
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PDMAEMA from α to ω chain ends: tools for elucidating the structure of poly(2-(dimethylamino)ethyl methacrylate)†
Maria Rosella
Telaretti Leggieri
 a,
Tahani
Kaldéus
ab,
Mats
Johansson
ab and
Eva
Malmström
*ab
a,
Tahani
Kaldéus
ab,
Mats
Johansson
ab and
Eva
Malmström
*ab
aDivision of Coating Technology, Department of Fibre and Polymer Technology, School of Engineering Science in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Teknikringen 56–58, SE-100 44 Stockholm, Sweden. E-mail: mavem@kth.se
bWallenberg Wood Science Center, Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56–58, SE-100 44 Stockholm, Sweden
First published on 23rd February 2023
Abstract
Poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) is currently used for a wide range of applications, often involving the synthesis of block copolymers. Here, an in-depth characterization of PDMAEMA prepared by atom transfer radical polymerization (ATRP) is reported, with a focus on end group analysis. The structure of the polymer was elucidated by one- and two-dimensional NMR spectroscopy, which assessed the presence of deactivated chains and allowed for a quantification of their fraction. Detailed characterization by MALDI-TOF MS further provided insightful information about the chain end fidelity. On this basis, termination by disproportionation was found to be the main mechanism for the loss of active chain ends. The detailed characterization allowed for an estimation of the preserved chain end functionality (CEF) of PDMAEMA. Additionally, a chain extension experiment was conducted, using PDMAEMA as a macroinitiator for the polymerization of methyl methacrylate (MMA) by ATRP. The results of chain extension supported the estimation of CEF based on the data provided by NMR and MS. Although assessing the degree of polymerization of a block copolymer proves challenging when the amount of the initial block able to act as a macroinitiator is not known a priori, an accurate estimation of the DP and Mn of the obtained block copolymer was possible by total nitrogen analysis. The tools here provided for the characterization of PDMAEMA and its block copolymer architectures allow the obtainment of essential information about the extent of control over the homo- and copolymerization. Therefore, they are of high importance when well-defined structures are aimed for.
Introduction
PDMAEMA is a polymethacrylate with attractive properties, the first being its solubility in aqueous media.1 Furthermore, due to the presence of dimethylamino pendant groups in its polymer chain (Chart 1), PDMAEMA is both pH- and temperature-responsive in water. At pH ≈ 8, the dimethylamino groups are partially protonated and the polymer displays a lower critical solution temperature (LCST) ranging between 32 and 57 °C. At pH ≈ 10, the contracted state of completely non-ionized chains results in a decrease of the LCST to as low as 25 °C. At pH ≈ 4, on the other hand, the polymer is fully protonated and not thermoresponsive.1–3 The exact values of pH and LCST are dependent on the molecular weight. Additionally, the introduction of permanent charges by methylation of the amines can systematically increase the LCST.4,5 Fully quaternized PDMAEMA is a strong polyelectrolyte whose solubility in water is no longer affected by temperature or pH. | ||
| Chart 1 Repeating unit of poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA). | ||
These tunable properties arising from the amine functionality, combined with PDMAEMA's biocompatibility, have opened up routes for applications in the biomedical field such as drug delivery with stimuli-driven controlled release6–8 and gene therapy, owing to its efficient DNA complexation.9,10 Moreover, the polymer is investigated for a wide spectrum of additional advanced applications such as water-dispersed nanolatexes,11,12 electrostatic adsorption and surface modification of negatively charged substrates,13,14 antimicrobial materials,15,16 biosensor and bioseparation systems based on stimuli-responsive grafted surfaces,17,18 water nanofiltration membranes,19,20 and stabilization of Pickering emulsions.21,22 The majority of these applications require a tailored design and a well-defined structure.
PDMAEMA as a homopolymer with narrow molecular weight distribution has been synthesized primarily by atom transfer radical polymerization (ATRP)23–25 and reversible addition–fragmentation chain-transfer (RAFT).26–28 The development of these reversible deactivation radical polymerization (RDRP) techniques has also led to the preparation of well-defined copolymers of PDMAEMA with various architectures, ranging from random, gradient, block and star copolymers29–31 to macromolecular brushes.27,32,33 Among these structures, amphiphilic block copolymers made of a hydrophilic PDMAEMA block and a hydrophobic block—often poly(methyl methacrylate) (PMMA), poly(butyl methacrylate) or polystyrene—have gained significant attention. Their most distinctive characteristic is the ability to spontaneously self-assemble in water, which has been extensively studied.34–36 PDMAEMA is an attractive choice of a hydrophilic corona in these micellar systems, not only promoting “smart” properties related to the response to varying pH values and temperatures,37–39 but also effective adsorption onto hydrophilic substrates.40–42
Well-defined PDMAEMA has an advantageous structure in the context of a wide variety of research fields over the past three decades. However, its synthesis entails specific challenges which are not always addressed in application-driven studies: (1) the amines in DMAEMA units may be involved in unwanted side reactions. In the context of RDRP techniques involving the use of metal catalysts, the pendant amine groups can compete with the selected ligands (usually amines) in metal complexation. Consequently, achieving good control over the polymerization is challenging and reducing the amount of the metal catalyst employed is hardly feasible, although it has been explored with tailored methods.24,43 Moreover, the amines may participate in nucleophilic substitutions and redox reactions. (2) Thermally induced self-initiated free radical polymerization of DMAEMA is known to take place, although with a low rate of initiation, as first elucidated by Shalati et al.44 (3) DMAEMA readily undergoes hydrolysis when polymerized in the presence of water, generating methacrylic acid which is then incorporated into the polymer chain, to an extent that can be limited by controlling the pH and quantified.45 PDMAEMA, on the other hand, is not sensitive to hydrolysis,46 unlike its acrylate counterpart poly(2-(dimethylamino)ethyl acrylate).47,48 (4) When synthesized in polar protic media, PDMAEMA's response to pH and temperature must be taken into account.
In studies where well-defined architectures are required, a thorough characterization of PDMAEMA and its copolymers is needed. However, there is a lack of systematic knowledge on how to elucidate PDMAEMA's structure with commonly available techniques. To the authors’ knowledge, during the past few decades only a limited number of studies have dwelled on PDMAEMA's detailed characterization by nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry techniques,26,49–51 which can provide useful information about the degree of polymerization, molecular weight distribution and chain end fidelity. In the context of mass spectrometry, both matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) and electrospray ionization mass spectrometry (ESI-MS) are widely used for the structural investigation of synthetic polymers, with different advantages and limitations,52,53 and the choice of one over the other is usually a matter of availability.54 However, neither of them has been intensely used and thus optimized for PDMAEMA. Size-exclusion chromatography (SEC), on the other hand, is often used for estimating the molecular weight of PDMAEMA, although it does not give highly representative results in terms of molecular weight,29,31 as discussed later.
In this work, the aim is to provide adequate tools for the characterization of PDMAEMA, with an in-depth investigation of its structure by one- and two-dimensional NMR spectroscopy and MALDI-TOF MS. The objective is not to optimize experimental parameters for the synthesis, but to highlight important information that can be deduced from widely available analysis techniques. With this, we aim to further strengthen the potential of PDMAEMA and its copolymer architectures.
For this purpose, a commonly used method for the RDRP of DMAEMA was applied to prepare a model PDMAEMA that was extensively characterized: a typical protocol for ATRP mediated by CuBr, with a tetradentate ligand, in acetone. Similar protocols are widely used for the synthesis of PDMAEMA in view of various applications,3,32,49,55 in some cases targeting its chain extension to yield block copolymers.30,42,56 Especially for the latter goal, as well as for the purpose of chain end functionalization post-polymerization,57 having information about the chain end fidelity is of primary importance. Traditional ATRP is most commonly mediated by CuBr. Copper bromide salts are preferred over copper chloride salts since alkyl bromides are more active than the corresponding alkyl chlorides.58 However, bromine is considered a rather labile end group due to the low dissociation energy of the C–Br bond and its loss has been observed during polymerization at medium to high conversions.59,60
Furthermore, samples of PDMAEMA were prepared by traditional ATRP mediated by CuCl (PDMAEMACl), as well as with a very different ATRP system mediated by a photoredox catalyst (PDMAEMAUV,EBiB and PDMAEMAUV,EBPA), to provide insights into the different impacts of side reactions when varying the parameters of the ATRP system.
In this study, characterization aspects providing important information about the control and livingness of the polymerization are highlighted. The investigated characterization tools allow the obtainment of essential information about the preservation of chain end functionality (CEF) i.e. the fraction of chains bearing the active ω-end group which enables controlled propagation. CEF is often considered the most important parameter for assessing the livingness of ATRP systems.61,62 Additionally, a chain extension experiment was conducted to add evidence with respect to the CEF of the synthesized PDMAEMA.
