
The role of tertiary amines as internal catalysts for disulfide exchange in covalent adaptable networks†
Kanta
Yamawake
and
Mikihiro
Hayashi
 *
*
Department of Life Science and Applied Chemistry, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya-city, Aichi 466-8555, Japan. E-mail: hayashi.mikihiro@nitech.ac.jp
First published on 10th January 2023
Abstract
Here we explore the true effects of internal tertiary amines on the bond exchange properties of cross-linked networks with a disulfide exchange mechanism. The creep and stress relaxation data are first compared for the cross-linked samples with and without internal tertiary amines, where the segmental mobility and cross-linking density are adjusted to be the same for a fair comparison. Much greater creep and faster stress relaxation are observed for the sample with internal tertiary amines. The role of tertiary amines in disulfide exchange is then discussed based on the result of electron spin resonance (ESR) spectroscopy focusing on the sulfide radicals during the bond exchange.
Great attention has been paid to healable materials for the realization of sustainable societies.1,2 The use of supramolecular chemistry has been considered to be effective in the preparation of healable materials, due to the reversible nature under appropriate stimuli.3,4 Supramolecular elastomers and gels cross-linked via hydrogen bonds, ionic association, and coordination bonds exhibit healability, where the external stimuli induce reversible solid–liquid transition based on the dissociation and re-association of supramolecular cross-links. However, the practical application of these materials has not been well achieved, which may be because their mechanical strength governed by their weak association strength at the cross-links is insufficient.5,6
For the simultaneous realization of healability and sufficient mechanical strength, the concept of covalent adaptable networks using dynamic covalent bonds has received great attention.7,8 As a representative example, the bond formed by the Diels–Alder reaction of furan and maleimide shows reversibility by heating and cooling.9,10 The incorporation of these bonds into network structures produces healable materials, where the network connectivity is lost at high temperatures and thus the materials can behave as thermoplastics. More recently, unlike the above dissociative bond exchange, a new type of dynamic covalent bond has been explored, where the bond exchanges pass through associative bond exchange mechanisms.11,12 Specifically, cross-linked materials operated via associative bond exchange pathways are termed vitrimers.13,14 Trans-esterification bond exchange has been frequently used for the preparation of vitrimers, where most of the reported designs need the addition of catalysts.15–17 The distinct feature of vitrimers is that the network connectivity cannot be lost during bond exchanges. Despite that, vitrimers can exhibit sustainable functions, such as healability and recyclability, similarly to materials with dissociative type bond exchange mechanisms.
For the practical application of the bond exchange concept of dynamic covalent bonds, various molecular designs with compatible bond exchange reactions have been reported. One of the important attempts is designing bond exchangeable materials without adding any catalysts. The addition of external catalysts causes some problems as follows:18,19 the conventional catalysts for bond exchange reactions are often toxic and corrosive. In addition, the catalyst concentration, which is a key parameter for accelerating the bond exchange rate, is limited due to the fear of unwanted aggregation at a high concentration of catalysts. More critically, the bond exchange ability is lost by aging or by leaching out of the added catalysts from the materials.
In this regard, the bond exchange reaction potentially operating without external catalysts should be ideal. As an alternative strategy, the incorporation of molecular units showing catalytic activity into network structures has been explored. These molecular units are called internal catalysts, which solves the above problem of the use of external catalysts.20 Similarly, the concept of neighboring group participation has been developed,21 where the activation energy of a bond exchange reaction is lowered due to substituent effects that stabilize the transition state by weak physical association to the reaction center.
