
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of the indeno[1,2-b]indole core of janthitrem B†
Marvin
Fresia
 a,
Alexandra
Dierks
a,
Peter G.
Jones
b and
Thomas
Lindel
*a
a,
Alexandra
Dierks
a,
Peter G.
Jones
b and
Thomas
Lindel
*a
aInstitute of Organic Chemistry, Technical University Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: th.lindel@tu-braunschweig.de
bInstitute of Inorganic and Analytical Chemistry, Technical University Braunschweig, Hagenring 30, 38106 Braunschweig, Germany
First published on 2nd November 2023
Abstract
The tetracyclic core structure of the majority of indole diterpenoids features a trans-hydrindane moiety that is fused to an indole unit. We report here a novel synthetic route that includes a photo-Nazarov cyclization of a 3-acylindole precursor initially providing the thermodynamically preferred cis-hydrindanone. After reduction and conversion to the cyclopentadiene, dihydroxylation and hydrogenation provided the indoline. The key step generated the trans-system by stereospecific hydride shift on a dioxaphospholane under Grainger's conditions, for the first time applied to an N-heterocycle. When starting from the corresponding indole, we observed the formation of hitherto unknown methanocyclohepta[b]indolones. Deoxygenation of the trans-hydrindanone was achieved after conversion to the 1,3-dithiolane, followed by RANEY® Ni reduction.
Introduction
Indole diterpenoids constitute a prominent class of fungal natural products.1 The core structure of most of the indole diterpenoids is characterized by an indole unit to which is anellated a tri- or tetracyclic terpenoid moiety. Since the indole diterpenoids exhibit versatile biological activities such as tremorgenic,2 anticancer,3 anti-insect,4 and antimicrobial5 effects, their synthesis is of prime importance for the development of new drugs. Much of the biological data need further analysis, which requires independent routes to the compounds themselves and to derivatives.Recently, we reported the synthesis of the ABCD tetracyclic part of janthitrem B (1), which required the functionalization of the benzene section of the indole core.6 That particular moiety also occurs in shearinine D, of which the Carreira group recently reported an elegant total synthesis.7 Common to a larger part of the indole diterpenoid family is the tetracyclic indeno[1,2-b]indole partial structure highlighted in Fig. 1, which is found frequently among the nodulisporic acids, paspalines, paxillines, terpendoles, shearinines, janthitrems, and lolitrems. A trans-indane moiety is present that seems to be necessary for biological activity. For instance, Giannis and coworkers8 showed that the cis-fused isomer of terpendole E lost the KSP inhibitory activity of the trans-fused natural product.
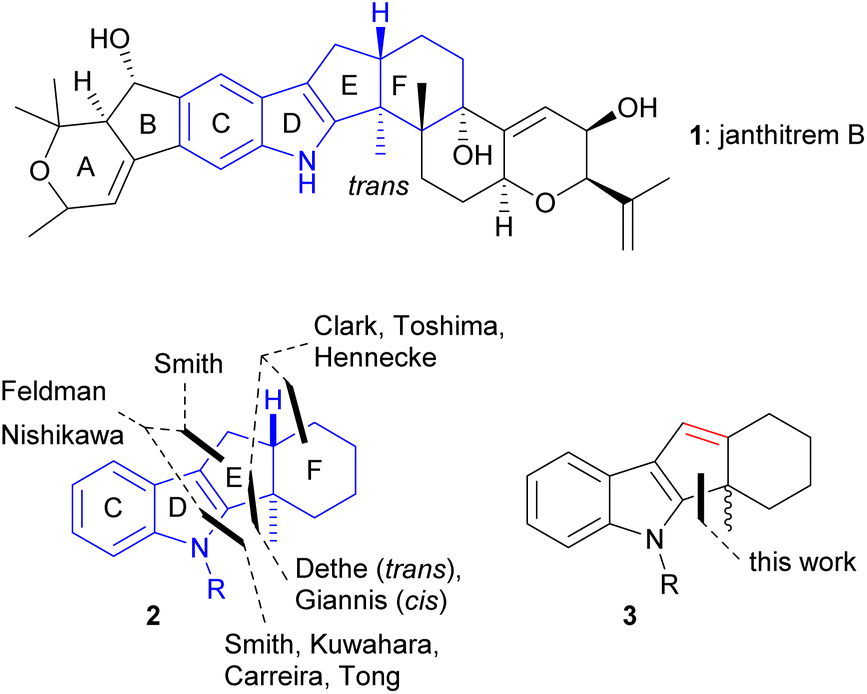 | ||
| Fig. 1 CDEF core structure present in the majority of indole diterpenoids, earlier approaches to the tetracycle, cyclopentadiene precursor investigated in this work. | ||
Most of the existing routes to indole diterpenoids assemble the pyrrole section of the indole part after having constructed trans-indane unit. In 1986, the Smith group reported the cyclization of an aldehyde function onto the indole 3-position affording a trans-fused indeno[1,2-b]indole by treatment with HNMe2 in dioxane, presumably via the corresponding iminium ion.9a In later work, the Smith group turned to Gassman, Madelung, or Barluenga indole syntheses, e.g. in the total syntheses of paspaline, nodulisporic acids, and penitrem.9b–d