Experimental
DMAEMA was polymerized by CuBr/HMTETA-mediated ATRP in acetone at 30 °C, using ethyl α-bromoisobutyrate (EBiB) as the initiator. The polymerization was conducted with the following molar ratios of monomer, initiator, catalyst, and ligand: [M]/[I]/[Cu(I)]/[L] = 65/1/1/2, in the absence of external stimuli. PDMAEMA was obtained at 52% conversion (t = 135 min); unless stated otherwise, the results of the characterization of this PDMAEMA sample were discussed. During the synthesis, purification and storage of PDMAEMA, heating was always avoided aiming to lower the risk of side reactions.In a second step, PDMAEMA was chain-extended with PMMA, producing a PDMAEMA-b-PMMA block copolymer. The polymerization of MMA was carried out by CuCl/HMTETA-mediated ATRP in acetone at 50 °C, with the following molar ratios of monomer, initiator, catalyst, and ligand: [M]/[I]/[Cu(I)]/[L] = 400/1/1/2. PDMAEMA-b-PMMA was obtained at 55% conversion (t = 280 minutes); unless stated otherwise, the results of the characterization of this PDMAEMA-b-PMMA sample were discussed.
The reaction steps are visualized in Scheme 1. Further experimental details, as well as the synthetic procedures for preparing PDMAEMACl, PDMAEMAUV,EBiB and PDMAEMAUV,EBPA, are reported in the ESI.†
 | ||
| Scheme 1 Synthesis of PDMAEMA and subsequent chain extension with MMA. X = Br or Cl. | ||
Results and discussion
DMAEMA polymerization kinetics
PDMAEMA was synthesized by applying a commonly used ATRP protocol employing a CuBr/HMTETA catalyst complex. The reaction kinetics was monitored by 1H-NMR and the polymerization was stopped at approximately 50% conversion, aiming for a limited extent of termination and for the preservation of the active chain end functionalities.60 The latter is specifically important to enable chain extension, from the perspective of designing well-defined polymer architectures for materials science applications. The 1H-NMR of the polymerization mixture at final conversion is reported in Fig. S1.†The polymerization kinetics is presented in Fig. 1A. The values of ln([M]0/[M]) were found to increase rather linearly with time after what could appear to be an induction period affecting the initial polymerization rate. A linear behavior of ln([M]0/[M]) is commonly considered a sign of good control over the reaction, indicating a stable concentration of active species. However, the slightly curved trend observed in the kinetics plot is likely to be the result of slow initiation, which is observed in the polymerization of methacrylates in traditional ATRP systems.63,64 The kinetics plot gives no direct indication of the chain end fidelity. Further investigations are essential to gain information about the fraction of chain end functionality (CEF) preserved when the polymerization is stopped.
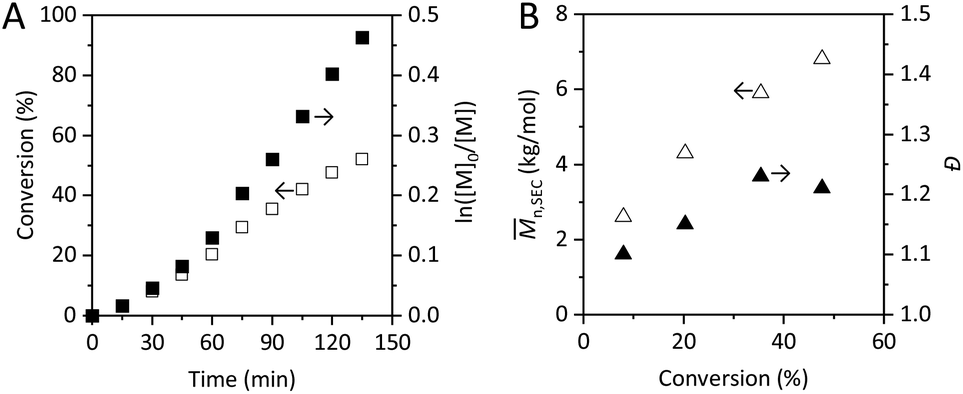 | ||
| Fig. 1 (A) Kinetic plot of ln([M]0/[M]) (■) and conversion (□) over time for the ATRP of DMAEMA. (B) Mn,SEC (△) and dispersity (▲) of PDMAEMA determined by SEC in DMF as a function of degree of conversion. | ||
The deviation from the linear behavior of the plot of number average molecular weights determined by SEC (Mn,SEC) as a function of conversion (Fig. 1B) can be correlated with slow initiation or side reactions that are hampering the control. Nonetheless, the evolution of Mn,SEC is not regarded as highly sensitive to termination reactions.65
An overlay of SEC traces revealing the evolution of molecular weight distributions (MWDs) with increasing monomer conversion is reported in Fig. S2.† To a certain extent, low molecular weight tailing was observed which may indicate loss of active chain ends in the course of the polymerization. The dispersity (Đ) values obtained, around 1.2 (Fig. 1B), are consistent with other reports of ATRP of DMAEMA at low temperatures.24,25 This Đ, rather high in the context of RDRP systems, indicates the presence of side reactions; it is usually higher for DMAEMA polymerizations performed at higher temperatures.23 However, SEC results are to be interpreted carefully, as discussed later.
From the final monomer conversion, the theoretical degree of polymerization of PDMAEMA was calculated as DPtheo = 34, resulting in an average molecular weight Mn,theo = 5500 g mol−1. This value was later confirmed by the end group analysis of PDMAEMA conducted by NMR after purification.
1D and 2D NMR analysis of PDMAEMA
A comprehensive and detailed interpretation of the NMR signals of PDMAEMA prepared by RDRP was not found in the literature. In fact, wrong interpretations have been reported because of this lack of accessible information. Here, an extensive investigation of the structure of the purified polymer was carried out by means of NMR spectroscopy, which allowed the determination of the experimental DPNMR from end group analysis and estimation of CEF.For the purpose of conducting an in-depth analysis of NMR spectra, acetone-d6 was preferred over CDCl3 and D2O, as it provided the highest peak resolution while allowing for high concentrations of PDMAEMA. The signals in the 1H-NMR spectrum (Fig. 2) and in the 13C-NMR spectrum (Fig. S3†) were identified with the aid of two-dimensional NMR spectroscopy—correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation (HMBC)—and distortionless enhancement by polarization transfer (DEPT). The resulting spectra, together with the respective assignments, are reported in Fig. S4–S9.†
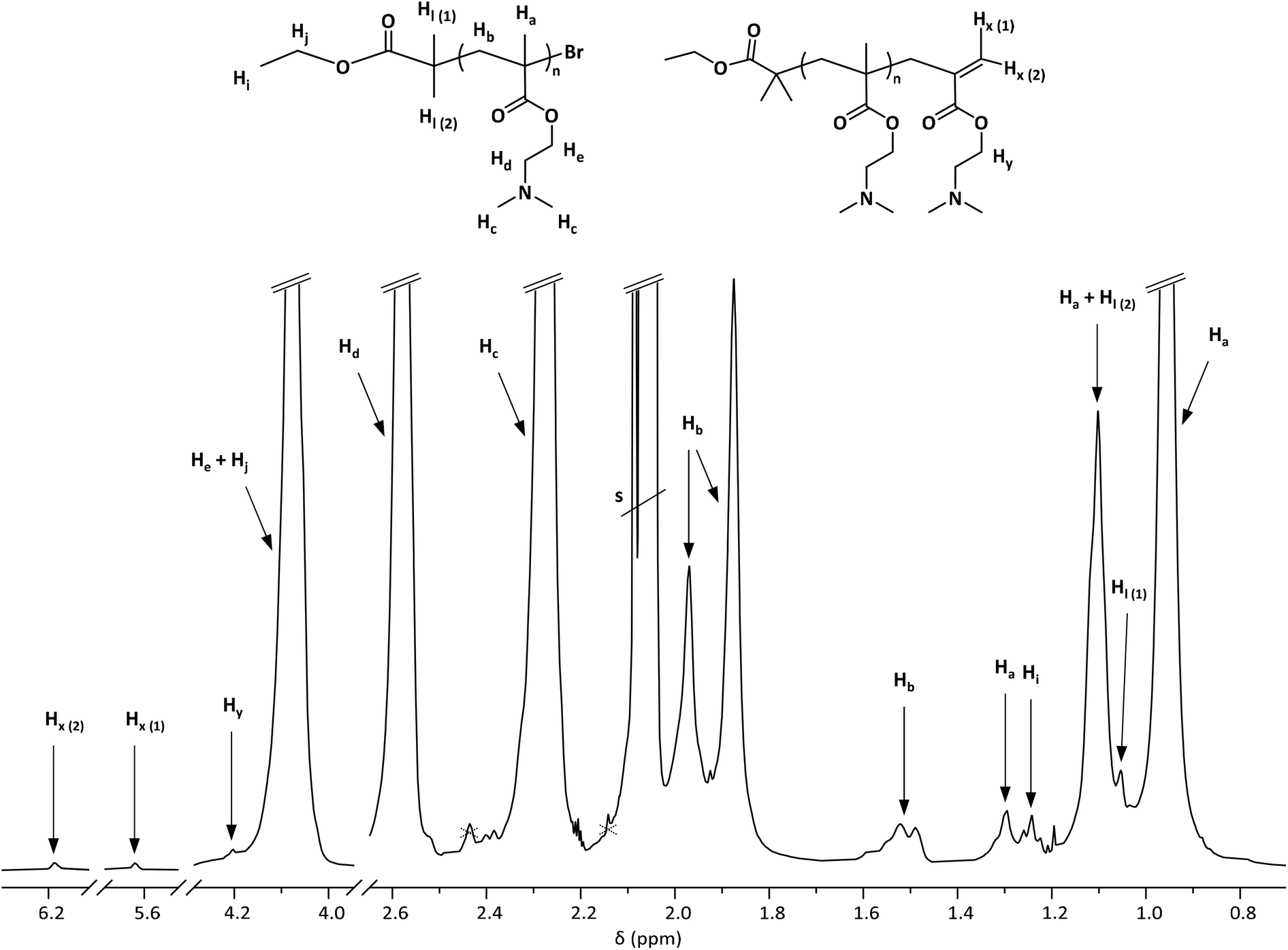 | ||
| Fig. 2 Enlargement of the 1H-NMR of PDMAEMA in acetone-d6. The full spectrum is reported in Fig. S11,† and the assignments are supported by 2D NMR spectra (Fig. S4–S9†). An “s” notation is used to mark the multiplet arising from acetone-d6 (2.05 ppm) along with the singlet arising from residual acetone (2.09 ppm). The signal at 2.44 ppm, marked with a dotted cross, is a satellite of the intense Hc signal, as it is evident in the HMBC spectrum (Fig. S8†); the second satellite overlaps with the solvent signal. | ||
In the 1H-NMR spectrum of PDMAEMA initiated by EBiB, the signals arising from the α chain end (Hi, Hj, Hl(1) and Hl(2)) were identified, providing useful information. The normalized integral ratio between the broad triplet arising from protons Hi and polymer signals, as shown in Fig. S10,† allowed the calculation of DPNMR = 34 and consequently Mn,NMR = 5500 g mol−1. This value, coinciding with the calculated Mn,theo, is an indication of overall good control over the polymerization; possible side reactions have not led to a deviation from the targeted DP. The occurrence of self-initiation (which has been shown to take place at higher polymerization temperature than that selected in this study44) would affect this calculation; however, the absence of self-initiation was confirmed by MALDI TOF-MS analysis.