The concept of internal catalysts and neighboring group participation has been mostly applied to bond exchange materials with trans-esterification mechanisms.22–25 As a recent example of the neighboring group participation system, the incorporation of a trifluoromethyl substituent into the ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group was revealed to activate the trans-esterification without external catalysts.26,27 This was due to the strong electron-withdrawing effect of the fluorine unit. As an internal catalyst concept, the incorporation of tertiary amines into the network structure has been reported to be effective in activating trans-esterification without external catalysts.28,29 Similarly, the attachment of tertiary or secondary amine units in the network structure helped to accelerate the bond exchange of boronic ester exchange, silyl ether exchange, and urethane exchange.30–32
O group was revealed to activate the trans-esterification without external catalysts.26,27 This was due to the strong electron-withdrawing effect of the fluorine unit. As an internal catalyst concept, the incorporation of tertiary amines into the network structure has been reported to be effective in activating trans-esterification without external catalysts.28,29 Similarly, the attachment of tertiary or secondary amine units in the network structure helped to accelerate the bond exchange of boronic ester exchange, silyl ether exchange, and urethane exchange.30–32
These catalyst-free designs are indeed important for the practical application of the covalent adaptable network concept. Disulfide exchange is another well-known exchange reaction used for the covalent adaptable network,33–41 which is actually operated without external catalysts. Unlike other exchangeable covalent bonds, the exchange of disulfide can be triggered by various stimuli, such as heat, light, and pH.42,43 Although the detailed mechanism of disulfide exchange may be under argument, the reported mechanisms have been classified into two, that is, a [2 + 2] metathesis reaction mechanism and a [2 + 1] radical-mediated mechanism.44,45 It should be noted that the mechanism is different when catalysts working as reducing agents are present. In the case of small molecule reactions, catalytic effects of amine-based molecules, such as diethylamine and 1,8-diazabicyclo[5.4.0]undec-7-ene,46–49 have been demonstrated, where the amines work as a reducing agent to generate active thiolates by the rupture of the disulfide bond. The disulfide bond was then attacked by the nucleophilic thiolate, promoting the bond exchange.50 A similar mechanism has also been reported for systems containing phosphine-based catalysts.51–53 In fact, these amine catalysts were sometimes incorporated into cross-linked materials with exchangeable disulfide networks to promote the bond exchange.54,55 On the other hand, the catalytic effectivity of chemically attached amino units as internal catalysts on the disulfide network has not been well explored so far, and thus the clarification of their effectivity must be important in the research field of covalent adaptable networks and for their practical application.
