Relative stereochemical determination of the C61–C83 fragment of symbiodinolide using a stereodivergent synthetic approach†‡
Received
5th September 2023
, Accepted 27th September 2023
First published on 29th September 2023
Abstract
Structural determination is required in the use of marine natural products to create novel drugs and drug leads in medicinal chemistry. Symbiodinolide, which is a polyol marine natural product with a molecular weight of 2860, increases the intracellular Ca2+ concentration and exhibits inhibitory activity against cyclooxygenase-1. Seventy percent of the structure of symbiodinolide has been stereochemically clarified. Herein, we report the elucidation of the relative configuration of the C61–C83 fragment, which is among the remaining thirty percent, using a stereodivergent synthetic strategy. We first assigned the relative configuration of the C61–C74 fragment. Two candidate diastereomers of the C61–C74 fragment were synthesized, and their NMR data were compared with those of the natural product, revealing the relative stereochemistry of this component. We then narrowed down the candidate compounds for the C69–C83 fragment from 16 possible diastereomers by analyzing the NMR data of the natural product, and we thus selected eight candidate diastereomers. Stereodivergent synthesis of the candidates for this fragment and comparison of the NMR data of the natural product and the eight synthetic products resulted in the relative stereostructural clarification of the C69–C83 fragment. These individually determined relative stereochemistries of the C61–C74 and C69–C83 fragments were connected via the common C69–C73 tetrahydropyran moiety of the fragments. Finally, the relative configuration of the C61–C83 fragment of symbiodinolide was determined. The stereodivergent synthetic approach used in this study can be extended to the stereochemical determination of other fragments of symbiodinolide.
Introduction
Marine natural products (MNPs) are critical in drug discovery and development.1 Structural determination of MNPs, including their planar structures and stereochemistries, is a fundamental research topic and a requirement in using MNPs to create novel drugs and drug leads in medicinal chemistry. Remarkable recent advancements in computational chemistry enhanced our ability to determine the configurations of structurally complex natural products.2 Thus, over the decades, computational modeling of the 1H and 13C nuclear magnetic resonance (NMR) chemical shifts of organic compounds resulted in marked increases in availability, accuracy, and application. Nevertheless, if the target natural product contains conformationally flexible acyclic motifs and/or remote stereogenic centers, a synergistic combination of spectroscopic analysis and chemical synthesis is effective in unambiguous structural elucidation.3
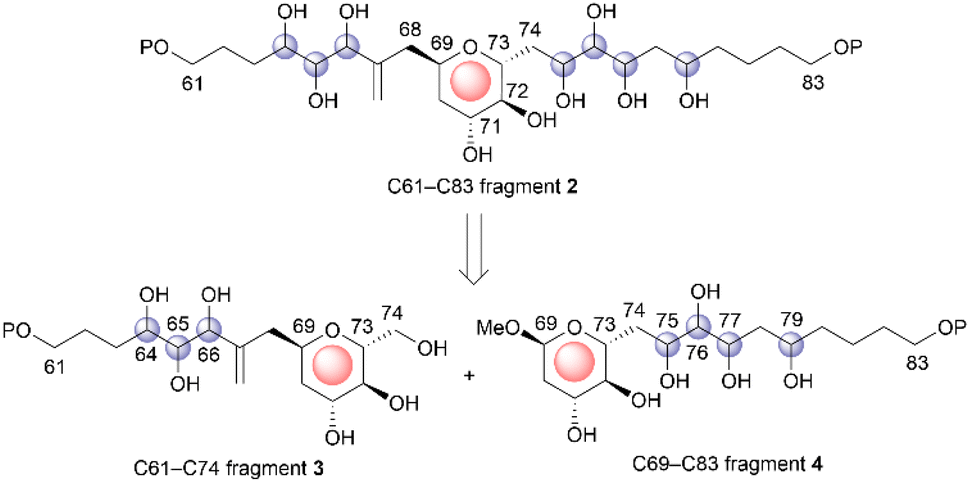
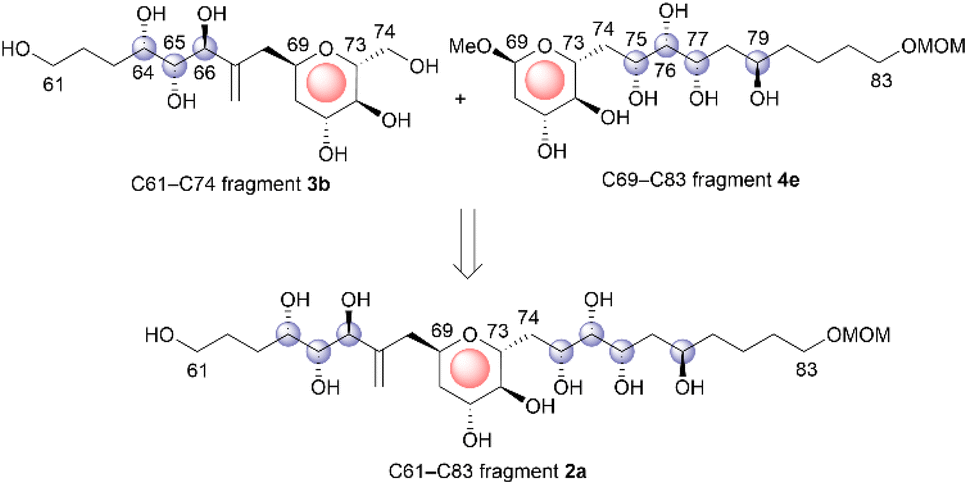
Symbiodinolide (1, Fig. 1) is a secondary metabolite that was isolated from the cultured dinoflagellate Symbiodinium sp. by Uemura et al.4 Natural product 1 increased the intracellular free Ca2+ concentration at 7 nM against IMR-32 human neuroblastoma cells and exhibited cyclooxygenase-1 inhibitory activity (65% inhibition) at 2 μM. Furthermore, 1 ruptured the tissue surface of the flatworm Amphiscolops sp., which is a host organism containing the symbiotic dinoflagellate Symbiodinium sp., at 2.5 μM. In an isolation study, detailed 2D NMR analysis and chemical degradation of 1 revealed its planar structure and partial stereochemistry. The structure of 1 displays a molecular weight of 2860, 61 chiral centers, and an extended carbon skeleton that is highly functionalized with oxygen atoms. The large, complicated chemical structure of 1 hampers its configurational assignment. Therefore, our group investigated the chemical synthesis of each fragment of 1, including the stereoisomers, leading to the stereochemical clarification of the moieties corresponding to 70% of the structure of 1.5 Herein, we report the stereodivergent synthesis6 of the C61–C74 and C69–C83 fragments (3 and 4), resulting in the determination of the relative stereochemistry of the C61–C83 fragment 2 that is among the remaining 30%.