The interpretation of the region of the 1H-NMR spectrum at a low chemical shift is often neglected when spectra of PDMAEMA are reported. However, since the signals of the α chain end are commonly found in this region, it is important to report a comprehensive analysis. The methyl and methylene protons in the backbone of PDMAEMA give rise to signals both in 1H-NMR and 13C-NMR spectra which are split in distinct peaks associated with different configuration sequences. In the 1H-NMR spectrum, in agreement with the peaks that have been described for PMMA,66 the methyl group on the polymer backbone (Ha) gives three signals: 0.95 ppm (syndiotactic triad, rr), 1.09 ppm (heterotactic triad, mr/rm), and 1.30 ppm (isotactic triad, mm). The content of triads with different tacticity configurations in the polymer chain is proportional to the areas of the respective peaks, when well resolved.67 Based on the signals arising from Ha, it is evident that isotactic triads are present only in low amounts, whereas syndiotactic triads are prevalent. The signals arising from the methylene protons in the backbone (Hb) appear at 1.50, 1.90 and 1.96 ppm, with a more complex configuration-based splitting compared to Ha.
Similarly, the 13C-NMR spectrum of PDMAEMA presented in Fig. S3† shows tacticity-split signals of methyl (Ca), methylene (Cb), quaternary (Cq) and carbonyl carbons in the backbone, with patterns that are typical of polymethacrylates.51,66
While the signals of the initiator segment at the α chain end were identified, ω-end group analysis by means of NMR was limited. Evidence of Br-terminated PDMAEMA chains is hard to find in NMR spectra because of overlapping signals. The protons of the methyl and methylene groups geminal to the Br-end group cannot be distinguished for this reason, as already pointed out in a previous study.25 Similarly, in the 13C-NMR spectrum the signal of the quaternary carbon at the chain end, expected at approximately 58 ppm in analogy to Br-terminated PMMA,68 overlaps with the Cd resonance (Fig. S3†).
On the other hand, small characteristic signals of ω-unsaturated polymer chains were detected in the NMR spectra of PDMAEMA, as a consequence of side reactions affecting the ω chain end. After proving that they did not originate from monomer residues, a quantitative analysis of CEF was conducted based on their integration, made possible by the selection of suitable parameters for the NMR analysis (i.e. the sample concentration and the number of scans).
Two signals of alkene protons were identified in the 1H-NMR spectrum at 5.61 and 6.18 ppm, denoted Hx(1) and Hx(2) in Fig. 2. These signals were found to correlate with each other in the COSY spectrum (Fig. S5†) and to correlate with carbon signals of low intensity in the HMBC spectrum, attributed to the ω chain end (Fig. S8†). Based on their chemical shift and on the HMBC long-range C–H correlations, the signals were attributed to the alkene protons of structure III shown in Scheme 2. Their chemical shift is extremely similar to that of the alkene protons of the DMAEMA monomer, but diffusion-ordered spectroscopy (DOSY) allowed their assignment to polymer units and not the residual monomer. In the DOSY spectrum of the polymer (Fig. S12†), protons Hx(1) and Hx(2) indeed have a diffusion coefficient of approximately 1 × 10−6 cm2 s−1, in contrast with the diffusion coefficient of 2 × 10−5 cm2 s−1 displayed by the monomer signals (Fig. S13†).
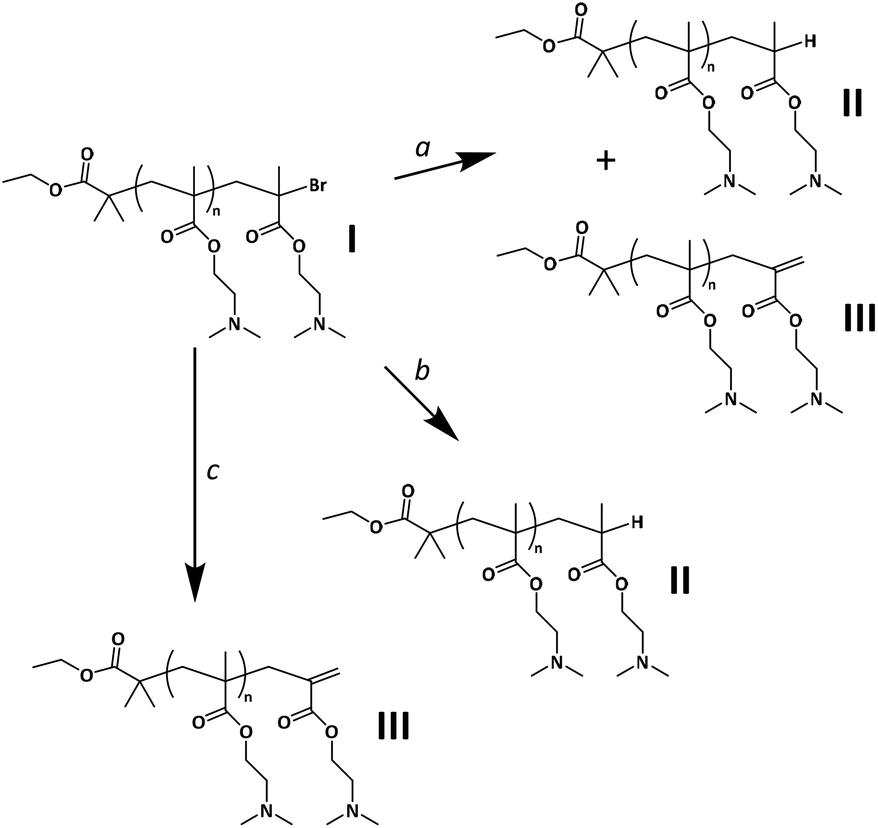 | ||
Scheme 2 Possible side reactions leading to active end group loss during the Cu(I)-mediated ATRP of DMAEMA. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Disproportionation. bChain transfer. cElimination of HBr. Disproportionation. bChain transfer. cElimination of HBr. | ||
Side reactions affecting the control over the polymerization are known to occur during polymerizations via traditional ATRP, especially when the reaction is stopped at medium to high conversions.60 In Scheme 2, the major possible pathways towards the formation of PDMAEMA “dead” chains, under the current experimental conditions, are summarized as follows: (a) termination via disproportionation, producing an ω-unsaturated chain (structure III)—via abstraction of a methyl proton from the propagating radical chain—and an ω-hydrogenated chain (structure II) in a 1:1 ratio. On the other hand, the elimination product originating from the abstraction of a methylene proton (structure III-A in Fig. S14A†), is commonly neglected,54,69,70 since structure III is largely favored due to lower steric hindrance.71 Nonetheless, the formation of structure III-A is not expected to alter the results of this study in terms of CEF assessment: in MS, the mass would be equal to that of structure III; in 1H-NMR, the signal of the resulting alkene proton is expected to overlap with the signal of proton Hx(2) of structure III, and therefore it would be accounted for. (b) Chain transfer via hydrogen radical abstraction, which has been shown to occur under certain conditions with the ligand acting as the hydrogen radical donor (usually at very high conversions or in the presence of excess ligand; however, in this study it cannot be excluded that DMAEMA amines may lead to a similar reaction).72–74 (c) Additionally, it has been reported that the elimination of HBr occurs during ATRP at medium to high conversion,60,75 also leading to structure III.
