
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphine-catalysed transformations of ortho- and para-quinone methides
Aitor
Maestro
 *ab and
Mercedes
Zurro
*c
*ab and
Mercedes
Zurro
*c
aDepartamento de Química Orgánica I, Centro de Investigación y Estudios Avanzados “Lucio Lascaray” – Facultad de Farmacia, University of the Basque Country, UPV/EHU Paseo de la Universidad 7, 01006 Vitoria-Gasteiz, Spain. E-mail: aitor.maestro@ehu.eus
bInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria
cDepartamento de Química Orgánica y Química Inorgánica, Universidad de Alcalá (IRYCIS), 28805-Alcalá de Henares, Madrid, Spain. E-mail: mercedes.zurro@uah.es
First published on 28th September 2023
Abstract
Organocatalytic methodologies for the derivatization of o-QM, p-QM and the analogous aza-QM have been recently developed and involve different catalytic systems such as phosphoric acids, thioureas, squaramides, NHC carbenes or chiral ammonium salts. Besides, phosphines, commonly used as ligands in metal-catalysed reactions, can be also used as organocatalysts. In this case, they are mainly involved as nucleophilic catalysts in reactions such as the Rauhut–Currier (RC) reaction. In this review, an analysis of the recent developments in racemic and enantioselective phosphine-catalysed transformations of o-QM, p-QM and aza-o-QM has been carried out.
 Aitor Maestro | Aitor Maestro obtained his PhD (2019) from the University of the Basque Country, where he worked with Prof. Francisco Palacios and Dr Javier Vicario on the synthesis of aminophosphonates and asymmetric organocatalysis. After that, he worked as a postdoctoral research associate on enantioselective Cu(I) catalysis with Prof. Syuzanna Harutyunyan (University of Groningen, 2020–2021) and on the mechanism and applications of CuAAC with Prof. Allan J. B. Watson (University of St. Andrews, 2021–2022). He is currently a postdoc in the group of Prof. Oliver Kappe (University of Graz), working on asymmetric catalysis in continuous flow. His research interests include the development of sustainable processes and asymmetric catalysis. |
 Mercedes Zurro | Mercedes Zurro completed her PhD in 2016 from Universität Münster under the supervision of Prof. Olga García Mancheño working on anion-binding catalysis. Afterwards, she pursued postdoctoral research in the groups of Prof. L. Sánchez, Prof. S. R. Harutyunyan and Prof. M. A. Pericàs. In 2022, she received a Talent Attraction Fellowship Type 2 at the University of Alcalá where she worked on photoredox catalysis and the synthesis of bioactive compounds. Recently, she was promoted to Assistant Professor at the same University. Her research interests include asymmetric catalysis, synthetic methodology and medicinal chemistry. |
1. Introduction
Quinone methides are very reactive species present in organisms and are involved in many chemical and biological processes such as enzyme inhibition, DNA alkylation and cross-linking.1 They have been known for more than a century,2 but their synthetic potential as versatile reactive molecules has not been exploited until recent years.In the last two decades, different authors have reviewed the chemistry of quinone methides, highlighting their underdeveloped study and versatility for their derivatization into a variety of valuable diarylmethine derivatives and oxa-heterocyclic structures.3–6
Quinone methides (QMs) consist of a cyclohexadiene moiety conjugated with a carbonyl and an exo-methylene component via para-conjugation (p-QM) or ortho-conjugation (o-QM) (Fig. 1). They are neutral molecules with an aromatic zwitterionic resonance structure, responsible for their electrophilicity toward the 1,4- and 1,6-positions, respectively.
 | ||
| Fig. 1 Resonance structures of o-QMs and p-QMs. | ||
Quinone methides can be generated from a variety of precursors involving an acidic or basic medium, oxidation, thermolysis, photolysis, etc., but it is also possible to synthesize stable quinone methides, whose reactivity is suitable for asymmetric transformations. 2,6-Substituted p-QM and o-QM bearing multiple electron-donating substituents are stable reactants used in asymmetric transformations (Scheme 1). While p-QM can undergo 1,6-addition of a nucleophile, the o-QM chemistry is richer and there are three typical reaction modes: 1,4-addition, [4 + n] cycloaddition (mainly [4 + 2] and [4 + 1]) and oxa-6π-electrocyclization. The [4 + 2] cycloaddition and oxa-6π-electrocyclization pathways provide chromane derivatives, while the [4 + 1] cycloaddition gives access to benzofuran derivatives.
 | ||
| Scheme 1 Stable p-QM and o-QM and typical reactivity. | ||
Asymmetric catalytic transformations for the derivatization of o-QM7,8 and p-QM9–11 and the analogous aza-QM12,13 have been developed recently. Among the organocatalysts used in asymmetric transformations of o-QM and p-QM, it is common to identify chiral phosphoric acids as Brønsted acids,14–24 chiral ammonium salts as phase transfer catalysts,25–27 NHC carbenes,28,29 and hydrogen bond donor systems such as thioureas30 or squarimides.31–36 In this review, we aim to focus on transformations of o-QM and p-QM where an organocatalyst bearing a phosphine moiety in its structure is employed. Phosphine catalysis mainly deals with nucleophilic catalysis, in which phosphine acts as a nucleophile for activating a substrate. Compared to the analogous tertiary amines, phosphines have higher nucleophilicity which gives them some clear advantages in reactions involving allenes or vinyl ketones, such as the Morita–Baylis–Hillman (MBH) or the Rauhut–Currier (RC) reactions.37–45
The enantioselective Morita–Baylis–Hillman reaction involving a 1,2-addition of activated alkenes to aldehydes, ketones, and imines has been well established.46–49 In contrast, the asymmetric Rauhut–Currier reaction, also called the vinylogous MBH reaction, was not carried out until 2011.50 Specifically, if we look at (asymmetric) Rauhut–Currier reactions with p-QM, only some examples are found in the literature involving vinyl ketones or activated allenes (Scheme 2). If we look at (asymmetric) phosphine-catalysed reactions with o-QM, different MBH carbonates or activated allenes have been used as nucleophiles but just a few works have been reported (Scheme 2).
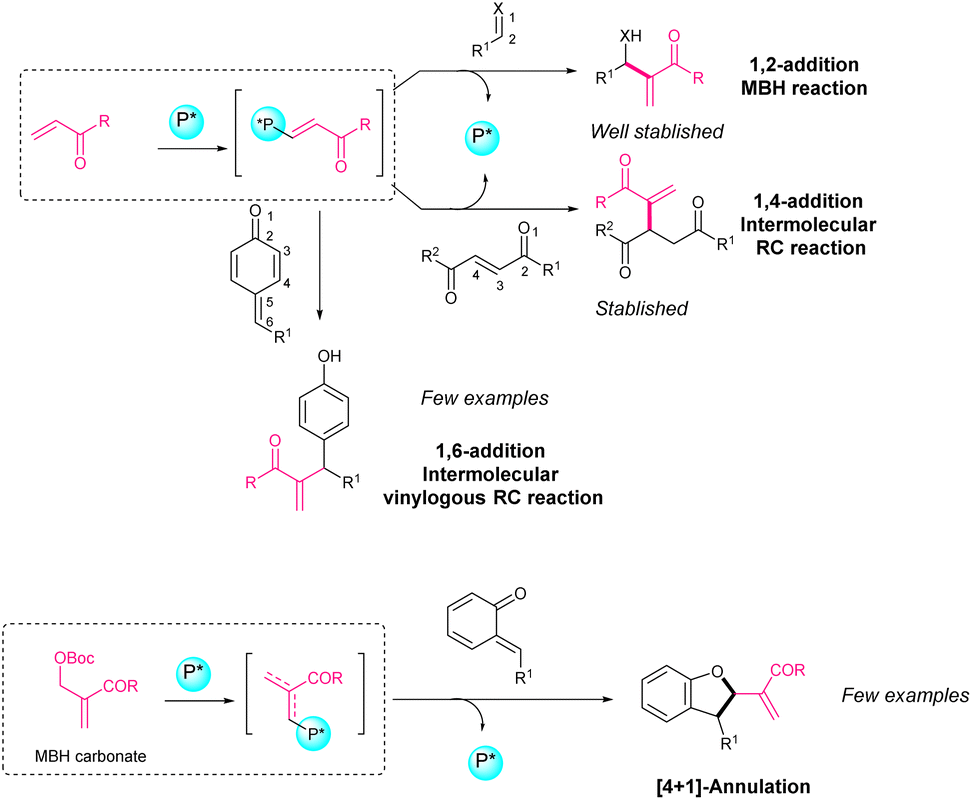 | ||
| Scheme 2 Phosphine-catalyzed enantioselective intermolecular 1,n-addition of vinyl ketones and [4 + 1] annulation of MBH carbonates. | ||
Along this review article, different works of Rauhut–Currier reactions involving activated alkenes, allenes or MBH carbonates and o-QM or p-QM will be described.
Besides, in the asymmetric reactions with quinone methides, the activation of the quinone methide derivatives is often required. Bifunctional organocatalysts bearing a phosphine moiety in addition to different functionalities such as thiourea, squaramide or peptidic residues are a perfect match for the reactions involving quinone methides by phosphine catalysis. These bifunctional catalysts are able to activate both quinone methide through hydrogen bonding and the nucleophile, as phosphine acts as a Lewis base or nucleophile. Many of the examples described in the review deal with the use of a bifunctional catalyst.
On the other hand, there are some catalytic reactions in which instead of phosphine, a phosphonium salt is used as the organocatalyst. In this case, the reactivity of the phosphorus atom is reversed and the phosphonium salts can activate negatively charged species, acting as phase transfer catalysts.
This review will provide a clear and chronological analysis of the literature related to phosphine-catalysed reactions of QM derivatives as well as a critical perspective of the future of the topic. Aiming to be a guide for researchers in the above-mentioned topics, this review will include not only enantioselective approaches but also racemic transformations, allowing the readers to obtain a clear picture of the existing literature when designing their future projects.
