
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substituted anilides from chitin-based 3-acetamido-furfural†
Cornelis H. M.
van der Loo
 a,
J. P.
Kaniraj
a,
Ting
Wang
d,
J. O. P.
Broekman
d,
Mark L. G.
Borst
b,
Kees
Pouwer
b,
André
Heeres
c,
Peter J.
Deuss
d and
Adriaan J.
Minnaard
*a
a,
J. P.
Kaniraj
a,
Ting
Wang
d,
J. O. P.
Broekman
d,
Mark L. G.
Borst
b,
Kees
Pouwer
b,
André
Heeres
c,
Peter J.
Deuss
d and
Adriaan J.
Minnaard
*a
aDepartment of Chemical Biology, Stratingh Institute for Chemistry, University of Groningen, Groningen, The Netherlands. E-mail: A.J.Minnaard@RUG.nl
bSymeres B.V., Kadijk 3, 9747 AT Groningen, The Netherlands
cHanze University of Applied Sciences, Zernikeplein 7, 9747 AS Groningen, The Netherlands
dDepartment of Chemical Engineering (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 5th October 2023
Abstract
The synthesis of aromatic compounds from biomass-derived furans is a key strategy in the pursuit of a sustainable economy. Within this field, a Diels–Alder/aromatization cascade reaction with chitin-based furans is emerging as a powerful tool for the synthesis of nitrogen-containing aromatics. In this study we present the conversion of chitin-based 3-acetamido-furfural (3A5F) into an array of di- and tri-substituted anilides in good to high yields (62–90%) via a hydrazone mediated Diels–Alder/aromatization sequence. The addition of acetic anhydride expands the dienophile scope and improves yields. Moreover, replacing the typically used dimethyl hydrazone with its pyrrolidine analogue, shortens reaction times and further increases yields. The hydrazone auxiliary is readily converted into either an aldehyde or a nitrile group, thereby providing a plethora of functionalized anilides. The developed procedure was also applied to 3-acetamido-5-acetylfuran (3A5AF) to successfully prepare a phthalimide.
Introduction
Chemical and pharmaceutical industries are increasingly acknowledging the importance of exploring the potential of biomass to align with future environmental objectives. The main goal is to address the challenges posed by depleting fossil resources and to reduce carbon dioxide emissions. In the context of striving for a more sustainable economy, the concept of biorefinery has gained prominence as a valuable approach, seeking to extract value from biomass waste streams for the production of chemicals.1,2 An important strategy in this field is the synthesis of substituted aromatics from bio-based furans. This can be achieved through either catalytic fast pyrolysis3–5 or Diels–Alder/aromatization strategies.6–8The majority of studies on the preparation of aromatics from bio-based furans is focused on substrates prepared from lignocellulosic biomass, such as furfural,9,10 5-hydroxymethylfurfural (HMF)11,12 and 2,5-dimethylfuran (DMF).13,14 However, many aromatics used in bulk chemicals, fine chemicals and pharmaceuticals are nitrogen substituted. Preparing these compounds, typically referred to as aniline derivatives, from lignocellulose-based furans requires a subsequent nitration step, which is not environmentally benign. From an environmental perspective, it is desirable to develop synthetic strategies for nitrogen-substituted aromatics from easily accessible biomass such as chitin. Chitin is a major component (15–40%)15 of non-edible crustacean shells waste and is readily converted to its monomer N-acetyl glucosamine (GlcNAc).16,17 From this amino-sugar the platform chemicals dihydroxyethyl acetamidofuran (Di-HAF)18 and 3-acetamido-5-acetylfuran (3A5AF)19–21 can be accessed through a dehydration process. These furans exhibit a rich reactivity profile22,23 and have been employed in rearrangement reactions,24,25 cyclo-additions26,27 and as synthetic building blocks for the preparation of various alkaloids.28,29
While the formation of an anilide side product was already identified during development of the synthesis of 3A5AF,19 it was only recently that the first targeted synthesis of anilides from a chitin-derived furan was published by Pastre et al. (Scheme 1A).30 A reduced analogue of 3A5AF, 3-acetamido-5-ethylfuran (3A5EF), was used to prepare several substituted anilides via an acetic anhydride mediated Diels–Alder-aromatization sequence under acidic conditions. Based on a DFT study, the authors concluded that reduction of the conjugated carbonyl functionality was required to overcome reactivity limitations. The electron withdrawing ketone function reduces the reactivity of the diene in the Diels–Alder reaction, and due to charge destabilization in a key transition state, hampers the subsequent ring-opening, a required step for aromatization. With 3A5EF, the dienophile scope remained limited primarily to maleimides, as poor yields were obtained with mono-activated dienophiles. Furthermore, the ethyl-group inherently present in the amido-phthalimide products can be restrictive for applications and a moiety that allows for further derivatization is desirable.
