
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating the shuttling motion of [2]rotaxanes built of p-xylylenediamine units through permethylation at the benzylic positions of the ring†
Julio
Puigcerver
 ,
Mateo
Alajarin
,
Alberto
Martinez-Cuezva
* and
Jose
Berna
*
,
Mateo
Alajarin
,
Alberto
Martinez-Cuezva
* and
Jose
Berna
*
Departamento de Química Orgánica, Facultad de Química, Regional Campus of International Excellence “Campus Mare Nostrum”, Universidad de Murcia, E-30100 Murcia, Spain. E-mail: amcuezva@um.es; ppberna@um.es
First published on 3rd November 2023
Abstract
In this study, we show the effect of the gem-dimethyl substitution at the four benzylic carbons of the ring on the internal dynamics of two-station [2]rotaxanes. Such structural modification of the polyamide macrocycles promotes a drastic change of the internal dynamics as shown by variable-temperature (VT) 1H NMR experiments. We determined that the shuttling rates of the octamethylated macrocycle along a series of symmetrical threads were significantly faster compared to the non-substituted ring. This effect was particularly pronounced in the fumaramide-based system, in which the rate was 27 times faster than that of the model system.
Introduction
Over recent decades, mechanically interlocked molecules (MIMs) have become a cutting-edge research topic, showcasing interesting breakthroughs in synthetic methodologies and in their potential applications.1,2 One particular area of attention lies in the design of these molecules, aiming to achieve precise control over the internal motions of their submolecular components, mimicking biological machinery,3 through the application of external stimuli.4 Consequently, the development of programmable systems capable of executing controlled movements on demand is highly desirable.5In order to achieve the precise assembly and control of sophisticated rotaxane-based molecular machines,6 it is particularly helpful to study simpler model systems that provide valuable information about their internal motions. In this regard, degenerate [2]rotaxanes, systems with two identical binding sites at the thread, serve as compelling candidates for such investigations. Within these structures, the macrocyclic component shuttles back and forth between the two stations, enabling easy measurement of its dynamics using simple techniques, such as NMR spectroscopy. The modulation of this motion, either by accelerating or decelerating the movement of the macrocycle, can be readily achieved by altering the solvent polarity (solvent variation)7 or introducing steric or electrostatic barriers along the thread between the two isoenergetic stations.8 Another strategy involving the modification of the backbone of the macrocycle has been less frequently implemented.9 For instance, in Leigh's polyamide-based rotaxanes, the incorporation of electron-withdrawing or donating groups into the isophthalamide units of the ring modifies the acidity of the NH protons in the macrocycle's secondary amides.10 As a result, the hydrogen-bonding ability of these units with the embedded binding sites is adjusted, leading to variations in the internal rotational and translational motions.11 Moreover, in this way the stability of the mechanical bond in kinetically stable [2]pseudorotaxanes can also be altered.12 It is worth noting that despite these strategies based on the electronic effects of substituents, other structural modifications of the macrocyclic component to modulate the internal dynamics in hydrogen-bonded [2]rotaxanes remains relatively unexplored.