2. Catalytic transformations of para-quinone methides
2.1. Covalent phosphine activation
In 2017, Liu, Zhang and coworkers reported a phosphine-catalysed asymmetric intermolecular cross-vinylogous Rauhut–Currier reactions of alkyl vinyl ketones with p-QM.51 The reaction of p-QM 1 and vinyl ketone 2 in the presence of 10 mol% of catalyst I, K2HPO4 as a base, and trifluoromethyl benzene as solvent at −20 °C provided facile access to a variety of diarylmethine stereogenic centres with excellent enantioselectivities and yields (Scheme 3). This work represents the first catalytic methodology of derivatization of p-QM through nucleophilic catalysis. The mechanism is depicted in Scheme 15: the phosphine catalyst is added to vinyl ketone leading to intermediate A, and then p-QM 1 undergoes addition with A forming intermediate B, which finally leads to the formation of addition product 3. The formation of intermediate B is the key step controlling the enantioselectivity of the reaction. The bifunctional phosphine catalysts stabilize the nucleophile through an intramolecular ion pairing effect combined with hydrogen bonding between the carbonyl group of nucleophile 2 and the amide of the catalyst. The bulky substituent on the catalysts favors the selective Re-face attack. | ||
| Scheme 3 Phosphine-catalysed asymmetric intermolecular cross-vinylogous Rauhut–Currier reactions of alkyl vinyl ketones with p-QM. | ||
Furthermore, a scale-up reaction was carried out leading to product 3e in 96% yield and 99% ee (Scheme 4) and the synthetic applicability of derivatives 3 was demonstrated. Firstly, the selective reduction using CeCl3 and NaBH4 provided alcohol 4 with retained enantioselectivity and low diastereoselectivity (Scheme 4). The sulfa-Michael addition of p-TolSH to 3e afforded product 5 in a good yield with excellent stereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 99% ee). Besides, the tert-butyl groups at the 2,6-position of p-QM were removed using a standard protocol providing derivatives 6d–f in good yields and without racemization.
:1 dr, 99% ee). Besides, the tert-butyl groups at the 2,6-position of p-QM were removed using a standard protocol providing derivatives 6d–f in good yields and without racemization.
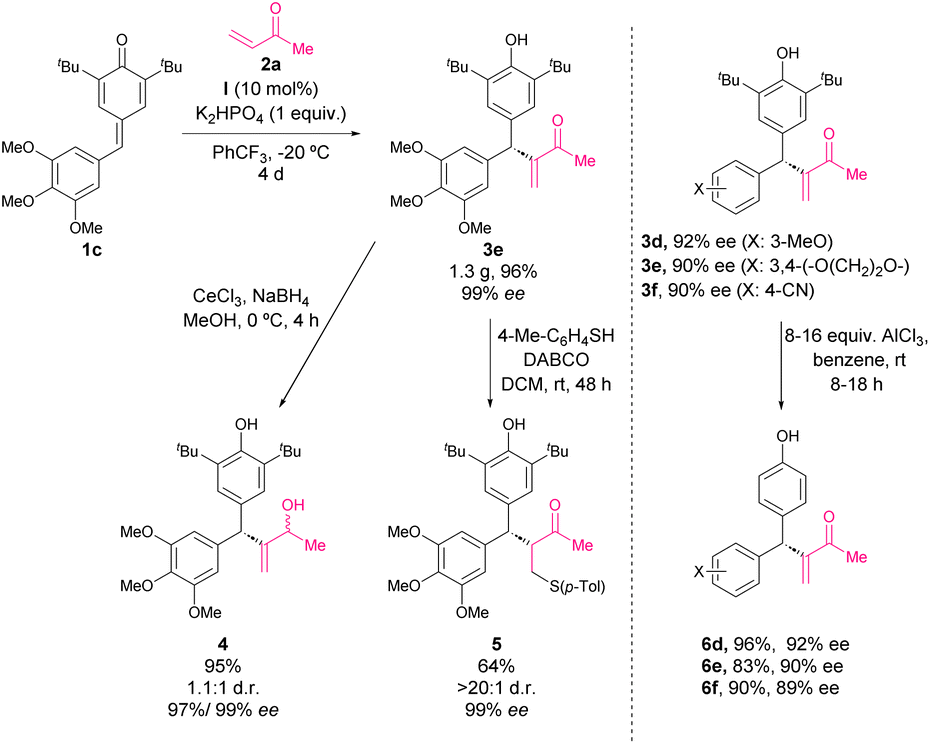 | ||
| Scheme 4 Synthetic applicability of 3. | ||
Also, at the beginning of 2017, the group of Wu developed a Rauhut–Currier reaction of methyl vinyl ketones and p-QM (Scheme 5).39 Firstly, a bifunctional phosphine–thiourea catalyst was tested in the reaction and 32% ee and 87% yield were obtained, which showed the feasibility of the reaction. Then, screening of the reaction conditions showed that catalyst II was suitable for the reaction affording chiral diarylmethines 8 with excellent yields and enantioselectivities. Scheme 5 shows a possible transition state which explains the high enantioselectivities in the reaction.
 | ||
| Scheme 5 Enantioselective vinylogous Rauhut–Currier 1,6-conjugate addition of methyl vinyl ketone to p-QM. | ||
In mid-2017, Fan and coworkers reported an enantioselective phosphine-catalysed intramolecular vinylogous Rauhut–Currier reaction of p-QM.52 With this methodology, chiral 4-aryl-3,4-dihydrocoumarins (10, X = O) and 4-aryl-3,4-dihydroquinolin-2-ones (10, X = N) were synthesized from p-QM esters (9, X = O) and p-QM amides (9, X = N), respectively, in excellent yields with moderate to excellent enantioselectivities (Scheme 6).
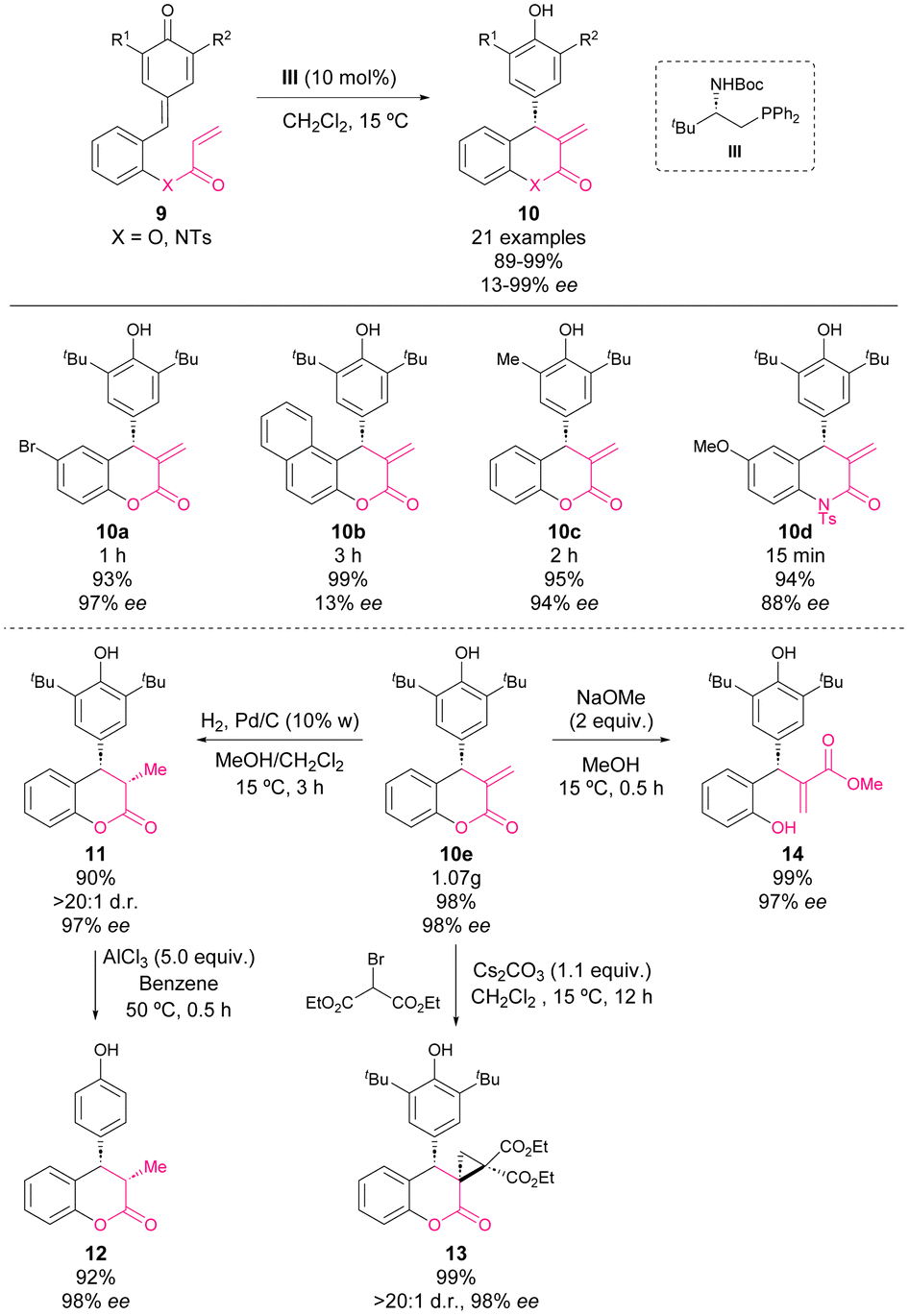 | ||
| Scheme 6 Enantioselective intramolecular vinylogous Rauhut–Currier reaction of p-QM. | ||
A scale-up reaction was carried out affording dihydrocoumarin 10e in 98% yield and 98% ee with only 5 mol% of the catalyst. Furthermore, in order to show the synthetic applicability, chiral 4-aryl-3,4-dihydrocoumarins 10 were subjected to several reaction conditions. Hydrogenation on Pd/C afforded derivative 11 in 90% yield, >20:1 dr, and 98% ee. Next, derivative 11 was treated with AlCl3 affording 12 in high yields with retained enantioselectivity. Besides, a cyclopropanation of 10e with 2-bromomalonic ester led to spirocycle 13 with an excellent yield and diastereoselectivity. The treatment of 10e with NaOMe promoted the opening of lactone to yield the acyclic derivative 14 in 99% yield and 97% ee.