Based on the above background, we here examine the internal catalytic effects of tertiary amines for network materials with disulfide cross-links (Fig. 1). Concretely, a polyacrylate-based polymer with epoxy side groups is used as the component, and elastomer samples are prepared by thermal cross-linking using a bis-carboxy terminated disulfide cross-linker. The main sample contains tertiary amines at the side groups, whereas the control sample does not. Overall, we demonstrate a dramatic increase in the rate of disulfide bond exchange with internal tertiary amino units and discuss the possible mechanism.
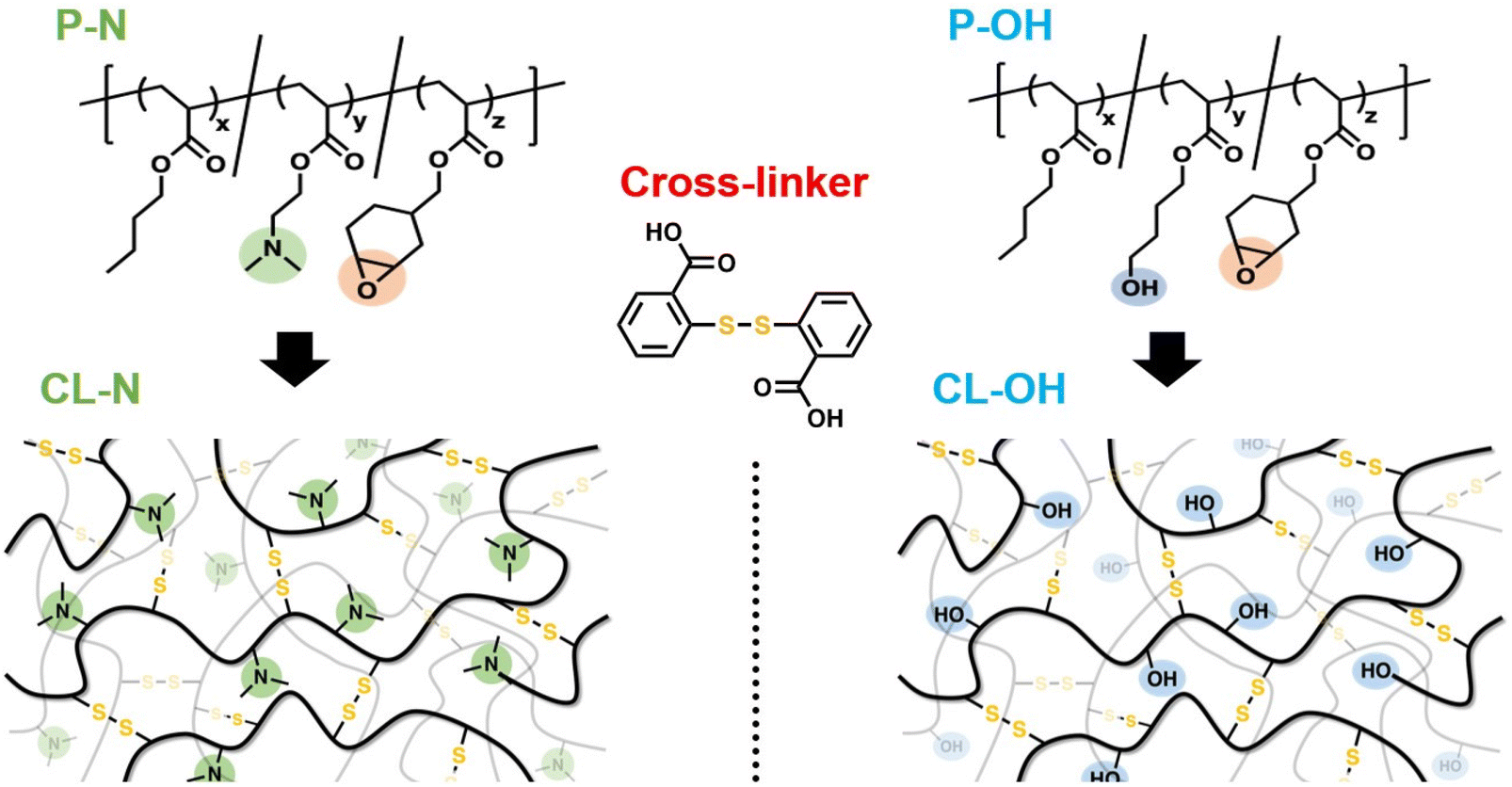 | ||
| Fig. 1 Schematic diagrams of the precursor polymers and network designs for exploring the effects of internal tertiary amines on the disulfide bond exchange in the network. | ||
The polyacrylate terpolymer bearing epoxy and tertiary amines was synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization (see the details in the ESI†) using butyl acrylate (BA), 2-(dimethylamino)ethyl acrylate (DMAEA), and (3,4-epoxycyclohexyl)methyl acrylate (ECHMA). It should be noted here that, in the preliminary experiment using small molecules, tertiary amines were confirmed to function as catalysts for disulfide exchange (Fig. S1 and S2†). The feed mole ratio and unit mole ratio in the obtained terpolymer were nearly identical, according to 1H-NMR (Fig. S3, see also the SEC curve in Fig. S5†). Concretely, the mole ratio of BA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMAEA:ECHMA was estimated to be 6.6:1.1:1. To avoid the unwanted side reaction of the RAFT residue in the chain end at high temperatures, the RAFT end group was chemically modified by aminolysis and the Michael addition reaction. The end group modification was confirmed by UV-vis spectroscopy (Fig. S6†). The size-exclusion chromatography (SEC) spectrum did not change by this modification (Fig. S5†), and the number average molecular weight (Mn) and dispersity index (Đ) was 21k and 1.3, respectively for the final product. Each polymer chain contained 18 epoxy groups, and thus the average molecular weight between the epoxy groups (Mepoxy) was estimated to be 1.2k, which was simply calculated by Mn/18. This terpolymer was coded as P–N in the following. Since the main monomer was BA, the Tg of P–N (∼−40 °C) was much lower than room temperature. The bis-carboxy terminated disulfide cross-linker, 2,2′-dithiodibenzoic acid, was then blended with P–N for cross-linking by solution casting using THF, where the epoxy:COOH ratio was set to be 1:1. After drying, the mixture was heated at 100 °C for 48 h, resulting in a self-standing elastomer film. The cross-linking formation was confirmed by Fourier transform infrared spectroscopy (FT-IR) (Fig. S7†). The signal from epoxy in the terpolymer disappeared, which was accompanied by the disappearance of the COOH signal in the disulfide cross-linker.56,57 These changes of characteristic signals indicate that the COOH-epoxy opening reaction progressed completely, and self-polymerization of the epoxy side group did not occur.
