polymers have been widely used to develop gene delivery systems due to their different desirable characteristics. In particular, poly(amido amine)s possess critical desirable characteristics, such as high cell transfection activity, that make them suitable and efficient for gene delivery. In this study, reducible poly(amido amine)s with different side chain lengths and glycopolymers have been developed to create polyplexes with self-amplifying RNA (saRNA). The cell transfection assay showed that sugar decorated poly(amido amine)s revealed better saRNA transfection activity than other non-decorated poly(amido amine)s. Nevertheless, no differences were obtained between the poly(amido amine)s with different side chain lengths. Overall, biodegradable poly(amido amine)s with different alcohols and sugars have been synthesized with high molecular weights and low molecular weight distributions to develop an optimal saRNA delivery system.
Introduction
In recent years, considerable attention has been drawn towards gene therapies which have shown promising results in the treatment of a variety of illnesses including diabetes, blindness, Parkinson's disease and many types of cancer.1,2 Gene therapies, however, often rely on the use of viral systems to deliver biomolecules such as DNA and mRNA and can trigger an immune response.1,3,4 Consequently, the drive to find safer alternatives to modified viral vectors has increased considerably over the last years. Polymers are generally inexpensive, easy to prepare, non-toxic and possess good cellular uptake.5,6Extensive research on poly(amido amine)s prepared via the Michael polyaddition reaction has shown that these polymers possess powerful gene delivery properties.3,7 For examples, poly(amido amines) have been demonstrated to readily form polyplexes with the negatively charged nucleic acid sequences since the nitrogen atoms present on the backbone undergo protonation under physiological conditions.6 Moreover, they possess excellent water solubility as well as stability against hydrolysis.3 This is an essential characteristic since the nucleic acid sequences can stay protected within the vehicles and arrive safely at the desired target(s).3,7–9
Overall, even though poly(amido amine)s are a promising class of gene delivery vehicles that have demonstrated high transfection efficiency, low cytotoxicity, and biodegradability, their suitability for specific gene delivery applications is limited due to their high charge density and molecular weight, which can result in toxicity and non-specific interactions with biological components such as cell membranes.
Lastly, poly(amido amine)s possess greater transfection efficiency compared to other polymers as the OH-containing side chains allow the polymers to form hydrogen bonds with the cell membrane resulting in enhanced cellular uptake.4,10 A very recent example of this is the preparation of poly(amido amine)s of up to 167 kDa from a reducible bisacrylamide (N,N′-bis(acryloyl)cystamine, BAC) and 4-amino-1-butanol.2 The large poly(amido amine)s were able to form complexations with self-amplifying RNA (saRNA) which has the potential to be an excellent vaccine platform for various illnesses, viruses and cancer.6 Additionally, it was shown that the saRNA/poly(amido amine) complexes led to very high protein expression in vivo as well as high percentage of saRNA expressing cells ex vivo (human skin),2 thus providing evidence that these polymers have real potential. Therefore, poly(amido amine)s-based glycopolymers with different carbohydrate moieties could advance this real potential in terms of their ability to efficiently transfect cells with low cytotoxicity, high stability and specific targeting properties.11,12
In this work, the conditions for the aza-Michael polyaddition of different poly(amido amines) are explored to find the best conditions for the efficient preparation of high molecular weight polymers. The poly(amido amine)s were prepared from a reducible bisacrylamide, N,N′-bis(acryloyl)cystamine (BAC) and a range of amines with different spacer lengths and also with mannose and galactose amines. The polymers were protonated to form polyplexes with saRNA at different N/P ratios. The cell transfection studies have been carried out via different cell lines. Additionally, poly(amido amine)s with carbohydrate were tested in terms of their binding activity with two human lectins via Surface plasmon resonance (SPR) spectrometer.13–16
Materials and methods
Materials
N,N′-Bis(acryloyl)cystamine (BAC), mannose amine and galactose amine were synthesised following literature procedures.40,41 Ethanolamine (≥98%), 3-amino-1-propanol (≥99%), 4-amino-1-butanol (98%), 5-amino-1-butanol (≥92%), propargylamine (98%) were purchased from Sigma Aldrich and used as received.Instruments and analysis
Proton nuclear magnetic resonance (1H NMR) was measured on a Bruker DPX-300. The NMR samples were in (CD3)2SO or CDCl3. For DMSO-d6, the resonance signal of residual DMSO at 2.50 ppm (1H) served as reference peaks for chemical shifts. The resonance signal of residual CHCl3 in CDCl3 is at 7.26 ppm. Gel permeation chromatography (GPC) measurements of polymers were carried out on an Agilent 1260 Infinity II-MDS instrument with two PLgel Mixed-D columns operating in DMF with 5 mM NH4BF4 or with PlAquagel Mixed-M columns in H2O with 0.5% NaN3 at 25 °C equipped with the following detectors: a refractive index (RI), viscometer, light scattering (LS), and variable wavelength detector (VWD). The instrument was calibrated with linear polyethylene glycol or poly(methyl methacrylate) standards (500–1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 g mol−1). The flow rate was 1 mL min−1. All samples were passed through 0.2 micron nylon filters prior to GPC measurements. Dynamic light scattering (DLS) measurements were carried out on a Malvern nano-series DLS instrument. The measurements were carried out in distilled water at 25 °C using disposable cuvettes and were repeated three times. Surface plasmon resonance (SPR) measurements were performed on a T200 BIAcore system (GE Healthcare). The lectins MBL and CLEC10A (5 μg mL−1) were immobilized on a gold CM5 chip via amino coupling reactions with N-hydroxysuccinimide and N-ethyl-N′-(dimethylaminopropyl)-carbodiimide at a flow rate of 5 μL min−1 for 5 min at ambient temperature. The measurements were carried out in buffer solution (10 mM HEPES, 150 mM NaCl, and 5 mM CaCl2) at pH = 7.4. The polymers were dissolved in the buffer, and their concentration varied between 10 and 0.63 μM. The regeneration was performed with the injection of a solution of 10 mM HEPES pH 7.4, 150 mM NaCl, 10 mM EDTA, and 0.01% Tween 20. The kinetic data of the bindings were determined from BIAevalulation 3.1 software. Z-average diameter and zeta potential of the polyplexes were analysed using a Zetasizer Nano ZS (Malvern Instruments, UK). Samples were equilibrated at room temperature and then diluted 1:100 in ultrapure H2O, in a total of 850 μL. DTS1070 cuvettes were used, and the parameters as follows: material refractive index of 1.529, absorbance of 0.010, dispersant viscosity of 0.8872 cP, refractive index of 1.330 and dielectric constant of 79.
500000 g mol−1). The flow rate was 1 mL min−1. All samples were passed through 0.2 micron nylon filters prior to GPC measurements. Dynamic light scattering (DLS) measurements were carried out on a Malvern nano-series DLS instrument. The measurements were carried out in distilled water at 25 °C using disposable cuvettes and were repeated three times. Surface plasmon resonance (SPR) measurements were performed on a T200 BIAcore system (GE Healthcare). The lectins MBL and CLEC10A (5 μg mL−1) were immobilized on a gold CM5 chip via amino coupling reactions with N-hydroxysuccinimide and N-ethyl-N′-(dimethylaminopropyl)-carbodiimide at a flow rate of 5 μL min−1 for 5 min at ambient temperature. The measurements were carried out in buffer solution (10 mM HEPES, 150 mM NaCl, and 5 mM CaCl2) at pH = 7.4. The polymers were dissolved in the buffer, and their concentration varied between 10 and 0.63 μM. The regeneration was performed with the injection of a solution of 10 mM HEPES pH 7.4, 150 mM NaCl, 10 mM EDTA, and 0.01% Tween 20. The kinetic data of the bindings were determined from BIAevalulation 3.1 software. Z-average diameter and zeta potential of the polyplexes were analysed using a Zetasizer Nano ZS (Malvern Instruments, UK). Samples were equilibrated at room temperature and then diluted 1:100 in ultrapure H2O, in a total of 850 μL. DTS1070 cuvettes were used, and the parameters as follows: material refractive index of 1.529, absorbance of 0.010, dispersant viscosity of 0.8872 cP, refractive index of 1.330 and dielectric constant of 79.
In vitro transcription of self-amplifying mRNA
Self-amplifying mRNA (saRNA) derived from VEEV alphavirus genome and encoding firefly luciferase (fLuc) was prepared by in vitro transcription. pDNA was linearised using MluI (New England BioLabs, UK) for 2 h at 37 °C, MluI was added again and incubated for another 1 h at 37 °C. Linearization was confirmed by agarose gel electrophoresis. For transcription into saRNA, 6 μl of linearised DNA template was synthesized into RNA transcripts via the mMessage Machine (Invitrogen, Thermo Fisher Scientific, UK) according to manufacturer's instructions. Transcripts were then purified by lithium chloride (LiCl) precipitation. Briefly, transcripts were frozen overnight at −20 °C and precipitated the next morning by centrifugation at 14000 rpm for 20 min at 4 °C. Pellets were resuspended in 70% ethanol and centrifuged at 14000 rpm for 5 min at 4 °C. The ethanol was removed, pellets were allowed to dry for 5 min, and transcripts were resuspended in ultrapure H2O. RNA quantification was done using a NanoDrop One (Thermo Fisher Scientific, UK) and RNA integrity was evaluated by RNA gel electrophoresis using a FlashGel™ System (Lonza, UK).