 |
| | Fig. 1 Structure of 1. | |
Results and discussion
Strategy for the stereochemical determination of fragment 2
The relative configuration of the tetrahydropyran domain at positions C69–C73 of 1 was verified via the 3JH,H coupling constants and nuclear Overhauser effect (NOE) observations.4 The large coupling constants (3J71,72 and 3J72,73 = 8.3 Hz) indicate that H-71, H-72, and H-73 are oriented axially. The stereochemistry at the C69 position was elucidated using the NOEs of Ha-68/H-71 and Ha-68/H-73. To determine the stereostructure of fragment 2, we divided it into two fragments, i.e., fragments 3 and 4 (Scheme 1). The C69–C73 tetrahydropyran ring, with a known relative stereochemistry, is a common structure in fragments 3 and 4. Therefore, if the relative configurations of 3 and 4 are established individually, these two stereostructures may be connected by this common tetrahydropyran moiety to finally reveal the relative stereochemistry of fragment 2.
 |
| | Scheme 1 Strategy for the configurational determination of fragment 2. | |
Fragment 3
The relative configurations of the three contiguous oxymethine stereogenic centers at the C64, C65, and C66 positions of 1 were determined via1H NMR data.4 The coupling constants 3J64,65 (5.5 Hz) and 3J65,66 (7.4 Hz) indicate that the respective relative relationships of C64/C65 and C65/C66 are syn and anti. The relative stereochemistries at the C66 and C69 positions are not correlated, and thus, we propose two candidate diastereomers, 3a and 3b, for fragment 3, as shown in Fig. 2.
 |
| | Fig. 2 Candidate diastereomers 3a and 3b for fragment 3. | |
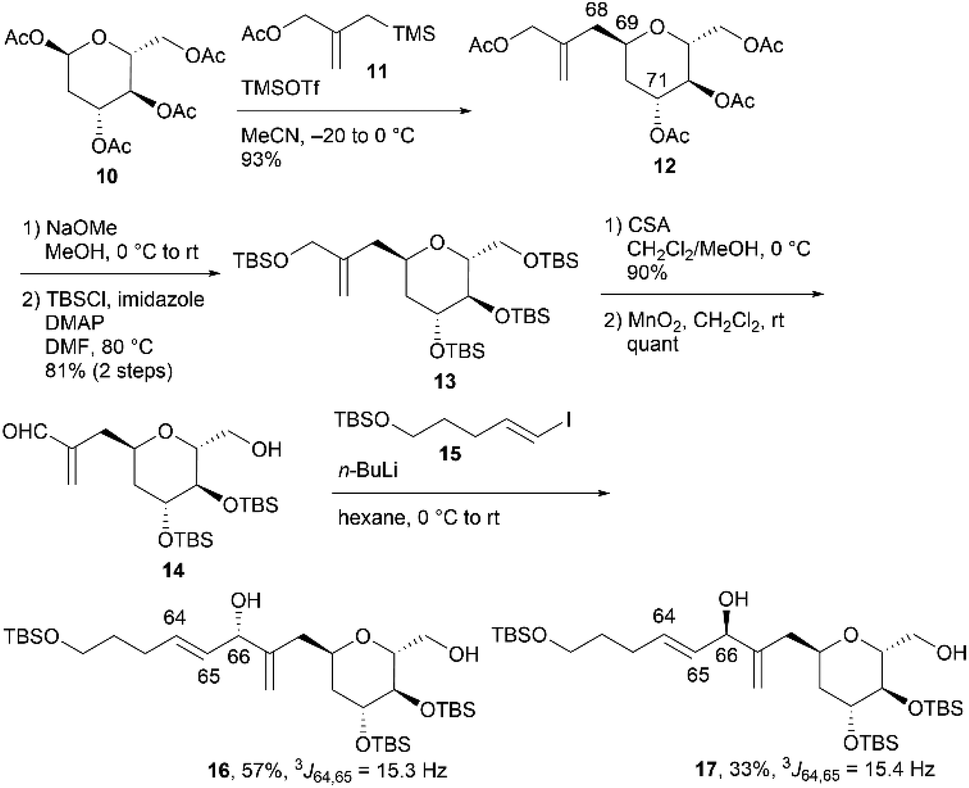
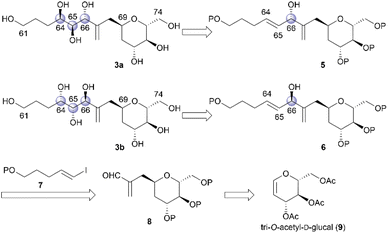
To stereochemically elucidate fragment 3 of 1, we planned to synthesize 3a and 3b and compare the NMR data of these synthetic products with those of natural product 1. In our retrosynthetic analysis, target molecules 3a and 3b may be synthesized via regio- and diastereoselective dihydroxylation at the C64 and C65 positions of dienes 5 and 6, respectively (Scheme 2). Alcohols 5 and 6, which are diastereomers at the C66 position, would be formed by coupling alkenyl iodide 7 and aldehyde 8. Aldehyde 8 would be synthesized by introducing a C4 side-chain into tri-O-acetyl-D-glucal (9) as a starting material.
 |
| | Scheme 2 Retrosynthetic analysis of candidate diastereomers 3a and 3b. | |
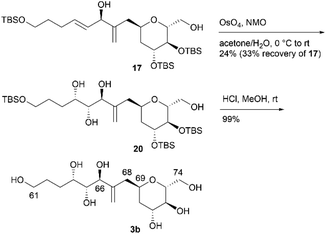
We first examined the stereoselective synthesis of aldehyde 14, which is a branching synthetic intermediate of 3a and 3b (Scheme 3). The reaction of the known tetraacetate 10, which was prepared using 9 in one step,7 with 2-[(acetoxymethyl)allyl]trimethylsilane (11)8 proceeded in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) as a Lewis acid to yield allylated product 12 as a single diastereomer.9 The stereochemistry at the C69 position was confirmed by the NOE correlations between H2-68 and H-71, which indicate that the introduced side-chain moiety is positioned axially. Global deprotection of 12via methanolysis and subsequent protection of the resulting tetraol using tert-butyldimethylsilyl chloride (TBSCl) afforded silyl ether 13 in 81% yield over two steps. The removal of the two primary tert-butyldimethylsilyl (TBS) groups of 13 with camphorsulfonic acid (CSA) in CH2Cl2/methanol (MeOH), followed by selective oxidation of the allylic alcohol component with MnO2, produced aldehyde 14. The coupling reaction of an alkenyl lithium species, which was generated via lithium–iodine exchange between alkenyl iodide 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and n-butyllithium (n-BuLi) in hexane,11 with aldehyde 14 generated diols 16 and 17 in respective yields of 57% and 33%.12 The C64/C65 geometries in 16 and 17 were confirmed as trans based on the coupling constants (3J64,65 = 15.3 Hz for 16 and 15.4 Hz for 17).