Termination via recombination, on the other hand, is known to have a minor impact on the polymerization of methacrylates, compared to disproportionation. To mention a largely studied example, approximately 70% of termination occurs via disproportionation for PMMA at 25 °C.69,76,77 However, disproportionation is known to be even more dominant for polymer chains with a bulkier ester alkyl side group, such as PDMAEMA;78 model studies have shown that the ratio of ktd/ktr (where ktd is the rate constant of termination via disproportionation and ktr is the rate constant of termination via recombination) is approximately twice as high for butyl methacrylate (BMA) compared to MMA.71 In this study, recombination was thus not expected to be a significant side reaction in the polymerization of DMAEMA, which was confirmed by MALDI-TOF MS.
From the ratio between the integrated signals of the Hx(1) or Hx(2) alkene protons at unsaturated ω chain ends and the signal of α chain end protons Hi (Fig. S10†), it was found that approximately 27% of all PDMAEMA chains are terminated with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (although small signals only allow for a rough estimation). If disproportionation was the main mechanism responsible for the formation of chains of structure III, a population of structure II should be present in an equal amount, resulting in CEF ≈ 46% (approximately 54% of all PDMAEMA chains are devoid of the active ω-end group). This assumption was further supported by the results of MALDI-TOF MS.
C double bond (although small signals only allow for a rough estimation). If disproportionation was the main mechanism responsible for the formation of chains of structure III, a population of structure II should be present in an equal amount, resulting in CEF ≈ 46% (approximately 54% of all PDMAEMA chains are devoid of the active ω-end group). This assumption was further supported by the results of MALDI-TOF MS.
The singlet of the terminal proton in structure II could not be identified in the 1H-NMR spectrum, therefore a direct quantification of structure II was not possible by means of NMR spectroscopy. Structure II was, however, detected by MALDI MS measurements.
Overall, highly valuable data were obtained from NMR analysis. The assignment of the signal of proton Hi at the α chain end allowed the determination of the Mn,NMR. Moreover, in combination with the unequivocal assignment of protons Hx(1) and Hx(2) at the ω chain end, it enabled a preliminary estimation of CEF.
When comparing the 1H-NMR spectrum of PDMAEMA shown in Fig. 2, obtained in acetone-d6, with a spectrum of the same polymer in D2O (Fig. S15†), it is evident that less information is provided by the latter.
The results highlight a rather poor chain end fidelity and a large impact of side reactions over the polymerization mechanism, despite the fairly good data obtained in terms of kinetics, the agreement between the targeted and experimental degree of polymerization, as well as dispersity of the polymer. In the current case, the end group loss was apparently slow enough for it not to alter the agreement between the targeted and experimental DP. Stopping the polymerization at higher conversion would most likely lead to more evident effects. However, a low CEF obviously has a detrimental effect on the possibility of chain-extending PDMAEMA, as confirmed later, and therefore it is important to highlight which tools are available to assess it.
The polymerization conditions may be tuned to improve the control over the side reactions, but this was beyond the scope of this work.
MALDI-TOF MS analysis of PDMAEMA
Structural investigation of PDMAEMA was further conducted by mass spectrometry, which can provide in-depth insight into the chain end fidelity. In this regard, soft ionization MS techniques are a powerful asset, since they can display specific populations of polymer chains. Quantitative analyses are, however, challenging due to limitations such as mass bias.52MS spectra of PDMAEMA have rarely been reported in the literature, and the use of this analysis should be further optimized. The interpretation of the spectra was not straightforward, and some important limitations were encountered when using MALDI-TOF MS for the characterization of PDMAEMA. The following discussion is expected to be a valuable basis for further studies.
An enlargement of the MALDI-TOF MS spectrum of PDMAEMA (isolated at 52% conversion) is presented in Fig. 3A. A population corresponding to structure I (Scheme 2) was not detected in the spectrum. Two main populations were observed, as Na+ adducts. The consecutive peaks in each of these populations are separated by a mass interval corresponding to the monoisotopic mass of DMAEMA repeating units (Δ = 157.1 ± 0.1), hence the peaks are to be attributed to polymer chains. This provides proof that no side reactions have affected the units in the main populations of polymer chains. Additionally, smaller peaks due to adducts with H+ and K+ are present.
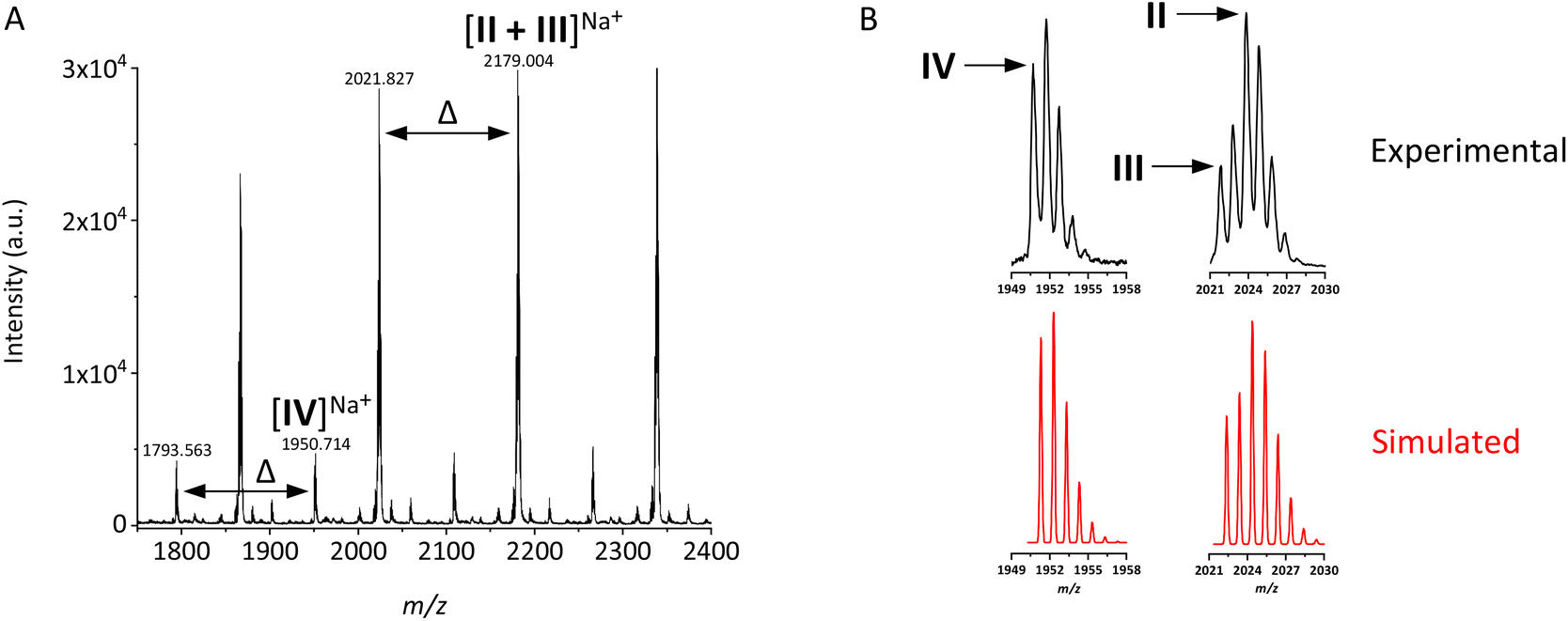 | ||
| Fig. 3 (A) Enlargement of the MALDI-TOF MS spectrum of PDMAEMA acquired in the reflector mode, where monoisotopic masses of different populations are reported. In the case of populations II and III, whose signals overlap, the monoisotopic mass of population III is reported. The full spectrum is shown in Fig. S17C.† (B) Comparison between the observed isotope distributions and those simulated via EnviPat,88 with arrows pointing at the monoisotopic masses of different populations for DP = 12. | ||
The masses of the peaks in the major series correspond to the masses of the overlapping isotopic distributions of structures II and III shown in Scheme 2. The presence of ω-hydrogenated and ω-unsaturated chains devoid of the Br-end group functionality is in agreement with the findings deduced from NMR analysis, although MALDI-TOF MS provides significantly more detailed information. The population of deactivated chains appears to be predominant in the mass spectrum.