THP-1 cells (ATCC, US) were routinely grown in RPMI-1640 Medium (Sigma, UK) supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (v/v) L-glutamine, and 1% (v/v) penicillin/streptomycin (Thermo Fisher, UK), at 37 °C under 5% CO2. When confluent, the whole cell suspension in culture media was centrifuged at 1750 rpm for 5 min, and the pellet was re-suspended in fresh RPMI-1640 medium for seeding in new culture flasks (Corning, US).
An immortalised cell line of human skeletal muscle cells (hSkMC) (PromoCell, UK) was routinely grown in Skeletal Muscle Cell Growth Medium (PromoCell, Germany), supplemented with SupplementMix (PromoCell, Germany). When confluent, cells were washed with DPBS 1X (Gibco, UK) and treated with trypsin. Neutralisation was done with DPBS 1X containing 10% FBS, and cells were centrifuged and re-suspended in the skeletal muscle cell growth medium for seeding in new culture flasks (Corning, US).
Results and discussion
Synthesis of polymers via the aza-Michael reaction
Poly(amido amine)s have been synthesized via the aza-Michael addition reaction (Scheme 1), involving a series of primary amines which acted as Michael donors, and a disulfide containing bisacrylamide (BAC) which acted as a Michael acceptor (Table 1).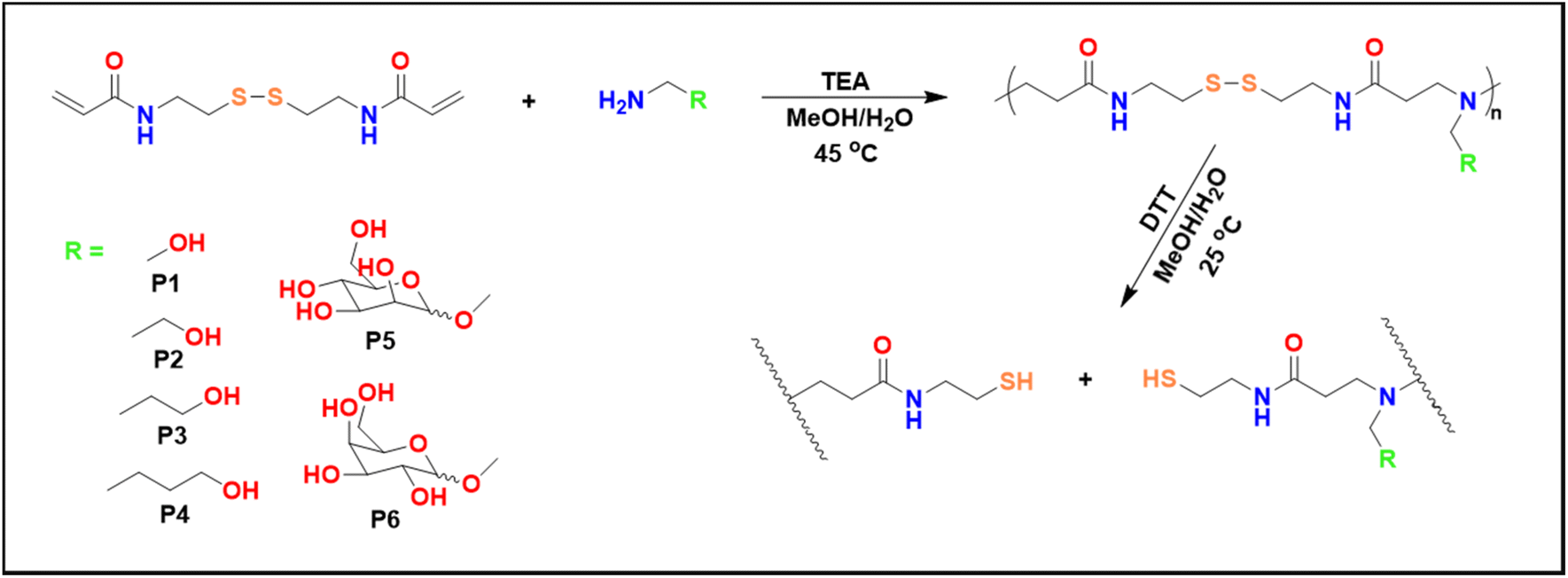 | ||
| Scheme 1 Reaction scheme of synthesis of the polymers via the aza-Michael addition. | ||
| Polymer | Amine | M n,GPC (Da) | M w,GPC (Da) | Đ |
|---|---|---|---|---|
| P1 | Ethanolamine | 3100 | 4500 | 1.4 |
| P2 | 3-Amino-1-propanol | 3900 | 5200 | 1.4 |
| P3 | 4-Amino-1-butanol | 4000 | 5400 | 1.4 |
| P4 | 5-Amino-1-pentanol | 5200 | 7100 | 1.4 |
| P5 | Mannosamine | 16000 |
46000 |
3.2 |
| P6 | Galactosamine | 12000 |
20000 |
2.4 |
Firstly, all reactions were performed at 45 °C in a mixture of MeOH/H2O (2:1) for about 16 h. It is worth noting, that even though a base is not required in the aza-Michael additions, triethylamine (TEA) was used to increase the rate of the reactions.17 Furthermore, a 1:1 molar ratio was aimed in all polymerizations. The reactions were monitored by 1H NMR which generally showed full consumption of the vinyl groups (at 5.5–6.5 ppm) as well as a shift of the amide group from 8.4 to 8.2 ppm. The amines used in these studies were all aminoalcohols with the spacer length between the primary amine and the hydroxyl group being the only difference between the amines (Scheme 1). Ethanolamine was the shortest aminoalcohol possessing two carbon atoms between the two functional groups. Conversely, 5-amino-1-propanol was the longest aminoalcohol with five carbon atoms between the amine and the hydroxyl groups. In all instances, the polymerizations were successful, and full consumption of the vinyl bonds (>99%) was observed in all reactions within 16 h. Furthermore, the formation of polymers was proven by the shift of the amide proton as well as the protons in the amines (Fig. 1B and Fig. S8†).
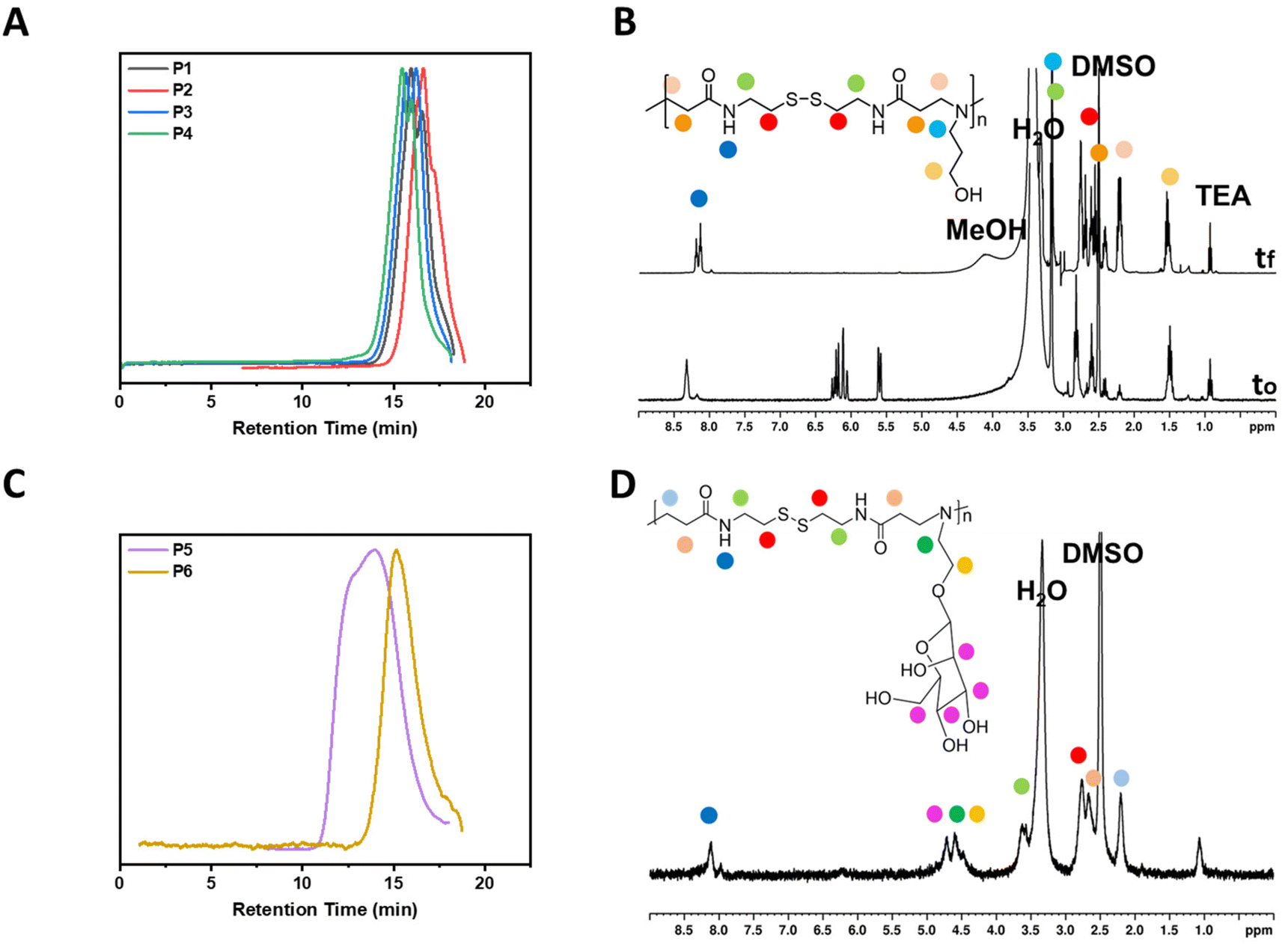 | ||
| Fig. 1 DMF GPC traces of P1–P4 (A), 1H NMR spectra of P2 (B), DMF GPC traces of P5–P6 (C), and 1H NMR spectra of P5 (D). | ||
The polymers prepared using aminoalcohols were also analysed by DMF GPC analysis (Fig. 1A). The MWs of the polymers were found to be proportional to their amine chain length. Thus, P1 which had the shortest chain had an Mn,GPC = 3100 gmol−1, followed by P2 with an Mn,GPC = 3900 g mol−1, P3 with an Mn,GPC = 4000 gmol−1, and P4 with an Mn,GPC = 5200 gmol−1(Table 1). Although, P1 resulted in the lowest Mn,GPC value and P4 provided the highest, the difference between all values was not significant. The small difference in the Mn,GPC values between the polymers is likely to be due only to the different chain lengths of the amines rather than the polymer size. Additionally, the dispersities of all polymers with aminoalcohols (1.4) were found to be similar. Furthermore, as depicted in Fig. S10,† the sizes derived by the DLS measurements of the polymers with aminoalcohols are consistent with the Mn and Mw values derived from the GPC. Therefore, P1 was found to be the smallest particle being 19.4 nm, followed by P2 (34.1 nm), P3 (36.3 nm) and P4 (38.8).