 | ||
| Scheme 1 (A) Acid catalysed Diels–Alder/aromatization of 3A5EF reported by Pastre30 (B) hydrazone mediated aromatization of furfural reported by Hailes33 (C) this work: hydrazone mediated Diels–Alder/aromatization of 3A5F. | ||
An alternative and established approach to suppress the deactivating effect of a carbonyl functionality, conjugated to a diene, has been demonstrated with furfural.31,32 Conversion of the carbonyl group to its dimethyl hydrazone effectively inverses the polarity (umpolung) to benefit the reactivity in the cycloaddition and facilitate subsequent ring-opening of the formed oxanorbornene. The reactivity of the dimethyl hydrazone of furfural remains modest, and thus the Diels–Alder reaction is usually carried out with reactive maleimides to furnish phthalimide derivatives (Scheme 1B).33 Mono-activated dienophiles remain challenging and typically give poor yields.34
In the course of our research on chitin-derived furans we developed a synthesis of enantiopure Di-HAF (Scheme 2).18 Malaprade oxidation of Di-HAF offers an easy route to 3-acetamido-furfural (3A5F). Notably, the viability of 3A5F as a building block has recently been demonstrated by Afonso et al.35 Given the structural similarity between 3A5F and furfural, and considering the resonance effect of the acetamide group that elevates the HOMO of the diene, thus promoting cycloaddition, we identified 3A5F as a starting material for the preparation of substituted anilides.
 | ||
| Scheme 2 Synthesis of Di-HAF16 and subsequent periodate oxidation to afford 3A5F. | ||
Herein we report a hydrazone mediated Diels–Alder aromatization using chitin-derived furan 3A5F for the efficient synthesis of di- and tri-functionalized anilides (Scheme 1C). The addition of acetic anhydride as trapping agent improved yields and enabled the use of mono-activated dienophiles. Replacing the typically used dimethyl hydrazone with its pyrrolidine analogue, shortened reaction times and further increased yields. The hydrazone auxiliary can readily be converted to either an aldehyde- or a nitrile group, to provide a plethora of functionalized anilides.
Results and discussion
Dimethyl hydrazone 1 was prepared by reacting 3A5F with dimethyl hydrazine (Scheme 3). Subsequently, the reaction with N-methylmaleimide was studied in ethanol at room temperature, and to our delight the corresponding phthalimide (2) was obtained in 56% yield. Next, we tried the reaction with the less reactive dienophile ethyl acrylate, which was initially unsuccessful. Based on our experience with Di-HAF,27 we assumed that the Diels–Alder reaction with ethyl acrylate was reversible and the retro reaction dominated. Reversibility in Diels–Alder reactions with furans is a common phenomenon.36 | ||
| Scheme 3 Synthesis of 1 and a Diels–Alder aromatization cascade with N-methylmaleimide to afford phthalimide 2. | ||
Inspired by Pastre et al.,30 we postulated that this retro Diels–Alder pathway could be suppressed by the addition of acetic anhydride. Acetic anhydride performs a dual role: (1) it traps intermediate 4, stopping the backward reaction that would restore the ether bridge (3), and thereby it prevents the retro Diels–Alder reaction to proceed. (2) It converts this alkoxide/hydroxyl group into a good leaving group to favor aromatization (Scheme 4).