The absence of the expected end group in the masses of at least one of the main populations detected by MALDI-TOF MS is often observed for polymethacrylates and polyacrylates prepared by RDRP techniques, confirming the presence of side reactions during polymerization such as those displayed in Scheme 2.72,74,79–81 It must be noted that dehalogenation via gas phase elimination of HBr or HCl during the MALDI ionization process, and thus not correlated with reactions occurring during polymerization, has also been proposed in this context.25,82–85 Nonaka et al. proved that under their experimental conditions, the appearance of peaks due to deactivated chains of poly(methyl methacrylate), poly(methyl acrylate) and polystyrene was linked to the high laser power and decomposition during the flight in the reflector mode.86 In this study, the minimum laser power required to generate a spectrum was used, and the ratio between the peak intensities of the two main populations observed when mass spectra were acquired in the reflector mode (Fig. 3A) is similar to that obtained in the linear mode (Fig. S16B†) with the latter displaying lower isotopic resolution. Moreover, complementary evidence of ω-unsaturated structures was found in the NMR spectra. Therefore, the mass populations assigned to structures II and III were believed to exist in the sample prior to analysis, at least partially; a contribution of gas phase HBr elimination during the MALDI process could not be excluded.
A MALDI-induced dehalogenation process involving cyclization at the ω-chain end is proposed here to account for the presence of the population of lower intensity identified in the mass spectrum, whose masses are equal to those of chains of structure IV, depicted in Scheme 3. It has been shown previously that the C–X (X = Br or Cl) bond in polymethacrylates may be unstable under the conditions of the MALDI ionization process, in contrast with polyacrylates.87 Irvine and co-workers demonstrated that decomposition via the loss of methyl bromide and formation of a five-membered lactone end group can occur for halogen-terminated PMMA.68 Evidence of PMMA with this end group in MALDI-TOF MS analysis has since been highlighted in several studies.89–91 Here, a similar mechanism is proposed for the end group decomposition of PDMAEMA during MALDI, with loss of 2-bromo-N,N-dimethylethanamine and cyclization as depicted in Scheme 3.
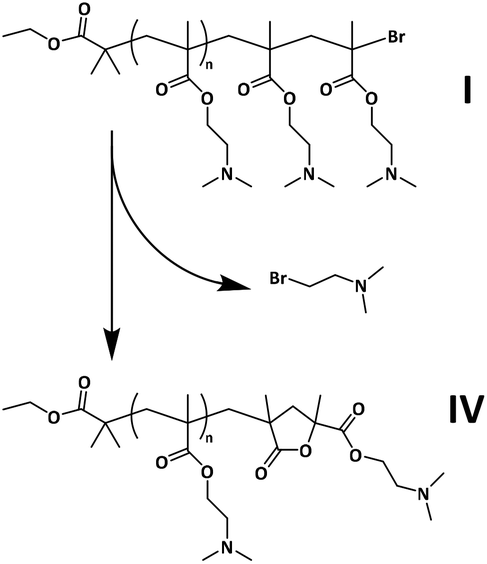 | ||
| Scheme 3 Proposed mechanism of PDMAEMA ω-end group decomposition during MALDI-TOF MS analysis. | ||
The interpretation of the masses allowed the identification of the main populations of PDMAEMA chains, with the support of well-resolved isotope distributions. The isotope distributions of the peaks assigned to populations IV and II + III were compared to simulated isotope distributions, as shown in Fig. 3B (where the simulated isotope distribution arising from population II + III was based on a 1:1 intensity ratio between the two populations). The similarity of the observed patterns with simulated patterns is a strong confirmation of the assignment of the peaks to the proposed structures.
The intensity ratio between peaks of populations IV and II + III was not used for an estimation of CEF, for two reasons. First, the mechanism shown in Scheme 3 might not be the only process of MALDI-induced end group decomposition occurring for PDMAEMA. Second, a strong effect of mass discrimination towards shorter chains was observed, especially when mass spectra were acquired in the reflector mode (e.g. the spectrum reported in Fig. 3A), but probably also affecting the spectra obtained in the linear mode (Fig. S16A†). For this reason, MS was not used for assessing the average molecular weight and dispersity of PDMAEMA. Mass discrimination towards shorter chains has been highlighted and thoroughly studied for more common polymethacrylates, such as PMMA.92–94 On the other hand, the data available on matrix selection and optimization of sample preparation for PDMAEMA are still very scarce. In this work, a limited screening of commonly used matrices was carried out before selecting 2,3-dihydrobenzoic acid (DHB) as the preferred matrix. Further studies may provide improvements in this regard.
The issue of mass discrimination obviously implies that the amount of terminated polymer chains is overestimated in the mass spectra. Accordingly, the intensity of the peaks assigned to population IV, even lower than expected on the basis of the NMR results, was not directly correlated to the amount of Br-terminated PDMAEMA chains in the sample prior to analysis.
The relative intensity of the signals of populations II versus III, on the other hand, provides information about the ratio between the different side reactions discussed above and displayed in Scheme 2. Since both populations II and III are present in the mass spectrum, and with a roughly 1:1 intensity ratio, it is reasonable to assume that disproportionation is the dominating side reaction leading to end group loss. A contribution from both elimination of HBr (mechanism c) and chain transfer (mechanism b) to the formation of populations III and II, respectively, could be additionally present. There are several studies investigating end group analysis of various polymers by mass spectrometry where only ω-hydrogenated chain ends have been detected,72,79,81,95 hence suggesting the occurrence of chain transfer reactions. In contrast, when populations of ω-unsaturated and ω-hydrogenated chains are detected with equal intensity, disproportionation is the most likely cause.80
Since MALDI-TOF MS spectra indicate that ω-hydrogenated polymer chains are present in a 1:1 ratio with respect to ω-unsaturated chains, the results corroborate the calculation of CEF ≈ 46% based on NMR analysis.
To gain some qualitative insight into the evolution of CEF with conversion of PDMAEMA, samples isolated at increasing monomer conversions were analyzed by MALDI-TOF MS. The mass spectrum of the polymer at 14% conversion (Fig. S17A†), when Mn,theo = 1400 g mol−1, shows a single distribution of peaks with masses corresponding to those of population IV, and therefore attributed to active chains with the preserved Br end group. It can be concluded that, at this early stage of the polymerization, termination was not significant. At 35% conversion (Fig. S17B†), when Mn,theo = 3600 g mol−1, active and deactivated chains generated two populations of roughly equal intensity. Finally, at 52% conversion (Fig. S17C†), when Mn,theo = 5500 g mol−1, the population of deactivated chains became predominant. Although the last two measurements were affected by mass discrimination to some extent, the results correlate with a gradual loss of bromine functionality during the synthesis, starting at rather low values of conversion.
The results of MALDI-TOF MS analysis on PDMAEMA confirmed the significant presence of deactivated chains, as first suggested by the NMR spectra. Furthermore, mass spectra allowed the obtainment of insightful information about the mode of end group loss and thus confirmed the estimation of CEF ≈ 46%. Finally, it is worth pointing out two additional considerations. First, a population with masses corresponding to self-initiated chains was not detected in the spectra, suggesting control over the initiation mechanism. Second, a population with masses corresponding to bimolecular termination via recombination was not present in the mass spectra acquired in the linear mode (less affected by mass discrimination), suggesting that this termination route is not plausible for PDMAEMA under the current experimental conditions.
Comparison with different ATRP systems
PDMAEMA synthesized with a traditional EBiB/CuBr/HMTETA ATRP system, and recovered at approximately 50% conversion, was herein used as a model PDMAEMA on which an extensive end group analysis was conducted. Since a variety of ATRP methods exist, with significant differences, it is relevant to apply the characterization tools investigated here to PDMAEMA synthesized using different ATRP systems, especially from the perspective of highlighting the value of CEF, and the impact of different side reactions by means of MALDI-TOF MS. For this reason, the analysis of PDMAEMA prepared using two additional ATRP systems is discussed here.First, PDMAEMACl was synthesized using CuCl while keeping all other experimental conditions previously used for the CuBr-mediated ATRP constant. Chlorine is regarded as a less labile RDRP end group compared to bromine, due to the higher dissociation energy of the C–Cl bond, therefore the use of CuCl is expected to result in less significant side reactions, as opposed to CuBr.96,97 The dispersity of PDMAEMACl, determined by SEC in DMF (Đ = 1.2) was similar to that observed for PDMAEMA synthesized by CuBr-mediated ATRP.
In the mass spectrum recorded for PDMAEMACl (Fig. S18A†), the same major populations observed for PDMAEMA synthesized with CuBr were detected (IV and II + III). However, the intensity ratio of the peaks of populations IV over II + III was found to be significantly higher. This supported the assumption that population IV corresponds to the population of chains with a preserved halogen end group: a population which is present in higher number in the case of PDMAEMACl due to the higher stability of the Cl end group. It is thus reasonable to propose that halogen-terminated PDMAEMA, under the current experimental conditions, undergoes the end group decomposition pathway presented in Scheme 3 during the MALDI process, both in the case of Br- and Cl-terminated chains. It is hypothesized that the pattern of isotope distribution attributed to populations II + III for PDMAEMACl (Fig. S18B†) differs from that shown in Fig. 3B for PDMAEMA prepared by CuBr-mediated ATRP due to a smaller contribution of the elimination of HCl, compared to HBr.
The 1H-NMR spectrum of PDMAEMACl, with integrated signals, is reported in Fig. S19.† Signals originating from protons in unsaturated chain ends were detected. By applying the same calculation carried out before, a CEF ≈ 34% was estimated for PDMAEMACl, lower than that of PDMAEMA synthesized by CuBr-mediated ATRP.