As carbohydrates play key roles in biological recognition processes such as cell–cell adhesion, but also the attachment of viruses and bacteria, glycopolymers can be considered excellent candidates for targeted gene delivery for the treatment of various genetic diseases that are currently untreatable.18–21 Furthermore, carbohydrates are overexpressed on the surface of all cancer cells which means glycopolymers are an ideal platform for anticancer thereapeutics.3,22 Therefore, sugar amines were synthesised from D-mannose and D-galactose and used to prepare glycopolymers (P5 and P6) with BAC via the aza-Michael polyaddition (Scheme 1).
The step growth polymerizations of D-mannosamine and D-galactosamine with BAC were carried out at 45 °C in a mixture of MeOH/H2O and the reaction mixtures were stirred for 15 days. Considering the results obtained for P2 using different reaction conditions, a slight excess of BAC (1.2 equiv.) was used for the polymerizations of P5 and P6 to achieve larger polymers. A lower reaction temperature (45 °C) was chosen, because sugar derived monomers are suspected to be more sensitive to higher temperatures for prolonged periods. Additionally, the polymerizations were monitored by GPC analysis (Fig. 1C) and stopped after 15 days.
Although the polymerizations of the sugar amines proceeded slowly, large glycopolymers, P5 and P6, were successfully obtained. P5, which was prepared from D-mannosamine, was the largest glycopolymer having an Mn,GPC = 16000 g mol−1. On the contrary, P6, prepared from D-galactosamine, was smaller than P5 and had an Mn,GPC = 12000 g mol−1. Additionally, the glycopolymers had larger polydispersity values (3.2 for P5 and 2.4 for P6) than those shown previously for the other step growth polymers (P1–P4). However, this was expected since an increase in the polydispersity values is directly proportional to an increase in the MW of polymers prepared following this method.
The polymerizations of poly(amido amine)s are generally carried out below 50 °C.3,7,23,24 However, even though good results are obtained using these conditions, longer reaction times (>24 h) are normally required. To decrease the long polymerization times, the Michael addition of 3-amino-1-propanol and BAC was carried out at 80 and 100 °C and the monomer conversion was monitored by 1H NMR and GPC analysis. During this investigation, it was observed that varying the temperature of the reaction had a massive influence in the polymerization rate but did not have an influence on any other aspect of the polymerization. Thus, the polymer size by GPC (Mn,GPC = ∼4000 g mol−1, Mw,GPC = ∼5000 g mol−1) remained constant and no real difference was observed between the reactions carried out at 45, 80 or 100 °C. The polymerization, however, proceeded significantly faster at 80 °C and was completed within 4 h, with 74% of the vinyl groups being consumed after only 1 h from the start of the reaction (Fig. 2A and B). The rate of the polymerization was further increased when the reaction was carried out at 100 °C, allowing a 98% reduction of the vinyl groups after 1 h from the beginning of the reaction and complete disappearance of the vinyl groups within 2 h (Fig. 2C).
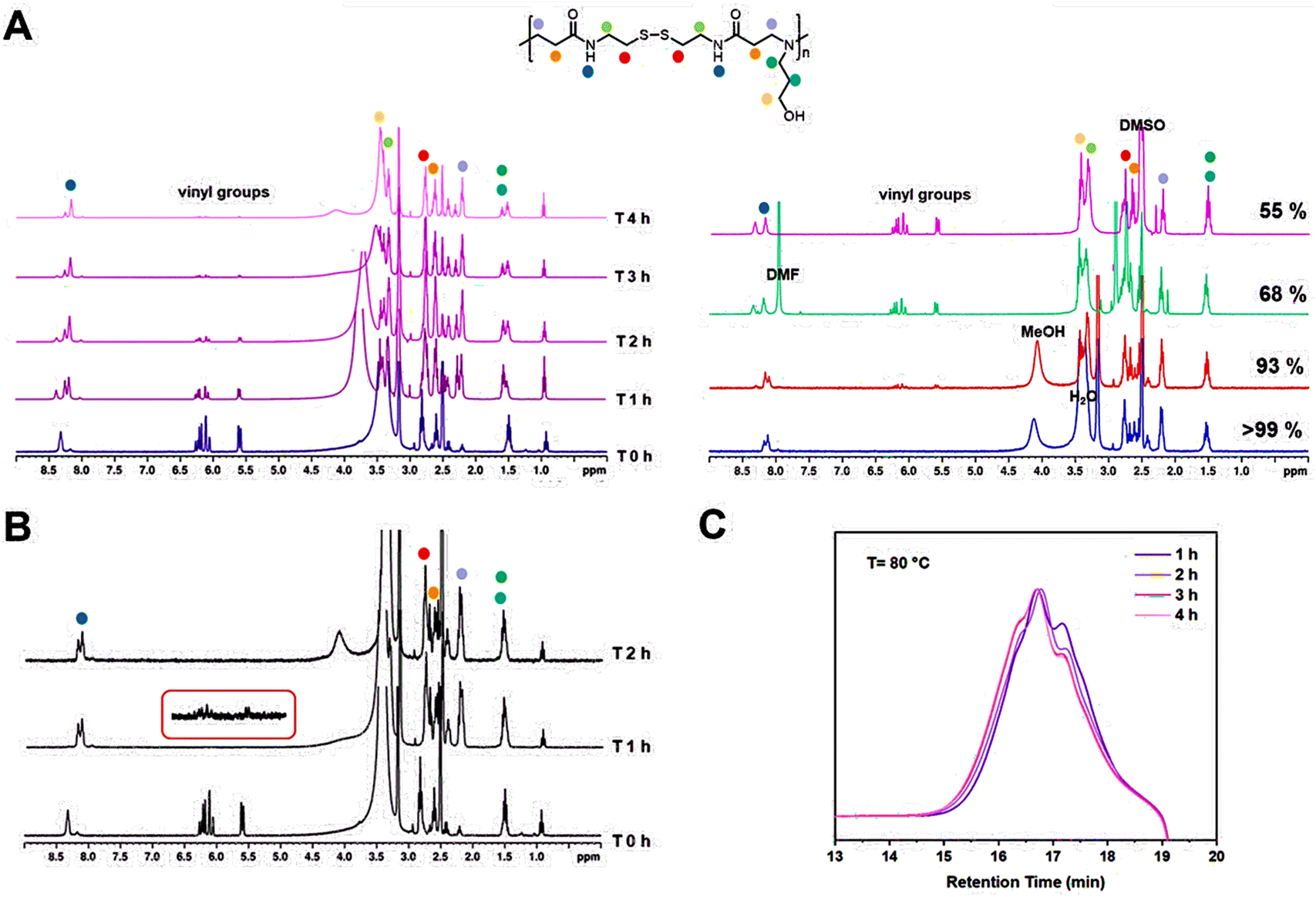 | ||
| Fig. 2 1H NMR spectra showing different phases of the polymerization of P2 at 80 °C (A, left), 1H NMR traces of the polymerization of P2 in different solvents after 16 h at 45 °C (A, right), DMF GPC traces of the polymerization of P2 at 80 °C (B), and 1H NMR spectra of the polymerization at 100 °C, which show nearly full conversion of the vinyl bonds after 1 h from the start of the reaction (C). | ||
The next variable explored during the investigation was the effect of the solvent in the polymerization. The solvents selected for this part of the study were DMSO, DMF and MeOH and were compared to the original solvent mixture (MeOH/H2O (2:1)). For the first part of this investigation, the reactions were carried out at 45 °C and were stopped after approximately 16 h from the start of the reaction. 1H NMR samples were taken at the end of the 16 h to quantify the reduction of the vinyl groups (Fig. 2A, right). The original solvent mixture MeOH/H2O afforded the best results and >99% reduction of the vinyl groups was observed after 16 h. Good results were also obtained for the polymerization in MeOH which led to a reduction of 93% after 16 h. Conversely, the polymerizations in the aprotic solvents, DMF and DMSO, were less efficient and led to lower values of 68% and 55%, respectively.