 | ||
| Scheme 4 Role of acetic anhydride as trapping agent of ring-opened intermediate 4 to prevent a retro Diels–Alder reaction. | ||
After switching the solvent from ethanol to acetonitrile and the addition of acetic anhydride, the yield of the reaction between 1 and N-methylmaleimide to afford 2, increased from 56% to 77%. Moreover, the reaction with 1 and ethyl acrylate at reflux furnished the desired aromatic product (7). Through reaction monitoring, it was observed that complete conversion to the acetylated ring-opened product 6 occurred within 2 h (Scheme 4). Subsequent aromatization occurred gradually over several hours, forming 7. The slow elimination reaction prompted us to add a base (triethylamine), resulting in a substantial increase in conversion from 31% to 76% after 4 h and ultimately furnished 7 in 49% yield.
The difference in the rate of aromatization between the reaction with N-methylmaleimide, which occurs readily at room temperature, and ethyl acrylate, which requires prolonged heating, is a clear reflection of the different reaction mechanisms involved, namely E1cb and E2 (Scheme 5). This difference in reactivity is related to the acidity of each proton that is abstracted.
 | ||
| Scheme 5 (A) Aromatization via an E1cb mechanism, (B) aromatization via an E2 mechanism. | ||
Further reaction optimization was performed with ethyl acrylate as dienophile (S3 in ESI†). Mesyl chloride and triflic anhydride were tested as alternatives to acetic anhydride, but both led to decomposition of the starting material. Increasing the number of equivalents of acetic anhydride to super-stoichiometric amounts did not improve the yield. Among the different bases examined, triethylamine (TEA) proved to be superior to diazabicycloundecene (DBU) and 1,4-diazabicyclo[2.2.2]octane (DABCO). However, it became evident that carrying out the reaction at a high substrate concentration was a requirement for good yield. For reactions with less reactive dienophiles at reflux temperature, 1 M was optimal. For reactions with maleimides at room temperature 0.5 M was sufficient. Higher concentrations exceeded the solubility limits of the reaction components. Finally, we varied the number of equivalents of the dienophile and no increase in yield was observed above 2 eq. However, in the case of less reactive dienophiles with respect to ethyl acrylate (e.g. diethyl phosphonate, vide infra) higher concentrations (5 eq.) were beneficial. Ultimately, two standard protocols were formulated. (A) For reactive dienophiles (maleimides and fumaronitrile) a substrate concentration of 0.5 M in acetonitrile with triethylamine (2.5 eq.) and acetic anhydride (1.1 eq.) at room temperature. (B) For less reactive dienophiles (maleates and mono-activated alkenes) a substrate concentration of 1 M in acetonitrile with triethylamine (2.5 eq.) and acetic anhydride (1.1 eq.) at reflux temperature. The addition of 2 eq. of dienophile was sufficient in most cases, but for less reactive dienophiles this was increased to 5 eq. With these optimized conditions, the Diels–Alder aromatization cascade with 1 and ethyl acrylate as dienophile afforded product 7 in an isolated yield of 64% (protocol B).
The advantage of a high concentration of the reactants and the need to use an excess of (less reactive) dienophiles both emphasized the importance of the reaction rate of the first step of the cascade—the cycloaddition—for the ultimate yield. We suspected decomposition of hydrazone 1 during the reaction, limiting the overall yield. Hence, we assessed the stability of 1 under the reaction conditions in the absence of a dienophile by quantitative NMR (S4 in ESI†). At room temperature (protocol A), degradation was negligible and 95% of the starting material remained after 18 h. However, under reflux conditions (81 °C, protocol B), the concentration of hydrazone 1 decreased by 20% in the first 2 h and after 18 h only 41% of the original concentration remained. Therefore, we investigated options to enhance the reactivity of the diene, with the aim of shortening reaction times and thus limiting decomposition of the starting material.