For the synthesis of the trans-indane unit itself, several routes exist. In their total synthesis of shearinine G and paspalicine, the Carreira and Tong groups, respectively, assemble the trans-indane moiety by one-electron reduction of hydrogenated cyclopropa[c]inden-2-ones, where a cyclopropane ring was installed trans to the bridge-head hydrogen. In both routes, the indole section is assembled by subsequent Pd-catalyzed anellation of a protected o-stannylaniline to an alkenyltriflate functionality.7,10 This strategy was developed by Kuwahara in 2012 during their total synthesis of paspalinine.11 A recent approach to the trans-system by Nishikawa and coworkers features a tandem palladium-catalyzed cyclization of an N-tosylated o-alkynylaniline precursor.12
Fewer approaches towards the trans-fused indeno[1,2-b]indole start from an intact indole. A biomimetic cascade by Toshima and coworkers provided a tetracyclic product in good yield by Lewis-acid-mediated cyclization of 3-(6,7-epoxygeranyl)indole. For sesqui- and diterpenoid analogs, mixtures of products were formed.13 A similar approach to the trans-indeno[1,2-b]indole was employed by the Hennecke group, who submitted a 3-geranyl-substituted indole to a bromocyclization with excellent diastereoselectivity and good yield.14 Feldman and coworkers prepared indeno[1,2-b]indole constructs, among them the unprotected indole, by a cyclization cascade of an alkenyl sulfide tethered to a 2-azido-1-allenylbenzene core or by cationic cyclization of a tethered alkenyl sulfide.15 Dethe and coworkers published the TMSOTf-mediated cyclization of N-free 3-allylated indoles to indeno[1.2-b]indoles.16
Another option is the use of a photo Nazarov reaction for the assembly of ring E that has been employed to the synthesis of 16-epi-terpendole E.8 The product obtained by Giannis and coworkers suffers from a cis-fusion of rings E and F.
In this work, we analyze how this route could be saved by converting the photo Nazarov product to a trans-fused indeno[1,2-b]indole via the cyclopentadiene intermediate 3 (Fig. 1). Our study was encouraged by findings by the Grainger group, who reported the construction of trans-hydrindane systems by intramolecular hydride transfer.17 Indole derivatives were not subjects of these studies.
Results and discussion
First, we had to develop a new, efficient route to the literature-known18 carboxylic acid 4, which was obtained in four steps (23% overall yield, 200 mmol scale) from cyclohexanone without requiring column chromatography (see the ESI†). Acid 4 was transformed to acyl benzotriazole 5, which was used for the selective acylation of indole in the 3-position,19 affording the desired, new Nazarov precursor 6 (Scheme 1). Dialkenyl ketone 6 underwent smooth photo-Nazarov cyclization (79%), yielding the cis-hydrindanone 7 diastereoselectively. In the crystal, two conformers are present in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The cis-hydrindanone is more stable than the desired trans-isomer.20 The photochemical conditions lead to an isomerization of the cyclohexene double bond to the strained (E)-alkene, which allows the subsequent 4π electrocyclization without the need for further activation of the carbonyl group.21 Surprisingly, it was not possible to reduce the keto function, a vinylogous amide, of 7 to the secondary alcohol, as had been achieved with LiBH4/i-PrOH in course of the total synthesis of epi-terpindole E.8 Since we suspected the indole NH to be responsible, we also synthesized the N-methylated analog of 7 (see the ESI†). Unfortunately, we could not functionalize this either.