The products obtained from the polymerizations in different solvents were also analysed by GPC (Fig. 3A). From the GPC traces, it appears that the reactions in DMF and in DMSO were only at partial conversion after the 16 h reaction time and the low molecular weight shoulder shows leftover BAC monomer, consistent with the NMR data. The Mw,GPC values from the polymerizations in DMF and DMSO are 2300 and 1900 g mol−1, respectively, and are significantly lower than that obtained in a mixture of MeOH/H2O (2:1) (Mw,GPC = 5200 g mol−1). Even though monodisperse polymer peaks can be observed for low molecular weight species within step growth polymers, it is likely that the species formed were oligomers and not polymers, which supports the obtained Mw,GPC values. On the contrary, the product from the polymerization in MeOH had an Mw,GPC = 3400 g mol−1 and the GPC traces indicate that larger species have started to form. This is because more species of different sizes are present in later stages of a step growth polymerization, resulting in asymmetrical peaks.
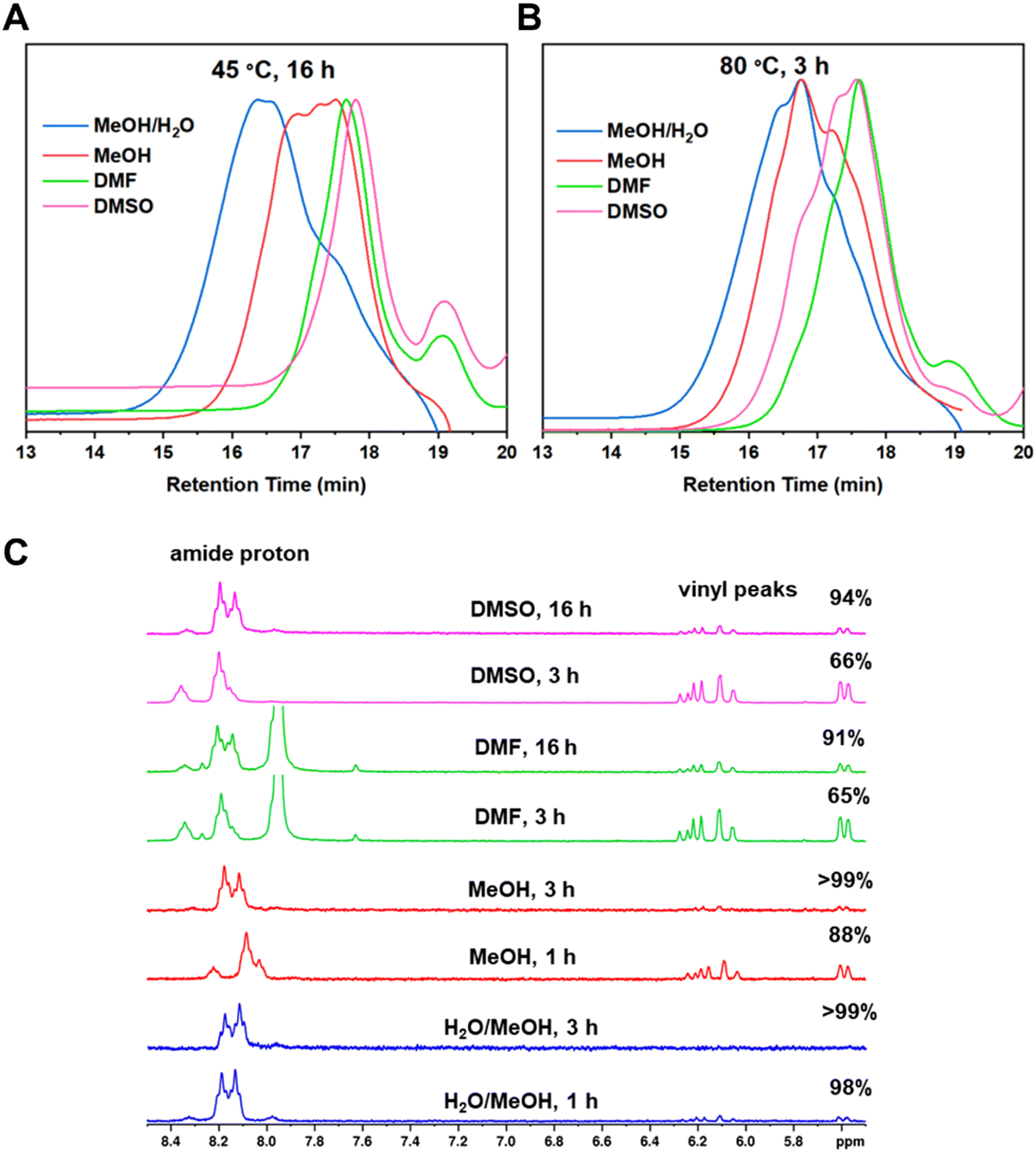 | ||
| Fig. 3 DMF GPC traces of the polymerization of P2 in different solvents at 45 °C (A) and at 80 °C (B), 1H NMR spectra of the area between 5–8.5 ppm of the Michael additions in different solvent at 80 °C show the consumption of the vinyl groups as well as the shift of the amide proton (C). | ||
To improve the polymerization in MeOH, DMF and DMSO, the temperature was increased to 80 °C. Again, the reactions were monitored by 1H NMR (Fig. 3C) to determine the monomer consumption. It was found that even though the reaction in MeOH/H2O was completed within 1 h, the reaction in pure MeOH went to full conversion after 3 h. As depicted in Fig. 3C, the full disappearance of the BAC vinyl groups and a complete shift of the amide proton from 8.2 to 8.1 ppm were observed after 3 h. It is worth noting that a small amide peak at 8.2 ppm is still visible in the 1H NMR spectrum after 1 h from the start of the polymerization, when the monomer conversion was 96%. Additionally, the Mw,GPC value (5000 g mol−1) and GPC trace (Fig. 3B) obtained in this reaction were similar to those obtained when water was used in the polymerization (Mw,GPC = 5100 g mol−1). Thus, the use of water in the reaction might not be necessary to achieve high conversions. However, water seems to accelerate the polymerization rate and its role as a potential catalyst in other Michael addition reactions has been previously reported.25 This is because water molecules allow for hydrogen bond formation which makes the amine more nucleophilic and the carbonyl group more electrophilic.26 However, because methanol is needed to solubilise the monomers, a combination of both solvents is ideal. The polymerizations in DMF and DMSO did not show a significant reduction of the vinyl groups at 80 °C after 3 h (Fig. 3C). Thus, the reactions were kept stirring for a total of 16 h. Increasing the temperature and the reaction time led to higher monomer conversions and Mw,GPC values. The two step growth polymers obtained in DMF and DMSO had an Mw,GPC = 3200 and 3900 g mol−1 with a monomer conversion of 91% and 94%, respectively.
Step-growth polymerization is a well-established process that is dependent on the concentration and stoichiometry of the reactants. This is because steric effects and incomplete reactions can limit the chain–chain addition steps, thereby affecting the polymerization process. It is possible to tune the molecular weight of polymers via adjusting the stoichiometry of the reactants. An equimolar ratio was used for the optimization reactions and only 2 reactions with a 20% excess of BAC were monitored at 45 °C for 16 and 48 hours. While the 16 hours reaction resulted in a 90% conversion, the obtained polymer was larger than those obtained at the equimolar ratio. The highest molecular weight in the synthesized polymers was obtained after 48 hours, with a dispersity of 1.2. The reason could be that using an excess of BAC may promote the formation of the polymer by shifting the equilibrium of the reaction towards the polymerization mechanism.