Mayr et al.37 determined that a pyrrolidine hydrazone is an order of magnitude more nucleophilic (N) than a dimethyl hydrazone (N = 7.84 vs. 6.98 for the hydrazones of formaldehyde). We prepared pyrrolidine hydrazone 12 (Scheme 6) and the higher electron density of the furan was evident from 1H-NMR analysis. An upfield shift of the signals corresponding to the aromatic protons was observed, indicating an elevated HOMO (S5 in ESI†). While a stability test displayed a degradation profile comparable to dimethyl hydrazone 1 (S4 in ESI†), the Diels–Alder aromatization cascade was considerably faster, thereby narrowing the timeframe during which decomposition of the starting material could occur. With pyrrolidine hydrazone 12, near complete conversion (97%) was observed after 4 h, whereas dimethyl hydrazone 1 showed 76% conversion at this timepoint. As a result, the product (13) was isolated with an improved yield (13: 71% vs.7: 64%).
 | ||
| Scheme 6 Preparation of pyrrolidine hydrazone 12 and Diels–Alder/aromatization with ethyl acrylate to afford 13. | ||
With the optimized conditions in hand, we demonstrated that this approach allows for considerable diversity in terms of the dienophile substrates (Scheme 7). Maleimides performed excellently and afforded the corresponding phthalimides (14–16) in high yields (88–90%). The products precipitated during the reaction and could be isolated in high purity by filtration. For acyclic bis-activated dienophiles, an interplay between electron withdrawing capability and steric hindrance of the substituents became apparent. Fumaronitrile, featuring two compact yet strongly electron withdrawing groups (EWG), afforded the di-cyano product (18) in 82% yield. In contrast, dimethyl maleate, incorporating two relatively larger and weaker EWGs, furnished the di-ester (17) in 62% yield. The same yield was achieved with its trans-isomer, dimethyl fumarate. Substitution of one EWG with a methyl group completely impaired the reaction and 24 could not be isolated. Mono-activated alkenes showed a similar trend. Acrylonitrile performed excellently and afforded 19 in 84% yield. Ethyl acrylate and tert-butyl acrylate furnished the corresponding esters 13 and 20 in 71% and 73% yield, respectively. A similar yield was obtained for sulfone 21 (75%). In the case of diethyl vinylphosphonate, initial tests indicated sluggish reactivity, leading to modest yields. However, the slow cycloaddition rate could be overcome by performing the reaction with 5 eq. of dienophile and ultimately phosphonate 22 was isolated in 67% yield. Throughout the substrate scope we observed full regioselectivity with mono-activated alkenes. The selectivity can be attributed to a combination of acetamide induced polarization of the diene and the inherent polarization of mono-activated alkenes. This was exemplified by methyl vinyl ketone, a highly polarized alkene, which did not give the cycloaddition product but led to Michael addition on the 2-position of the furan (S6 in ESI†).