10 and n-butyllithium (n-BuLi) in hexane,11 with aldehyde 14 generated diols 16 and 17 in respective yields of 57% and 33%.12 The C64/C65 geometries in 16 and 17 were confirmed as trans based on the coupling constants (3J64,65 = 15.3 Hz for 16 and 15.4 Hz for 17).
 |
| | Scheme 3 Synthesis of alcohols 16 and 17. | |
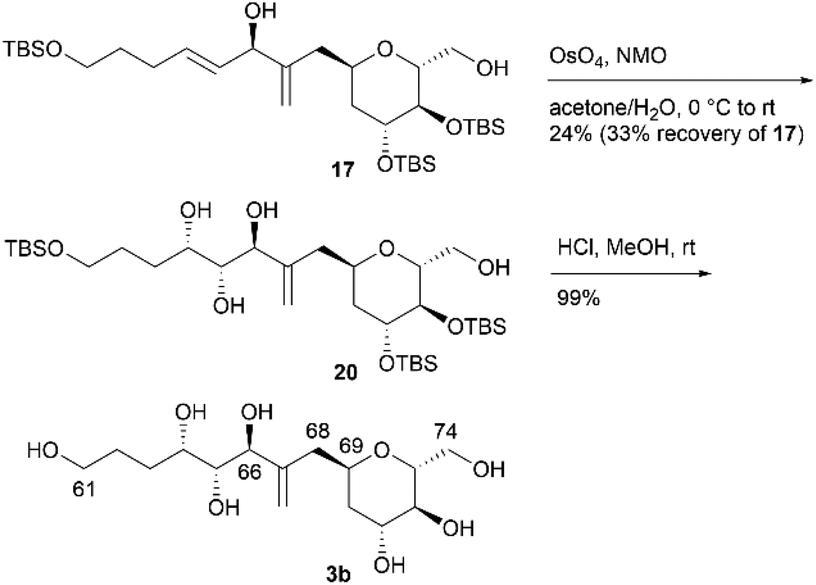
We then attempted to convert 16 to the first target molecule 3a (Scheme 4). Treatment of allylic alcohol 16 with OsO4/N-methylmorpholine N-oxide (NMO) in acetone/H2O13 produced the desired tetraol 18, along with its structural isomer, wherein the exo-olefin moiety was dihydroxylated.14 This obtained mixture could not be easily separated, and thus, the primary hydroxy group of 18 was protected with TBSCl/imidazole to furnish tetrakis-TBS ether 19, which was purified using silica gel column chromatography. The use of AD-mix-α or AD-mix-β15 in dihydroxylation did not improve the regioselectivity or chemical yield. The four TBS protective groups of 19 were removed using HCl in MeOH to quantitatively obtain the first target molecule, 3a. The second target molecule, 3b, was synthesized using 17via a transformation similar to that employed in synthesizing 3a using 16 (Scheme 5). Thus, dihydroxylation of the disubstituted alkene moiety of 17 was conducted using OsO4/NMO13 to afford the desired tetraol 2014 in 24% yield, along with recovered 17 in 33% yield. When the reaction time was extended, dihydroxylation of the exo-olefin component of 20 was observed. Finally, tris-TBS ether 20 was deprotected using HCl in MeOH to furnish the second target molecule, 3b. Although we obtained candidate compounds 3a and 3b, the regioselectivities in the dihydroxylation of 16 and 17 were uncontrolled, and the chemical yields of 18 and 20 were low. Therefore, alternative synthetic routes to 3a and 3b were investigated.
 |
| | Scheme 4 Synthesis of candidate compound 3a. | |
 |
| | Scheme 5 Synthesis of candidate compound 3b. | |
In an alternative synthetic route, we temporarily and regioselectively protected the exo-olefin moieties of dienes 16 and 17 prior to dihydroxylation. The second synthesis of 3a is shown in Scheme 6. After protecting diol 16 using TBSCl, regioselective hydroboration of the obtained diene was examined by changing the borane reagent. The use of BH3·SMe2 (SMe2 = dimethylsulfide) or thexylborane was ineffective in the regioselective hydroboration of the exo-olefin moiety. When we treated the obtained diene with 9-borabicyclo[3.3.1]nonane (9-BBN) and performed an oxidative work-up, the desired alcohol 21 was obtained in 76% yield over two steps. Tosylation of 21, iodination of the tosylate with NaI, and treatment of the iodoalkane with thiophenol (PhSH)/1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) afforded sulfide 22 in 68% yield over three steps. Diastereoselective dihydroxylation of alkene 22 with a stoichiometric amount of OsO4 in pyridine13,16 afforded the desired diol 23 and its C64,C65-epimer (dr = 6.2:1). The alkene component of 24 was regenerated via the following sequence: (1) meta-chloroperoxybenzoic acid (mCPBA) oxidation of sulfide 23 to the corresponding sulfoxide and (2) elimination of the sulfoxide with NaHCO3 in xylene at reflux. Diol 24 and its C64,C65-epimer were separated, and 24 was obtained in 56% yield in three steps from 22. Finally, pentakis-TBS ether 24 was deprotected to afford 3a. The respective total number of steps and overall yield of 3a from 16 in the second synthesis, as shown in Scheme 6, were 9 and 29%. Although the number of steps was increased compared to that in the first synthesis (3 steps from 16, as shown in Scheme 4), the overall yield was improved compared to that in the first synthesis (23% from 16, as shown in Scheme 4). We then investigated the second synthesis of 3b using a conversion similar to that shown in Scheme 6 (Scheme 7). TBS protection of diol 17 and subsequent regioselective hydroboration with 9-BBN afforded alcohol 25 in 75% yield over two steps. Tosylation, nucleophilic iodination, and thioetherification afforded sulfide 26. Alkene 26 reacted with OsO4 in pyridine13,16 to produce diol 27 (dr = 4.0:1), which was then converted to alkene 28via oxidation with mCPBA and elimination of the resulting sulfoxide. Global removal of the TBS protecting groups from 28 provided 3b. As in the case of 3a, although the transformation from 17 to 3b in the second synthesis (9 steps, Scheme 7) was lengthy, the overall yield of the second synthesis (29%) was higher than that of the first synthesis (24%, Scheme 5).
 |
| | Scheme 6 Improved synthesis of candidate compound 3a. | |
 |
| | Scheme 7 Improved synthesis of candidate compound 3b. | |
After obtaining candidate compounds 3a and 3b, we analyzed their 2D NMR spectra. The differences in the 1H NMR chemical shifts representing the C66, C68, and C69 positions of 1 and the synthetic products 3a and 3b are graphically illustrated in Fig. 3.17 The chemical shift deviations of 3b were smaller than those of 3a, as the sums of the magnitudes of the differences in the 1H NMR chemical shifts representing the C66, C68, and C69 positions were 0.23 (3a) and 0.15 (3b). Therefore, the relative configuration of fragment 3 of 1 is that of 3b.
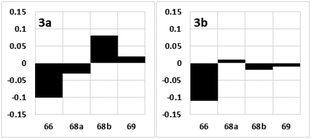 |
| | Fig. 3 Deviations in the 1H NMR chemical shifts of the synthetic products 3a and 3b (Δδ = δ1 − δ3 in ppm) relative to those of 1. The x and y axes show the carbon number and Δδ, respectively. | |
Fragment 4
We then focused on the stereochemical clarification of fragment 4. This fragment contains four unidentified chiral centers at positions C75, C76, C77, and C79, and thus, 16 diastereomers are possible. Prior to commencing the synthetic approach for structural determination, we analyzed the NMR data of natural product 1 to identify candidate compounds. The NMR chemical shifts of positions C75–C77 of 1, which are reported in an isolation study,4 are shown in Table 1. A detailed comparison of these chemical shifts with data reported in the Kishi universal NMR database for 1,2,3-triols18 clarified the relative relationships of C75–C76/C76–C77 as syn/syn or anti/anti. Therefore, eight hexaols, 4a–4h, as shown in Fig. 4, were used as candidate compounds for fragment 4.