At the end of 2017, Li, Tang and coworkers reported an asymmetric intermolecular Rauhut–Currier reaction of vinyl ketones 2 and p-QM derived from isatins 15.53 This methodology allowed the formation of 3,3-disubstituted oxindoles 16 with quaternary centres in good yields and selectivities by using a bifunctional thiourea–phosphine organocatalyst IV (Scheme 7).
 | ||
| Scheme 7 Asymmetric intermolecular Rauhut–Currier reaction of vinyl ketones and p-QM derived from isatins. | ||
In order to show the synthetic potential of 3,3-disubstituted oxindoles, different transformations were carried out. Firstly, the tert-butyl groups were removed by using AlCl3 in benzene, affording derivatives 17e–g in good yields. Next, a Chan–Lam cross-coupling between 17e and PhB(OH)2 afforded derivative 18 in a good yield. The sulfa-Michael addition of thiol p-ClC6H4SH to derivative 16 led to the addition product 20 in a good yield with excellent diastereoselectivity. Finally, the addition of phenyl magnesium bromide to derivative 16 yielded alcohol 19 in a good yield with retained enantioselectivity.
In 2022, Wang and coworkers reported the enantioselective Rauhut–Currier reaction with p-QM 21 and a variety of α,β-unsaturated carbonyl compounds (Scheme 8).58 The mechanism involves the initial 1,4-addition to the carbonyl compound 22. The formed phosphonium salt stabilizes the enolate as the nucleophile for 1,6-addition to p-QM. The presence of an ester moiety in quinone methide may indicate a crucial role of the carbonyl group in the activation of the electrophile (A). The reaction tolerates the use of ketones, esters and aldehydes as nucleophiles, obtaining the reaction products 23 with high yields and enantiocontrol. Although with lower yields, the presence of amides or thioesters in p-QM is also well tolerated. Moreover, the authors explored the use of chiral esters derived from natural products and chiral drugs in the p-QM moiety, obtaining high yields and stereocontrol.
 | ||
| Scheme 8 Enantioselective phosphine-catalysed Rauhut–Currier reaction. | ||
In 2022, Rachwalski and coworkers reported an intramolecular Rauhut–Currier reaction catalysed by a chiral phosphine/aziridine catalyst VI (Scheme 9).59 The reaction works through a similar mechanism shown in previous examples, affording optically active chroman-2-ones 25. Although the reaction yields and the enantiocontrol of the process are high, the scope is rather limited.
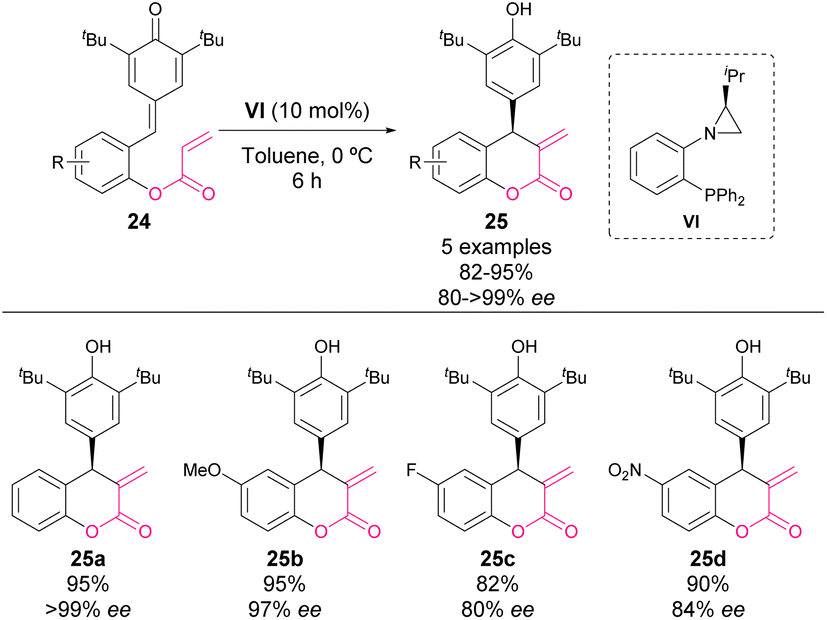 | ||
| Scheme 9 Enantioselective intramolecular Rauhut–Currier reaction. | ||
In 2019, Waser and coworkers reported an enantioselective phosphine-catalysed [4 + 1]-cyclization of o-hydroxy p-QM with allenoates.54 In the first attempt, the reaction between 26 and 27 using PPh3 (VII) as the catalyst afforded dihydrobenzofuran 28 in 90% yield (Scheme 8). Then, a screening of chiral phosphine catalysts was performed, showing that phosphine VIII was suitable for the reaction. Under the optimized conditions, a variety of dihydrobenzofuran derivatives were obtained with good yields and selectivities (Scheme 10).
 | ||
| Scheme 10 Phosphine-catalysed [4 + 1]-cyclization of o-hydroxy p-QM with allenoates. | ||
A proposed mechanism of the reaction based on NMR studies is depicted in Scheme 11. Firstly, the addition of phosphine to allenoate leads to the zwitterionic form B after proton transfer of the addition product A. Next, the addition of B to quinone methide 21 leads to intermediate C. The formation of this intermediate is also the key step controlling the enantioselectivity of the reaction. However, there are no further studies shedding light on the detailed interactions between 21 and the chiral intermediate B. After proton transfer, C is converted into D. Next, the isomerization of D through proton transfer occurs to generate E, which finally can react through an SN2′ reaction to yield the dihydrobenzofuran derivative 23. This last step seems to be the rate-determining step, which also explains why slightly larger amounts of the catalyst were needed to obtain a good catalyst turnover. The elimination of phosphine in intermediate E is also the key step controlling the diastereoselectivity of the reaction, which seems to be directed by the steric demand of the molecule.
 | ||
| Scheme 11 Enantioselective catalytic [4 + 1]-cyclization of hydroxyl-substituted p-QM with allenoates. | ||
In 2019, Shi and coworkers reported a phosphine-catalysed synthesis of chromanes through a formal [4 + 2] cyclization of p-QM 29 and allenes 30 (Scheme 12).55 The initial reaction of phosphine with allene forms a vinylogous enolate as a nucleophile as shown in the addition model A. Then, an intramolecular oxa-Michael addition (B) followed by a subsequent elimination of phosphine leads to the regeneration of the phosphine catalysts. The reaction works with a broad variety of substituted benzyl allenes, affording chromane derivatives 31 in yields up to 97% with excellent E/Z selectivity. However, when aliphatic allenes are used, a drastic decrease of the reactivity is observed.
 | ||
| Scheme 12 Phosphine-catalysed reaction of allenes 30 and p-QM. | ||
At the same time, Huang and coworkers reported another synthesis of chromanes through a phosphine-mediated addition of allenes 33 to p-QM 32 (Scheme 13).56 In this case, they obtained chromane derivatives 34 containing two chiral centers with high yields and diastereocontrol. Moreover, they also applied the methodology for the synthesis of 3,4-disubstituted tetrahydroquinolines in high yields but with limited stereocontrol.
 | ||
| Scheme 13 Synthesis of chromane derivatives 34 from allenes and p-QM. | ||
Regarding the reaction mechanism, phosphine has a double function (Scheme 14). Initially, it reacts with the allene, favouring the elimination of the acetate moiety in order to form a diene intermediate B. A subsequent base-mediated elimination allows the regeneration of phosphine as well as the formation of an acrylate derivative 35. The phospha-Michael addition to this intermediate led to nucleophile C, which reacts with p-QM. Next, an H-shift followed by intramolecular cyclization (E) affords the chromane or tetrahydroquinoline products 34. The fact that bulky esters (R = iPr, tBu) resulted in higher diastereocontrol than less sterically hindered substituents (R = Bn, Et) might indicate a crucial role of the steric demand in the selectivity of the reaction.
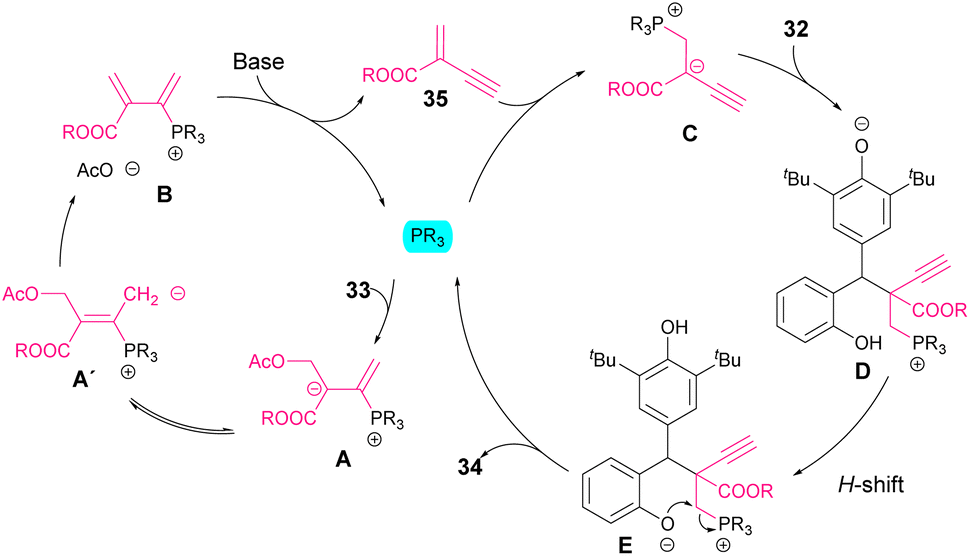 | ||
| Scheme 14 Proposed catalytic cycle for the synthesis of chromane derivatives 34. | ||
In 2021, Wang et al. reported the reaction between alkynes 37 and p-QM 36 (Scheme 15).57 The reaction, catalysed by a combination of 2-nitrobenzoic acid (38) and tributylphosphine (IX), affords dienylation products 39 in moderate to high yields as a single diastereomer. Moreover, they also reported the reaction with some isatin-derived p-QM, forming in this case an all-carbon quaternary center with excellent stereocontrol. Concerning the mechanism, the initial conjugate addition of phosphine to propiolic acid esters and amides 37, followed by an intramolecular isomerization of the formed alkene leads to the nucleophile as shown in Scheme 15 (A). The elimination of phosphine leads to product 39 with the consequent recovery of the phosphine catalyst. The high flexibility of the phosphonium salt obtained after the initial 1,6-addition allows the formation of a thermodynamically most stable product as the major isomer.