:1 ratio. The cis-hydrindanone is more stable than the desired trans-isomer.20 The photochemical conditions lead to an isomerization of the cyclohexene double bond to the strained (E)-alkene, which allows the subsequent 4π electrocyclization without the need for further activation of the carbonyl group.21 Surprisingly, it was not possible to reduce the keto function, a vinylogous amide, of 7 to the secondary alcohol, as had been achieved with LiBH4/i-PrOH in course of the total synthesis of epi-terpindole E.8 Since we suspected the indole NH to be responsible, we also synthesized the N-methylated analog of 7 (see the ESI†). Unfortunately, we could not functionalize this either.
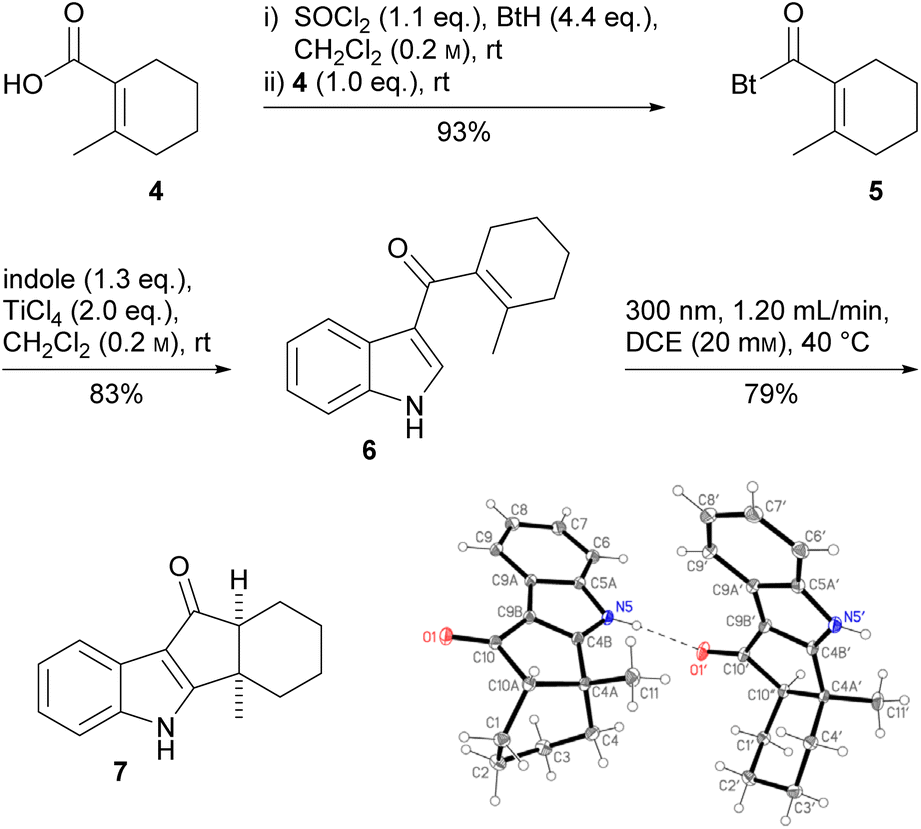 | ||
| Scheme 1 Synthesis of cis-hydrindanone 7 and its crystal structure. | ||
We synthesized a number of 3-allylated indoles that did not contain the vinylogous amide present in precursor 6. Unfortunately, all our attempts to induce cyclization to the desired tetracyclic systems were unsuccessful (see the ESI† for details).