Herein we disclose the impact of the geminal disubstitution13,14 at the methylene units of the polyamide macrocycle on the internal motions of a series of degenerate [2]rotaxanes (Fig. 1). The geminal dialkyl substitution at a methylene carbon atom, also known as the Thorpe–Ingold effect, has been widely used in organic synthesis to promote cyclization reactions as the driving force accelerating the ring closure of the substrates.15
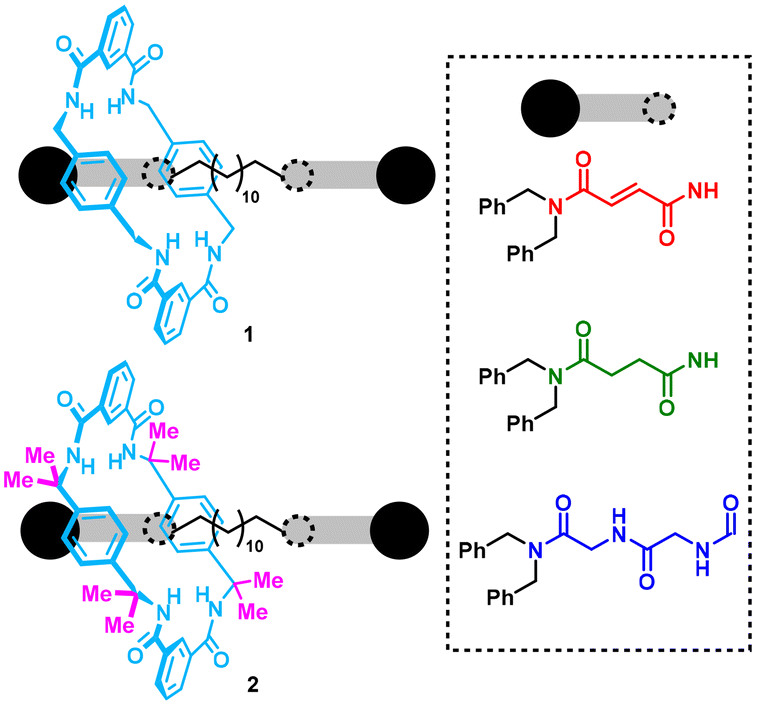 | ||
| Fig. 1 Degenerate polyamide-based [2]rotaxanes 1 and 2 designed for this study. | ||
For this task, we conducted a comparative analysis by synthesizing two types of rotaxanes: rotaxanes 1, featuring the original non-substituted Leigh-type tetrabenzylic macrocycle, and compounds 2, incorporating an interlocked octamethylated ring bearing two methyl groups at each of the four benzylic carbon atoms of the p-xylylenediamine fragments (Fig. 1, methyl groups are highlighted in magenta). These two rings were combined with three different threads, each with two identical binding sites: fumaramide (red), succinamide (green), and glycylglycine (dark blue) units.
For giving account of the impact of the geminal disubstitution on the internal dynamics, we performed variable-temperature (VT) 1H NMR experiments, allowing us to calculate the activation energy barriers for the translational motion of each macrocycle along the respective thread.
Results and discussion
Hydrogen-bonding directed assembly of the octamethylated fumaramide-based [2]rotaxane
As starting point, we synthesized the p-xylylenediamine 3, not commercially available, bearing two methyl groups at each of the four benzylic positions. The synthesis started from diol 4 and was accomplished in two steps (Scheme 1a). Diol 4 smoothly reacted with sodium azide in the presence of trifluoroacetic acid, resulting in the diazide 5. Subsequently, catalytic hydrogenation of 5 quantitatively produced the diamine 3. We then assessed the suitability of 3 for the assembly of benzylic amide rotaxanes using the fumaramide-based thread 6, previously identified as the most effective template for synthesizing this type of systems (Scheme 1b).16 By following a five-component clipping reaction with thread 6, isophthaloyl chloride, and diamine 3 in the presence of Et3N, we successfully obtained the [2]rotaxane 7 in 11% yield. Previous reports have shown the benefits of the Thorpe–Ingold effect in related [2 + 2] macrocyclization processes.17 However, this low yield, compared to the one previously reported by using the parent p-xylylenediamine (97%) suggests the instability of the supramolecular intermediates leading to the rotaxane 7 and the apparently moderate affinity of the octamethylated macrocycle for the fumaramide binding site. The introduction of the methyl groups at the ring could affect the electronics of its amide functions playing a key role during the supramolecular recognition between the rotaxane precursors. The smooth electron-donating character of the methyl groups probably makes slightly less acidic the NH protons of the amides at the macrocycle precursor. In fact, a pKa = 15.2 was computationally estimated for a dimethylated amide surrogate, while the value of the unsubstituted model was pKa = 15.0 (see ESI, Scheme S5†).18 The comparable calculated pKa values suggest that an additional significant factor is contributing to the diminished binding affinity between the thread and the macrocyclic counterpart. The steric hindrance imposed by the methyl groups can distort the optimal conformation of the macrocycle precursors, thereby precluding an ideal interaction with the thread. During the purification of rotaxane 7, we also isolated the octamethylated macrocycle, which turned out to be soluble in chlorinated solvents due to its methyl groups (see ESI for further details†), in stark contrast to the significant insolubility of the parent macrocycle.19 | ||