 | ||
| Scheme 15 Phosphine-catalysed addition of alkynes 37 to p-QM 36. | ||
2.2. Non-covalent phosphine activation
In 2016, Lin, Yao and coworkers reported the first asymmetric bifunctional thiourea–phosphine-catalyzed 1,6-conjugate addition of dicyanoolefins 41 to p-QMs 40 (Scheme 16).60 This reaction provided chiral diarylmethines 42 in good yields with good to excellent enantioselectivity and diastereoselectivity. To gain insight into the reaction mechanism, a mass spectrometry study was carried out. When organocatalyst XI, 40 and Et3N were mixed in Et2O for 40 minutes, a new peak was detected (m/z = 885.5), which corresponded to a complex existing between 40 and XI. This finding indicates the existence of hydrogen bonding between thiourea and p-QM. The authors propose that the phosphine moiety may act as a Lewis base activating dicyanoolefin. Based on this finding, the transition state depicted in Scheme 16 was proposed to explain the enantioselectivity of the reaction.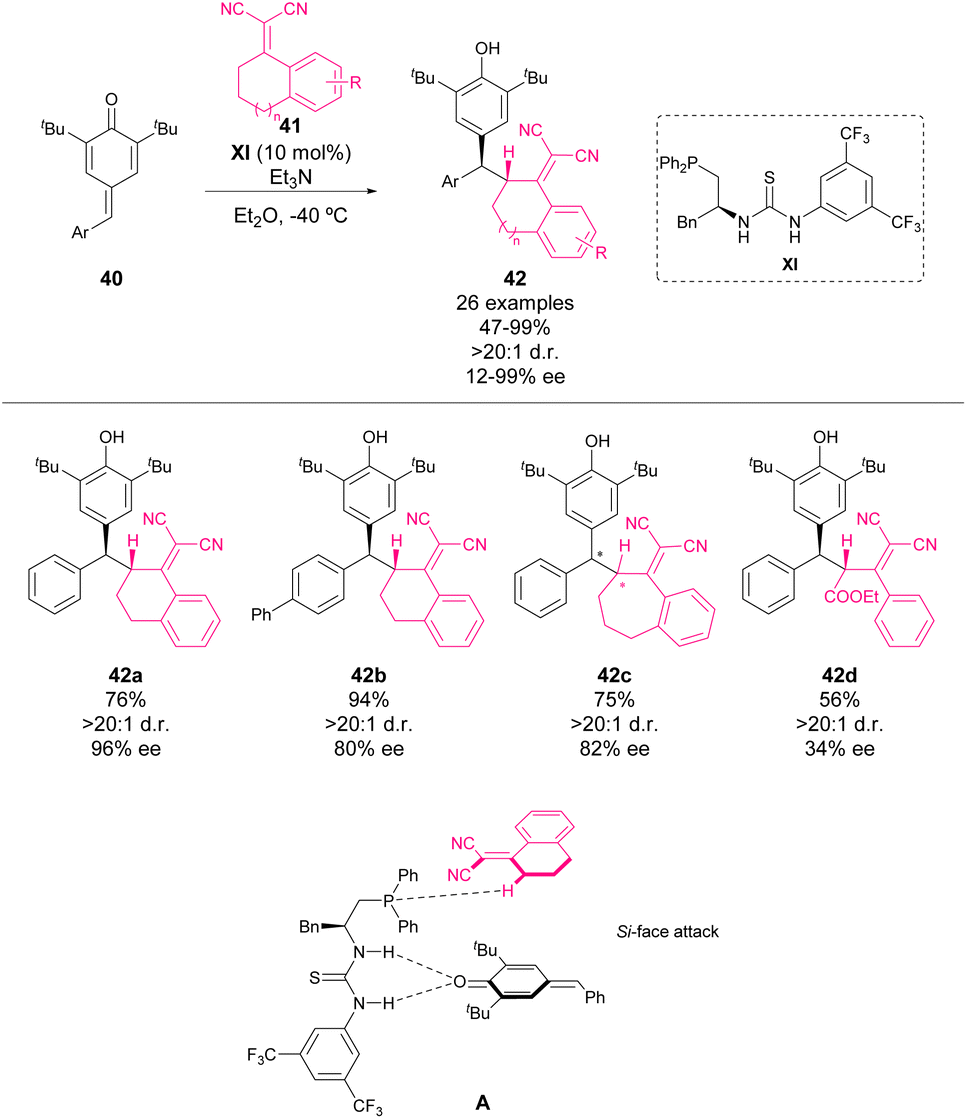 | ||
| Scheme 16 Asymmetric 1,6-conjugate addition of p-QM with dicyanoolefins. | ||
In 2017, Zhang and coworkers reported one of the pioneering works on phosphine-mediated additions to p-QM 43 (Scheme 17).61,62 In particular, they achieved the addition of 2-naphthols 44 mediated by a catalytic amount of phosphine XII and methyl acrylate 45. The initial phospha-Michael addition of phosphine to acrylate forms the phosphonium salt 47 as the active catalyst of the reaction. This catalyst acts as a base by stabilizing the enolate of naphthol (A) as the nucleophile for 1,6-addition to p-QM. The reaction works with a broad scope of aryl and heteroaryl p-QM, affording yields from 55 to 99%.
 | ||
| Scheme 17 Phosphine-catalysed addition of naphthols to p-QM. | ||
Also in 2016, Wu, Cao and coworkers reported an asymmetric 1,6-addition of ester malonates to p-QM 48 by using an amide-phosphonium salt as a bifunctional phase transfer catalyst (Scheme 18).63 The reaction between stable p-QM and ester malonate 49 in the presence of 1 to 5 mol% of chiral phosphonium salt XIII and K2CO3 as a base provided chiral diarylmethine stereocenters with high yields and stereoselectivities. The amide moiety seems to be crucial for the enantioinduction as the reaction with the organocatalyst bearing an N-methyl substituent in amide led to only 19% ee in the reaction. N–H can form hydrogen bonds between p-QM stabilizing the transition state A, which leads to the major enantiomer.
 | ||
| Scheme 18 Asymmetric bifunctional phosphonium salt-catalysed 1,6-addition of malonate esters to p-QM. | ||
Also in 2019, Wang and coworkers reported an enantioselective phosphonium salt-catalysed formal [4 + 1] annulation of hydroxyl-substituted p-QM with α-halogenated ketones.64 The reaction between hydroxyl-p-QM 51 and α-halogenated ketone 52 in the presence of dipeptide-derived phosphonium salt catalyst XIV and CsCO3 as a base afforded functionalized 2,3-dihydrobenzofurans 53 in high yields with excellent stereoselectivities (Scheme 19). In order to understand the function of the chiral phosphonium salt, DFT calculations were carried out revealing the presence of multiple H-bonding interactions between the catalyst and the enolate intermediate, which determine the transition state of the reaction and the transfer of the chirality to the 2,3-dihydrobenzofuran products 53. Also, mechanistic studies were carried out, revealing that the reaction occurred through a nucleophilic substitution and subsequent intramolecular 1,6-addition pathway.
 | ||
| Scheme 19 Bifunctional phosphonium salt-catalysed enantioselective formal [4 + 1] annulation of hydroxyl-substituted p-QM with α-halogenated ketones. | ||
Although it is not strictly a phosphine-catalysed reaction, in 2021, Wang and coworkers reported the enantioselective addition of secondary phosphine oxides 55 to p-QM 54 in the presence of a chiral phosphonium salt catalyst XV.65 Based on the DFT studies, it was proposed that after the removal of the acidic proton by the base, the amide and carbamate moieties as hydrogen donors activate the phosphoryl group while the phosphonium salt stabilizes the formed anion as shown in Scheme 20 (A). The key transition state involves the 1,6-addition of phosphine oxide to p-QM (B). The products 56, bearing a tetrasubstituted chiral centre, are obtained with excellent yields and enantiocontrol. Moreover, the authors explored the possibility of using chiral phosphine oxides in the reaction, obtaining in this case low diastereocontrol but high enantioselectivity for both isomers.