:DMAEA:ECHMA was estimated to be 6.6:1.1:1. To avoid the unwanted side reaction of the RAFT residue in the chain end at high temperatures, the RAFT end group was chemically modified by aminolysis and the Michael addition reaction. The end group modification was confirmed by UV-vis spectroscopy (Fig. S6†). The size-exclusion chromatography (SEC) spectrum did not change by this modification (Fig. S5†), and the number average molecular weight (Mn) and dispersity index (Đ) was 21k and 1.3, respectively for the final product. Each polymer chain contained 18 epoxy groups, and thus the average molecular weight between the epoxy groups (Mepoxy) was estimated to be 1.2k, which was simply calculated by Mn/18. This terpolymer was coded as P–N in the following. Since the main monomer was BA, the Tg of P–N (∼−40 °C) was much lower than room temperature. The bis-carboxy terminated disulfide cross-linker, 2,2′-dithiodibenzoic acid, was then blended with P–N for cross-linking by solution casting using THF, where the epoxy:COOH ratio was set to be 1:1. After drying, the mixture was heated at 100 °C for 48 h, resulting in a self-standing elastomer film. The cross-linking formation was confirmed by Fourier transform infrared spectroscopy (FT-IR) (Fig. S7†). The signal from epoxy in the terpolymer disappeared, which was accompanied by the disappearance of the COOH signal in the disulfide cross-linker.56,57 These changes of characteristic signals indicate that the COOH-epoxy opening reaction progressed completely, and self-polymerization of the epoxy side group did not occur.
As a control sample, we synthesized another terpolymer without tertiary amines but with hydroxyl (OH) groups (coded as P–OH). In this synthesis, 4-hydroxybutyl acrylate (HBA) was used, instead of DMAEA. The terpolymer was again synthesized via RAFT polymerization, and the RAFT residue in the end group was modified in the same way. Mn and Đ of this control terpolymer was 19k and 1.3, respectively, and the mole fraction of BA:HBA:ECHMA was estimated to be 4.7:2.5:1 (see 1H-NMR, SEC, and UV-vis data in Fig. S4–S6†). More specifically, each polymer chain contains 17 epoxy groups, and Mepoxy was estimated to be 1.1k. Thus, the molecular characteristics were almost identical to those of P–N. The same disulfide cross-linker was blended with P–OH by solution casting, and an elastomeric film was obtained by heating the dried mixture. In the FT-IR data (Fig. S7†), the disappearance of the epoxy signal and COOH signal was observed. Although OH-epoxy reaction was also possible in this system, this type of reaction may not be dominant in the presence of COOH groups. This was supported by our preliminary experiment, where P–OH after being heated solely (without cross-linkers) remained sticky and soluble, indicating no occurrence of cross-linking. Henceforth, the cross-linked samples using P–N and P–OH have been coded as CL–N and CL–OH. It should be noted that the weight fraction of disulfide cross-linkers was almost identical in the samples, which is important since the fraction of disulfide affects the bond exchange properties.42
The gel fraction (ϕgel) and swelling ratio (Rs) were estimated by swelling tests using THF. ϕgel was ∼100% for both samples, and Rs was 237% for CL–N and 230% for CL–OH. The similar Rs values indicate that the cross-linking density was nearly the same in the two samples. Differential scanning calorimetry (DSC) revealed that the Tg (ca. 3 °C) of CL–N and CL–OH was similar (Fig. S8†). The cross-link density and the segmental relaxation were then evaluated by temperature-sweep rheology. Fig. 2 presents the variation of storage modulus (E′) and loss tangent (tanδ) as a function of temperature. The tanδ peak originating from the segmental relaxation was observed at nearly the same temperature for CL–N and CL–OH, which was consistent with the DSC data. More importantly, the plateau modulus (EP) was the same (ca. 6.0 MPa) for CL–N and CL–OH. From these results, it was evident that the restriction degree of the segmental movement and the cross-linking density were similar to each other. These points are actually critical for the fair discussion of the bond exchange properties, since these parameters (i.e., the cross-link density and Tg) were known to affect the bond exchange efficiency at a given temperature.58–60 As another thermal property, the decomposition temperature was evaluated by TGA (Fig. S9†), where a 5 wt% loss was observed at ≫250 °C for both samples.
 | ||
| Fig. 2 Temperature-sweep rheology data for CL–N and CL–OH. | ||
For more quantitative analysis, the experimental EP (EP,ex) was compared with the theoretical EP (EP,theo), following the phantom network model (eqn (1)):61
 | (1) |
In the equation, ρ, R, and T represent the polymer density (1.1 g cm−3), gas constant, and the absolute temperature, respectively. The f in eqn (1) represents the functionality at the cross-linking point, which reflects the fluctuations of the cross-linking points. In the present design, f = 4 was assumed, according to the past reports dealing with the elastomers cross-linked by the side groups using short cross-linkers.62,63MX represents the molecular weight between cross-linking points, and Mepoxy was incorporated as MX since Mepoxy was much smaller than the entanglement molecular weight (∼18k) of poly(butyl acrylate)64 and thus dominates the elastic modulus. Combining these information, EP,theo was estimated to be ca. 4.5 MPa for both CL–N and CL–OH. These values were nearly consistent with EP,ex for both samples. Based on the above analysis, it was evident that the network structure of CL–N and CL–OH were nearly identical, and thus suitable for the exploration of effects of tertiary amines on bond exchange properties.