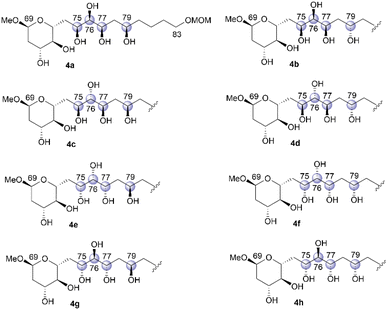 |
| | Fig. 4 Candidate diastereomers 4a–4h for fragment 4. | |
Table 1 NMR chemical shifts of 1 in CD3ODa
|
|
| Position |
1H NMR |
13C NMR |
|
Data reported in ref. 4. Chemical shifts are reported in ppm with reference to the internal residual solvent (1H NMR: 3.30 ppm, 13C NMR: 49.0 ppm).
|
|
75
|
3.89 |
69.6 |
|
76
|
3.18 |
78.5 |
|
77
|
3.95 |
70.5 |
The stereodivergent synthetic strategy6 for candidate compounds 4a–4h is outlined in Scheme 8. The reaction of aldehyde 29 and alkyne 30 would give two propargylic alcohols, 31 and 32, which are stereoisomeric at the C75 position (Scheme 8a). This coupling reaction is the first step in stereodivergence. Respective trans- and cis-alkenes 33 and 34 could be synthesized via the stereoselective reduction of alkyne 31, which is the second stereodivergence step (Scheme 8b). The dihydroxylation of trans-alkene 33 would afford hexaol 4a, and hexaol 4b, which is the C79-epimer of 4a, could be synthesized via stereoinversion at the C79 position, which is the third stereodivergence step, after the dihydroxylation of 33. Similarly, hexaols 4c and 4d, which are the respective C76-stereoisomers of 4a and 4b, could be produced using cis-alkene 34. Furthermore, the other four candidates, 4e–4h, could be prepared using propargylic alcohol 32, with the respective trans- and cis-alkenes 35 and 36 as the key synthetic intermediates, via a similar transformation. We then commenced the stereodivergent synthesis of candidate compounds 4a–4h.
 |
| | Scheme 8 Stereodivergent synthetic strategy of candidate compounds 4a–4h. | |
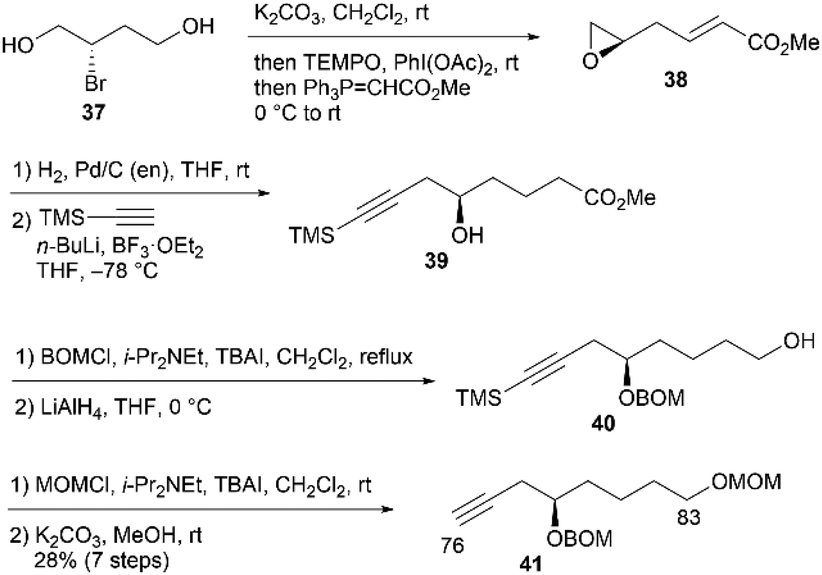
First, we examined the synthesis of alkyne 41, which corresponds to the C76–C83 fragment (Scheme 9). The synthesis commenced with (S)-2-bromobutane-1,4-diol (37), which was prepared in two steps using L-aspartic acid.19 Compound 37 was treated with K2CO3 in CH2Cl2 to yield a known epoxy alcohol,19 which underwent oxidation with 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO)/PhI(OAc)220 and subsequent Wittig olefination with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Me in one-pot to afford unsaturated ester 38. The alkene moiety of 38 was chemoselectively hydrogenated with a Pd/C-ethylenediamine complex catalyst in tetrahydrofuran (THF)21 to provide the saturated ester, with the epoxide structure retained. The resulting epoxide was reacted with trimethylsilylacetylene/n-BuLi/BF3·OEt222 (OEt2 = diethyl ether) to produce alcohol 39. The hydroxy group of 39 was protected as the benzyloxymethyl (BOM) ether, and the ester group was reduced using LiAlH4 to yield alcohol 40. Protection of 40 with methoxymethyl chloride/N,N-diisopropylethylamine/tetra-n-butylammonium iodide (MOMCl/i-Pr2NEt/TBAI), followed by removal of the trimethylsilyl (TMS) moiety with K2CO3 in MeOH, afforded alkyne 41 in 28% yield in seven steps from 37.
CHCO2Me in one-pot to afford unsaturated ester 38. The alkene moiety of 38 was chemoselectively hydrogenated with a Pd/C-ethylenediamine complex catalyst in tetrahydrofuran (THF)21 to provide the saturated ester, with the epoxide structure retained. The resulting epoxide was reacted with trimethylsilylacetylene/n-BuLi/BF3·OEt222 (OEt2 = diethyl ether) to produce alcohol 39. The hydroxy group of 39 was protected as the benzyloxymethyl (BOM) ether, and the ester group was reduced using LiAlH4 to yield alcohol 40. Protection of 40 with methoxymethyl chloride/N,N-diisopropylethylamine/tetra-n-butylammonium iodide (MOMCl/i-Pr2NEt/TBAI), followed by removal of the trimethylsilyl (TMS) moiety with K2CO3 in MeOH, afforded alkyne 41 in 28% yield in seven steps from 37.