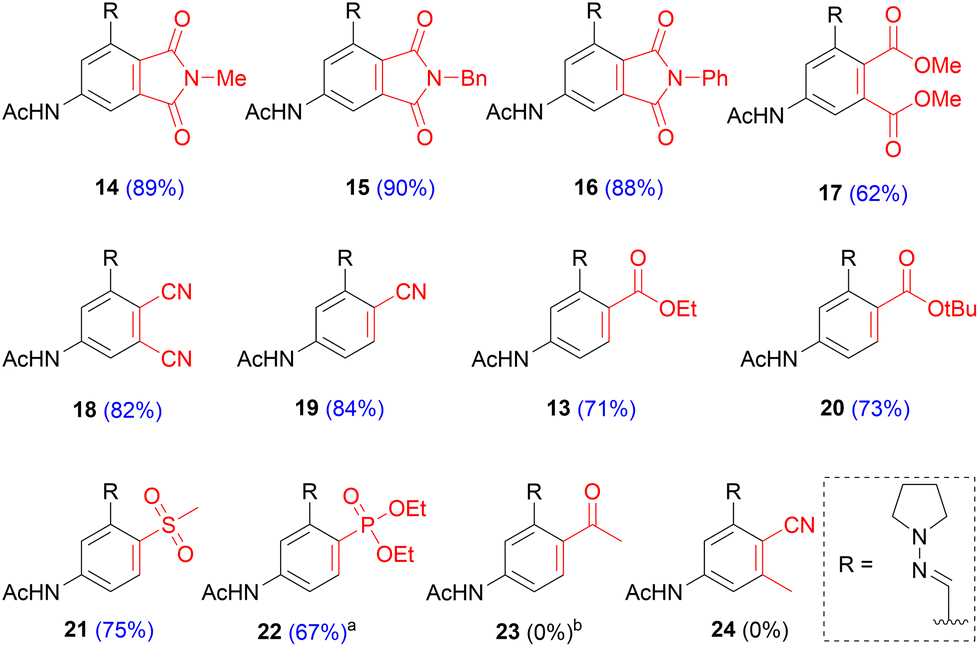 | ||
Scheme 7 Substrate scope. In red: the appropriate dienophile employed; a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) reaction performed with 5 eq. of dienophile; ba 1,4-addition product was formed. reaction performed with 5 eq. of dienophile; ba 1,4-addition product was formed. | ||
The product scope of this approach can readily be expanded further by conversion of the hydrazone into various other functional groups. To demonstrate this, compound 20 was subjected to ozonolysis and treatment with mCPBA to afford aldehyde 25 and nitrile 26 in respectively 67% and 79% yield (Scheme 8).38
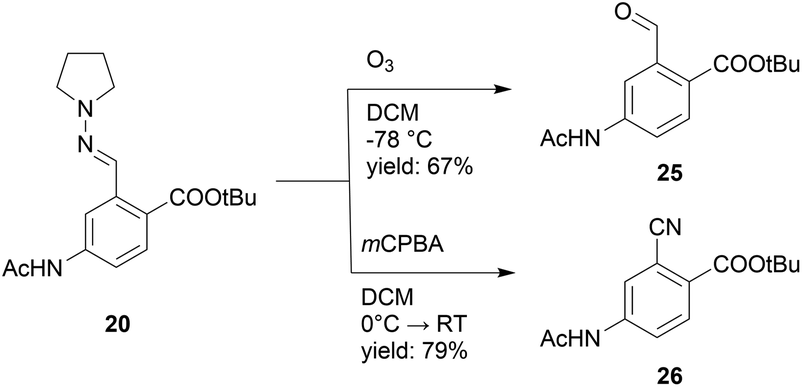 | ||
| Scheme 8 Ozonolysis and mCPBA oxidation of 20 to afford aldehyde 25 and nitrile 26. | ||
Returning to the original work of Pastre et al.,30 we wondered whether this new procedure would allow the same strategy for ketone 3A5AF. Avoiding the necessity to reduce the carbonyl- to the ethyl function prior to the Diels–Alder/aromatization cascade would considerably improve the applicability of the products. Both dimethyl hydrazone 27 and pyrrolidine hydrazone 28 were prepared (Scheme 9). To compensate for the lower electrophilicity of a ketone, an increased temperature (78 °C) and acid catalysis were required. Compared to aldo-hydrazones 1 and 12, a dramatic decrease in the rate of ring-opening of the cycloaddition product was observed in the reaction of keto-hydrazones 27 and 28 with both N-methylmaleimide and fumaronitrile. We attribute this to a steric clash between the methyl groups (or the pyrrolidine ring) on the hydrazone nitrogen and the methyl group of the keto hydrazone, as indicated in Scheme 10. For the ring-opening to be feasible, optimal overlap of the lone pair on the dimethyl/pyrrolidine nitrogen and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety is essential. This necessitates sp2-hybridization of both nitrogen atoms, placing the substituents on the nitrogen in the same plane, as the indicated methyl in 31. This arrangement poses no challenge for aldo-hydrazones 1 and 12, where the dimethylamine or pyrrolidine can readily orient itself due to the presence of the less sterically demanding hydrogen in place of the methyl. However, this conformation is problematic with keto-hydrazones 27 and 28 due to steric hindrance, in fact an appearance of allylic strain. To overcome this thermodynamic barrier the reaction needs to be performed at higher temperatures. Unfortunately, retro cycloaddition becomes more pronounced at higher temperatures, leading to the decomposition of reaction components or their recombination through 1,4-addition. The latter undesired side reaction was particularly present with pyrrolidine hydrazone (28), where 1,4-addition dominated as the primary reaction pathway (S6 in ESI†).