For comparison of bond exchange properties, temperature-ramp creep and stress relaxation tests were performed. In the temperature-ramp creep test, the activation of bond exchange was evaluated by the softening behavior. Concretely, a constant small stress was applied during the temperature increase, and the softening point, indicated by the deviation from the linear temperature dependence of the length change, is simply defined as the activation temperature of the bond exchange (Tact). In the past reports, the Tact value was revealed to vary by the variation of the sample modulus and applied stress.65 The moduli in the present samples were similar to each other, and thus, a fair comparison could be conducted.
Fig. 3a shows the variation of sample length with the increase in temperature, where the Y-axis was normalized by the length at 100 °C. A softening was observed at ca. 130 °C (= Tact) for both samples, whereas at higher temperatures, the deviation from the linear dependence was significantly larger for CL–N. Such creep behaviors of vitrimers were reported to be governed by the characteristic time for bond exchange reaction (i.e., bond exchange rate) and also for molecular diffusion.66,67 Since the segmental mobility was similar for CL–N and CL–OH, the difference of creep behaviors reflects these characteristic time.
 | ||
| Fig. 3 (a) Temperature-ramp creep data for CL–N and CL–OH. The Y-axis represents the sample length normalized by the length at 100 °C. The solid curves indicate the experimental data, whereas the dotted line represents the approximation straight line extrapolated from the low temperature region. (b) Stress relaxation curves for CL–N and CL–OH at 160 °C. In the Y-axis, the stress (σ) was normalized by the initial stress (σ0). The dotted curves indicate the fitting curves based on the KWW function (see main text). (c) Temperature dependence of 〈τ〉 for CL–N and CL–OH. The dotted line indicates the approximate straight line between the points. The values of R2 represents the decision coefficient for the approximation. | ||
The difference of bond exchange rates could be more directly evaluated by stress relaxation tests. For a clear comparison, the relaxation spectra were recorded at 160 °C as shown in Fig. 3b. CL–N showed fast relaxation, whereas the relaxation was very slow for CL–OH. Importantly, another control sample cross-linked with the simple diacid molecule bearing no disulfide bonds did not show any relaxation (Fig. S10†), indicating that the observed relaxation for CL–N and CL–OH was simply due to the bond exchange of disulfide bonds. For a more quantitative comparison, we estimated the average relaxation time 〈τ〉 based on the Kohlrausch–Williams–Watts (KWW) function fitting (2) and gamma function (3),68,69
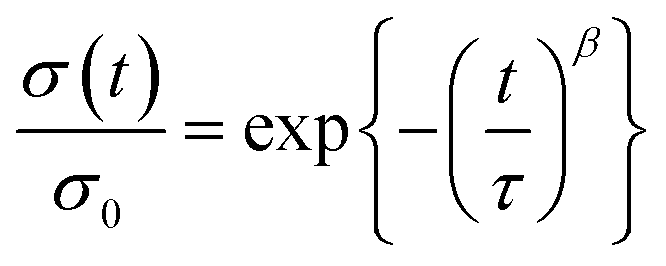 | (2) |
 | (3) |
100 s, and thus the incorporation of tertiary amines in the present fraction (ca. 0.1 mol fraction per chain) could bring about nearly 20-fold acceleration of the bond exchange. We performed an additional measurement for CL–OH in the presence of external tertiary amine catalysts (Fig. S11†), where the mole ratio of the amines to disulfide bonds was the same as that in CL–N. The relaxation rate was faster for this sample with external amine catalysts than CL–N, which may be due to the larger mobility of the amine molecules.
We performed the stress relaxation tests at various high temperatures (Fig. S12†) to estimate the activation energy (Ea) for the bond exchange. The Arrhenius dependence of 〈τ〉 was confirmed in Fig. 3c as usually observed for vitrimers, where Ea estimated by the slope was 45 kJ mol−1 for CL–N and 127 kJ mol−1 for CL–OH. The result supports the effect of internal amines on the bond exchange efficiency. In addition, the Ea value of CL–N was somewhat lower than the reported values (60–90 kJ mol−1) of elastomeric vitrimers prepared using side-group functionalized low Tg polymers by the reaction with disulfide cross-linkers, again indicating catalytic effects of tertiary amines in the network.70–72 To further support the importance of tertiary amines, we prepared another sample containing less fraction of tertiary amines by varying the initial feed mole ratio (see the characterization studies in Fig. S13–S15†). The fraction of tertiary amines was reduced to be half of CL–N. In this case, the relaxation rate was slower than CL–N (Fig. S16†) but faster than CL–OH, and Ea (∼85 kJ mol−1) was located in the middle between CL–N and CL–OH. These results clearly demonstrate that the tertiary amine is the key to control the bond exchange properties.