We then returned to our successful Nazarov cyclization providing cis-hydrindanone 7. We were able to tosylate indole 7, rendering the vinylogous amide more electrophilic (Scheme 2). Gratifyingly, reduction of 8 with DIBAL-H afforded a mixture of diastereomeric alcohols in high yield. Elimination (MsCl/NEt3) finally gave cyclopentadiene 9 (86% from 8).
 | ||
| Scheme 2 Synthesis and reactivity probing of the key tetracyclic cyclopentadiene 9. | ||
We considered structure 9 as a versatile synthetic intermediate and probed its reactivity. Exposure of 9 to bromine in water afforded bromohydrin 10. The bromine is introduced from the same face as the methyl group of 9 (NOESY analysis). Reaction of cyclopentadiene 9 with tetracyanoethylene afforded cyclobutane 11 in a formal [2 + 2] cycloaddition, which probably proceeds via radical intermediates and has been described for 3-vinylindoles.22 The tetracyanoethylene is added on the side of the methyl group (NOESY analysis). Importantly, these reactions identify the sterically less hindered face of the tetracyclic indeno[1,2-b]indole 9, predicting cis-diastereoselectivity of other additions to that double bond.
The cycloaddition forming 11 puts the methine hydrogen in a position opposite to the methyl group. Therefore, to achieve the desired trans-fusion, we turned to a [1,2]-hydride shift strategy. The methine hydrogen of other cycloaddition products of 9 could potentially be shifted as hydride to a carbenium ion to be formed at the neighboring bridge-head (12, Scheme 3), leading to formation of a trans-hydrindane moiety (13). Precursors of that carbenium ion would be cyclic sulfites or dioxaphospholanes.
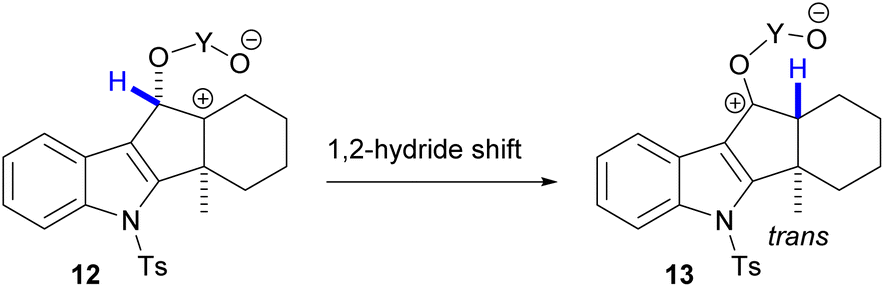 | ||
| Scheme 3 Envisaged access to the trans-hydrindane moiety from a pentacyclic precursor by 1,2-hydride shift. | ||
Os-catalyzed dihydroxylation afforded the cis-diol 14 with both hydroxy groups pointing to the same face as the methyl group (Scheme 4). In their work on the synthesis of dictyoxetane, Grainger and coworkers17 reported an interesting phosphorane-mediated, pinacol-like rearrangement of a cis-diol via a formal 1,2-hydride shift, affording a trans-hydrindane. As intermediate, a dioxaphospholane was formed, which opened thermally to the carbenium ion. The method was later used by Baran and coworkers in their synthesis of (+)-calcipotriol.23 However, when we applied these conditions to indole 14, we obtained an intractable product mixture.
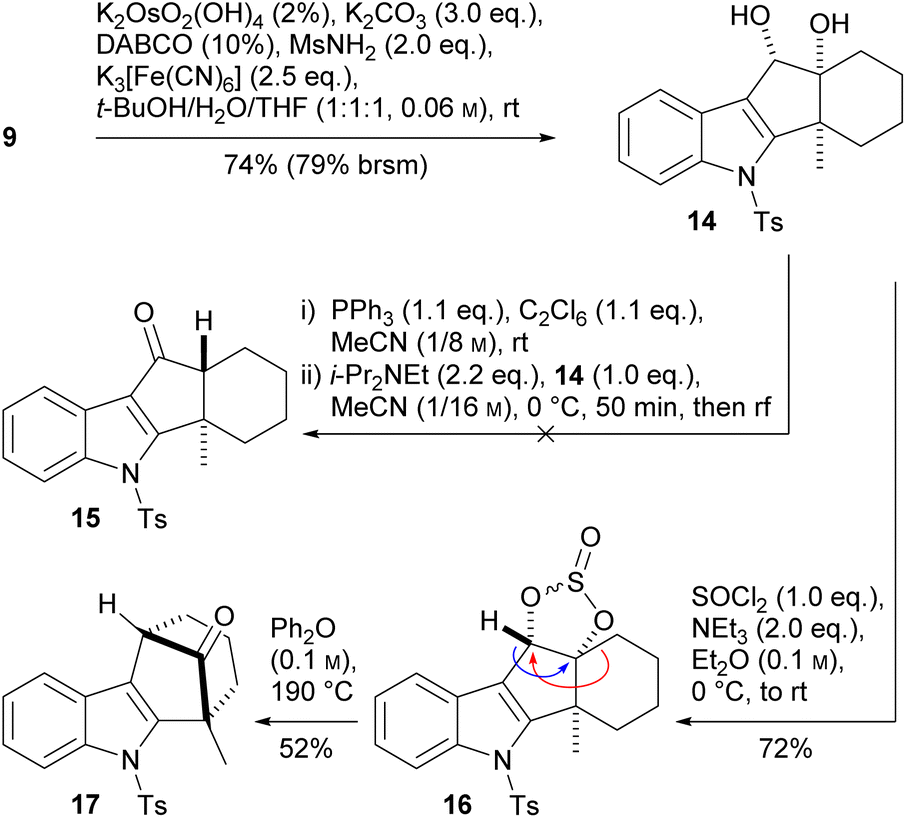 | ||
| Scheme 4 Unexpected pinacol rearrangement starting from cyclic sulfite 16. | ||
As alternative, the Grainger group published the use of cyclic sulfites, which were employed for the synthesis of bridged bicyclic lactams.24 We converted diol 14 to the cyclic sulfite 16 by treatment with SOCl2/NEt3. Surprisingly, heating the product in diphenyl ether (190 °C) did not result in the desired 1,2-hydride shift (blue arrow) but instead in ring expansion with loss of SO2.