| Scheme 1 (a) Synthesis of methylated diamine 3; (b) synthesis of the octamethylated rotaxane 7. Reaction conditions: (i) NaN3, TFA, CHCl3, 0 °C to 25 °C, 6 h, 89%; (ii) H2, Pd/C 10%, MeOH, 25 °C, overnight, 99%; (iii) isophthaloyl dichloride, diamine 3, Et3N, CHCl3, 25 °C, 4 h, 11%. | ||
The structure of rotaxane 7 was elucidated through single-crystal X-ray diffraction (SCXRD). Interestingly, the carbonyl groups of the amides in the ring adopt an alternating in/out/in/out conformation, leading to a significant distortion of the macrocycle (Fig. 2 and Fig. S20, S21†). As a result, the NH groups of the amides with an out-facing carbonyl group establish hydrogen bonds with the oxygen atoms of the fumaramide moiety (N3H3⋯O1, 2.12 Å), while the carbonyl groups with in conformation form hydrogen bonds with the NH groups of the fumaramide (N1H1⋯O2, 2.02 Å). Furthermore, this in/out conformation also favours the establishment of intermolecular hydrogen-bonding interactions between neighbouring rotaxanes, thereby enhancing the stability of the crystalline network (Fig. S21†). This solid-state conformation differs greatly from that observed in Leigh's rotaxane and other systems of this type, where the ring adopts a chair-like conformation, with all its carbonyl groups oriented outward from the cavity, and the NH groups of the amides establishing four bifurcated hydrogen bonds with the two oxygens of the fumaramide thread.1–3,20 A similar ring distortion and in/out/in/out conformation of its carbonyl groups were also observed in solid-state systems that only bear four methyl groups, one at each benzylic position of the ring.21
 | ||
| Fig. 2 X-ray structure of rotaxane 7 in: (a) inclined view; (b) front view. For clarity, selected hydrogen atoms and solvent molecules have been deleted. Colour code: grey: carbon atoms at the thread; light blue: carbon atoms at the macrocycle; red: oxygen atoms; dark blue: nitrogen atoms; magenta: methyl groups at the macrocycle; white: hydrogen atoms. Intramolecular hydrogen-bond lengths [Å] (and angles [°]): N3–H3⋯O1 2.12 (172); N1–H1⋯O2 2.02 (178). | ||
The lower affinity of the octamethylated macrocycle for the fumaramide station in rotaxane 7, in comparison to its non-methylated counterpart, is unveiled by the 1H NMR spectra of thread 6 and rotaxane 7 measured in CDCl3 (Fig. 3). In solution, where the individual molecules are isolated from each other, the components in both rotaxanes must adopt intercomponent hydrogen bonding patterns to lower their energy, which differs from the conformation of 7 at the solid state. In the previously reported non-methylated rotaxanes, the signals corresponding to the NHs of the amide within the embedded fumaramide (Hb) are significantly shifted to lower fields (Δδ = +2.5 ppm), in comparison with the isolated thread, whereas in the methylated rotaxane 7, the shift is less pronounced (Δδ = +1.5 ppm). Similarly, the signals associated with the four NH groups of the macrocyclic amides (HD) are in the aromatic region (between 7.10–7.40 ppm) in rotaxane 7, whereas in the non-methylated derivative, they appear at higher chemical shifts (∼7.83 ppm). This signal at the free methylated macrocycle (HD) appears at lower chemical shift (6.59 ppm), indicating the lack of any intercomponent hydrogen bonding interactions (Fig. 3c). All these observations indicate a weaker interaction between the fumaramide binding-site and the octamethylated macrocycle, compared to its non-methylated analogous. To gain quantitative data regarding the rotational motion of the methylated macrocycle around the thread, we conducted variable-temperature experiments on rotaxane 7. However, we could not obtain suitable data for analysis due to the complex NMR spectra observed at low temperatures, probably due to additional conformational changes around the amide functions of the thread (see ESI for additional details, Fig. S1†).