N moiety is essential. This necessitates sp2-hybridization of both nitrogen atoms, placing the substituents on the nitrogen in the same plane, as the indicated methyl in 31. This arrangement poses no challenge for aldo-hydrazones 1 and 12, where the dimethylamine or pyrrolidine can readily orient itself due to the presence of the less sterically demanding hydrogen in place of the methyl. However, this conformation is problematic with keto-hydrazones 27 and 28 due to steric hindrance, in fact an appearance of allylic strain. To overcome this thermodynamic barrier the reaction needs to be performed at higher temperatures. Unfortunately, retro cycloaddition becomes more pronounced at higher temperatures, leading to the decomposition of reaction components or their recombination through 1,4-addition. The latter undesired side reaction was particularly present with pyrrolidine hydrazone (28), where 1,4-addition dominated as the primary reaction pathway (S6 in ESI†).
 | ||
| Scheme 9 Synthesis of dimethyl hydrazone 27 and pyrrolidine hydrazone 28. | ||
 | ||
| Scheme 10 Steric hindrance between the methyl substituent (red) and the methyl substituents of the hydrazone hampers optimal orbital overlap required for ring-opening. | ||
In the case of dimethyl hydrazone 27, 1,4-addition was less prominent and could be further mitigated by using a reactive dienophile (e.g. a maleimide) and excluding the use of a base. Reaction of 27 with N-methylmaleimide afforded the aromatic product as a mixture of hydrazone 32 and ketone 33 in a ratio of approximately 1:1, in a high total yield of 88% (Scheme 11). Subsequently, 32 could successfully be converted to 33 by ozonolysis in a yield of 61%.
 | ||
| Scheme 11 Reaction of 27 and N-methylmaleimide affords a mixture of 32 and 33. Ozonolysis of 32 furnishes 33. | ||
Conclusions
In this study we show the efficient synthesis of di- and tri-substituted anilides from chitin-derived aldo-furan 3A5Fvia a hydrazone mediated Diels–Alder/aromatization cascade reaction. By using acetic anhydride as trapping agent, the retro Diels–Alder pathway is suppressed which increases yields and broadens the dienophile-scope, to include mono-activated alkenes. Furthermore, replacing the commonly used dimethyl hydrazone with its pyrrolidine analogue, shortens reaction times and further improves yields by approximately 6–12%. The product scope of this method is wide, and the hydrazone auxiliary is readily converted into an aldehyde or a nitrile function. Moreover, after small adjustments to mitigate a competing 1,4-addition reaction, we demonstrated extension of the method to 3A5AF. This keto-furan can be directly obtained from chitin derived N-acetyl glucosamine by dehydration, and aromatization of its Diels–Alder products was previously reported to be unsuccessful.26The methodology provides access to a diverse range of aromatics, featuring commonly encountered substitution patterns found in important classes of compounds (Scheme 12). These encompass pharmaceutical compounds like blood coagulation inhibitor 40,39 vasopressin receptor antagonist 41,40 and drug release linker 42.41 Moreover, their relevance extends to other fields of chemistry, such as polymer thermosetting resin 43,42 pigment precursor 44,43 fluorescent DNA base substitute 4544 and 46, a key intermediate in the synthesis of anti-depressant citalopram.45 Furthermore, this method provides access to entirely novel building blocks, such as 47 and 48, further amplifying its importance in diversifying the range of attainable chemical structures.