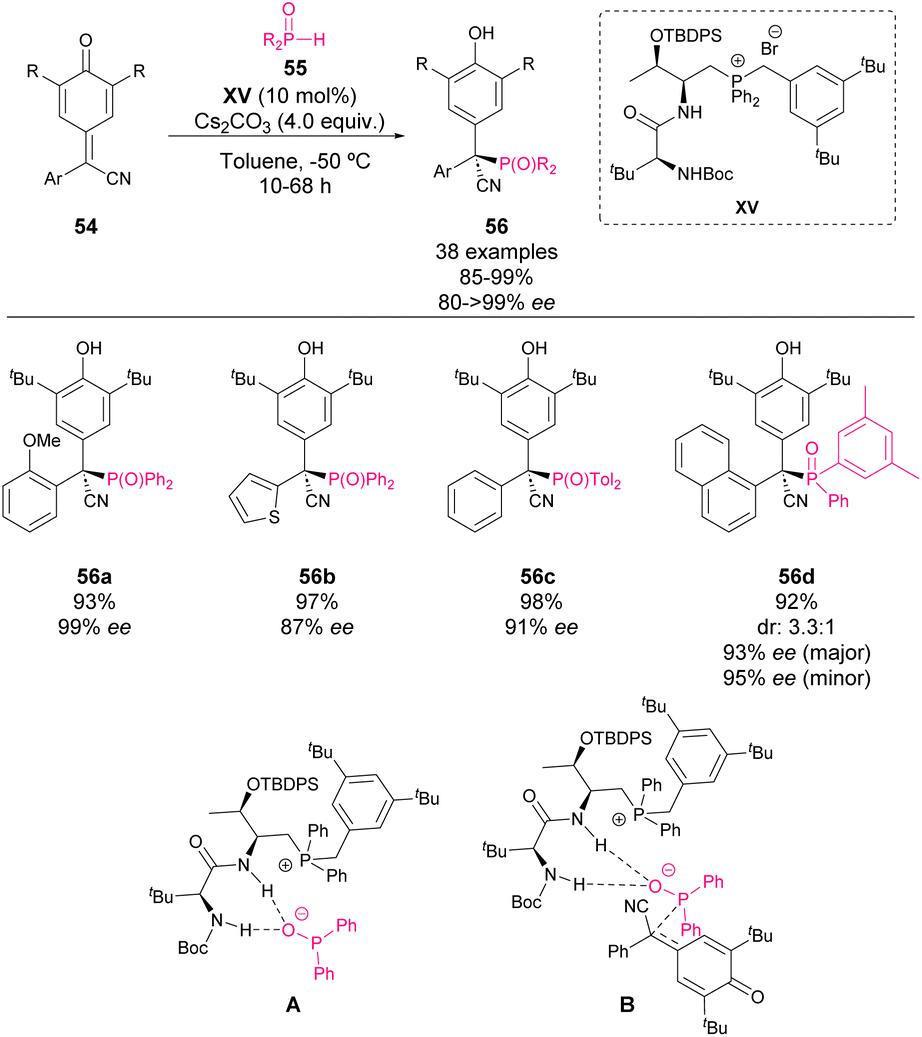 | ||
| Scheme 20 Phosphonium salt-catalysed asymmetric construction of chiral phosphonates. | ||
In 2022, Wang's group reported the 1,6-cyanation of p-QM 57 (Scheme 21).66 Similar to the mechanism shown in previous examples, a combination of phosphine XVI and acrylate 59 is used as the precatalyst. In this case, the TMS protection of the ester enolate is proposed as the key step to activate the nucleophilicity of the cyanide anion (A). The reaction affords phenol derivatives 60 in excellent yields and can also be applied to the synthesis of tetrasubstituted products. The authors explored the possibility of inducing some enantiocontrol in the process by using chiral phosphines and acrylate derivatives, but only the racemic cyanation products 60 were obtained.
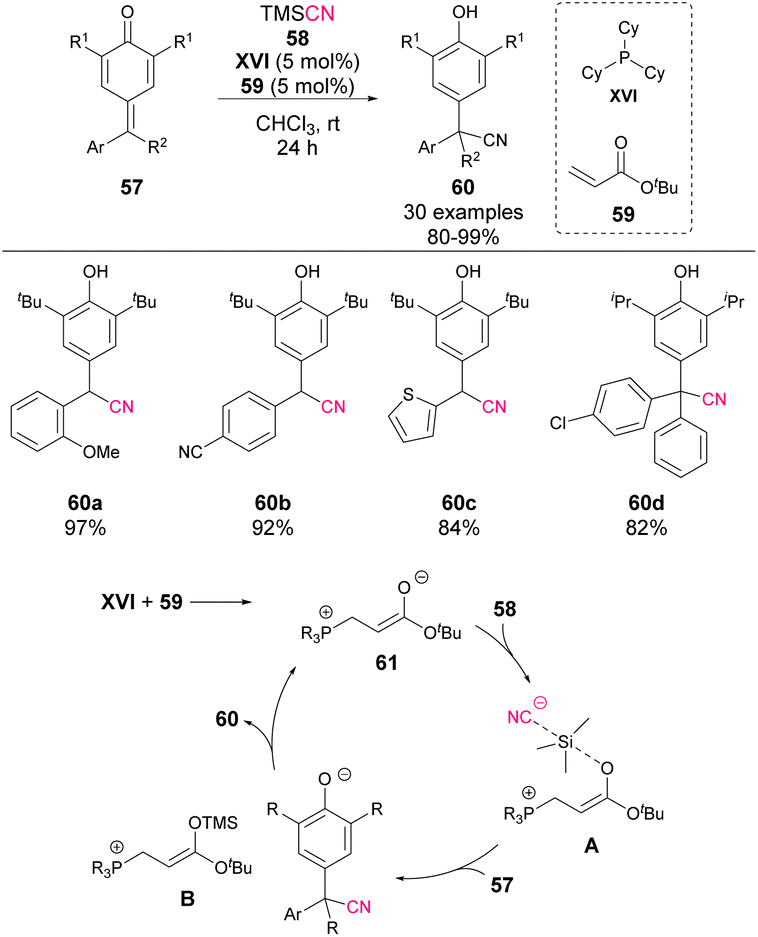 | ||
| Scheme 21 1,6-Cyanation of p-QM. | ||
In 2022, Mohanan and coworkers reported the addition of fluorinated 1,3-dicarbonyl compounds 63 to p-QM derived from isatin 62.67 The obtained products 64 were isolated in yields up to 98% but with moderate to low diastereocontrol. Concerning the mechanism, the authors proposed a double function of the QM derivatives. The initial 1,6-addition of phosphine VII to p-QM could lead to the corresponding phosphonium salts (VII′) as the active catalyst of the reaction. V′ salts are basic enough to activate 1,3-dicarbonyls 63 (A). After the reaction, the phosphonium salt VII′ could be recovered by an intermolecular H-shift as shown in Scheme 22.
 | ||
| Scheme 22 Phosphine-catalysed addition of fluorinated 1,3-dicarbonyl compounds 63. | ||
3. Catalytic transformations of ortho-quinone methides
3.1. Covalent phosphine activation
In 2017 Lu, Wang and coworkers reported the first phosphine-catalysed [4 + 2] cycloaddition of o-QM 65 with allene ketones 66 to synthesize chromane derivatives 67 in high yields with excellent enantioselectivity.68 The most effective catalysts were found to be a t-Leu-derived phosphine–amide XVII or XVIII (in case of vinyl-substituted o-QMs), affording enantioselectivities of up to 99% ee in the reaction (Scheme 23). The mechanism is depicted in Scheme 23: firstly, phosphine is added to the allene forming the zwitterionic form A, which is then added to o-QM leading to intermediate B, which yields chiral phosphine as the product of the reaction.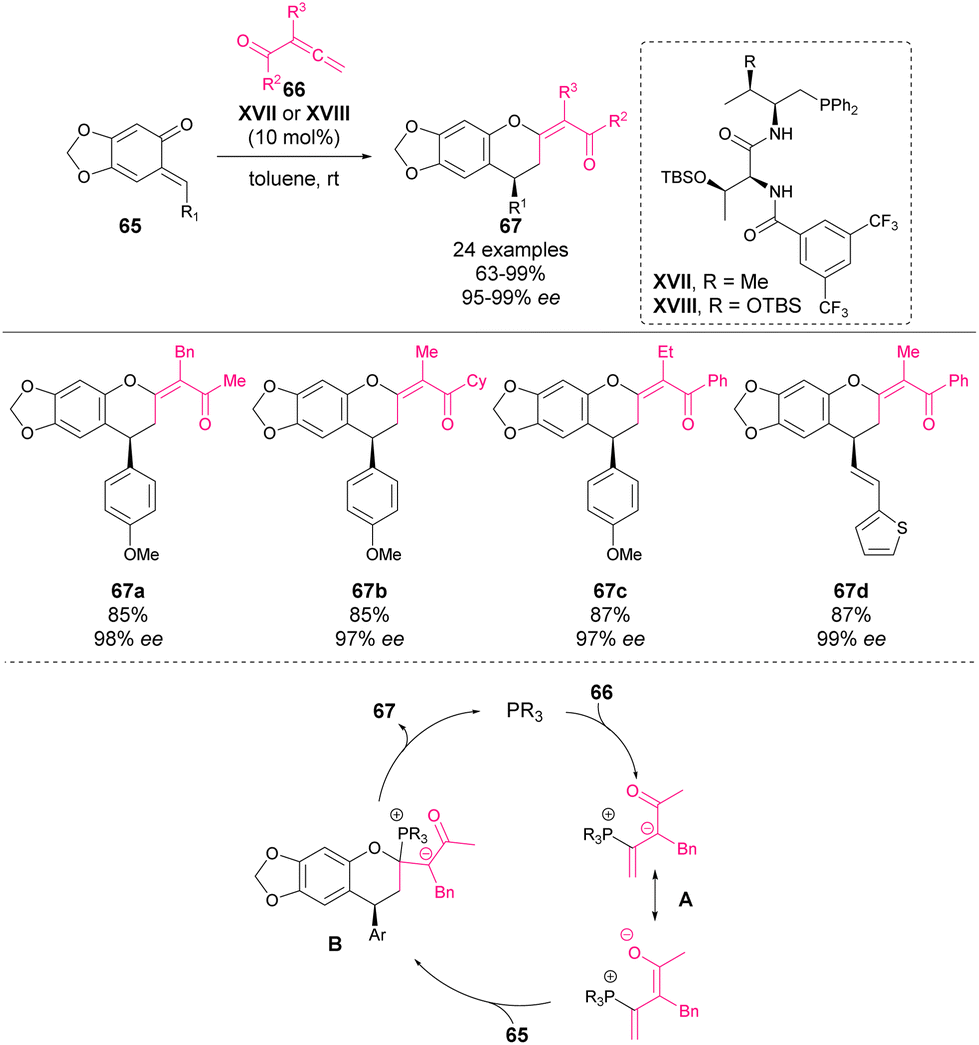 | ||
| Scheme 23 Phosphine-catalysed enantioselective [4 + 2] annulation of o-QM with allene ketones. | ||
In 2018, Shi and coworkers reported an organocatalytic [4 + 1] cyclization of o-QMs 65 with MBH carbonates 68 for the construction, in this case, of 2,3-dihydrobenzofuran scaffolds (Scheme 24).69 Although the mechanism is not fully disclosed, two potential pathways are proposed. In both of them, o-QM acts as a 4C synthon and MBH carbonate as a 1C synthon, making the generation of 5-membered heterocycles possible. In this work, a novel naphthylindole-derived phosphine organocatalyst was used for the first time.70 The 2,3-dihydrobenzofuran derivatives 69 were obtained in good to excellent yields. Furthermore, different chiral naphthylindole-derived phosphines were tested under the optimized conditions observing low enantiomeric ratios in all cases (<26% ee).