The degradation was carried out at room temperature using an excess of DTT which allowed for a rapid reduction of the polymer disulfide groups. This resulted in polymer fragmentation into smaller molecules with thiol end groups and the oxidation of DTT to form a stable cyclic molecule.32 The reaction was monitored by 1H NMR and GPC which showed degradation within 2 h from the start of the reaction. Fig. 4A shows a shift to the left of the amide proton around the 8.0–8.3 ppm area, but also a shift to the left of the protons on the polymer side chains in the area between 1.2–1.6 ppm, indicating successful degradation. Degradation was also observed in the GPC traces in Fig. 4B, which shows a massive shift towards the right (lower apparent molecular weight) representing the degraded polymer. The degraded product had an Mn,GPC = 1900 g mol−1 and an Mw,GPC = 2300 g mol−1 and thus, P2 was disintegrated into a much smaller molecule. It is worth noting that the degradation was monitored for 3 days, but no further shifts in the GPC were observed after the initial 2 h sample.
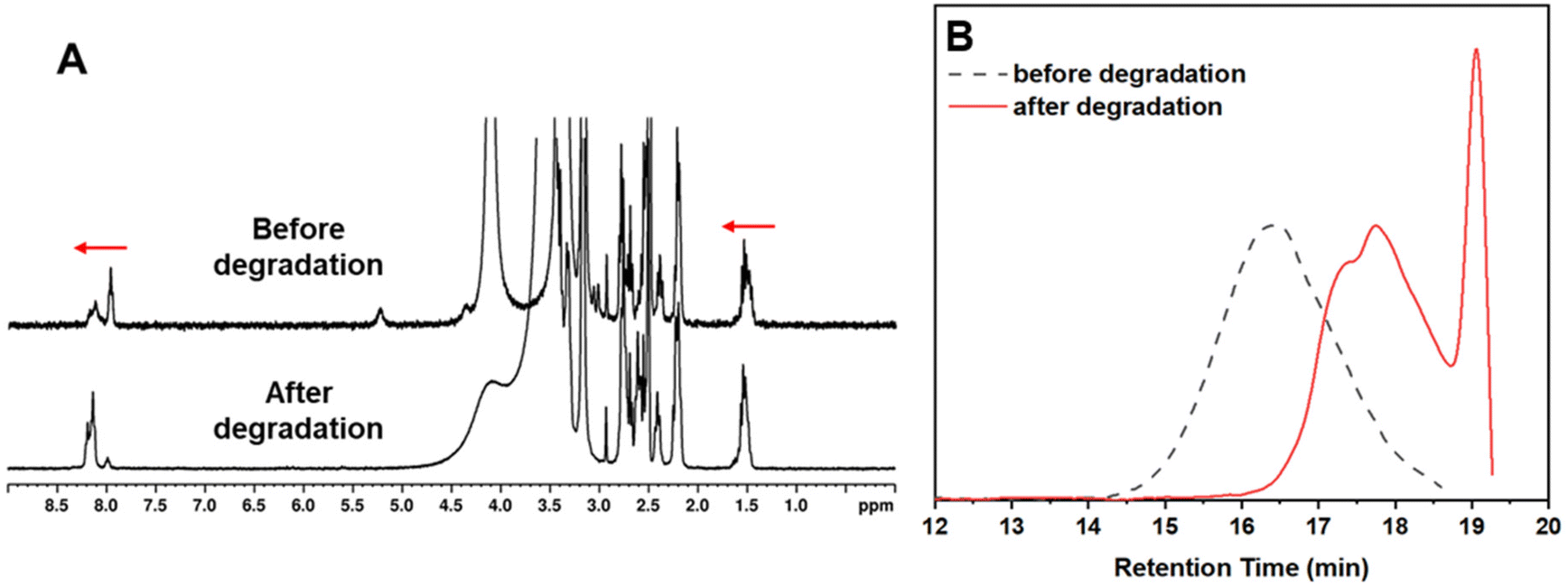 | ||
| Fig. 4 1H NMR of the reaction mixture before and after degradation had occurred (A), DMF GPC traces of P2 before and after degradation (B). | ||
The different concentrations of P5 and P6 between 20 μM and 1.25 μM were measured to determine the binding effectiveness between the glycopolymers and the lectins (Fig. 5). As expected, MBL only showed specificity towards P5 at all concentrations used, whereas it did not show any binding with P6 at its highest concentration (20 μM). Conversely, CLEC10A showed interactions with P6 at all concentrations, but no interactions with P5 at 20 μM were observed. Generally, P5 and P6 showed high affinity towards MBL and CLEC10A, respectively, and bound rapidly with the lectins. Moreover, the binding of P5 with MBL occurred in a more gradual manner compared to that of P6 with CLEC10A. For this reason, the SPR curves obtained from the binding of P6 with CLEC10A are sharper than those obtained from the measurements of P5 with MBL. As shown in Table S2,† this is also confirmed by the association constant, ka, which is significantly higher for P6 (3.05 × 103) than for P5 (122). Although it is important to clarify that in both cases, strong interactions with the lectins were obtained and that the binding showed saturation within 5 minutes from the injection of the polymers. However, the dissociation constant (kd) was higher for P6 (1.51 × 10−3) than P5 (6.79 × 10−5), indicating that not all binding between P6 and CLEC10A was retained with the injection of buffer. This is also visible in Fig. 5B, where the SPR curves appear to decline rapidly at 250 s which is the point in which buffer was injected. Nevertheless, interactions between P6 and CLEC10A are still existent after that point, despite the decline.
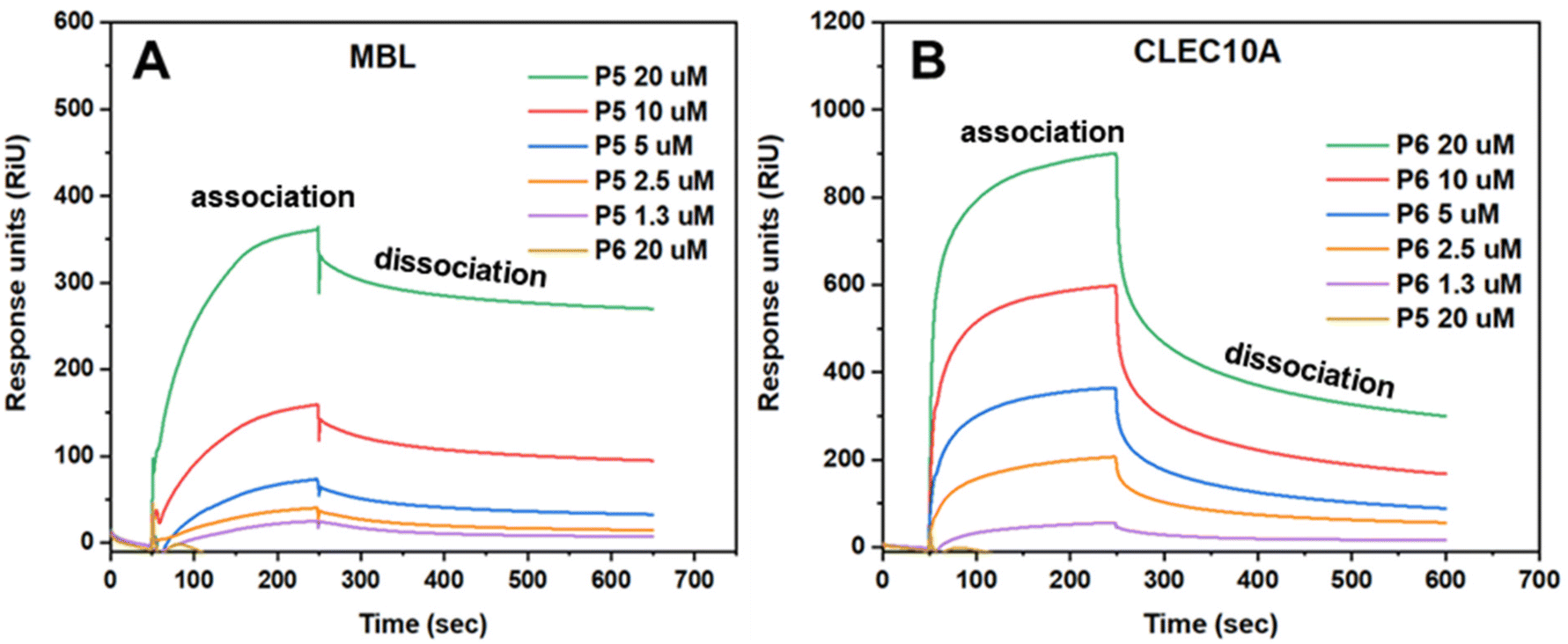 | ||
| Fig. 5 SPR measurements of P6 with MBL (A) and P5 with CLEC10A (B). | ||
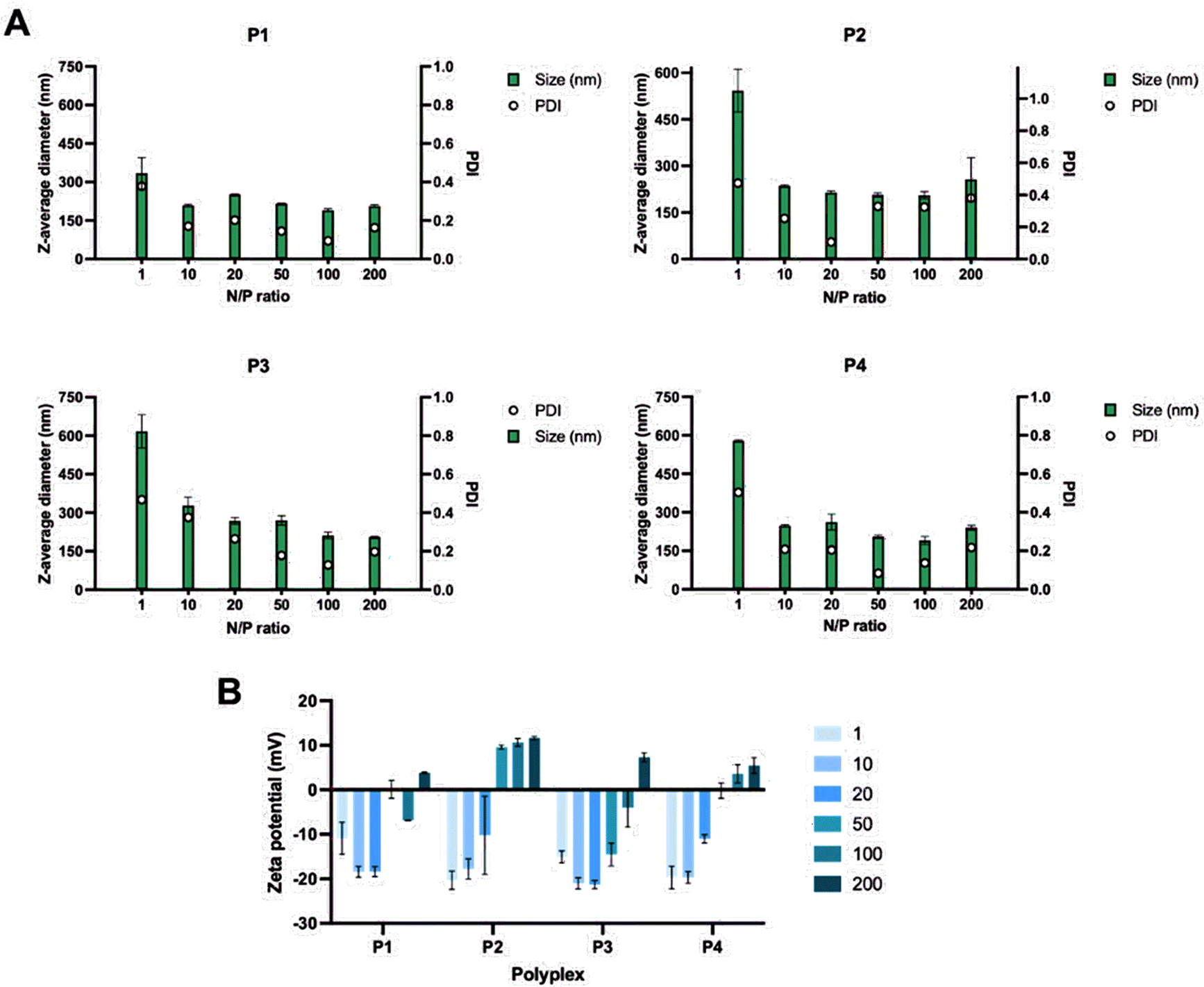 | ||
| Fig. 6 Size of the polyplexes formed using different N/P ratios of P1–P4 (A) and zeta potential measurements of the polyplexes at different N/P ratios (B). | ||
While the size of the polyplexes ranged between 200 and 300 nm, the polyplexes formed at N/P = 1 were around 600 nm for P2, P3 and P4. The reason could be that higher quantities of negative charges result in greater electrostatic repulsion which affects the size of the polyplexes. This was also confirmed by the zeta potential measurements (Fig. 6B) which showed that the N/P ratios of 1–50 resulted in more negatively charged polyplexes, indicating that more positively charged polymer was needed. For this reason, the N/P = 100–200 were more suitable for the formulations of polyplexes using P1–P4. Cell transfection studies of the polyplexes at all N/P ratios were performed in HEK 293/T17 cells to investigate their potential in gene delivery (Fig. 7). The results obtained from the cell transfection studies are comparable to those reported in the literature.17 The polyplexes made from P1–P4 showed similar transfection efficacy implying that different side chain lengths do not have a significant impact on cell transfection activity.
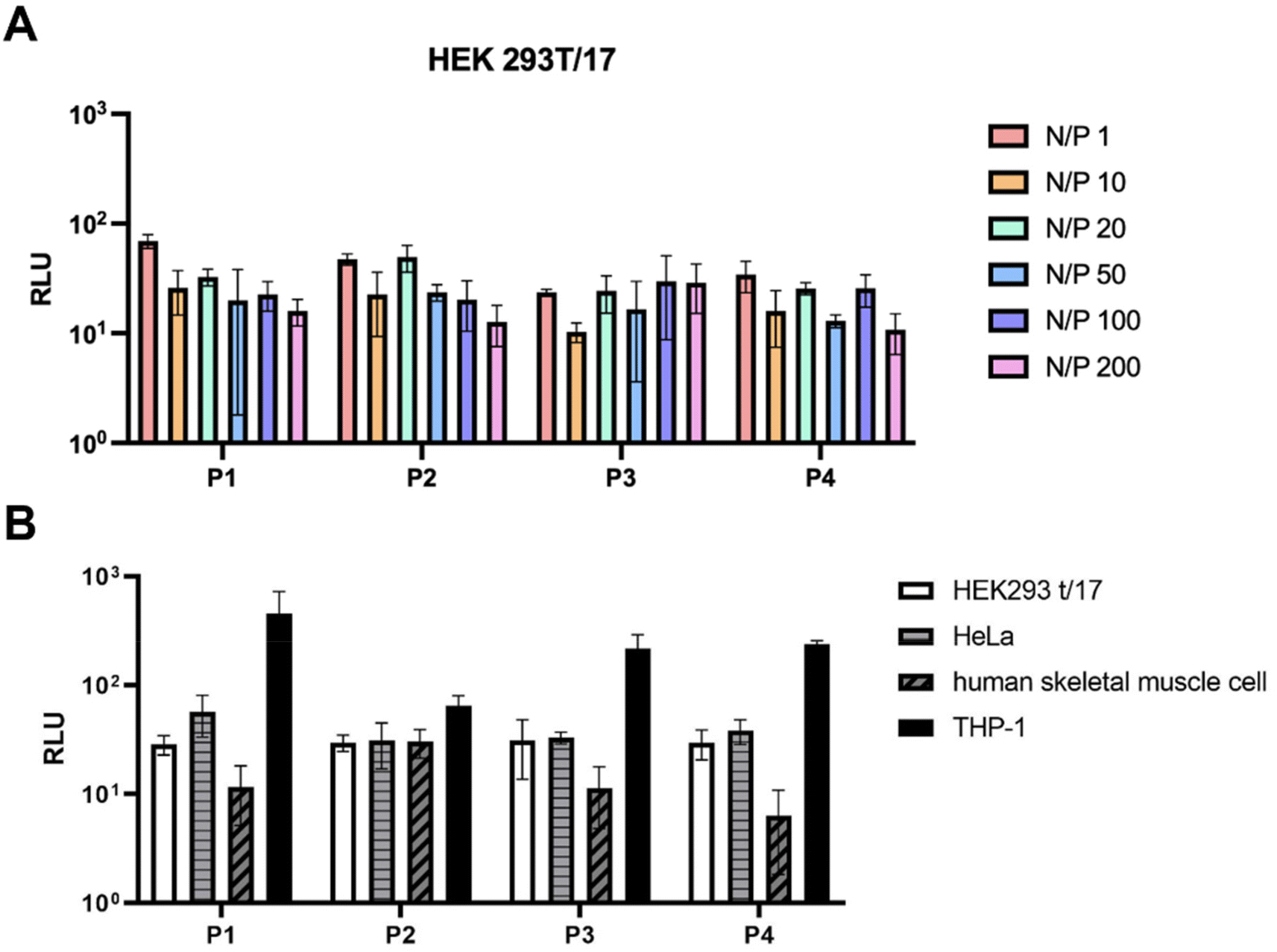 | ||
| Fig. 7 Cell transfection efficiency in HEK 293T/17 cells of the polyplexes prepared with fLuc-saRNA and P1–P4 at different N/P ratios (A), cell transfection efficiency in different cell lines at N/P = 100 (B). RLU: relative luminescence unit. | ||
Further studies with different cell lines were performed using P1–P4 showing a variety of results. The polyplexes were tested for transfection activities in HeLa cells, THP-1 cells and human skeletal muscle cells (Fig. 7B). Whilst the results with the HeLa cells were similar to those obtained with HEK293 t/17 cells, the polyplexes showed higher levels of transfection in THP-1 cells, but a decrease in transfection activity in human skeletal muscle cells.