To provide an additional comparison, PDMAEMAUV,EBiB and PDMAEMAUV,EBPA were prepared by photomediated ATRP using 10-phenylphenothiazine (PTH) as a photoredox catalyst, according to a method adapted from that described by Treat et al.98 (however, a different light source was used). The two polymers were prepared by using two different initiators, EBiB and ethyl α-bromophenylacetate (EBPA), respectively. A high dispersity was determined by SEC in DMF when analyzing PDMAEMAUV,EBiB and PDMAEMAUV,EBPA (Đ = 2.4, in both cases), indicating very low control over the polymerization.
In the MALDI-TOF MS spectra of the polymers (Fig. S20 and S21†), two major populations are present. The population with the highest intensity corresponds to terminated chains of structure II + III. Interestingly, both for PDMAEMAUV,EBiB and PDMAEMAUV,EBPA the masses of the signals of the population of lower intensity correspond to those of self-initiated PDMAEMA chains devoid of the Br end group. It is proposed that, under the current experimental conditions, the amine group of DMAEMA is involved in a photoredox reaction resulting in the abstraction of a hydrogen atom from the carbon adjacent to the amine group, with a mechanism analogous to that reported by Allushi et al. for tertiary amines used as “co-initiators” for photomediated ATRP in the presence of thioxanthone photocatalysts.99 Under these conditions, the resulting DMAEMA radical (Fig. S14B†) is thus able to initiate polymerization, competing with the initiator. It is evident that the choice of experimental set-up for photomediated ATRP (e.g. choice of photocatalyst, wavelength of the light source and light intensity) is particularly important, to achieve polymers with narrow MWD and to control the initiation mechanism. Since self-initiated chains are present in the samples of PDMAEMAUV,EBiB and PDMAEMAUV,EBPA, it is not possible to calculate CEF on the basis of the ratio of integrated signals of ω and α chain ends. However, since neither a population of chains having the Br end group nor a population of chains corresponding to structure IV were identified in the mass spectra, CEF can be expected to be extremely low.
It is evident that the results of mass spectrometry conducted on PDMAEMA samples prepared by these additional ATRP systems raise insightful considerations, although more extensive investigations are needed to draw conclusions with respect to the polymerization mechanisms. CuCl can ensure higher control over the polymerization, compared to CuBr. Photomediated ATRP of DMAEMA, on the other hand, can lead to a notable extent of self-initiation of DMAEMA and a high dispersity.
SEC analysis of PDMAEMA
Size-exclusion chromatography, performed using N,N-dimethylformamide (DMF) as the eluent and poly(methyl methacrylate) standard calibration, was used to provide information about the polymerization kinetics, but its use is not relevant for giving representative values of molecular weight of PDMAEMA. SEC analysis on purified PDMAEMA determined Mn,SEC values significantly deviating from Mn,theo. When the analysis was repeated, traces with varying Mn,SEC values were obtained, ranging from 7000 to 9600 g mol−1. Polymers containing amine functionalities such as PDMAEMA are difficult to characterize using conventional SEC systems. Many authors have highlighted that the plausible interaction of PDMAEMA with the columns may lead to a broadening of the MWD, especially at low molecular weights, and additionally the Mn,SEC obtained is highly affected by different hydrodynamic volumes in relation with the standards.29,31,100 Addition of triethylamine in the mobile phase may help in avoiding interactions with the columns101 and other existing solutions (e.g. converting the polymer into poly(methyl methacrylate) for the purpose of the analysis3 and using more complex SEC systems26).Chain extension of PDMAEMA
A chain extension experiment produced additional evidence of the extent of chain end fidelity of PDMAEMA. PDMAEMA was used as a macroinitiator for the synthesis of PDMAEMA-b-PMMA by ATRP mediated by CuCl. CuCl was used for chain extension since the high activity of the Br end group of the macroinitiator, more labile than Cl which is expected to become predominant in the propagation step, should ensure initiation efficiency.102 The kinetics plot of the reaction, reported in Fig. 4A, reveals a trend similar to that observed in the polymerization of DMAEMA. The plots of Mn,SEC and Đ over conversion are shown in Fig. S22.† The rather linear trend observed in the ln([M]0/[M]) might be the result of a balance between slow initiation and termination, since it is not corroborated by the evidence of high control over the polymerization. | ||
| Fig. 4 (A) Kinetic plot of ln([M]0/[M]) over time during ATRP of MMA initiated by PDMAEMA. (B) Overlay of MWDs obtained by SEC in DMF: PDMAEMA macroinitiator (black, solid line); samples from the PDMAEMA-b-PMMA reaction mixture at t = 150 min, 34% conversion (blue, dotted line) and at t = 210 min, 43% conversion (blue, dashed line); purified PDMAEMA-b-PMMA isolated at t = 280 min, 55% conversion (blue, solid line). | ||
In agreement with the presence of deactivated chains revealed by NMR and MS, the MWDs of aliquots withdrawn from the polymerization mixture at 34% and 43% conversion, plotted in dotted and dashed blue lines in Fig. 4B, show bimodality. The peak at lower molecular weight, overlapping with the trace of the PDMAEMA macroinitiator, corresponds to the fraction of PDMAEMA dead chains, unable to initiate copolymerization.
During copolymerization, the presence of dead PDMAEMA, unable to initiate chain extension, could also be immediately detected by NMR, using a DOSY experiment. In Fig. S23,† the DOSY spectrum of the sample withdrawn from the polymerization mixture at 34% conversion is reported. The spectrum reveals the presence of the PDMAEMA homopolymer, whose protons show distinctive signals with a lower diffusion coefficient compared to the protons of the copolymer.
The selected method of purification of the copolymer (precipitation in MeOH) ensured the complete removal of unreacted PDMAEMA, as it is evident from the molecular weight distribution of the purified copolymer plotted in a solid blue line in Fig. 4B. The block copolymer showed Đ = 1.2, suggesting that a functional material can be obtained despite the non-ideal character of the polymerization.
When discussing the results of chain extension of PDMAEMA, an additional consideration can be elaborated. The consequence of a significant amount of disproportionation is that the ω-unsaturated chains are not truly dead; in fact, they can be involved in chain transfer reactions103 and may even react further as macromonomers (although with low reactivity) and thus be incorporated into the copolymer structure. This mechanism might contribute to the chain extension; however, further investigations should be conducted in this regard.
Molecular weight determination of PDMAEMA-b-PMMA
Assessing the molecular weight of the PDMAEMA-b-PMMA copolymer proved challenging. The presence of deactivated chains affects the monomer/macroinitiator ratio, hindering the correlation between targeted and experimental DP. Since the amount of the initial block able to act as the macroinitiator was not known a priori, the DPtheo of PMMA initiated by PDMAEMA (and, consequently, Mn,theo) was an underestimated value. Moreover, after purification of the block copolymer, determining Mn,NMR based on the ratio between integrated signals of the extension block and of the initiating block was not possible, due to the different solubility of the two blocks in the NMR solvent leading to the overestimation of one over the other. The PMMA/PDMAEMA integrated signal ratio in the 1H-NMR spectrum of the copolymer obtained in acetone-d6, reported in Fig. S24,† would lead to a DPNMR of 1470 for PMMA, resulting in a Mn,NMR of approximately 150000 g mol−1 which is not a reliable estimation.
To determine the molecular weight of PDMAEMA-b-PMMA, the amine in the repeating units of PDMAEMA was an advantageous asset. Elemental analysis (EA) was conducted on the copolymer using a total nitrogen analyzer. With this method, the ratio of molDMAEMA per gcopolymertomolMMA per gcopolymer was determined, from which it was possible to estimate the DPEA = 553 of PMMA and, consequently, the average molecular weight Mn,EA as presented in Table 1. The obtained value of Mn,EA was considered highly representative of the average molecular weight of the purified copolymer, in contrast with Mn,SEC which is highly affected by the difference in the hydrodynamic volume compared to the standards and thus not accurate.
M n,EA strongly deviates from Mn,theo, confirming that deactivated PDMAEMA chains were largely present in the polymerization mixture. Since PMMA's DPEA is 2.5 times the DP calculated from the conversion of MMA assessed by 1H-NMR (DPtheo = 219), an estimation of CEF ≈ 40% is deduced. This value is slightly lower than that calculated on the basis of NMR combined with MS analysis (CEF ≈ 46%), but very close to it. This shows that the estimation of CEF based on the data provided by NMR and MS is rather reliable. When designing the block copolymerization of PDMAEMA, the calculation of CEF may allow the adjustment of the ratio of the monomer to PDMAEMA for a better prediction of the final degree of polymerization.
Conclusions
In response to a lack of information available in the literature, an in-depth characterization of PDMAEMA prepared by ATRP was carried out by using NMR and MALDI-TOF MS techniques. The detailed investigation of PDMAEMA's structure, especially the chain ends, provides insightful information about future studies where PDMAEMA-based architectures are prepared.A comprehensive one- and two-dimensional NMR characterization allowed the assignment of the signals originating from PDMAEMA and consequent verification of the experimental degree of polymerization of the polymer, as well as the presence of side reactions lowering the fraction of chain end functionality (CEF).