 | ||
| Scheme 24 Naphthylindole-derived phosphine-catalysed [4 + 1] cyclizations of o-QM with Morita–Baylis–Hillman carbonates. | ||
In the same year, Li and coworkers reported an enantioselective variant of this reaction.71 This work represents a facile strategy for the synthesis of enantioenriched 2,3-dihydrobenzofurans 72. The mechanism proposed by the authors is in agreement with the reaction pathway B proposed by Shi and coworkers and is depicted in Scheme 25: phosphine is added to MBH carbonate provoking the release of tBuOH and CO2 and formation of intermediate A, Then, A is added to o-QM affording intermediate B, which then reacts through an intramolecular nucleophilic substitution reaction affording the product of the reaction.
 | ||
| Scheme 25 Phosphine-catalysed enantioselective [4 + 1] annulations between o-QM and Morita–Baylis–Hillman carbonates. | ||
Also in 2018, Zhang, Liu and coworkers reported an enantioselective phosphine-catalyzed [4 + 1] annulation of o-QM 73 with MBH carbonates 74.72 The main difference between this work and the report by Li and coworkers, relies on the nature of the catalyst and the use of an aryl-substituted MBH carbonate, which allows the formation of a tetrasubstituted stereocenter. In this work, a diphosphine ligand bearing a 3,5-bistrifluoromethyl benzoyl amide moiety provides the highest enantioselectivities in the catalytic reaction (Scheme 26), which might be explained through the H-bonding properties of the NH of the amide. The mechanism of the reaction is analogous to previous work. The authors reported the synthesis of several cyclization products 75 with high yields and enantiocontrol but, in contrast to the previous reports from Li and Shi, low diastereoselectivity.
 | ||
| Scheme 26 Phosphine-catalysed enantioselective [4 + 1] annulations between o-QM and Morita–Baylis–Hillman carbonates. | ||
4. Catalytic transformations of aza-ortho-quinone methides
4.1. Covalent phosphine activation
In 2015, Chen and coworkers reported an unprecedented [4 + 3] cycloaddition reaction with bromosubstituted Morita–Baylis–Hillman adducts of isatins 77 and N-(ortho-chloromethyl)aryl amides 76 for the construction of a challenging azaspirocycloheptane oxindole scaffold 78 (Scheme 27).73 The use of a stoichiometric amount of phosphine XXIII enables the formation of a reactive allylic ylide (B). Simultaneously, transient aza-o-quinone methide was generated from precursor 76 in the presence of Cs2CO3 (A). The allylic ylide acted as a nucleophile in the addition to aza-o-QM and followed by cyclization, led to the azaspirocycloheptane oxindole derivative 78 in moderate to good yields with a wide substrate scope.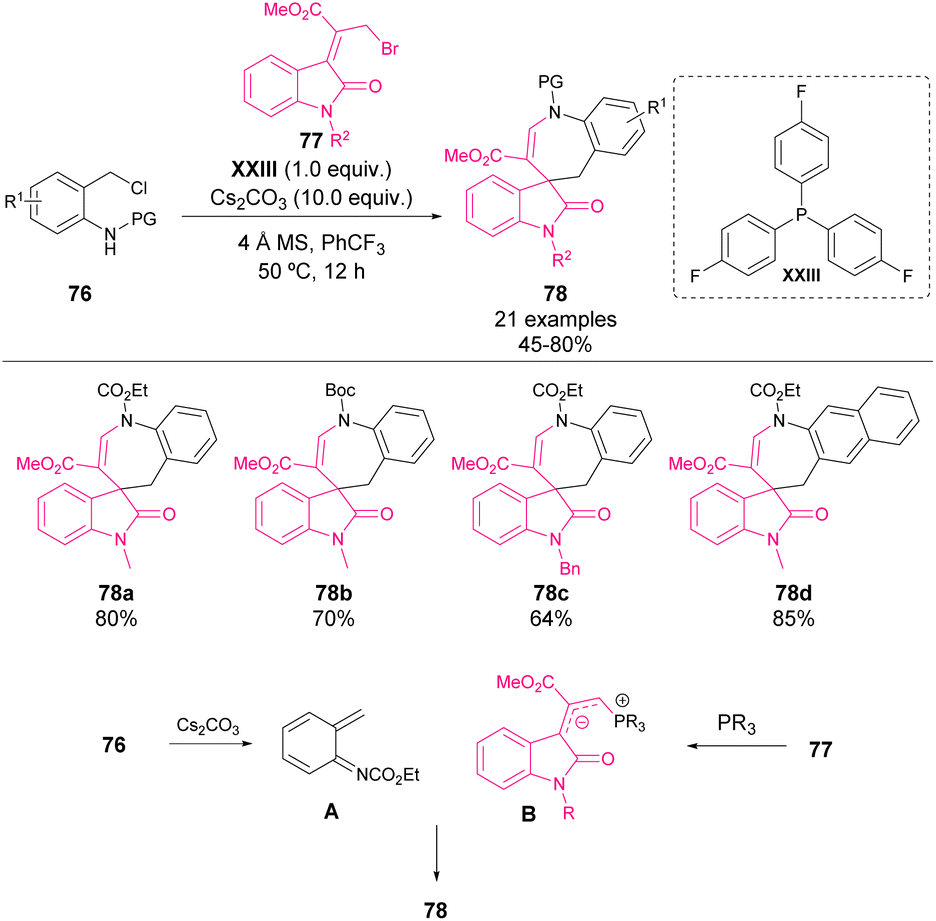 | ||
| Scheme 27 [4 + 3] Cycloadditions with bromo-substituted Morita–Baylis–Hillman adducts of isatins and N-(ortho-chloromethyl)aryl amides. | ||
4.2. Non-covalent phosphine activation
Recently, in 2022, Li's group reported the first example of phosphine-catalyzed reaction of isocyanates with aza-o-QM.74 Inspired by the chemistry of benzoxazinanones and following their research program on phosphine catalysis with Morita–Baylis–Hillman (MBH) adducts, they developed a new type of MBH adducts bearing a benzoxazine motif 79 that could serve for the in situ generation of polarized aza-o-QM by phosphine catalysis. This methodology enables the synthesis of chiral dihydroquinazolinones 81 in good yields with good to excellent enantioselectivities (Scheme 28). The proposed mechanism based on 31P-NMR analysis is depicted in Scheme 28: the phosphine catalyst is added to the vinyl moiety, generating the polarized aza-o-QM A. Then, the negatively charged nitrogen atom of aza-o-QM attacks isocyanate generating intermediate B, which upon intramolecular nucleophilic addition of the N atom of isocyanate leads to intermediate C, and finally releases the phosphine catalyst and yields dihydroquinazolinone 81. | ||
| Scheme 28 Asymmetric phosphine-catalysed reaction of isocyanates with aza-o-QM intermediates. | ||
5. Non-catalytic transformations with quinone methides
Aiming to give a more general overview of the field, different examples involving a stoichiometric use of phosphines will be summarized in this section.In 2018, Waser and coworkers reported a phosphine-mediated addition of allene-derived nucleophiles to o-QM 82 (Scheme 29).75 Following Lu's work68 concerning formal [4 + 2] reactions with stabilised o-QM, they aimed to mimic the reactivity with in situ formed o-QM. However, unlike in the previous precedents, dihydrobenzofurans 84 were observed in this case. The reaction involves an initial base-mediated elimination to afford the o-QM intermediate as the electrophile, while phosphine forms the nucleophile through a nucleophilic addition to the allene 83. After the initial addition (A), an intramolecular oxa-Michael addition (B) leads to the final product 84 as a single isomer in yields ranging from 28 to 90%.
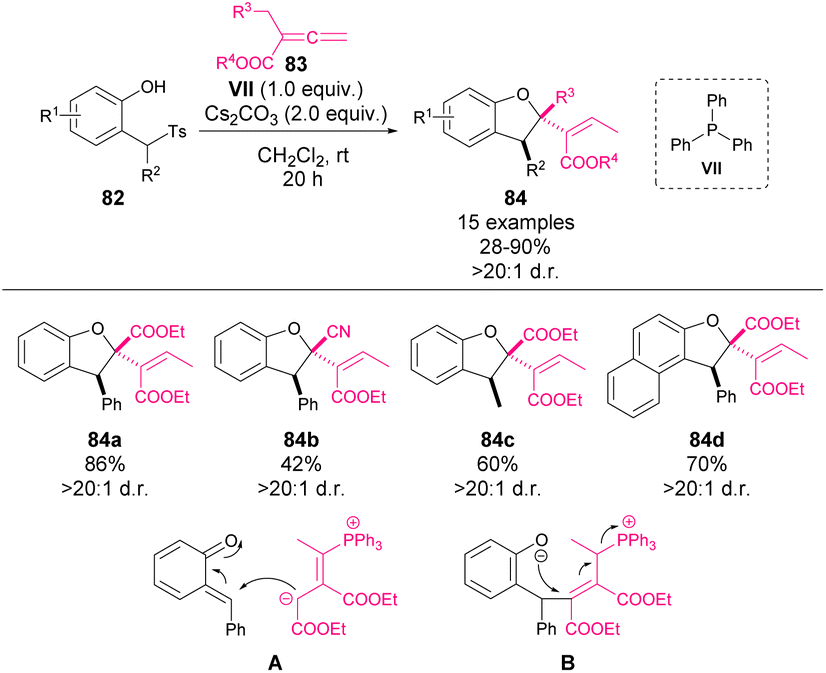 | ||
| Scheme 29 Phosphine-mediated synthesis of chiral dihydrobenzofuran derivatives. | ||
In 2019, Lin and coworkers reported a methodology for accessing 2,3-disubstituted benzofurans through a phosphine-catalysed reaction of acyl chlorides 86 and p-QM 85 (Scheme 30).76 Although the reaction mechanism is not fully elucidated, it may involve an initial protection of the phenol with the acyl chloride. Then, a 1,6-addition of phosphine to p-QM forms the phosphonium salt intermediate for an intramolecular Wittig reaction. However, the initially developed conditions required a stoichiometric amount of phosphine, which is oxidized to phosphine oxide during the Wittig reaction. For this reason, the authors developed an alternative method for regenerating in situ the phosphine catalyst. Both methods allowed the formation of benzofuran products 87 in high yields.
 | ||
| Scheme 30 Phosphine-catalysed synthesis of benzofurans 87. | ||
In 2021, Xiong et al. reported the addition of sodium sulfinates 89 to p-QM 88 mediated by a combination of sulfuric acid and triphenylphosphine VII (Scheme 31).77 Although the mechanism is not clear, phosphine is involved as the reductant of sulfur oxides. One of the tentative reaction pathways involves the reduction of sulfinates 89 to form sulfanyl radicals as nucleophiles for the 1,6-addition. Another possible route is the partial reduction of sulfinates, leading to sulfide oxides as the initial reaction products, which are reduced to their corresponding thioethers 90 by phosphine. The authors reported a broad scope of nucleophiles and electrophiles, which are highly selective towards p-QM in the presence of other reactive groups such as aldehydes or alcohols.