Owing to the enhanced bond exchange rate, macroscopic functions were exhibited more efficiently for CL–N. Bond exchangeable materials, including vitrimers, generally show reprocessability on being kept above Tact and then cooled, where the elasticity for the recovery is lost during the bond exchange at high temperatures. Fig. 4 shows the reprocessability for CL–N and CL–OH under the same processing conditions. The thin film samples were wrapped with metal bars and fixed using Kapton tapes, and the series of samples were kept at 150 °C for 30 min. After cooling and removal of tapes, only CL–N was re-shaped into a spiral film, whereas CL–OH recovered the original shape due to insufficient stress relaxation.
 | ||
| Fig. 4 Difference of reprocessability between CL–N and CL–OH during the process with A: fixing the sample with kapton tapes, B: heating in an oven (150 °C, 30 min), and C: cooling to 25 °C (room temperature). | ||
Finally, we discuss the bond exchange mechanism in the presence of tertiary amines. Disulfide exchange is known to be a radical-mediated mechanism.44,45 To assess this point, we performed electron spin resonance (ESR) measurements at 160 °C for both CL–N and CL–OH (Fig. 5). An ESR signal from the sulfide radical44,73 with a g-value of 2.005 was observed for both samples. It should be noted that the signal was less obvious at a lower temperature (∼120 °C) (Fig. S17†), supporting the temperature dependence of the bond exchange rate. The intensity of the signal at 160 °C was not so different between the samples, indicating that a radical-mediated mechanism was operated in a similar manner. Based on this result, an additional bond exchange path could be operated for CL–N, considering the dramatic change of the creep and stress relaxation data. In a past report describing the reaction path of disulfide exchange for small molecules,44 a thiolate anion was generated in the presence of a nucleophilic agent, and the disulfide exchange was promoted by the nucleophilic attack of the thiolate anion on the disulfide bond.50 Assuming that this fact could be applied to the present cross-linked system, the thiolate anion-mediated mechanism would additionally work in the bond exchange in the CL–N network owing to the attachment of tertiary amines (see the schematic diagrams in Fig. 5b and c).
 | ||
| Fig. 5 (a) ESR spectroscopy data for CL–N and CL–OH measured at 160 °C. The arrows represent the signal from the sulfide radical, and the distinct outside signals were from Mn2+ used for the magnetic field calibration. (b) The bond exchange mechanism of the disulfide bond via a radical-mediated mechanism and (c) a thiolate anion-mediated mechanism in the presence of tertiary amines. | ||
In summary, we have shed light on the true effects of internal tertiary amines on the bond exchange properties of cross-linked networks with disulfide exchange, where the network parameters, including segmental mobility and cross-linking density, were nearly identical to extract the intrinsic effects. The finding of this study is that the operation of the radical-mediated mechanism is not changed in the absence and presence of tertiary amines, whereas the additional thiolate anion-mediated mechanism should occur in the presence of tertiary amines attached in the network, resulting in a significant enhancement of the overall bond exchange efficiency. The present knowledge is thus valuable for the creation of high functional materials with more efficient bond exchanging. In the future full article, we will further investigate the universality of effects of tertiary amines for various disulfide networks with different molecular architectures.
Author contributions
Conceptualization: M. H.; formal analysis: K. Y. and M. H.; funding acquisition: M. H.; investigation: K. Y.; supervision: M. H.; validation: K. Y.; visualization: K. Y. and M. H.; writing – original draft: K. Y. and M. H.; writing – review & editing: M. H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. K. Yamamoto in Nagoya institute of technology for his assistance on the ESR measurements. We also thank Prof. A. Takasu for his assistance on the SEC measurements. We also thank Prof. K. Nagata for his assistance in performing TGA. The temperature-ramp rheology, stress relaxation tests, and ESR were conducted in the Equipment Sharing Division, Organization for Co-Creation Research and Social Contributions, Nagoya Institute of Technology. This work was supported by JSPS KAKENHI Grant Number JP 22K05210 (M. H.).References
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- Q. Chen, Nihon Reoroji Gakkaishi, 2019, 47, 197–205 CrossRef CAS.