An interpretation of the peaks in MALDI-TOF MS spectra was given, with the conclusion that a MALDI-induced fragmentation—with the formation of a lactone at the ω chain end—affects the observed masses of halogen-terminated PDMAEMA chains (both Br- and Cl-terminated). Based on the mass spectra, the loss of the active end group was found to be mainly due to disproportionation during polymerization. MALDI-TOF MS also enabled a qualitative assessment of the evolution of the loss of the active end group throughout the course of the polymerization. It was observed that Br-terminated chains were gradually converted into chains devoid of the expected end group, already with a significant end group loss at low values of conversion.
The selected analyses allowed the examination of aspects of the non-ideal character of the conducted polymerization of DMAEMA and subsequent chain extension. Additionally, the combination of the data collected from NMR and MS led to estimate CEF ≈ 46%. Providing tools for conducting these analyses will be useful especially in view of the synthesis of block copolymers of PDMAEMA by ATRP.
By varying the ATRP system used, different samples of PDMAEMA were analyzed and compared. MALDI-TOF MS and NMR analyses provided evidence that replacing CuBr with CuCl leads to higher control over the ω chain end. Furthermore, when samples of PDMAEMA synthesized by photomediated ATRP were analyzed, MALDI-TOF MS allowed the assessment of the presence of a self-initiated polymer.
The chain extension of PDMAEMA with MMA confirmed the poor chain end fidelity highlighted by NMR and MALDI-TOF MS; only a fraction of the PDMAEMA chains initiated the block copolymerization, as shown by SEC and DOSY. The obtained copolymer was, however, successfully purified from any unreacted PDMAEMA. The challenge of determining the DP of the extension block, not knowing a priori the percentage of active PDMAEMA chains, was overcome by means of elemental analysis.
PDMAEMA is a polymer with highly tunable properties, which are attractive in the context of a variety of advanced applications. It is often employed in the synthesis of tailor-made copolymer architectures. The characterization tools provided in this work are important in view of verifying the extent of control over DMAEMA polymerization and chain extension, and highlighting how well-defined the polymer structures are. Therefore, these tools are believed to strengthen the versatility of PDMAEMA and its copolymer architectures, especially in the contexts where well-defined structures are needed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Wilhelm Beckers Jubileumsfond (Stockholm, Sweden), SSAB (Stockholm, Sweden) and the Knut and Alice Wallenberg Foundation (KAW) through the Wallenberg Wood Science Center (Stockholm, Sweden). M. R. Telaretti Leggieri would like to thank Dr Jamie Godfrey for the valuable discussions and the important support with MALDI-TOF MS spectrum interpretation.References
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- G. M. Liu, D. Wu, C. C. Ma, G. Z. Zhang, H. F. Wang and S. H. Yang, ChemPhysChem, 2007, 8, 2254–2259 CrossRef CAS PubMed.
- F. A. Plamper, M. Ruppel, A. Schmalz, O. Borisov, M. Ballauff and A. H. E. Muller, Macromolecules, 2007, 40, 8361–8366 CrossRef CAS.
- M. Vamvakaki, G. F. Unali, V. Butun, S. Boucher, K. L. Robinson, N. C. Billingham and S. P. Armes, Macromolecules, 2001, 34, 6839–6841 CrossRef CAS.
- R. Yanez-Macias, I. Alvarez-Moises, I. Perevyazko, A. Lezov, R. Guerrero-Santos, U. S. Schubert and C. Guerrero-Sanchez, Macromol. Chem. Phys., 2017, 218, 7 Search PubMed.
- C. L. Peng, H. M. Tsai, S. J. Yang, T. Y. Luo, C. F. Lin, W. J. Lin and M. J. Shieh, Nanotechnology, 2011, 22, 11 Search PubMed.
- A. Car, P. Baumann, J. T. Duskey, M. Cham, N. Bruns and W. Meier, Biomacromolecules, 2014, 15, 3235–3245 CrossRef CAS PubMed.
- M. Farshbaf, S. Davaran, A. Zarebkohan, N. Annabi, A. Akbarzadeh and R. Salehi, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1872–1891 CAS.
- P. van de Wetering, J. Y. Cherng, H. Talsma, D. J. A. Crommelin and W. E. Hennink, J. Control. Release, 1998, 53, 145–153 CrossRef CAS PubMed.
- S. Agarwal, Y. Zhang, S. Maji and A. Greiner, Mater. Today, 2012, 15, 388–393 CrossRef CAS.
- B. M. Reis, S. P. Armes, S. Fujii and S. Biggs, Colloids Surf., A, 2010, 353, 210–215 CrossRef CAS.
- J. Engström, F. L. Hatton, L. Wågberg, F. D'Agosto, M. Lansalot, E. Malmström and A. Carlmark, Polym. Chem., 2017, 8, 1061–1073 RSC.
- M. Wåhlander, F. Nilsson, A. Carlmark, U. W. Gedde, S. Edmondson and E. Malmström, Nanoscale, 2016, 8, 14730–14745 RSC.
- T. Kaldéus, M. R. Telaretti Leggieri, C. Cobo Sanchez and E. Malmström, Biomacromolecules, 2019, 20, 1937–1943 CrossRef.
- A. Munoz-Bonilla and M. Fernandez-Garcia, Prog. Polym. Sci., 2012, 37, 281–339 CrossRef CAS.
- L. A. B. Rawlinson, S. M. Ryan, G. Mantovani, J. A. Syrett, D. M. Haddleton and D. J. Brayden, Biomacromolecules, 2010, 11, 443–453 CrossRef CAS PubMed.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Biomacromolecules, 2008, 9, 1340–1347 CrossRef CAS.
- O. Schepelina and I. Zharov, Langmuir, 2008, 24, 14188–14194 CrossRef CAS PubMed.
- A. R. Roudman and F. A. DiGiano, J. Membr. Sci., 2000, 175, 61–73 CrossRef CAS.
- R. H. Du and J. S. Zhao, J. Membr. Sci., 2004, 239, 183–188 CrossRef CAS.
- J. T. Tang, M. F. X. Lee, W. Zhang, B. X. Zhao, R. M. Berry and K. C. Tam, Biomacromolecules, 2014, 15, 3052–3060 CrossRef CAS.
- T. Saigal, H. C. Dong, K. Matyjaszewski and R. D. Tilton, Langmuir, 2010, 26, 15200–15209 CrossRef CAS PubMed.
- X. Zhang, J. H. Xia and K. Matyjaszewski, Macromolecules, 1998, 31, 5167–5169 CrossRef CAS PubMed.
- H. Dong and K. Matyjaszewski, Macromolecules, 2008, 41, 6868–6870 CrossRef CAS.
- R. A. Cordeiro, N. Rocha, J. P. Mendes, K. Matyjaszewski, T. Guliashvili, A. C. Serra and J. F. J. Coelho, Polym. Chem., 2013, 4, 3088–3097 RSC.
- M. Sahnoun, M. T. Charreyre, L. Veron, T. Delair and F. D'Agosto, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3551–3565 CrossRef CAS.
- A. Zengin, G. Karakose and T. Caykara, Eur. Polym. J., 2013, 49, 3350–3358 CrossRef CAS.
- Q. F. Xiong, P. H. Ni, F. Zhang and Z. Q. Yu, Polym. Bull., 2004, 53, 1–8 CrossRef CAS.
- A. P. Narrainen, S. Pascual and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 439–450 CrossRef CAS.
- S. B. Lee, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2003, 4, 1386–1393 CrossRef CAS PubMed.
- C. Bruce, I. Javakhishvili, L. Fogelström, A. Carlmark, S. Hvilsted and E. Malmström, RSC Adv., 2014, 4, 25809 RSC.
- J. Pietrasik, B. S. Sumerlin, R. Y. Lee and K. Matyjaszewski, Macromol. Chem. Phys., 2007, 208, 30–36 CrossRef CAS.
- Y. Y. Xu, S. Bolisetty, M. Drechsler, B. Fang, J. Y. Yuan, M. Ballauff and A. H. E. Müller, Polymer, 2008, 49, 3957–3964 CrossRef CAS.
- V. V. de Souza, M. L. D. Noronha, F. L. A. Almeida, C. A. R. Prado, A. C. Doriguetto and F. H. Florenzano, Polym. Bull., 2011, 67, 875–884 CrossRef CAS.
- G. L. Xiao, Z. B. Hu, G. P. Zeng, Y. Q. Wang, Y. Q. Huang, X. L. Hong, B. L. Xia and G. Y. Zhang, J. Appl. Polym. Sci., 2012, 124, 202–208 CrossRef CAS.
- F. L. Baines, S. P. Armes, N. C. Billingham and Z. Tuzar, Macromolecules, 1996, 29, 8151–8159 CrossRef CAS.
- B. P. Binks, R. Murakami, S. P. Armes and S. Fujii, Angew. Chem., Int. Ed., 2005, 44, 4795–4798 CrossRef CAS PubMed.