 | ||
| Fig. 3 1H NMR spectra (400 MHz, 2 mM, CDCl3, 298 K) of: (a) thread 6; (b) rotaxane 7 and (c) free macrocycle. Colours code: red: signals referred to fumaramide binding site; light blue: signals referred to the macrocycle. For lettering see Scheme 1b. | ||
Synthesis of the permethylated and non-methylated degenerate [2]rotaxanes and evaluation of the internal dynamics
We next synthesized the degenerate [2]rotaxanes 1 and 2, by combining the two rings with the three different two-station threads (Scheme 2), as ideal candidates for measuring the internal translational kinetics of their macrocycles. The five-component reactions involving the fumaramide-based thread 8a (in red), succinamide 8b (in green), or glycylglycine 8c (in dark blue), along with isophthaloyl chloride and either p-xylylenediamine or diamine 3, resulted in the formation of the respective rotaxanes 1a–c and 2a–c, (see ESI† for detailed synthetic procedures). For rotaxanes 1, we employed 4 equiv. of the macrocyclic precursors, while for rotaxanes 2, a higher excess of isophthaloyl dichloride and diamine 3 (8 equiv.) was used to ensure good conversions, considering the low yield obtained in the preparation of the one-station rotaxane 7. Additionally, in most of these reactions the corresponding [3]rotaxanes, with one macrocycle at each of the two stations, were also isolated and characterized (see ESI† for further details).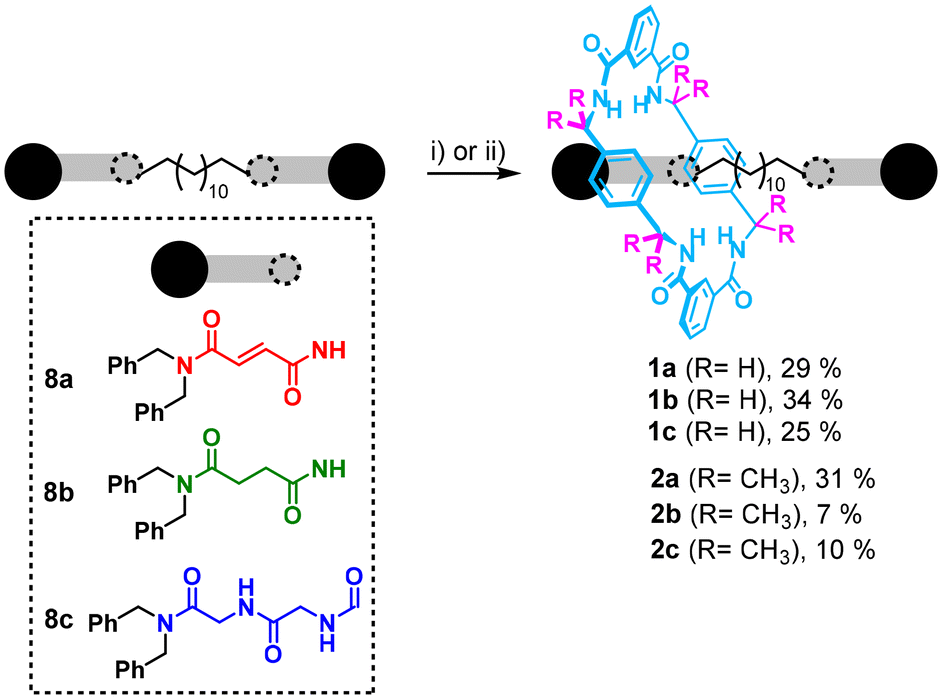 | ||
| Scheme 2 Synthesis of degenerate rotaxanes 1 (without methyl groups, R = H) and 2 (with methyl groups, R = CH3) in the presence of different threads 8. Reaction conditions: (i) isophthaloyl dichloride (4 equiv.), p-xylylenediamine (4 equiv.), Et3N (12 equiv.), CHCl3, 25 °C, 4 h; (ii) isophthaloyl dichloride (8 equiv.), diamine 3 (8 equiv.), Et3N (24 equiv.), CHCl3, 25 °C, 4 h. Yields are referred to NMR yields due to the difficulty of some purification steps. | ||
We then investigated the macrocycle-shuttling dynamics in the synthesized degenerate [2]rotaxanes 1 and 2. As discussed above, the methyl groups in 2 should slightly decrease the acidity of the amide-NHs at the macrocyclic backbone thus weakening the intercomponent hydrogen bonds between rings and threads, and expectedly accelerating the internal shuttling dynamics. In order to confirm this assumption, we analysed the translational motion of the ring along the thread in systems 1 and 2 by VT 1H-NMR experiments (Table 1 and Fig. S2–S7, Table S1†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1/2 |
T
c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (K) a (K) |
Δν (Hz) | k c (s−1) | ΔGc‡b (kcal·mol−1) |
K (s−1, 298 K) |
| a NMR temperature calibration was performed using a pure methanol sample. b Calculated value ±0.2. | ||||||
| 1 | 1a | 288 | 580 | 1288 | 12.7 | 235 |
| 2 | 2a | 248 | 405 | 900 | 11.2 | 6350 |
| 3 | 1b | 253 | 76 | 169 | 12.1 | 1099 |
| 4 | 2b | 233 | 230 | 511 | 10.6 | 2085 |
| 5 | 1c | 243 | 180 | 400 | 11.6 | 4213 |
| 6 | 2c | 228 | 160 | 355 | 10.6 | 4621 |