It seems that formation of a benzylic carbenium ion outcompetes that of a tertiary carbocation, which undergoes pinacol rearrangement to the [3.2.1] bicyclic partial structure of ketone 17.
To solve this problem, we switched to indolines. Indole 14 was hydrogenated employing Crabtree's catalyst affording indoline 18 in perfect diastereoselectivity, with both bridgehead hydrogens introduced on the side of the hydroxy groups (Scheme 5). The relative configuration of 18 is based on NOESY analyses, which revealed a U-shaped conformation of the tetracyclic core. Diol 18 was converted to the cyclic sulfite 19. Upon heating of 19 in diphenyl ether (190 °C), conversion was significantly slower than in the case of sulfite 16. Even after 12 h we recovered 43% of the starting material. We obtained hydrindanone 20 in 37% yield, but, depressingly, with cis-fused rings E and F. Apparently, complete epimerization occurred at the α-carbon under the harsh reaction conditions.
 | ||
| Scheme 5 Synthesis of cis- and trans-hydrindanones 20 and 21 under Grainger's conditions. | ||
Thus, we returned to the use of dioxaphospholane intermediates, which can be removed under milder conditions, even in refluxing MeCN. Treatment of diol 18 first with PPh3/C2Cl6 in MeCN in the presence of Hünig's base and then refluxing (Grainger's conditions) afforded a mixture of cis- and trans-hydrindanones 20 and 21. We were able to separate them to some extent by column chromatography and obtained pure trans-hydrindanone 21 (43%). Epimerization during silica chromatography could mostly be suppressed by the addition of 1% NEt3 to the mobile phase.
We studied the deoxygenation of both diastereomeric tetracycles 20 and 21 employing a Barton–McCombie sequence starting from the corresponding secondary alcohols obtained smoothly by reduction with DIBAL-H. In the cis case we achieved deoxygenation, but not in the trans case (see the ESI† for details). We were neither able to transform the sterically hindered ketone 21 into an alkenyl triflate (LDA/PhNTf2) or a tosyl hydrazone (TsNHNH2).
Deoxygenation of trans-compound 21 required the synthesis of a dithioketal. Upon exposure to ethylene dithiol and BF3·OEt2, ketone 21 was converted to dithiolane 22 (Scheme 6). For the sake of diastereospecifity, the reaction was stopped after 15 h, which allowed the isolation of diastereomerically pure dithiolane 22 in 28% yield. We re-isolated 54% of unreacted starting material (dr = 20:1), which could be used in further batches (82% brsm). Finally, desulfurization with RANEY® nickel afforded the desired trans-hydrindane 23.
 | ||
| Scheme 6 Conversion of trans-hydrindanone 21 to the target compound trans-hydrindane 24. | ||
The relative configuration of trans-hydrindanone 23 was determined by NOESY analysis and proved by single crystal X-ray diffraction analysis (Fig. 2). Tosylindoline 23 was converted to the corresponding indole 24 by detosylation (Na/naphthalene) and dehydrogenation (Pd/C) in 31% yield.