- J. C. P. de Souza, A. F. Naves and F. H. Florenzano, Colloid Polym. Sci., 2012, 290, 1285–1291 CrossRef.
- V. Chrysostomou and S. Pispas, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 598–610 CrossRef CAS.
- H. Walter, C. Harrats, P. Muller-Buschbaum, R. Jerome and M. Stamm, Langmuir, 1999, 15, 1260–1267 CrossRef CAS.
- J. Engström, T. Benselfelt, L. Wågberg, F. D'Agosto, M. Lansalot, A. Carlmark and E. Malmström, Nanoscale, 2019, 11, 4287–4302 RSC.
- E. Larsson, C. Cobo Sanchez, C. Porsch, E. Karabulut, L. Wågberg and A. Carlmark, Eur. Polym. J., 2013, 49, 2689–2696 CrossRef CAS.
- I. Zaborniak, M. Sroka and P. Chmielarz, Polymer, 2022, 254, 9 CrossRef.
- M. D. Shalati and R. M. Scott, Macromolecules, 1975, 8, 127–130 CrossRef CAS.
- L. Carlsson, A. Fall, I. Chaduc, L. Wågberg, B. Charleux, E. Malmströrn, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063–8068 CrossRef CAS.
- P. Cotanda, D. B. Wright, M. Tyler and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3333–3338 CrossRef CAS.
- N. P. Truong, Z. F. Jia, M. Burges, N. A. J. McMillan and M. J. Monteiro, Biomacromolecules, 2011, 12, 1876–1882 CrossRef CAS PubMed.
- L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, W. Van Camp, O. Coulembier, P. Degee, R. Flammang, F. Du Prez and P. Dubois, React. Funct. Polym., 2008, 68, 990–1003 CrossRef CAS.
- V. Barboiu, G. L. Ailiesei, A. G. Grigoras, E. Rusu and E. S. Dragan, Int. J. Polym. Anal. Charact., 2019, 24, 610–618 CrossRef CAS.
- J. Niskanen, C. Wu, M. Ostrowski, G. G. Fuller, S. Hietala and H. Tenhu, Macromolecules, 2013, 46, 2331–2340 CrossRef CAS.
- T. Gruendling, S. Weidner, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 599–617 RSC.
- A. H. Soeriyadi, M. R. Whittaker, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1475–1505 CrossRef CAS.
- M. Buback, F. Günzler, G. T. Russell and P. Vana, Macromolecules, 2009, 42, 652–662 CrossRef CAS.
- K. Matyjaszewski, H. C. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- A. Boujemaoui, C. Cobo Sanchez, J. Engström, C. Bruce, L. Fogelström, A. Carlmark and E. Malmström, ACS Appl. Mater. Interfaces, 2017, 9, 35305–35318 CrossRef CAS PubMed.
- D. Zhou, L.-W. Zhu, B.-H. Wu, Z.-K. Xu and L.-S. Wan, Polym. Chem., 2022, 13, 300–358 RSC.
- P. Krys and K. Matyjaszewski, Eur. Polym. J., 2017, 89, 482–523 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- J. F. Lutz and K. Matyjaszewski, Macromol. Chem. Phys., 2002, 203, 1385–1395 CrossRef CAS.
- M. Zhong and K. Matyjaszewski, Macromolecules, 2011, 44, 2668–2677 CrossRef CAS.
- F. Lorandi and K. Matyjaszewski, Isr. J. Chem., 2020, 60, 108–123 CrossRef CAS.
- S. Karanam, H. Goossens, B. Klumperman and P. Lemstra, Macromolecules, 2003, 36, 3051–3060 CrossRef CAS.
- A. K. Nanda and K. Matyjaszewski, Macromolecules, 2003, 36, 8222–8224 CrossRef CAS.
- J. Qiu, B. Charleux and K. Matyjaszewski, Polimery, 2001, 46, 453–460 CAS.
- A. S. Brar, G. Singh and R. Shankar, J. Mol. Struct., 2004, 703, 69–81 CrossRef CAS.
- G. Greyling and H. Pasch, Anal. Chem., 2015, 87, 3011–3018 CrossRef CAS PubMed.
- C. D. Borman, A. T. Jackson, A. Bunn, A. L. Cutter and D. J. Irvine, Polymer, 2000, 41, 6015–6020 CrossRef CAS.
- Y. Nakamura and S. Yamago, Macromolecules, 2015, 48, 6450–6456 CrossRef CAS.
- Y. Nakamura, T. Ogihara and S. Yamago, ACS Macro Lett., 2016, 5, 248–252 CrossRef CAS PubMed.
- G. Moad and D. H. Solomon, in The Chemistry of Radical Polymerization, Elsevier Science Ltd, Amsterdam, 2nd edn, 2005, pp. 233–278 Search PubMed.
- M. Bednarek, T. Biedron and P. Kubisa, Macromol. Chem. Phys., 2000, 201, 58–66 CrossRef CAS.
- F. Nystrom, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5313–5321 CrossRef.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672–2675 RSC.
- J. F. Lutz and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 897–910 CrossRef CAS.
- C. Bamford, G. Eastmond and D. Whittle, Polymer, 1969, 10, 771–783 CrossRef CAS.
- T. Kashiwagi, A. Inaba, J. E. Brown, K. Hatada, T. Kitayama and E. Masuda, Macromolecules, 1986, 19, 2160–2168 CrossRef CAS.
- G. Odian, in Principles of polymerization, John Wiley & Sons, 4th edn, 2004, pp. 236–237 Search PubMed.
- K. Matyjaszewski, S. M. Jo, H. J. Paik and D. A. Shipp, Macromolecules, 1999, 32, 6431–6438 CrossRef CAS.
- E. Frick, A. Anastasaki, D. M. Haddleton and C. Barner-Kowollik, J. Am. Chem. Soc., 2015, 137, 6889–6896 CrossRef CAS PubMed.
- M. Bednarek, T. Biedron and P. Kubisa, Macromol. Rapid Commun., 1999, 20, 59–65 CrossRef CAS.
- S. Coca, C. B. Jasieczek, K. L. Beers and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1417–1424 CrossRef CAS.
- K. Matyjaszewski, Y. Nakagawa and C. B. Jasieczek, Macromolecules, 1998, 31, 1535–1541 CrossRef CAS.
- A. Can, E. Altuntas, R. Hoogenboom and U. S. Schubert, Eur. Polym. J., 2010, 46, 1932–1939 CrossRef CAS.
- X. D. Liu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2016, 7, 689–700 RSC.
- H. Nonaka, M. Ouchi, M. Kamigaito and M. Sawamoto, Macromolecules, 2001, 34, 2083–2088 CrossRef CAS.
- L. Charles, Mass Spectrom. Rev., 2014, 33, 523–543 CrossRef CAS PubMed.
- M. Loos, C. Gerber, F. Corona, J. Hollender and H. Singer, Anal. Chem., 2015, 87, 5738–5744 CrossRef CAS PubMed.
- N. K. Singha, S. Rimmer and B. Klumperman, Eur. Polym. J., 2004, 40, 159–163 CrossRef CAS.
- A. T. Jackson, A. Bunn, I. M. Priestnall, C. D. Borman and D. J. Irvine, Polymer, 2006, 47, 1044–1054 CrossRef CAS.
- N. K. Singha and A. L. German, J. Appl. Polym. Sci., 2007, 103, 3857–3864 CrossRef CAS.
- B. C. Guo, H. Chen, H. Rashidzadeh and X. Liu, Rapid Commun. Mass Spectrom., 1997, 11, 781–785 CrossRef CAS.
- S. J. Wetzel, C. M. Guttman and J. E. Girard, Int. J. Mass Spectrom., 2004, 238, 215–225 CrossRef CAS.
- G. Montaudo, F. Samperi and M. S. Montaudo, Prog. Polym. Sci., 2006, 31, 277–357 CrossRef CAS.
- A. Herberg, X. Q. Yu and D. Kuckling, Polymers, 2019, 11, 23 CrossRef PubMed.
- D. A. Shipp, J. L. Wang and K. Matyjaszewski, Macromolecules, 1998, 31, 8005–8008 CrossRef CAS.
- K. Matyjaszewski, D. A. Shipp, J. L. Wang, T. Grimaud and T. E. Patten, Macromolecules, 1998, 31, 6836–6840 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- A. Allushi, C. Kutahya, C. Aydogan, J. Kreutzer, G. Yilmaz and Y. Yagci, Polym. Chem., 2017, 8, 1972–1977 RSC.
- X. Zhang and K. Matyjaszewski, Macromolecules, 1999, 32, 1763–1766 CrossRef CAS.
- S. Creutz, P. Teyssié and R. Jérôme, Macromolecules, 1997, 30, 6–9 CrossRef CAS.
- C. H. Peng, J. Kong, F. Seeliger and K. Matyjaszewski, Macromolecules, 2011, 44, 7546–7557 CrossRef CAS.
- C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1996, 29, 7717–7726 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01604d |
| This journal is © The Royal Society of Chemistry 2023 |