At elevated temperatures (>290 K), the macrocycles exhibited a rapid back and forth motion between the two identical binding sites, leading to an averaged co-conformation of the systems. As the temperature gradually decreased below the coalescence temperatures (Tc), different co-conformations emerged. In the case of the fumaramide-based rotaxanes 1a and 2a, a clear splitting of the NMR signals attributed to the fumaramide Hb+c protons (see Scheme 1 for lettering) was observed below 288 K for 1a and 248 K for 2a. A similar behaviour was observed for the succinamide derivatives, with the signal splitting occurring at 253 K for 1b and below 233 K for 2b, in this case by observing the signals of the succinamide methylenes and the NH-linked methylenes of the C10-alkyl chain. Similarly, the glycylglycine-based rotaxanes 1c and 2c exhibited splitting of the alkyl chain methylene signals below 243 K and 228 K, respectively.
By analyzing the separation of the different signals (Δν) at low temperatures and the coalescence temperature (Tc) for each system, we determined the energy barriers (ΔG‡) of the macrocycle shuttling (Table 1). A clear correlation was observed between the data and the structure of the ring as, in all cases, the non-methylated rotaxanes 1 exhibited higher translational barriers compared to their methylated analogues 2. Furthermore, we calculated the exchange rates at different temperatures by fitting the Lorentzian line of the peak above the coalescence temperature, enabling the determination of the exchange constants for each system, 1 and 2, at 298 K (Table 1 and see ESI, Table S2, Fig. S8–S19†). Remarkably, the methylated macrocycle in 2a exhibited a shuttling speed at 298 K that is 27 times faster than that of the non-methylated macrocycle in 1a (Table 1, entries 1 and 2). For the succinamide-derived systems 1b and 2b (Table 1, entries 3 and 4), the difference in shuttling speed decreased to only 2 times faster in favour of the permethylated system. In the case of the glycylglycine-based rotaxanes 1c and 2c, the experimental exchange rates were very similar (Table 1, entries 5 and 6). Not unexpectedly, as the shuttling rates of the non-methylated macrocycles increase (the rates in 1b and 1c are nearly 5 and 20 times faster than in 1a respectively, as shown in Table 1), the differences with its permethylated analogous 2b and 2c decrease in accordance. Moreover, the large difference in the shuttling rates between rotaxanes 1a and 2a (27 times faster in 2a) can be attributed to the rigidity of the fumaramide binding site, which is not able to establish strong hydrogen bonds with the methylated macrocycle, required to stabilize the in/out/in/out conformation as that observed at the crystalline state. In contrast, this stabilization is more feasible in the case of rotaxane 2b–c, with more flexible binding sites (succinamide and glycylglycine) that can be better accommodated in the inner of the permethylated macrocycle, increasing the hydrogen bonds between components. In addition, the increase of the hydrogen bonding donor and acceptor atoms in the glyglylglycine stations is also beneficial for this purpose, and therefore, no difference in the kinetics is observed between systems 1c and 2c.