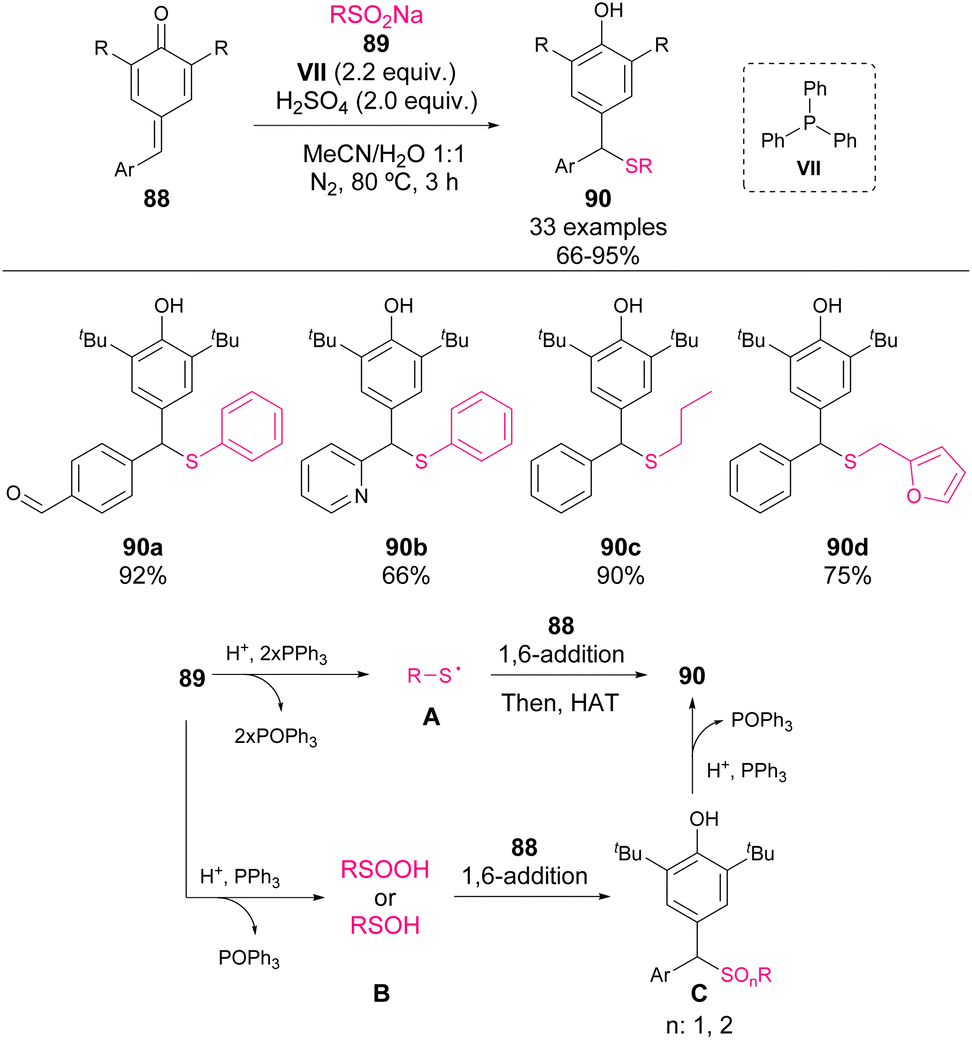 | ||
| Scheme 31 Phosphine-mediated thiolation of p-QM. | ||
In 2022, Singh and coworkers reported the synthesis of 2-quinolone derivatives 93 from isatins 92 and p-QM 91 (Scheme 32).78 The activation of isatin as indole derivatives by XXVI allows an initial 1,6-addition to p-QM (A). The overall reaction is a two-step reaction involving an initial 1,2-phospha-Brook rearrangement. Once the cyclopropane intermediate C is formed, a stoichiometric amount of BF3·Et2O is added as a Lewis acid (D), activating the cyclopropane ring opening and leading to the 6-membered N-heterocycle 93 in moderate to high yields (30–92%).
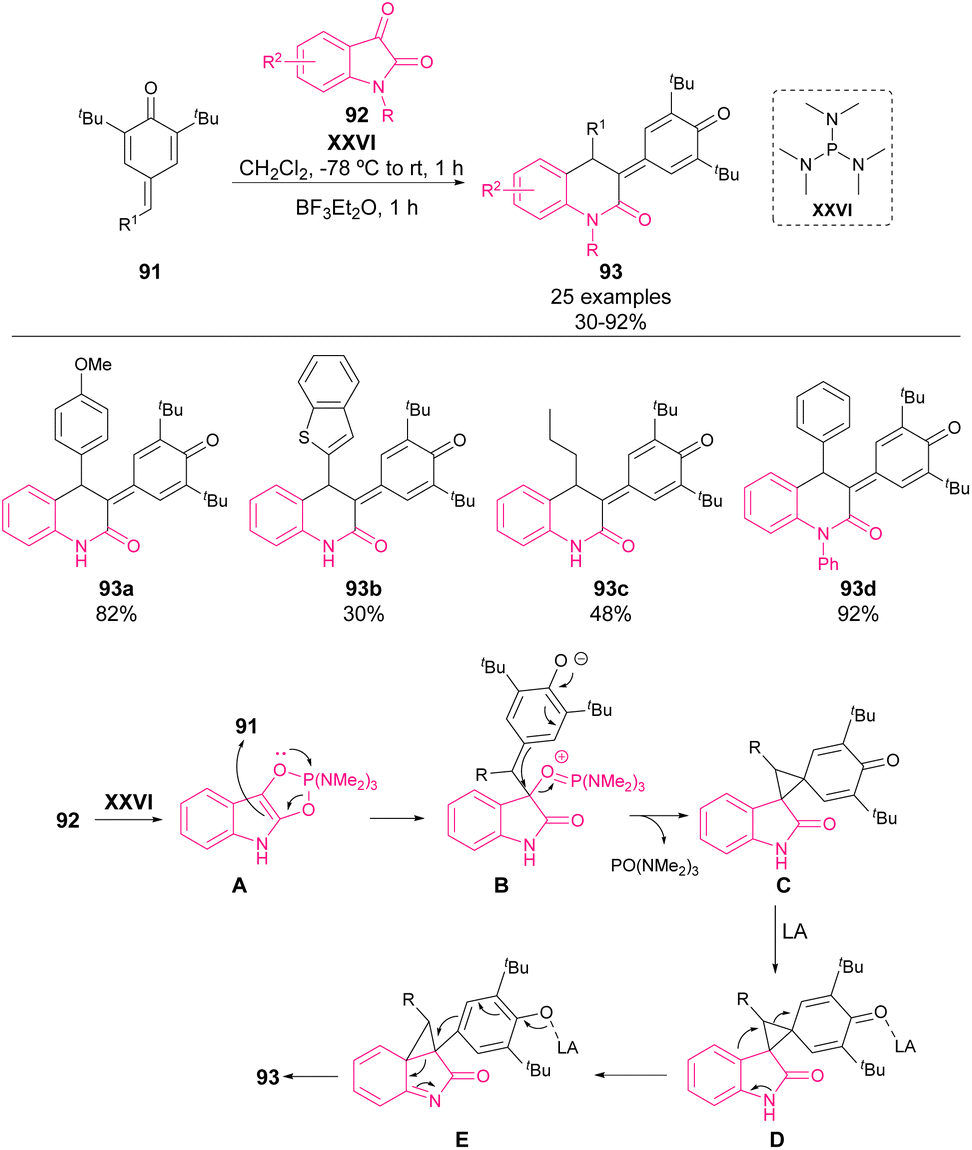 | ||
| Scheme 32 Phosphine/Lewis acid-mediated one-pot construction of 2-quinolones 93. | ||
6. Conclusions
In this review, an analysis of the recent developments in phosphine-catalysed transformations of o-QM, p-QM and aza-o-QM has been carried out. It should be noted that most of the reported examples are related to p-QM, and the analogous reactions with o-QM or aza-QM are still almost unexplored.Among the nucleophiles involved in the covalent phosphine-catalyzed reactions, we can find activated alkenes, allenes, and MBH carbonates, which intervene in cycloaddition reactions or Rauhut–Currier reactions with o-QM or p-QM providing a wide variety of addition products such as chiral diaryl methine stereocenters and cyclization derivatives, such as chiral dihydrobenzofurans and chromanes. On the other hand, reactions involving non-covalent interactions and phosphonium salts have also been disclosed. This type of phosphonium salt catalyst stabilizes the corresponding nucleophiles. In this case, anionic nucleophiles are typically used, in contrast to allenoates and MBH carbonates. Among the involved nucleophiles, alkoxy or phosphoryl groups have been employed.