 | ||
| Scheme 12 Examples of important compounds with aromatic motifs (in red) that have a substitution pattern that in principle can be accessed via a Diels–Alder aromatization cascade reaction with 3A5F. | ||
Experimental
Protocol A (maleimides and fumaronitrile)
To a mixture of hydrazone (1 mmol) in acetonitrile (2 mL) was added triethylamine (2.5 mmol, 2.5 eq.), acetic anhydride (1.1 mmol, 1.1 eq.) and the appropriate dienophile (2 mmol, 2 eq.). The suspension was stirred for 18 h at room temperature. The product was isolated by filtration, washed with EtOAc (5 mL) and dried in vacuo to afford the target compound (82–90%).Protocol B (maleates and mono-activated alkenes)
To a mixture of hydrazone (1 mmol) in acetonitrile (1 mL) was added triethylamine (2.5 mmol, 2.5 eq.), acetic anhydride (1.1 mmol, 1.1 eq.) and the appropriate dienophile (2 mmol, 2 eq.). The suspension was stirred for 18 h at 81 °C. The volatiles were removed in vacuo and the crude product was purified by automated column chromatography to afford the target compound (62–84%).Author contributions
All authors took part in the conceptualization of this study. C. van der Loo carried out the experimental work, C. van der Loo and A. Minnaard wrote the original draft of the manuscript. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCC Carbobased program and the Topconsortium voor Kennis en Innovatie Chemie (TKI Chemie) (Chemie.PGT.2018.003) are acknowledged for funding. For analytical support are acknowledged J. L. Sneep (University of Groningen) and H. van der Mel (Symeres Groningen).References
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–555 RSC.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- A. I. Nekhaev and A. L. Maksimov, Pet. Chem., 2021, 61, 15–34 CrossRef CAS.
- A. Heeres, N. Schenk, I. Muizebelt, R. Blees, B. De Waele, A. J. Zeeuw, N. Meyer, R. Carr, E. Wilbers and H. J. Heeres, ACS Sustainable Chem. Eng., 2018, 6, 3472–3480 CrossRef CAS PubMed.
- M. B. Figueirêdo, I. Hita, P. J. Deuss, R. H. Venderbosch and H. J. Heeres, Green Chem., 2022, 24, 4680–4702 RSC.
- J. M. J. M. Ravasco and R. F. A. Gomes, ChemSusChem, 2021, 14, 3047–3053 CrossRef CAS PubMed.
- F. A. Kucherov, L. V. Romashov, G. M. Averochkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2021, 9, 3011–3042 CrossRef CAS.
- Z. Li, Y. Jiang, Y. Li, H. Zhang, H. Li and S. Yang, Catal. Sci. Technol., 2022, 12, 1902–1921 RSC.
- I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. de Oliveira Vigier, R. Wischert and F. Jérôme, Angew. Chem., Int. Ed., 2018, 57, 10510–10514 CrossRef CAS PubMed.
- F. Xu, Z. Li, L. L. Zhang, S. Liu, H. Li, Y. Liao and S. Yang, Green Chem., 2023, 25, 3297–3305 RSC.
- A. J. Kumalaputri, C. Randolph, E. Otten, H. J. Heeres and P. J. Deuss, ACS Sustainable Chem. Eng., 2018, 6, 3419–3425 CrossRef CAS PubMed.
- S. Zheng, Z. Wei, B. Wozniak, F. Kallmeier, E. Baráth, H. Jiao, S. Tin and J. G. de Vries, Nat. Sustain., 2023 DOI:10.1038/s41893-023-01190-w.
- C. L. Williams, C. C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- C. C. Chang, H. Je Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- J. H. Kuk, W. J. Jung, G. H. Jo, Y. C. Kim, K. Y. Kim and R. D. Park, Appl. Microbiol. Biotechnol., 2005, 68, 384–389 CrossRef CAS PubMed.
- F. A. Cardozo, J. M. Gonzalez, V. A. Feitosa, A. Pessoa and I. N. G. Rivera, World J. Microbiol. Biotechnol., 2017, 33, 1–11 CrossRef CAS PubMed.
- C. H. M. Van Der Loo, M. L. G. Borst, K. Pouwer and A. J. Minnaard, Org. Biomol. Chem., 2021, 19, 10105–10111 RSC.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073–20080 RSC.
- C. Wang, C. Wu, A. Zhang, K. Chen, F. Cao and P. Ouyang, ChemistrySelect, 2022, e20210457 Search PubMed.
- T. T. Pham, A. C. Lindsay, S. W. Kim, L. Persello, X. Chen, N. Yan and J. Sperry, ChemistrySelect, 2019, 4, 10097–10099 CrossRef CAS.