 |
| | Scheme 9 Synthesis of alkyne 41. | |
With coupling precursor 41 in hand, we then synthesized aldehyde 44, which is the coupling partner of 41 (Scheme 10). Deacetylation of the known methyl glucoside 42,23 TBS protection of the resulting triol, and selective deprotection of the primary TBS ether provided alcohol 43 in 76% yield over three steps. One-carbon elongation from 43 was conducted via a combination of tosylation and cyanation to yield a nitrile, which was reduced with diisobutylaluminum hydride (DIBAL-H) to afford aldehyde 44. Deprotonation of alkyne 41 using n-BuLi and subsequent addition of the resulting anion to aldehyde 44 afforded the desired alcohols 45 and 46 in respective yields of 28% and 57%.24
 |
| | Scheme 10 Synthesis of alcohols 45 and 46. | |
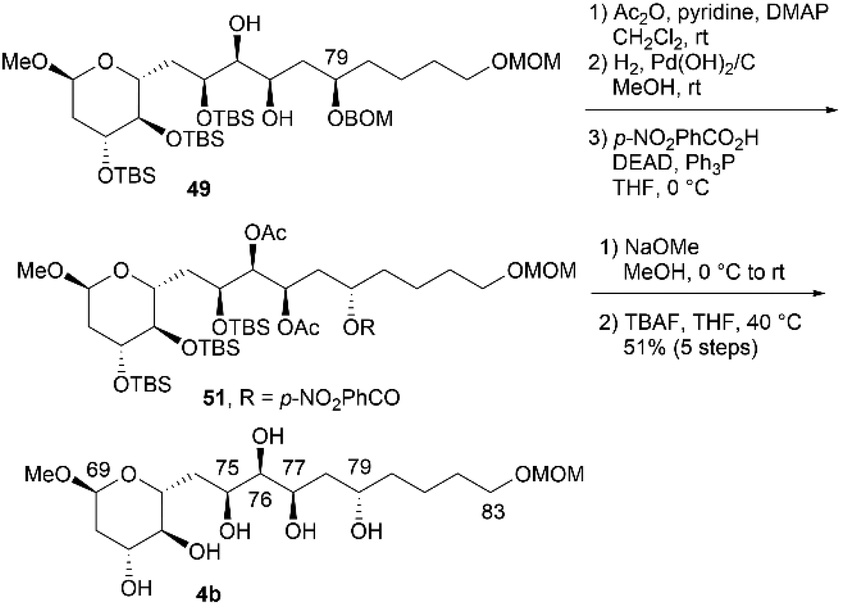
We then converted 45 to 4a (Scheme 11). Propargylic alcohol 45 was reduced using Red-Al25 to yield trans-allylic alcohol 47. The trans-geometry of 47 was determined based on the observed coupling constant (3J76,77 = 15.4 Hz). After protection of 47 with tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf)/2,6-lutidine, trans-alkene 48, with an allylic siloxy group, was dihydroxylated with OsO4 in pyridine13,16 to afford the desired diol 49 in 28% yield and its C76,C77-epimer in 55% yield (dr = 1:2.0). These products could be separated via silica gel column chromatography.14,26 The hydrogenation of BOM ether 49 with Pd(OH)2/C in MeOH generated triol 50. The three TBS groups of 50 were removed using tetra-n-butylammonium fluoride (TBAF) to produce candidate compound 4a. We then focused on the synthesis of candidate compound 4b, which is the C79-stereoisomer of 4a (Scheme 12). Diol 49, which is the dihydroxylation product in the synthesis of 4a, was acetylated to afford the diacetate, and the obtained BOM ether was deprotected to yield the corresponding C79-alcohol. The stereochemistry at the C79 position was inverted under Mitsunobu conditions27 using p-nitrobenzoic acid/diethyl azodicarboxylate (DEAD)/triphenylphosphine (Ph3P)28 to produce p-nitrobenzoate 51. The simultaneous removal of the acetyl and benzoyl groups of triester 51via methanolysis afforded the triol, which was reacted with TBAF to afford candidate compound 4b in 51% yield in five steps. The 1H and 13C NMR data of 4b differed from those of 4a, and thus, the C79-stereochemistry was inverted in the Mitsunobu reaction, leading to the formation of 51.
 |
| | Scheme 11 Synthesis of candidate compound 4a. | |
 |
| | Scheme 12 Synthesis of candidate compound 4b. | |
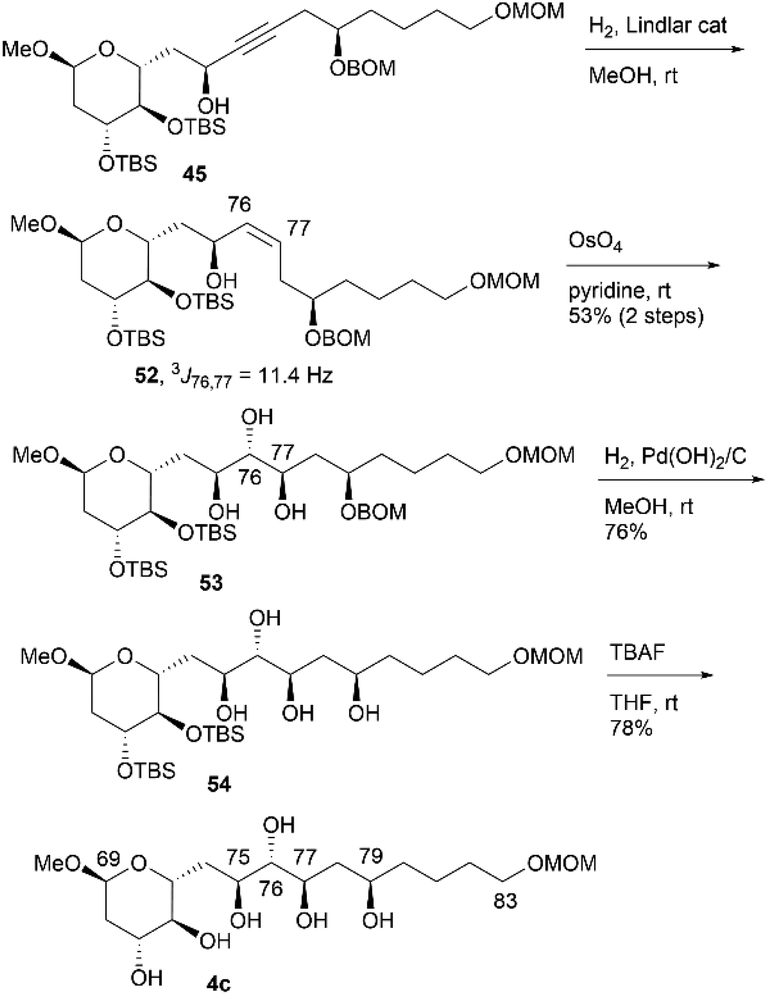
We then investigated the synthesis of candidate compound 4c, which is the C76-epimer of 4a (Scheme 13). The hydrogenation of alkyne 45 with a Lindlar catalyst produced cis-alkene 52, with the geometry confirmed by the coupling constant 3J76,77 = 11.4 Hz. The osmylation of cis-allylic alcohol 52 with OsO4 in pyridine13,16 generated the desired triol 53 in 53% yield in two steps,14 with the C76,C77-epimer of 53 obtained in 44% yield (dr = 1.2:1). The two diastereomers were separated using silica gel column chromatography. The deprotection of BOM ether 53via hydrogenation provided tetraol 54, which was desilylated using TBAF to afford candidate compound 4c. Candidate compound 4d, which is the C79-epimer of 4c, was synthesized via a conversion similar to that used in synthesizing 4b (Scheme 14). Thus, acetyl protection of triol 53, which is a synthetic intermediate of 4c, removal of the BOM moiety, and Mitsunobu esterification27,28 with configurational inversion provided the desired p-nitrobenzoate 55. The methanolysis of tetraester 55 afforded tetraol 56 in 59% yield in four steps from 53. The stereochemical inversion from 53 to 55via Mitsunobu esterification was confirmed by the difference in the NMR data of 56 and 54. Finally, bis-TBS ether 56 was deprotected to produce candidate compound 4d.