Conclusions
We successfully synthesized a p-xylylenediamine surrogate tetramethylated at the benzylic positions, which was utilized for preparing a series of tetraamide-based [2]rotaxanes. The assembly of a one-station rotaxane with a fumaramide thread was a proof of the low affinity between the octamethylated macrocycle, or its precursors, and the thread. The synthesis of three degenerate rotaxanes with two identical binding sites by combining the same tetramethylated diamine with three symmetrical two-station threads (with two fumaramide, succinamide, or glycylglycine fragments) allowed its comparison with those containing non-substituted macrocycles and the same threads. These results revealed the faster shuttling rates of the octamethylated macrocycle along the symmetrical threads in comparison with those of the respective non-substituted analogous. Such acceleration effect is particularly pronounced in the pair of fumaramide-based systems, with the methylated macrocycle showing an impressive rate, 27 times faster than its unsubstituted partner, whereas the differences in shuttling rate are smaller with the succinamide thread, but still considerable (×2), and very small (×1.1) between the two glycylglycine stations. Thus, the adequate selection of the ring-thread pair allows the modulation of the shuttling rate in two-station [2]rotaxanes between a wide range of values, from 235 to 6350 s−1. These findings contribute to a better understanding of the principles governing the internal dynamics of rotaxanes and hopefully they would be useful for tailoring their properties in order to facilitate the discovery of new potential applications of this type of MIMs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministry of Science and Innovation (project PID2020-113686GB-I00/MICINN/AEI/10.13039/501100011033) and the Fundacion Seneca-CARM (project 21907/PI/22). J. P. also thanks Ministerio de Universidades for his predoctoral contract (FPU19/05419).References
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley-VCH, Weinheim, 2016 Search PubMed.
- (a) Y. C. Tse and H. Y. Au-Yeung, Chem. – Asian J, 2023, 18, e202300290 CrossRef CAS PubMed; (b) A. Saura-Sanmartin and C. A. Schalley, Chem, 2023, 9, 823 CrossRef CAS; (c) A. W. Heard, J. M. Suárez and S. M. Goldup, Nat. Rev. Chem., 2022, 6, 182 CrossRef PubMed; (d) A. Saura-Sanmartin, A. Pastor, A. Martinez-Cuezva, G. Cutillas-Font, M. Alajarin and J. Berna, Chem. Soc. Rev., 2022, 51, 4949 RSC; (e) L. Chen, X. Sheng, G. Li and F. Huang, Chem. Soc. Rev., 2022, 51, 7046 RSC; (f) J. Riebe and J. Niemeyer, Eur. J. Org. Chem., 2021, 5106 CrossRef CAS; (g) K. Yang, S. Chao, F. Zhang, Y. Pei and Z. Pei, Chem. Commun., 2019, 55, 13198–13210 RSC.
- (a) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25 CrossRef CAS PubMed; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed; (c) F. Lancia, A. Ryabchun and N. Katsonis, Nat. Rev. Chem., 2019, 3, 536 CrossRef CAS; (d) M. N. Tasbas, E. Sahin and S. Erbas-Cakmak, Coord. Chem. Rev., 2021, 443, 214039 CrossRef CAS; (e) S. R. Beeren, C. T. McTernan and F. Schaufelberger, Chem, 2023, 9, 1378 CrossRef CAS.