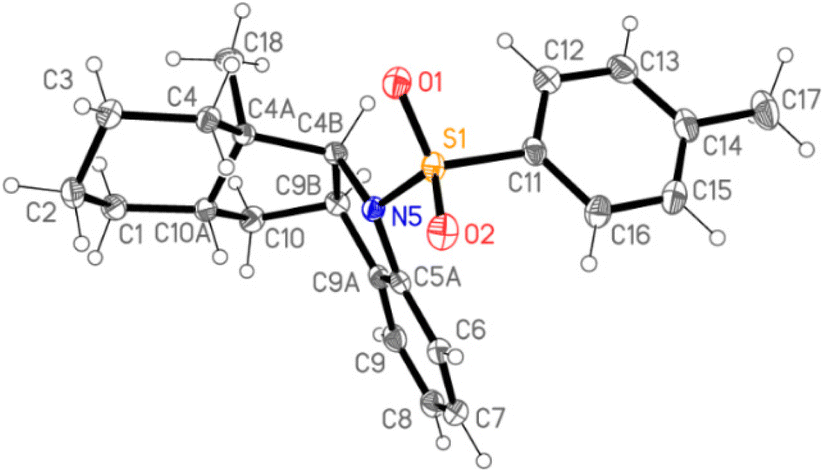 | ||
| Fig. 2 Crystal structure of trans-hydrindane 23. | ||
Conclusions
In summary, we have shown that the photo-Nazarov route is a viable approach to trans-fused indeno[1,2-b]indoles. We were able to overcome the cis-selectivity of the initial cyclization of alkenyl indolyl ketone 6 to the cyclopentenone 7 by conversion to the cyclopentadiene, dihydroxylation, reduction to the indoline, and Grainger's hydride shift, for the first time applied to an N-heterocyclic system. When starting from the corresponding indole, we observed the formation of hitherto unknown methanocyclohepta[b]indolones. The deoxygenation of trans-indanone 21 was possible after conversion to the 1,3-dithiolane, followed by RANEY® Ni reduction. Finally, trans-hydrindanone 23 was deprotected by sodium naphthalide and dehydrogenated by palladium on charcoal, giving the trans-fused indeno[1,2-b]indole 24 (2.5% yield, 12 steps starting from indole).Conflicts of interest
There are no conflicts to declare.Acknowledgements
TU Braunschweig is thanked for continuous support of this work.References
- For recent comprehensive reviews, see (a) P. Reddy, K. Guthridge, S. Vassiliadis, J. Hemsworth, I. Hettiarachchige, G. Spangenberg and S. Rochfort, Toxins, 2019, 11, 302 CrossRef CAS PubMed; (b) J. Niu, J. Qi, P. Wang, C. Liu and J.-M. Gao, Nat. Prod. Bioprospect., 2023, 13, 3 CrossRef CAS PubMed.
- R. T. Gallagher and A. D. Hawkes, Experienta, 1986, 42, 823 CrossRef CAS PubMed.
- L.-T. Dai, L. Yang, F.-D. Kong, Q.-Y. Ma, Q.-Y. Xie, H.-F. Dai, Z.-F. Yu and Y.-X. Zhao, Mar. Drugs, 2021, 19, 613 CrossRef CAS PubMed.
- J. V. Babu, A. J. Popay, C. O. Miles, A. L. Wilkins, M. E. di Menna and S. C. Finch, J. Agric. Food Chem., 2018, 66, 13116 CrossRef CAS PubMed.
- J.-C. Zhao, Y.-L. Wang, T.-Y. Zhang, Z.-J. Chen, T.-M. Yang, Y.-Y. Wu, C.-P. Sun, X.-C. Ma and Y.-X. Zhang, Phytochemistry, 2018, 148, 21 CrossRef CAS PubMed.
- M. Fresia and T. Lindel, Eur. J. Org. Chem., 2022, e202101454 CrossRef CAS.