 |
| | Scheme 13 Synthesis of candidate compound 4c. | |
 |
| | Scheme 14 Synthesis of candidate compound 4d. | |
Having synthesized candidate compounds 4a–4d by branching from the common synthetic intermediate 45 with the 75S absolute configuration, we then attempted to synthesize the other four candidate compounds, 4e–4h, using propargylic alcohol 46, with 75R stereochemistry, as the common intermediate. The transformation of 46 into candidate compound 4e is shown in Scheme 15. Propargylic alcohol 46 was treated with Red-Al25 to produce trans-allylic alcohol 57 in 75% yield, and the trans-structure of alkene 57 was elucidated using the coupling constant 3J76,77 = 15.6 Hz. Allylic alcohol 57 was subjected to the directed dihydroxylation in accordance with the protocol reported by Donohoe et al.29 Thus, the reaction of 57 with OsO4/tetramethylethylenediamine (TMEDA) in CH2Cl2 afforded the desired triol 58 in 47% yield14 and its C76,C77-stereoisomer in 39% yield (dr = 1.2:1). The two triols were separated using silica gel column chromatography. The removal of the BOM protecting group via hydrogenation and the two TBS groups using TBAF provided candidate compound 4e. Triol 58 was then converted to candidate compound 4fvia stereoinversion at the C79 position (Scheme 16). The acetylation of 58, removal of the BOM group, and Mitsunobu esterification27,28 produced inverted p-nitrobenzoate 60, which underwent methanolysis to afford tetraol 61 in 59% yield over four steps. The configurational inversion under Mitsunobu conditions was verified by comparing the NMR data of 61 and its C79-epimer 59. The deprotection of bis-TBS ether 61 afforded candidate compound 4f in 66% yield.
 |
| | Scheme 15 Synthesis of candidate compound 4e. | |
 |
| | Scheme 16 Synthesis of candidate compound 4f. | |
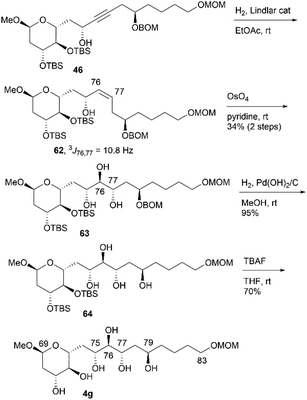
We then attempted to convert 46 to candidate compound 4g, which is the C76-epimer of 4e (Scheme 17). Alkyne 46 was hydrogenated using a Lindlar catalyst to yield cis-alkene 62, with the geometry elucidated using the coupling constant 3J76,77 = 10.8 Hz. Treatment of cis-allylic alcohol 62 with OsO4 in pyridine13,16 afforded the desired triol 63 and its C76,C77-stereoisomer in respective yields of 34% and 26% in two steps (dr = 1.3:1).14 Synthetic 63 and its diastereomer were separated via silica gel column chromatography. The removal of the BOM group of 63, followed by desilylation, yielded candidate compound 4g. Finally, we investigated the synthesis of candidate compound 4h, which is the C79-stereoisomer of 4g (Scheme 18). Triol 63 was converted to p-nitrobenzoate 65via the following sequence: (1) acetylation, (2) deprotection of the BOM ether, and (3) Mitsunobu esterification.27,28 Methanolysis of tetraester 65 and subsequent deprotection of bis-TBS ether 66 produced candidate compound 4h. The difference in the NMR data of 66 and 64 confirmed the stereoinversion in the Mitsunobu reaction during the synthesis of 65 using 63.
 |
| | Scheme 17 Synthesis of candidate compound 4g. | |
 |
| | Scheme 18 Synthesis of candidate compound 4h. | |
With candidate compounds 4a–4h in hand, we compared the NMR data of these eight synthetic products with those of natural 1. Deviations in the 1H NMR chemical shifts of natural 1 and the synthesized samples 4a–4h at the C75, C76, C77, and C79 positions were calculated, as shown in Fig. 5.17 The 1H NMR characteristics of 4e and 4f were more similar to those of the natural product than those of the other candidate compounds. The total magnitudes of the differences in the 1H NMR chemical shifts at the C75, C76, C77, and C79 positions are shown in Table 2, and those of 4e and 4f were 0.21. We then compared the 13C NMR chemical shifts of the eight synthetic candidates with those of the natural product. As shown in Fig. 6, candidate compound 4e exhibited the most similar characteristics of candidate compounds 4a–4h to those of the natural product.17 The sum of the magnitudes of the deviations in the 13C NMR chemical shifts of 4e was 2.1, as shown in Table 2, which was the smallest of those calculated for 4a–4h. Therefore, the relative stereochemistry of fragment 4 is shown in 4e.
 |
| | Fig. 5 Deviations in the 1H NMR chemical shifts of the synthetic products 4a–4h (Δδ = δ1 − δ4 in ppm) relative to those of 1. The x and y axes show the carbon number and Δδ, respectively. | |
 |
| | Fig. 6 Deviations in the 13C NMR chemical shifts of the synthetic products 4a–4h (Δδ = δ1 − δ4 in ppm) relative to those of 1. The x and y axes show the carbon number and Δδ, respectively. | |
Table 2 Sums of the magnitudes of the differences in the NMR chemical shifts (ppm) of natural 1 and the synthetic products 4a–4h at the C75, C76, C77, and C79 positions
| Compound |
1H NMR |
13C NMR |
|
4a
|
0.33 |
8.0 |
|
4b
|
0.26 |
5.4 |
|
4c
|
0.35 |
7.5 |
|
4d
|
0.25 |
4.9 |
|
4e
|
0.21 |
2.1 |
|
4f
|
0.21 |
3.1 |
|
4g
|
0.37 |
5.2 |
|
4h
|
0.30 |
3.2 |
Relative stereochemistry of fragment 2
We determined the relative configurations of fragments 3 and 4 in 1 as those described by 3b and 4e, respectively, by synthesizing all candidate compounds for each fragment and comparing their NMR chemical shifts with those of the natural product (Scheme 19). The C69–C73 tetrahydropyran moiety is a common structure in both fragments. Therefore, by connecting the relative stereostructures 3b and 4evia the common C69–C73 component, we elucidated the relative stereochemistry of fragment 2 in 1 as that of 2a.
 |
| | Scheme 19 Relative configuration of fragment 2. | |
Conclusions
Fragment 2 is among the 30% of the structure of 1 that has not been previously clarified. To determine the stereochemistry of fragment 2, we divided it into fragments 3 and 4 and elucidated the relative configurations of these two fragments using a stereodivergent synthetic approach. For fragment 3, the first synthesis of candidate compounds 3a and 3b was performed by coupling aldehyde 14 and alkenyl iodide 15, with the dihydroxylation of dienes 16 and 17, wherein the regioselectivities were uncontrolled. To overcome this problem, in the second synthesis of 3a and 3b, the exo-olefin components of dienes 16 and 17 were temporally and regioselectively protected prior to dihydroxylation. The overall yields of 3a and 3b in the second synthesis were improved compared to those in the first synthesis. A comparison of the NMR chemical shifts of these two synthetic products and the natural product revealed that natural product 1 displayed the relative stereochemistry of 3b in fragment 3. For fragment 4, we first analyzed the NMR data of positions C75–C77 of the natural product and selected candidate compounds 4a–4h from 16 possible diastereomers. The stereodivergent, unified synthesis of target molecules 4a–4h was performed via the coupling of alkyne 41 and aldehyde 44, stereoselective reduction of alkynes 45 and 46, and Mitsunobu inversion as the stereodivergence steps. The NMR chemical shifts of the synthetic products 4a–4h were compared with those of natural product 1, which clarified that the relative configuration in this domain of 1 was that of 4e. The tetrahydropyran component at positions C69–C73 is a common structure in fragments 3 and 4. Therefore, by overlapping the common tetrahydropyran moieties in 3b and 4e, the relative stereochemistry of fragment 2 of 1 is shown as 2a. The stereodivergent synthetic strategy used in this study can be employed for the configurational determination of other fragments of 1. Further study for the complete structural clarification of 1 is currently underway in our group.