Thereafter, polyplexes of the glycopolymers P5 and P6 with saRNA were also prepared. The size and zeta potential of polyplexes prepared from P5 and P6 at N/P = 100 are similar to those obtained from P1–P4 at this concentration and are shown in Fig. 8A and B. Nonetheless, the cell transfection results obtained with P5 and P6 showed that they have greater transfection efficacy than P1-P4 (Fig. 8C). This might be due to a much greater number of hydroxyl groups present in P5 and P6 which may help with the formation of hydrogen bonding on the cell surface for better uptake of polyplexes of P5 and P6. In addition, the Mw,GPC of P5 and P6 is significantly higher than that obtained from the other polymers, which might enhance the encapsulation efficiency of heavy genes like saRNA (∼9500 nt).3 Finally, the HEK 293/T17 cells did not show any specificity between P5 and P6 which performed equally well in these studies, despite containing different sugars. However, more studies using multiple cells would need to be carried out to determine any specificity achieved from using different sugars.
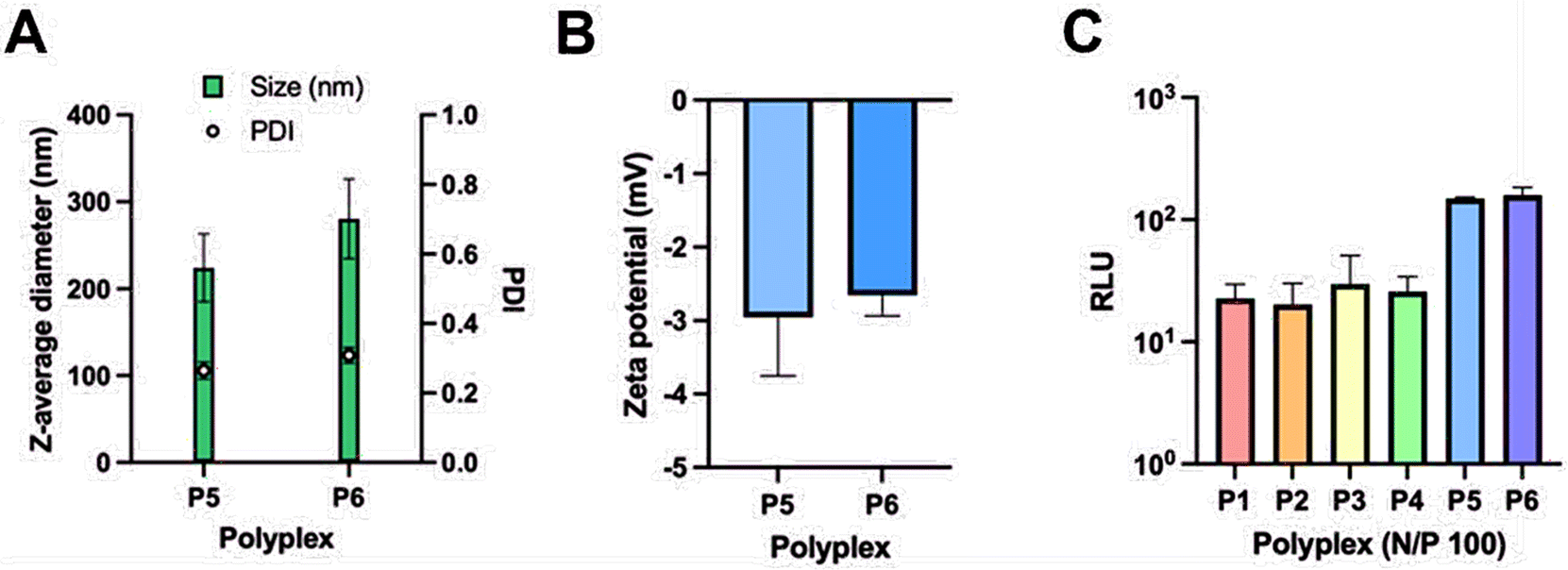 | ||
| Fig. 8 DLS measurements showing the size of the polyplexes formed from P5 and P6 with fLuc-saRNA (A), zeta potential measurements of the P5 and P6 polyplexes (B) and cell transfection results in HEK 293/T17 cells obtained from all polymers (P1–P6) at N/P = 100 (C). RLU: relative luminescence unit. | ||
Conclusions
The step growth polymerization via the Michael polyaddition of different aminoalcohols and a reducible monomer, BAC, was succeeded in this work. Firstly, different aminoalcohols in terms of chain length were used to produce reducible step growth polymers. The polymerization reactions were monitored by 1H NMR and the obtained polymers were characterized by DMF GPC. Secondly, different reaction conditions by changing temperature, solvent and stoichiometry have been studied to understand and improve the Michael addition process more. It was found that a temperature increase accelerates the reaction rate drastically, reducing the time of the polymerization from 16 h to 2–4 h. Furthermore, even though the reaction was demonstrated to work more efficiently in protic solvents, the presence of water accelerates the polymerization rate. Conversely, the reaction in aprotic solvents such as DMF and DMSO was significantly slower than protic solvents. Varying the stoichiometry by adding a slight excess of BAC allowed the formation of larger polymers. Additionally, the obtained polymers were degraded by reduction of the disulfide bonds using DTT. A very rapid degradation was observed, which is a highly desirable characteristic of polymeric gene carriers.Furthermore, mannose and galactose amines were synthesized to prepare glycopolymers via the aza-Michael polyaddition. The incorporation of sugar is desirable for targeted gene delivery which could help the treatment of specific types of cancer. The high MW biodegradable glycopolymers were synthesized successfully and then their binding affinities towards two human lectins have been investigated.
Finally, the step growth polymers were used to prepare polyplexes with saRNA which is a promising vaccine platform. The cell transfection studies of the polyplexes showed that while there is not a considerable effect of different aminoalcohols as a side chain, glycopolymers exhibited slightly higher transfection levels than aminoalcohol based polymers in HEK 293/T17 cells. Although, no difference was obtained between glycopolymers due to sugar type in terms of selective uptake, the results demonstrated that the addition of these molecules to polyplexes may lead to more efficient gene delivery systems and warrants further research.
Overall, biodegradable poly(amido amine)s with different sugars and alcohols with high molecular weights and molecular weight distributions have been synthesized to develop saRNA delivery system. The polymerization conditions were improved, and it can be easily scaled up with a small batch-batch variability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
BDB has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 859416.References
- T. K. Georgiou, Star Polymers for Gene Delivery, Polym. Int., 2014, 63(7), 1130–1133 CrossRef CAS.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, The Current Landscape of Nucleic Acid Therapeutics, Nat. Nanotechnol., 2021, 16(6), 630–643 CrossRef CAS PubMed.
- A. K. Blakney, Y. Zhu, P. F. Mckay, C. R. Bouton, J. Yeow, J. Tang, K. Hu, K. Samnuan, C. L. Grigsby, R. J. Shattock and M. M. Stevens, Big Is Beautiful: Enhanced SaRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer, ACS Nano, 2020, 14, 5711–5727 CrossRef CAS PubMed.
- Y.-W. Won, M. Ankoné, J. F. J. Engbersen, J. Feijen and S. W. Kim, Poly(Amido Amine)s Containing Agmatine and Butanol Side Chains as Efficient Gene Carriers, Macromol. Biosci., 2016, 16(4), 619–626 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Non-Viral Vectors for Gene-Based Therapy, Nat. Rev. Genet., 2014, 541–555 CrossRef CAS PubMed.
- T. Zhao, R. Terracciano, J. Becker, A. Monaco, G. Yilmaz and C. R. Becer, Hierarchy of Complex Glycomacromolecules: From Controlled Topologies to Biomedical Applications, Biomacromolecules, 2022, 23(3), 543–575 CrossRef CAS PubMed.
- D. Wang, Y. Liu, Z. Hu, C. Hong and C. Pan, Michael Addition Polymerizations of Trifunctional Amines with Diacrylamides, Polymer, 2005, 46(10), 3507–3514 CrossRef CAS.
- C. Lin, Z. Zhong, M. C. Lok, X. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, Linear Poly(Amido Amine)s with Secondary and Tertiary Amino Groups and Variable Amounts of Disulfide Linkages: Synthesis and in Vitro Gene Transfer Properties, J. Controlled Release, 2006, 116(2 spec. iss.), 130–137 CrossRef CAS PubMed.
- W. Cheng, D. Wu and Y. Liu, Michael Addition Polymerization of Trifunctional Amine and Acrylic Monomer: A Versatile Platform for Development of Biomaterials, Biomacromolecules, 2016, 17(10), 3115–3126 CrossRef CAS PubMed.
- T. Lovato, V. Taresco, A. Alazzo, C. Sansone, S. Stolnik, C. Alexander and C. Conte, Rapid Formulation of Redox-Responsive Oligo-b-Aminoester Polyplexes with SiRNA via Jet Printing, J. Mater. Chem. B, 2018, 6, 6550 RSC.
- A. K. Blakney, R. Liu, G. Yilmaz, Y. Abdouni, P. F. McKay, C. R. Bouton, R. J. Shattock and C. R. Becer, Precisely Targeted Gene Delivery in Human Skin Using Supramolecular Cationic Glycopolymers, Polym. Chem., 2020, 11(22), 3768–3774 RSC.
- A. K. Blakney, Y. Abdouni, G. Yilmaz, R. Liu, P. F. McKay, C. R. Bouton, R. J. Shattock and C. R. Becer, Mannosylated Poly(Ethylene Imine) Copolymers Enhance SaRNA Uptake and Expression in Human Skin Explants, Biomacromolecules, 2020, 21(6), 2482–2492 CrossRef CAS PubMed.