- (a) V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542 RSC; (b) M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200 CrossRef CAS PubMed; (c) S. Borsley, D. A. Leigh and B. M. W. Roberts, Nat. Chem., 2022, 14, 728 CrossRef CAS PubMed; (d) J.-X. Liu, K. Chen and C. Redshaw, Chem. Soc. Rev., 2023, 52, 1428 RSC.
- (a) C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491 RSC; (b) J. S. W. Seale, Y. Feng, L. Feng, R. D. Astumian and J. F. Stoddart, Chem. Soc. Rev., 2022, 51, 8450 RSC.
- (a) C. Dietrich-Buchecker, M. C. Jimenez-Molero, V. Sartor and J.-P. Sauvage, Pure Appl. Chem., 2003, 75, 1383 CAS; (b) A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117 CrossRef CAS PubMed; (c) Y. Qiu, B. Song, C. Pezzato, D. Shen, W. Liu, L. Zhang, Y. Feng, Q.-H. Guo, K. Cai, W. Li, H. Chen, M. T. Nguyen, Y. Shi, C. Cheng, R. D. Astumian, X. Li and J. F. Stoddart, Science, 2020, 368, 1247 CrossRef CAS PubMed; (d) S. Amano, S. D. P. Fielden and D. A. Leigh, Nature, 2021, 594, 529 CrossRef CAS PubMed; (e) M. Canton, J. Groppi, L. Casimiro, S. Corra, M. Baroncini, S. Silvi and A. Credi, J. Am. Chem. Soc., 2021, 143, 10890 CrossRef CAS PubMed; (f) L. Binks, C. Tian, S. D. P. Fielden, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2022, 144, 15838 CrossRef CAS PubMed.
- (a) D. B. Amabilino, P. R. Ashton, V. Balzani, C. L. Brown, A. Credi, J. M. J. Frechet, J. W. Leon, F. M. Raymo, N. Spencer, J. F. Stoddart and M. Venturi, J. Am. Chem. Soc., 1996, 48, 12012 CrossRef; (b) J. G. Cao, M. C. T. Fyfe, J. F. Stoddart, G. R. L. Cousins and P. T. Glink, J. Org. Chem., 2000, 7, 1937 CrossRef PubMed; (c) S. M. Goldup, D. A. Leigh, P. J. Lusby, R. T. McBurney and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 3381 CrossRef CAS PubMed.
- (a) A. S. Lane, D. A. Leigh and A. Murphy, J. Am. Chem. Soc., 1997, 119, 11092 CrossRef CAS; (b) S. Kang, S. A. Vignon, H.-R. Tseng and J. F. Stoddart, Chem. – Eur. J., 2004, 10, 2555 CrossRef CAS PubMed; (c) L. Jiang, J. Okano, A. Orita and J. Otera, Angew. Chem., Int. Ed., 2004, 43, 2121 CrossRef CAS PubMed; (d) P. Ghosh, G. Federwisch, M. Kogej, C. A. Schalley, D. Haase, W. Saak, A. Lützen and R. M. Gschwind, Org. Biomol. Chem., 2005, 3, 2691 RSC; (e) A. Coskun, D. C. Friedman, H. Li, K. Patel, H. A. Khatib and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 2493 CrossRef CAS PubMed; (f) M. Hmadeh, A. C. Fahrenbach, S. Basu, A. Trabolsi, D. Benitez, H. Li, A.-M. Albrecht-Gary, M. Elhabiri and J. F. Stoddart, Chem. – Eur. J., 2011, 17, 6076 CrossRef CAS PubMed; (g) J. Berná, M. Alajarín, C. Marín-Rodríguez and C. Franco-Pujante, Chem. Sci., 2012, 3, 2314 RSC; (h) D. D. Günbaş and A. M. Brouwer, J. Org. Chem., 2012, 77, 5724 CrossRef PubMed; (i) M. Douarre, V. Martí-Centelles, C. Rossy, I. Pianet and N. D. McClenaghan, Eur. J. Org. Chem., 2020, 5820 CrossRef CAS.