Author contributions
H. T. conceived and directed the study. K. H., T. Ohashi, and T. Otsu conducted the syntheses and collected the data. All authors analyzed the collected data. H. T. prepared the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to Mr Soichiro Inoue (Okayama University, Okayama, Japan) for experimental support. We appreciate the assistance of the Division of Instrumental Analysis at Okayama University with NMR spectroscopy and high-resolution mass spectrometry. This study was supported by a Japan Society for the Promotion of Science KAKENHI grant (grant number: JP21H01938).
References
- For selected reviews, see:
(a) D. J. Newman and G. M. Cragg, Drugs and drug candidates from marine sources: An assessment of the current “state of play”, Planta Med., 2016, 82, 775–789 CrossRef CAS PubMed;
(b) K.-H. Altmann, Drugs from the oceans: Marine natural products as leads for drug discovery, Chimia, 2017, 71, 646–652 CrossRef CAS PubMed;
(c) C. Alves, J. Silva, S. Pinteus, H. Gaspar, M. C. Alpoim, L. M. Botana and R. Pedrosa, From marine origin to therapeutics: The antitumor potential of marine algae-derived compounds, Front. Pharmacol., 2018, 9, 777 CrossRef PubMed;
(d) A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Marine natural products, Nat. Prod. Rep., 2023, 40, 275–325 RSC.
- For selected reviews, see:
(a) M. Elyashberg, A. J. Williams and K. Blinov, Structural revisions of natural products by Computer-Assisted Structure Elucidation (CASE) systems, Nat. Prod. Rep., 2010, 27, 1296–1328 RSC;
(b) M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed;
(c) N. Grimblat and A. M. Sarotti, Computational chemistry to the rescue: Modern toolboxes for the assignment of complex molecules by GIAO NMR calculations, Chem. – Eur. J., 2016, 22, 12246–12261 CrossRef CAS PubMed;
(d) R. B. Nazarski, Summary of DFT calculations coupled with current statistical and/or artificial neural network (ANN) methods to assist experimental NMR data in identifying diastereomeric structures, Tetrahedron Lett., 2021, 71, 152548 CrossRef CAS.
- For selected reviews, see:
(a) K. C. Nicolaou and S. A. Snyder, Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation, Angew. Chem., Int. Ed., 2005, 44, 1012–1044 CrossRef CAS PubMed;
(b) M. E. Maier, Structural revisions of natural products by total synthesis, Nat. Prod. Rep., 2009, 26, 1105–1124 RSC;
(c) T. L. Suyama, W. H. Gerwick and K. L. McPhail, Survey of marine natural product structure revisions: A synergy of spectroscopy and chemical synthesis, Bioorg. Med. Chem., 2011, 19, 6675–6701 CrossRef CAS PubMed;
(d) T. F. Molinski and B. I. Morinaka, Integrated approaches to the configurational assignment of marine natural products, Tetrahedron, 2012, 68, 9307–9343 CrossRef CAS PubMed;
(e) H. Fuwa, Structure determination, correction, and disproof of marine macrolide natural products by chemical synthesis, Org. Chem. Front., 2021, 8, 3990–4023 RSC;
(f) M. W. Ha, J. Kim and S.-M. Paek, Recent achievements in total synthesis for integral structural revisions of marine natural products, Mar. Drugs, 2022, 20, 171 CrossRef CAS PubMed.
- M. Kita, N. Ohishi, K. Konishi, M. Kondo, T. Koyama, M. Kitamura, K. Yamada and D. Uemura, Symbiodinolide, a novel polyol macrolide that activates N-type Ca2+ channel, from the symbiotic marine dinoflagellate Symbiodinium sp., Tetrahedron, 2007, 63, 6241–6251 CrossRef CAS.
-
(a) H. Takamura, J. Ando, T. Abe, T. Murata, I. Kadota and D. Uemura, Stereocontrolled synthesis of the C79–C96 fragment of symbiodinolide, Tetrahedron Lett., 2008, 49, 4626–4629 CrossRef CAS;
(b) T. Murata, M. Sano, H. Takamura, I. Kadota and D. Uemura, Synthesis and structural revision of symbiodinolide C23–C34 fragment, J. Org. Chem., 2009, 74, 4797–4803 CrossRef CAS PubMed;
(c) H. Takamura, T. Murata, T. Asai, I. Kadota and D. Uemura, Stereoselective synthesis and absolute configuration of the C1′–C25′ fragment of symbiodinolide, J. Org. Chem., 2009, 74, 6658–6666 CrossRef CAS PubMed;
(d) H. Takamura, Y. Kadonaga, Y. Yamano, C. Han, Y. Aoyama, I. Kadota and D. Uemura, Synthesis and structural determination of the C33–C42 fragment of symbiodinolide, Tetrahedron Lett., 2009, 50, 863–866 CrossRef CAS;
(e) H. Takamura, Y. Kadonaga, Y. Yamano, C. Han, I. Kadota and D. Uemura, Stereoselective synthesis and absolute configuration of the C33–C42 fragment of symbiodinolide, Tetrahedron, 2009, 65, 7449–7456 CrossRef CAS;
(f) H. Takamura, Y. Kadonaga, I. Kadota and D. Uemura, Stereoselective synthesis of the C14–C24 degraded fragment of symbiodinolide, Tetrahedron Lett., 2010, 51, 2603–2605 CrossRef CAS;
(g) H. Takamura, Y. Kadonaga, I. Kadota and D. Uemura, Stereocontrolled synthesis and structural confirmation of the C14–C24 degraded fragment of symbiodinolide, Tetrahedron, 2010, 66, 7569–7576 CrossRef CAS;
(h) H. Takamura, K. Tsuda, Y. Kawakubo, I. Kadota and D. Uemura, Stereoselective synthesis of the C94–C104 fragment of symbiodinolide, Tetrahedron Lett., 2012, 53, 4317–4319 CrossRef CAS;
(i) H. Takamura, T. Fujiwara, I. Kadota and D. Uemura, Stereoselective synthesis of the C79–C97 fragment of symbiodinolide, Beilstein J. Org. Chem., 2013, 9, 1931–1935 CrossRef PubMed;
(j) H. Takamura, H. Wada, M. Ogino, T. Kikuchi, I. Kadota and D. Uemura, Stereodivergent synthesis and relative stereostructure of the C1–C13 fragment of symbiodinolide, J. Org. Chem., 2015, 80, 3111–3123 CrossRef CAS PubMed;
(k) H. Takamura, T. Fujiwara, Y. Kawakubo, I. Kadota and D. Uemura, Stereoselective synthesis of the proposed C79–C104 fragment of symbiodinolide, Chem. – Eur. J., 2016, 22, 1979–1983 CrossRef CAS PubMed;
(l) H. Takamura, T. Fujiwara, Y. Kawakubo, I. Kadota and D. Uemura, Stereodivergent synthesis and stereochemical reassignment of the C79–C104 fragment of symbiodinolide, Chem. – Eur. J., 2016, 22, 1984–1996 CrossRef CAS PubMed.