- S. L. Wu and Q. Chen, Macromolecules, 2022, 55, 697–714 CrossRef CAS.
- N. Roy, B. Bruchmann and J. M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- Y. L. Liu and T. W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- T. Liu, B. M. Zhao and J. W. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- M. Hayashi, Polymer, 2020, 12, 1322 CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- J. R. Han, T. Liu, C. Hao, S. Zhang, B. H. Guo and J. W. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
- S. P. Wang, N. Teng, J. Y. Dai, J. K. Liu, L. J. Cao, W. W. Zhao and X. Q. Liu, Polymer, 2020, 210, 123004 CrossRef CAS.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- F. Cuminet, S. Caillol, E. Dantras, E. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS.
- C. Hao, T. Liu, S. Zhang, W. C. Liu, Y. F. Shan and J. W. Zhang, Macromolecules, 2020, 53, 3110–3118 CrossRef CAS.
- S. Debnath, S. Kaushal and U. Ojha, ACS Appl. Polym. Mater., 2020, 2, 1006–1013 CrossRef CAS.
- M. Hayashi and T. Inaba, ACS Appl. Polym. Mater., 2021, 3, 4424–4429 CrossRef CAS.
- M. Hayashi, ACS Appl. Polym. Mater., 2020, 2, 5365–5370 CrossRef CAS.
- D. Berne, F. Cuminet, S. Lemouzy, C. Joly-Duhamel, R. Poli, S. Caillol, E. Leclerc and V. Ladmiral, Macromolecules, 2022, 55, 1669–1679 CrossRef CAS.
- D. Berne, G. Coste, R. Morales-Cerrada, M. Boursier, J. Pinaud, V. Ladmiral and S. Caillol, Polym. Chem., 2022, 13, 3806–3814 RSC.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2019, 113, 297–304 CrossRef CAS.
- X. M. Feng and G. Q. Li, ACS Appl. Mater. Interfaces, 2021, 13, 53099–53110 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. B. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. B. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- A. Hernandez, H. A. Houck, F. Elizalde, M. Guerre, H. Sardon and F. E. Du Prez, Eur. Polym. J., 2022, 168, 111100 CrossRef CAS.
- A. Trejo-Machin, L. Puchot and P. Verge, Polym. Chem., 2020, 11, 7026–7034 RSC.
- J. C. Dong, B. Y. Liu, H. N. Ding, J. B. Shi, N. Liu, B. Dai and I. Kim, Polym. Chem., 2020, 11, 7524–7532 RSC.
- S. Huang, Y. K. Shen, H. K. Bisoyi, Y. Tao, Z. C. Liu, M. Wang, H. Yang and Q. Li, J. Am. Chem. Soc., 2021, 143, 12543–12551 CrossRef CAS PubMed.
- H. L. Wang, Y. J. Huang, Z. Shi, X. P. Zhou and Z. G. Xue, ACS Macro Lett., 2022, 11, 991–998 CrossRef CAS PubMed.
- H. H. Wu, X. D. Liu, D. K. Sheng, Y. Zhou, S. B. Xu, H. P. Xie, X. X. Tian, Y. L. Sun, B. R. Shi and Y. M. Yang, Polymer, 2021, 214, 123261 CrossRef CAS.
- Z. Xiang, C. Z. Chu, H. Xie, T. Xiang and S. B. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 1463–1473 CrossRef CAS PubMed.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS PubMed.
- H. Gao, Y. C. Sun, M. M. Wang, B. Wu, G. Q. Han, L. Jin, K. Zhang and Y. Y. Xia, Polymer, 2021, 212, 123132 CrossRef CAS.
- F. Ji, Y. M. Zhou and Y. M. Yang, J. Mater. Chem. A, 2021, 9, 7172–7179 RSC.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS PubMed.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- J. M. Matxain, J. M. Asua and F. Ruiperez, Phys. Chem. Chem. Phys., 2016, 18, 1758–1770 RSC.
- D. Kandemir, S. Luleburgaz, U. S. Gunay, H. Durmaz and V. Kumbaraci, Macromolecules, 2022, 55, 7806–7816 CrossRef CAS.
- A. M. Belenguer, T. Friscic, G. M. Day and J. K. M. Sanders, Chem. Sci., 2011, 2, 696–700 RSC.