- M. Hartweg, Y. Jiang, G. Yilmaz, C. M. Jarvis, H. V.-T. Nguyen, G. A. Primo, A. Monaco, V. P. Beyer, K. K. Chen, S. Mohapatra, S. Axelrod, R. Gómez-Bombarelli, L. L. Kiessling, C. R. Becer and J. A. Johnson, Synthetic Glycomacromolecules of Defined Valency, Absolute Configuration, and Topology Distinguish between Human Lectins, JACS Au, 2021, 1(10), 1621–1630 CrossRef CAS PubMed.
- G. Yilmaz, V. Uzunova, R. Napier and C. R. Becer, Single-Chain Glycopolymer Folding via Host-Guest Interactions and Its Unprecedented Effect on DC-SIGN Binding, Biomacromolecules, 2018, 19(7), 3040–3047 CrossRef CAS PubMed.
- A. K. Blakney, G. Yilmaz, P. F. McKay, C. R. Becer and R. J. Shattock, One Size Does Not Fit All: The Effect of Chain Length and Charge Density of Poly(Ethylene Imine) Based Copolymers on Delivery of PDNA, MRNA, and RepRNA Polyplexes, Biomacromolecules, 2018, 19(7), 2870–2879 CrossRef CAS PubMed.
- C. Lavilla, G. Yilmaz, V. Uzunova, R. Napier, C. R. Becer and A. Heise, Block-Sequence-Specific Glycopolypeptides with Selective Lectin Binding Properties, Biomacromolecules, 2017, 18(6), 1928–1936 CrossRef CAS PubMed.
- P. Gurnani, A. K. Blakney, J. Yeow, C. R. Bouton, R. J. Shattock, M. M. Stevens and C. Alexander, An Improved Synthesis of Poly(Amidoamine)s for Complexation with Self-Amplifying RNA and Effective Transfection, Polym. Chem., 2020, 11(36), 5861–5869 RSC.
- R. Sunasee and R. Narain, Glycopolymers and Glyco-Nanoparticles in Biomolecular Recognition Processes and Vaccine Development, Macromol. Biosci., 2013, 13(1), 9–27 CrossRef CAS PubMed.
- S. Saxena and B. Kandasubramanian, Glycopolymers in Molecular Recognition, Biomimicking and Glycotechnology: A Review, Int. J. Polym. Mater. Polym. Biomater., 2021, 1–17 Search PubMed.
- Q. Qin, S. Lang and X. Huang, Synthetic Linear Glycopolymers and Their Biological Applications., J. Carbohydr. Chem., 2021, 40(1–3), 1–44 CrossRef CAS PubMed.
- T. Lipinski, P. I. Kitov, A. Szpacenko, E. Paszkiewicz and D. R. Bundle, Synthesis and Immunogenicity of a Glycopolymer Conjugate, Bioconjugate Chem., 2010, 22, 274–281 CrossRef PubMed.
- A. L. Parry, N. A. Clemson, J. Ellis, S. S. R. Bernhard, B. G. Davis and N. R. Cameron, “Multicopy Multivalent” Glycopolymer-Stabilized Gold Nanoparticles as Potential Synthetic Cancer Vaccines, J. Am. Chem. Soc., 2013, 135(25), 9362–9365 CrossRef CAS PubMed.
- J. Chen, C. Wu and D. Oupický, Bioreducible Hyperbranched Poly(Amido Amine)s for Gene Delivery, Biomacromolecules, 2009, 10(10), 2921–2927 CrossRef CAS PubMed.
- M. Piest, C. Lin, M. A. Mateos-Timoneda, M. C. Lok, W. E. Hennink, J. Feijen and J. F. J. Engbersen, Novel Poly(Amido Amine)s with Bioreducible Disulfide Linkages in Their Diamino-Units: Structure Effects and in Vitro Gene Transfer Properties, J. Controlled Release, 2008, 130(1), 38–45 CrossRef CAS PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Michael Addition Reactions in Macromolecular Design for Emerging Technologies, Prog. Polym. Sci., 2006, 31(5), 487–531 CrossRef CAS.
- B. C. Ranu and S. Banerjee, Significant Rate Acceleration of the Aza-Michael Reaction in Water, Tetrahedron Lett., 2007, 48(1), 141–143 CrossRef CAS.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Glomerular Barrier Behaves as an Atomically Precise Bandpass Filter in a Sub-Nanometre Regime, Nat. Nanotechnol., 2017, 12(11), 1096–1102 CrossRef CAS PubMed.
- S. Son, R. Namgung, J. Kim, K. Singha and W. J. Kim, Bioreducible Polymers for Gene Silencing and Delivery, Acc. Chem. Res., 2011, 45(7), 1100–1112 CrossRef PubMed.
- H. Y. Nam, K. Nam, M. Lee, S. W. Kim and D. A. Bull, Dendrimer Type Bio-Reducible Polymer for Efficient Gene Delivery, J. Controlled Release, 2012, 160(3), 592–600 CrossRef CAS PubMed.
- T. I. Kim and S. W. Kim, Bioreducible Polymers for Gene Delivery, React. Funct. Polym., 2011, 71(3), 344–349 CrossRef CAS PubMed.
- L. V. Christensen, C.-W. Chang, W. J. Kim, S. W. Kim, Z. Zhong, C. Lin, J. F. J. Engbersen and J. Feijen, Reducible Poly(Amido Ethylenimine)s Designed for Triggered Intracellular Gene Delivery, Bioconjugate Chem., 2006, 17(5), 1233–1240 CrossRef CAS PubMed.
- K. Subramonia Iyer, W. A. Klee and M. Wooi, Direct Spectrophotometric Measurement of the Rate of Reduction of Disulfide Bonds. The reactivity of the disulfide BONDS of bovine Cr-lactalbumin, J. Biol. Chem., 1973, 248(2), 707–710 CrossRef.
- M. W. Turner, The Role of Mannose-Binding Lectin in Health and Disease, Mol. Immunol., 2003, 40(7), 423–429 CrossRef CAS PubMed.
- L. Heger, S. Balk, J. J. Lühr, G. F. Heidkamp, C. H. K. Lehmann, L. Hatscher, A. Purbojo, A. Hartmann, F. Garcia-Martin, S. I. Nishimura, R. Cesnjevar, F. Nimmerjahn and D. Dudziak, CLEC10A Is a Specific Marker for Human CD1c+Dendritic Cells and Enhances Their Toll-like Receptor 7/8-Induced Cytokine Secretion, Front. Immunol., 2018, 9(Apr), 744 CrossRef PubMed.
- H. Ytting, I. J. Christensen, R. Steffensen, J. Alsner, S. Thiel, J. C. Jensenius, U. Hansen and H. J. Nielsen, Mannan-Binding Lectin (MBL) and MBL-Associated Serine Protease 2 (MASP-2) Genotypes in Colorectal Cancer, Scand. J. Immunol., 2011, 73(2), 122–127 CrossRef CAS PubMed.
- A. St Swierzko, A. Szala, S. Sawicki, J. Szemraj, M. Sniadecki, A. Sokolowska, A. Kaluzynski, D. Wydra and M. Cedzynski, Mannose-Binding Lectin (MBL) and MBL-Associated Serine Protease-2 (MASP-2) in Women with Malignant and Benign Ovarian Tumours, Cancer Immunol. Immunother., 2014, 3, 1129–1140 CrossRef PubMed.
- A. Swierzko, Mannan-Binding Lectin (MBL) in Women with Tumours of the Reproductive System Related Papers, Cancer Immunol. Immunother., 2007, 56, 957–971 CrossRef PubMed.
- A.-K. Kurze, S. Buhs, D. Eggert, L. Oliveira-Ferrer, V. Müller, A. Niendorf, C. Wagener and P. Nollau, Immature O-Glycans Recognized by the Macrophage Glycoreceptor CLEC10A (MGL) Are Induced by 4-Hydroxy-Tamoxifen, Oxidative Stress and DNA-Damage in Breast Cancer Cells, Cell Commun. Signaling, 2019, 17, 1–17 CrossRef CAS PubMed.
- J. He, L. Jiang, J. Wang, G. Min, X. Wang, W. Chen, H. Wang and N. Yao, CLEC10A Is a Potential Prognostic Biomarker and Immune Regulator of Tumor Microenvironment in Breast Cancer, Res. Sq., 2021, 1–16 Search PubMed.
- Z.-Q. Yu, X.-M. Xu, C.-Y. Hong, D.-C. Wu and Y.-Z. You, A Responsive Hyperbranched Polymer Not Only Can Self-Immolate but Also Can Self-Cross-Link, Macromolecules, 2014, 47(13), 4136–4143 CrossRef CAS.
- S. Liu and K. J. Edgar, Staudinger Reactions for Selective Functionalization of Polysaccharides: A Review, Biomacromolecules, 2015, 16(9), 2556–2571 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis procedures for BAC, mannose and galactose amine, including Fig. S1–S10, Tables S1 and S2. See DOI: https://doi.org/10.1039/d3py00260h |
| This journal is © The Royal Society of Chemistry 2023 |