- For selected reviews, see:
(a) H. Takamura, Recent topics of the stereodivergent synthesis of natural products, Tetrahedron Lett., 2018, 59, 955–966 CrossRef CAS;
(b) C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Stereodivergent routes in organic synthesis: Carbohydrates, amino acids, alkaloids and terpenes, Org. Biomol. Chem., 2020, 18, 1232–1278 RSC;
(c) C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Stereodivergent routes in organic synthesis: Marine natural products, lactones, other natural products, heterocycles and unnatural compounds, Org. Biomol. Chem., 2020, 18, 1279–1336 RSC.
- S. N. Lam and J. Gervay-Hague, Efficient route to 2-deoxy β-O-aryl-D-glycosides via direct displacement of glycosyl iodides, Org. Lett., 2003, 5, 4219–4222 CrossRef CAS PubMed.
-
(a) B. M. Trost and D. M. T. Chan, New conjunctive reagents. 2-Acetoxymethyl-3-allyltrimethylsilane for methylenecyclopentane annulations catalyzed by palladium(0), J. Am. Chem. Soc., 1979, 101, 6429–6432 CrossRef CAS;
(b) B. M. Trost, M. Buch and M. L. Miller, Convenient alternative approach to 2-(acetoxymethyl)-3-(trimethylsilyl)propene, J. Org. Chem., 1988, 53, 4887–4888 CrossRef CAS.
-
(a) M. Morimoto, H. Matsukura and T. Nakata, Total synthesis of hemibrevetoxin B, Tetrahedron Lett., 1996, 37, 6365–6368 CrossRef CAS;
(b) I. Kadota, H. Takamura, H. Nishii and Y. Yamamoto, Total synthesis of brevetoxin B, J. Am. Chem. Soc., 2005, 127, 9246–9250 CrossRef CAS PubMed.
- D. P. Curran, Q. Zhang, C. Richard, H. Lu, V. Gudipati and C. S. Wilcox, Total synthesis of a 28-member stereoisomer library of murisolins, J. Am. Chem. Soc., 2006, 128, 9561–9573 CrossRef CAS PubMed.
- T. Yokoo, H. Shinokubo, K. Oshima and K. Utimoto, A room temperature preparation of alkenyllithiums by lithium-halogen exchange between alkenyl iodides and n-BuLi in hydrocarbon solvents, Synlett, 1994, 645–646 CrossRef CAS.
- For the stereochemical determination at the C66 position of 17, see ESI.‡.
-
(a) J. K. Cha, W. J. Christ and Y. Kishi, On stereochemistry of osmium tetraoxide oxidation of allylic alcohol systems. Empirical rule, Tetrahedron, 1984, 40, 2247–2255 CrossRef CAS;
(b) J. K. Cha and N.-S. Kim, Acyclic stereocontrol induced by allylic alkoxy groups. Synthetic applications of stereoselective dihydroxylation in natural product synthesis, Chem. Rev., 1995, 95, 1761–1795 CrossRef CAS.
- For the stereostructural determination, see ESI.‡.
- For a review, see: H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Catalytic asymmetric dihydroxylation, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- J. S. Baran, Notes – Method for the cleavage of osmate esters, J. Org. Chem., 1960, 25, 257 CrossRef CAS.
- The chemical shifts are described in ESI.‡.
- S. Higashibayashi, W. Czechtizky, Y. Kobayashi and Y. Kishi, Universal NMR databases for contiguous polyols, J. Am. Chem. Soc., 2003, 125, 14379–14393 CrossRef CAS PubMed.
- C. D. Donner, The divergent asymmetric synthesis of kalafungin, 5-epi-frenolicin B and related pyranonaphthoquinone antibiotics, Tetrahedron, 2013, 69, 377–386 CrossRef CAS.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, A versatile and highly selective hypervalent iodine (III)/2,2,6,6-tetramethyl-1-piperidinyloxyl-mediated oxidation of alcohols to carbonyl compounds, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS.
- H. Sajiki, K. Hattori and K. Hirota, Highly chemoselective hydrogenation with retention of the epoxide function using a heterogeneous Pd/C-ethylenediamine catalyst and THF, Chem. – Eur. J., 2000, 6, 2200–2204 CrossRef CAS PubMed.
- M. Yamaguchi and I. Hirao, An efficient method for the alkynylation of oxiranes using alkynyl boranes, Tetrahedron Lett., 1983, 24, 391–394 CrossRef CAS.
-
(a) C. J. France, I. M. McFarlane, C. G. Newton, P. Pitchen and M. Webster, Straightforward homochiral synthesis of the lactone moiety of mevinic acids, Tetrahedron Lett., 1993, 34, 1635–1638 CrossRef CAS;
(b) H. Fuwa, Y. Okamura and H. Natsugari, Synthetic studies on antascomicin A: Construction of the C18–C34 fragment, Tetrahedron, 2004, 60, 5341–5352 CrossRef CAS.
- For the stereochemical determination at the C75 position of 46, see ESI.‡.
- S. E. Denmark and T. K. Jones, (E)-3-(Trimethylsilyl)-2-propen-1-ol. An improved preparation, J. Org. Chem., 1982, 47, 4595–4597 CrossRef CAS.
- When we treated allylic alcohol 47 with OsO4/TMEDA in CH2Cl2,29 the desired triol and its C76,C77-epimer were obtained as an inseparable mixture in respective yields of 38%. Our attempts to derivatize and separate this mixture failed.
- For selected reviews, see:
(a) O. Mitsunobu, The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products, Synthesis, 1981, 1–28 CrossRef CAS;
(b) K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. V. P. P. Kumar, Mitsunobu and related reactions: Advances and applications, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed.
- S. F. Martin and J. A. Dodge, Efficacious modification of the mitsunobu reaction for inversions of sterically hindered secondary alcohols, Tetrahedron Lett., 1991, 32, 3017–3020 CrossRef CAS.
-
(a) T. J. Donohoe, N. J. Newcombe and M. J. Waring,
syn Stereocontrol in the directed dihydroxylation of acyclic allylic alcohols, Tetrahedron Lett., 1999, 40, 6881–6885 CrossRef CAS;
(b) T. J. Donohoe, K. Blades, P. R. Moore, M. J. Waring, J. J. G. Winter, M. Helliwell, N. J. Newcombe and G. Stemp, Directed dihydroxylation of cyclic allylic alcohols and trichloroacetamides using OsO4/TMEDA, J. Org. Chem., 2002, 67, 7946–7956 CrossRef CAS PubMed.
Footnotes |
| † We dedicate this work to the memory of Prof. Daisuke Uemura, who sadly passed away on 13th April 2021. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01420g |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Kosuke
Hattori
,
Takumi
Ohashi
,
Taichi
Otsu
and
Isao
Kadota
*,
Kosuke
Hattori
,
Takumi
Ohashi
,
Taichi
Otsu
and
Isao
Kadota