- K. Hirose, Y. Shiba, K. Ishibashi, Y. Doi and Y. Tobe, Chem. – Eur. J., 2008, 14, 3427 CrossRef CAS PubMed.
- S. Y. Chang, H. S. Kim, K.-J. Chang and K.-S. Jeong, Org. Lett., 2004, 6, 181 CrossRef CAS PubMed.
- (a) P. Farràs, E. C. Escudero-Adán, C. Viñas and F. Teixidor, Inorg. Chem., 2014, 53, 8654 CrossRef PubMed; (b) J. Berna, M. Alajarin, J. S. Martinez-Espin, L. Buriol, M. A. P. Martins and R.-A. Orenes, Chem. Commun., 2012, 48, 5677 RSC; (c) A. Saura-Sanmartin, J. S. Martinez-Espin, A. Martinez-Cuezva, M. Alajarin and J. Berna, Molecules, 2017, 22, 1078 CrossRef PubMed; (d) J. de Maria Perez, M. Alajarin, A. Martinez-Cuezva and J. Berna, Org. Chem. Front., 2022, 9, 2690–2696 RSC.
- (a) A. Martinez-Cuezva, L. V. Rodrigues, C. Navarro, F. Carro-Guillen, L. Buriol, C. P. Frizzo, M. A. P. Martins, M. Alajarin and J. Berna, J. Org. Chem., 2015, 80, 10049 CrossRef CAS PubMed; (b) A. Martinez-Cuezva, F. Morales, G. R. Marley, A. Lopez-Lopez, J.-C. Martinez-Costa, D. Bautista, M. Alajarin and J. Berna, Eur. J. Org. Chem., 2019, 3480 CrossRef CAS.
- For selected examples of mechanically interlocked molecules with polyamide macrocycles disubstituted at germinal positions: (a) C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS; (b) R. Jager and F. Vogtle, Angew. Chem., Int. Ed. Engl., 1997, 36, 930 CrossRef CAS.
- The presence of a cyclohexyl group at the methylene carbon of a polyamide macrocycle increases the solubility of the key intermediates: C. A. Hunter, J. Chem. Soc., Chem. Commun., 1991, 749 RSC.
- M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735 CrossRef CAS PubMed.
- F. G. Gatti, D. A. Leigh, S. A. Nepogodiev, A. M. Z. Slawin, S. J. Teat and J. K. Y. Wong, J. Am. Chem. Soc., 2001, 123, 5983 CrossRef CAS PubMed.
- (a) S. Madhu, E. V. Rashmi, R. H. G. Gonnade and G. J. Sanjayan, New J. Chem., 2017, 41, 8721 RSC; (b) N. Nazari, S. Bernard, D. Fortin, T. Marmin, L. Gendron and Y. L. Dory, Chem. – Eur. J., 2023, 29, e202203717 CrossRef CAS PubMed.
- The calculations were carried with the software included in the package Marvin Suite version 5.11.5 and Chemicalize.org, both developed by the ChemAxom Company: M. Swain, J. Chem. Inf. Model., 2012, 52, 613–615 CrossRef CAS.
- A. G. Johnston, D. A. Leigh, A. Murphy, J. P. Smart and M. D. Deegan, J. Am. Chem. Soc., 1996, 118, 10662 CrossRef CAS.
- F. F. S. Farias, G. H. Weimer, S. K. Kunz, P. R. S. Salbego, T. Orlando and M. A. P. Martins, J. Mol. Liq., 2023, 385, 122291 CrossRef CAS.
- C. Lopez-Leonardo, A. Martinez-Cuezva, D. Bautista, M. Alajarin and J. Berna, Chem. Commun., 2019, 55, 6787 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic and mass spectrometry data for all new compounds, and full crystallographic details. CCDC 2279619 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01611k |
| This journal is © The Royal Society of Chemistry 2023 |