- T. T. Pham, A. C. Lindsay, X. Chen, G. Gözaydin, N. Yan and J. Sperry, Sustainable Chem. Pharm., 2019, 13, 100143 CrossRef.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Green Chem., 2020, 22, 1978–1984 RSC.
- T. T. Pham, G. Gözaydın, T. Söhnel, N. Yan and J. Sperry, Eur. J. Org. Chem., 2019, 2019, 1355–1360 CrossRef CAS.
- J. G. Pereira, J. M. J. M. Ravasco, J. R. Vale, F. Queda and R. F. A. Gomes, Green Chem., 2022, 2022, 7131–7136 RSC.
- C. H. M. van der Loo, R. Schim van der Loeff, A. Martín, P. Gomez-Sal, M. L. G. Borst, K. Pouwer and A. J. Minnaard, Org. Biomol. Chem., 2022, 21, 1888–1894 RSC.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- J. C. Neville, M. Y. Lau, T. Söhnel and J. Sperry, Org. Biomol. Chem., 2022, 20, 6562–6565 RSC.
- C. S. Santos, R. Rodini Mattioli, J. Soares Baptista, V. H. Menezes da Silva, D. L. Browne and J. C. Pastre, Green Chem., 2023, 25, 5059–5067 RSC.
- K. T. Potts and E. B. Walsh, J. Org. Chem., 1984, 4099–4101 CrossRef CAS.
- K. T. Potts and E. B. Walsh, J. Org. Chem., 1988, 53, 1199–1202 CrossRef CAS.
- S. Higson, F. Subrizi, T. D. Sheppard and H. C. Hailes, Green Chem., 2016, 18, 1855–1858 RSC.
- R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, ChemSusChem, 2022, 15, e202201139 CrossRef CAS PubMed.
- R. F. A. Gomes, B. M. F. Gonçalves, K. H. S. Andrade, B. B. Sousa, N. Maulide, G. J. L. Bernardes and C. A. M. Afonso, Angew. Chem., Int. Ed., 2023, 62, e202304449 CrossRef CAS PubMed.
- R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994–8002 CrossRef CAS PubMed.
- B. Maji, K. Troshin and H. Mayr, Angew. Chem., Int. Ed., 2013, 52, 11900–11904 CrossRef CAS PubMed.
- R. Fernández and J. M. Lassaletta, Synlett, 2000, 1228–1240 Search PubMed.
- X. Zhang, P. W. Glunz, J. A. Johnson, W. Jiang, S. Jacutin-Porte, V. Ladziata, Y. Zou, M. S. Phillips, N. R. Wurtz, B. Parkhurst, A. R. Rendina, T. M. Harper, D. L. Cheney, J. M. Luettgen, P. C. Wong, D. Seiffert, R. R. Wexler and E. S. Priestley, J. Med. Chem., 2016, 59, 7125–7137 CrossRef CAS PubMed.
- Y. Iwasaki, N. Okada, N. Ito, Y. Uchino, Y. Nagasaka and H. Masaki, PatentJP2021014406A, 2021 Search PubMed.
- B. Tan, X. Zhang, X. Quan, G. Zheng, X. Li, L. Zhao, W. Li and B. Li, Bioorg. Med. Chem. Lett., 2020, 30, 1–5 Search PubMed.
- K. Zhang, X. Yu, Y. Wang and Y. Liu, ACS Appl. Polym. Mater., 2019, 1, 2713–2722 CrossRef CAS.
- T. Wang, X. Wang, J. Zhang, C. Wang, J. Shao, Z. Jiang and Y. Zhang, Dyes Pigm., 2018, 154, 75–81 CrossRef CAS.
- M. Weinberger, F. Berndt, R. Mahrwald, N. P. Ernsting and H. A. Wagenknecht, J. Org. Chem., 2013, 78, 2589–2599 CrossRef CAS PubMed.
- H. S. Yathirajan, B. Nagaraj, S. L. Gaonkar, R. S. Narasegowda, B. Prabhuswamy and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, 343–344 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01461d |
| This journal is © The Royal Society of Chemistry 2023 |