- R. J. Sarma, S. Otto and J. R. Nitschke, Chem. – Eur. J., 2007, 13, 9542–9546 CrossRef CAS PubMed.
- T. Eom and A. Khan, Chem. Commun., 2020, 56, 7419–7422 RSC.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- R. Caraballo, M. Rahm, P. Vongvilai, T. Brinck and O. Ramstrom, Chem. Commun., 2008, 48, 6603–6605 RSC.
- R. Caraballo, M. Sakulsombat and O. Ramstrom, Chem. Commun., 2010, 46, 8469–8471 RSC.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- M. A. Alraddadi, V. Chiaradia, C. J. Stubbs, J. C. Worch and A. P. Dove, Polym. Chem., 2021, 12, 5796–5802 RSC.
- S. J. Tonkin, C. T. Gibson, J. Campbell, D. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- L. M. Cao, Z. Gong, C. A. H. Xu and Y. K. Chen, ACS Sustainable Chem. Eng., 2021, 9, 9409–9417 CrossRef CAS.
- K. Tangthana-umrung, Q. A. Poutrel and M. Gresil, Macromolecules, 2021, 54, 8393–8406 CrossRef CAS.
- A. Gablier, M. O. Saed and E. M. Terentjev, Soft Matter, 2020, 16, 5195–5202 RSC.
- R. Hajj, A. Duval, S. Dhers and L. Averous, Macromolecules, 2020, 53, 3796–3805 CrossRef CAS.
- K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 48682–48690 RSC.
- P. C. Himenz and T. P. Lodge, Polymer Chemistry, CRC Press, 2nd edn, 2007 Search PubMed.
- M. Hayashi, Y. Oba, T. Kimura and A. Takasu, Polym. J., 2021, 53, 835–840 CrossRef CAS.
- I. H. Syed, P. Stratmann, G. Hempel, M. Kluppel and K. Saalwachter, Macromolecules, 2016, 49, 9004–9016 CrossRef CAS.
- L. G. D. Hawke, M. Ahmadi, H. Goldansaz and E. van Ruymbeke, J. Rheol., 2016, 60, 297–310 CrossRef CAS.
- S. Kaiser, P. Novak, M. Giebler, M. Gschwandl, G. Pilz, M. Morak and S. Schlogl, Polymer, 2020, 204, 122804 CrossRef CAS.
- A. M. Hubbard, Y. X. Ren, C. R. Picu, A. Sarvestani, D. Konkolewicz, A. K. Roy, V. Varshney and D. Nepal, ACS Appl. Polym. Mater., 2022, 4, 4254–4263 CrossRef CAS.
- M. Hubbard, Y. X. Ren, D. Konkolewicz, A. Sarvestani, C. R. Picu, G. S. Kedziora, A. Roy, V. Varshney and D. Nepal, ACS Appl. Polym. Mater., 2021, 3, 1756–1766 CrossRef.
- M. Hayashi and R. Yano, Macromolecules, 2020, 53, 182–189 CrossRef CAS.
- M. Hayashi, R. Yano and A. Takasu, Polym. Chem., 2019, 10, 2047–2056 RSC.
- Y. X. Yang, L. Y. Huang, R. Y. Wu, Z. Niu, W. F. Fan, Q. Q. Dai, L. Cui, J. Y. He and C. X. Bai, ACS Appl. Mater. Interfaces, 2022, 14, 3344–3355 CrossRef CAS PubMed.
- L. Q. Zhu, L. Xu, S. Y. Jie and B. G. Li, Polymer, 2021, 13, 4157 CAS.
- T. Y. Gong, L. H. Guo, J. Ye, L. F. He, T. Qiu and X. Y. Li, J. Polym. Sci., 2021, 59, 1807–1820 CrossRef CAS.
- M. Hernandez, A. M. Grande, W. Dierkes, J. Bijleveld, S. van der Zwaag and S. J. Garcia, ACS Sustainable Chem. Eng., 2016, 4, 5776–5784 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, analytical methods, synthesis protocols, blending and cross-linking reaction, preliminary experiments using small molecules, 1H-NMR spectra, SEC curves, UV-vis spectra, FT-IR spectra, DSC data, TGA thermograms, relaxation data for the sample without disulfide, relaxation data for CL-OH in the presence of external amine catalysts, temperature dependence of the stress relaxation for CL-N and CL-OH, relaxation data for the sample with a smaller fraction of tertiary amines, and ESR spectra at 120 °C. See DOI: https://doi.org/10.1039/d2py01406h |
| This journal is © The Royal Society of Chemistry 2023 |