- N. Hauser, M. A. Imhof, S. S. Eichenberger, T. Kündig and E. M. Carreira, Angew. Chem., Int. Ed., 2022, 61, e202112838 CrossRef CAS PubMed.
- F. Churruca, M. Fousteris, Y. Ishikawa, M. von Wantoch Rekowski, C. Hounsou, T. Surrey and A. Giannis, Org. Lett., 2010, 12, 2096 CrossRef CAS PubMed.
- (a) A. B. Smith III, M. Visnick, J. N. Haseltine and P. A. Sprengeler, Tetrahedron, 1986, 42, 2957 CrossRef; (b) A. B. Smith III, T. Sunazuka, T. L. Leenay and J. Kingery-Wood, J. Am. Chem. Soc., 1990, 112, 8197 CrossRef; (c) A. B. Smith III, N. Kanoh, H. Ishiyama, N. Minakawa, J. D. Rainier, R. A. Hartz, Y. Shin Cho, H. Cui and W. H. Moser, J. Am. Chem. Soc., 2003, 125, 8228 CrossRef PubMed; (d) Y. Zou, X. Li, Y. Yang, S. Berritt, J. Melvin, S. Gonzales, M. Spafford and A. B. Smith III, J. Am. Chem. Soc., 2018, 140, 9502 CrossRef CAS PubMed.
- L.-D. Guo, Z. Xu and R. Tong, Angew. Chem., Int. Ed., 2022, 61, e202115384 CrossRef CAS PubMed.
- M. Enomoto, A. Morita and S. Kuwahara, Angew. Chem., Int. Ed., 2012, 51, 12833 CrossRef CAS PubMed.
- Y. Ono, K. Higuchi, M. Yamaguchi, K. Sugino, A. Nakazaki, M. Adachi and T. Nishikawa, Synlett, 2023, 34, 364 CrossRef CAS.
- T. Isaka, M. Hasegawa and H. Toshima, Biosci. Biotechnol. Biochem., 2011, 75, 2213 CrossRef CAS PubMed.
- J. Bock, C. G. Daniliuc and U. Hennecke, Org. Lett., 2019, 21, 1704 CrossRef CAS PubMed.
- (a) K. S. Feldman, I. Y. Gonzalez and C. M. Glinkerman, J. Am. Chem. Soc., 2014, 136, 15138 CrossRef CAS PubMed; (b) K. S. Feldman, I. Y. Gonzalez and C. M. Glinkerman, J. Org. Chem., 2015, 80, 11849 CrossRef CAS PubMed.
- D. H. Dethe, S. K. Sau and S. Mahapatra, Org. Lett., 2016, 18, 6392 CrossRef CAS PubMed.
- B. Defaut, T. B. Parsons, N. Spencer, L. Male, B. M. Kariuki and R. S. Grainger, Org. Biomol. Chem., 2012, 10, 4926 RSC.
- H. Christol, A. Donche and F. Plenat, Bull. Soc. Chim. Fr., 1966, 8, 2535 Search PubMed.
- A. R. Katritzky, K. Suzuki, S. K. Singh and H.-Y. He, J. Org. Chem., 2003, 68, 5720 CrossRef CAS PubMed.
- N. L. Allinger and M. T. Tribble, Tetrahedron, 1972, 28, 1191 CrossRef CAS.
- (a) J. Leitich, I. Heise, S. Werner, C. Krüger and K. Schaffner, J. Photochem. Photobiol., A, 1991, 57, 127 CrossRef CAS; (b) J. Leitich, I. Heise, J. Rust and K. Schaffner, Eur. J. Org. Chem., 2001, 2719 CrossRef CAS.
- J. D. Lambert and Q. N. Porter, Aust. J. Chem., 1981, 34, 1483 CrossRef CAS.
- J. Gu, K. X. Rodriguez, Y. Kanda, S. Yang, M. Ociepa, H. Wilke, A. V. Abrishami, L. Jørgensen, T. Skak-Nielsen, J. S. Chen and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2200814119 CrossRef CAS PubMed.
- R. S. Grainger, M. Betou, L. Male, M. B. Pitak and S. J. Coles, Org. Lett., 2012, 14, 2234 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2289675 and 2289676. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01566a |
| This journal is © The Royal Society of Chemistry 2023 |