Among the phosphine catalysts involved in the asymmetric reactions with o-QM and p-QM, we can find bifunctional phosphine catalysts that possess a hydrogen donor such as a thiourea or peptidic residue enabling the activation of quinone methide and at the same time the nucleophile, which provides a preferred transition state ensuring high enantioinduction in the reactions.
Although phosphine catalysis has a broad scope, the reactions involving asymmetric MBH or RC-type processes are quite scarce. Moreover, the enantioselective phosphine-catalysed reactions of quinone methide intermediates involving allenes, alkynes or Michael acceptors are rather limited. Therefore, we expect further developments in enantioselective phosphine-catalysed reactions to o-QM and p-QM in the following years. In particular, the development of novel methodologies involving o-QM, o-hydroxy p-QM and aza-o-QM would be of great interest as an alternative to the actual synthetic methodologies for the construction of optically active chromane and tetrahydroquinoline derivatives. Furthermore, we expect novel phosphine-catalysed cyclization strategies of quinone methide species to appear soon in the literature, which would be useful approaches for the synthesis of novel oxa-heterocycles and N-heterocycles.
Author contributions
All authors contributed equally to this work. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M. acknowledges the funding from the Department of Education of the Basque Government (Postdoctoral Program). M. Z. acknowledges the funding from the Talent Attraction Modality 2 Program from Comunidad de Madrid (2020-T2/BMD-20391). Open Access funding provided by University of Basque Country.Notes and references
- H.-U. Wagner and R. Gompper, in The Chemistry of the Quinonoid Compounds, ed. S. Patai, Wiley-VCH, New York, 1974, pp. 1145–1178 Search PubMed.
- M. Freccero, Mini-Rev. Org. Chem., 2004, 1, 403–415 CrossRef CAS.
- R. W. Van de Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367–5405 CrossRef CAS.
- M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924–55959 RSC.
- N. J. Willis and C. D. Bray, Chem. – Eur. J., 2012, 18, 9160–9173 CrossRef CAS PubMed.
- W. Bai, J. G. David, Z. Feng, M. G. Weaver, K. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655–3664 CrossRef CAS PubMed.
- L. Caruana, M. Fochi and L. Bernardi, Molecules, 2015, 20, 11733–11764 CrossRef CAS PubMed.
- Y. H. Ma, X. Y. He, Q. Q. Yang, A. Boucherif and J. Xuan, Asian J. Org. Chem., 2021, 10, 1233–1250 CrossRef CAS.
- A. Parra and M. Tortosa, ChemCatChem, 2015, 7, 1524–1526 CrossRef CAS.
- W. Li, X. Xu, P. Zhang and P. Li, Chem. – Asian J., 2018, 13, 2350–2359 CrossRef CAS PubMed.
- J. Y. Wang, W. J. Hao, S. J. Tu and B. Jiang, Org. Chem. Front., 2020, 7, 1743–1778 RSC.
- H. H. Liao, S. Miñoza, S. C. Lee and M. Rueping, Chem. – Eur. J., 2022, 28, e202201112 CrossRef CAS PubMed.
- M. Zurro and A. Maestro, ChemCatChem, 2023, e202300500 CrossRef CAS.
- Y. F. Wong, Z. Wang and J. Sun, Org. Biomol. Chem., 2016, 14, 5751–5754 RSC.
- X. Q. Li, H. Yang, J. J. Wang, B. B. Gou, J. Chen and L. Zhou, Chem. – Eur. J., 2017, 23, 5381–5385 CrossRef CAS PubMed.
- M. Chen and J. Sun, Angew. Chem., Int. Ed., 2017, 56, 11966–11970 CrossRef CAS PubMed.
- N. Dong, Z. P. Zhang, X. S. Xue, X. Li and J. P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1460–1464 CrossRef CAS PubMed.
- X. Y. Yu, J. R. Chen, Q. Wei, H. G. Cheng, Z. C. Liu and W. J. Xiao, Chem. – Eur. J., 2016, 22, 6774–6778 CrossRef CAS PubMed.
- L. Zhang, Y. Liu, K. Liu, Z. Liu, N. He and W. Li, Org. Biomol. Chem., 2017, 15, 8743–8747 RSC.
- Z. P. Zhang, K. X. Xie, C. Yang, M. Li and X. Li, J. Org. Chem., 2018, 83, 364–373 CrossRef CAS PubMed.
- T. Z. Li, C. A. Geng, X. J. Yin, T. H. Yang, X. L. Chen, X. Y. Huang, Y. B. Ma, X. M. Zhang and J. J. Chen, Org. Lett., 2017, 19, 429–431 CrossRef CAS PubMed.
- Q. Wu, J. Zhao, S. Sun, M. Tu and F. Shi, Acta Chim. Sin., 2016, 74, 576–581 CrossRef CAS.
- C. C. Hsiao, S. Raja, H. H. Liao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 5762–5765 CrossRef CAS PubMed.
- D. Qian, L. Wu, Z. Lin and J. Sun, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- W. D. Chu, L. F. Zhang, X. Bao, X. H. Zhao, C. Zeng, J. Y. Du, G. B. Zhang, F. X. Wang, X. Y. Ma and C. A. Fan, Angew. Chem., Int. Ed., 2013, 52, 9229–9233 CrossRef CAS PubMed.
- X. Z. Zhang, Y. H. Deng, X. Yan, K. Y. Yu, F. X. Wang, X. Y. Ma and C. A. Fan, J. Org. Chem., 2016, 81, 5655–5662 CrossRef CAS PubMed.
- X. L. Lian, A. Adili, B. Liu, Z. L. Tao and Z. Y. Han, Org. Biomol. Chem., 2017, 15, 3670–3673 RSC.
- W. Li, H. Yuan, Z. Liu, Z. Zhang, Y. Cheng and P. Li, Adv. Synth. Catal., 2018, 360, 2460–2464 CrossRef CAS.
- X. Chen, R. Song, Y. Liu, C. Y. Ooi, Z. Jin, T. Zhu, H. Wang, L. Hao and Y. R. Chi, Org. Lett., 2017, 19, 5892–5895 CrossRef CAS PubMed.
- K. Zhao, Y. Zhi, T. Shu, A. Valkonen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 12104–12108 CrossRef CAS PubMed.
- K. Zhao, Y. Zhi, A. Wang and D. Enders, ACS Catal., 2016, 6, 657–660 CrossRef CAS.
- Y. H. Deng, X. Z. Zhang, K. Y. Yu, X. Yan, J. Y. Du, H. Huang and C. A. Fan, Chem. Commun., 2016, 52, 4183–4186 RSC.
- L. Zhang, X. Zhou, P. Li, Z. Liu, Y. Liu, Y. Sun and W. Li, RSC Adv., 2017, 7, 39216–39220 RSC.
- X. Liu, K. Wang, Y. Liu and C. Li, Chem. – Eur. J., 2021, 27, 735–739 CrossRef CAS PubMed.
- B. Wu, Z. Yu, X. Gao, Y. Lan and Y. G. Zhou, Angew. Chem., Int. Ed., 2017, 56, 4006–4010 CrossRef CAS PubMed.
- J. Zhou, M. L. Wang, X. Gao, G. F. Jiang and Y. G. Zhou, Chem. Commun., 2017, 53, 3531–3534 RSC.
- Y. I. N. Wei and M. I. N. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed.
- S. X. Wang, X. Han, F. Zhong, Y. Wang and Y. Lu, Synlett, 2011, 2766–2778 CAS.
- T. C. Kang, L. P. Wu, Q. W. Yu and X. Y. Wu, Chem. – Eur. J., 2017, 23, 6509–6513 CrossRef CAS PubMed.
- Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588–11619 RSC.
- L. W. Xu, ChemCatChem, 2013, 5, 2775–2784 CrossRef CAS.
- Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927–2940 RSC.
- Y. Wei and M. Shi, Chem. – Asian J., 2014, 9, 2720–2734 CrossRef CAS PubMed.
- Y. Xiao, Z. Sun, H. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089–2121 CrossRef PubMed.
- W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657–1677 RSC.
- Y. Wei and M. Shi, Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, Wiley-VCH, Weinheim, Germany, 2013, vol. 3 Search PubMed.
- Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed.
- V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS PubMed.
- D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed.
- Q. Y. Zhao, C. K. Pei, X. Y. Guan and M. Shi, Adv. Synth. Catal., 2011, 353, 1973–1979 CrossRef CAS.
- S. Li, Y. Liu, B. Huang, T. Zhou, H. Tao, Y. Xiao, L. Liu and J. Zhang, ACS Catal., 2017, 7, 2805–2809 CrossRef CAS.
- X. Z. Zhang, K. J. Gan, X. X. Liu, Y. H. Deng, F. X. Wang, K. Y. Yu, J. Zhang and C. A. Fan, Org. Lett., 2017, 19, 3207–3210 CrossRef CAS PubMed.
- H. Wang, K. Wang, Y. Man, X. Gao, L. Yang, Y. Ren, N. Li, B. Tang and G. Zhao, Adv. Synth. Catal., 2017, 359, 3934–3939 CrossRef CAS.
- K. Zielke, O. Kováč, M. Winter, J. Pospíšil and M. Waser, Chem. – Eur. J., 2019, 25, 8163–8168 CrossRef CAS PubMed.
- F. R. Yuan, F. Jiang, K. W. Chen, G. J. Mei, Q. Wu and F. Shi, Org. Biomol. Chem., 2019, 17, 2361–2369 RSC.
- Y. Zhu, D. Wang and Y. Huang, Org. Lett., 2019, 21, 908–912 CrossRef CAS PubMed.
- Z. Song, W. Wang, Z. Liu, Y. Lu and D. Wang, J. Org. Chem., 2021, 86, 8590–8599 CrossRef CAS PubMed.
- Y. Lu, N. He, X. Miao and D. Wang, Org. Chem. Front., 2022, 9, 4840–4845 RSC.
- A. Buchcic-Szychowska, S. Leśniak and M. Rachwalski, Symmetry, 2022, 14, 4–11 CrossRef.
- X. Li, X. Xu, W. Wei, A. Lin and H. Yao, Org. Lett., 2016, 18, 428–431 CrossRef CAS PubMed.
- T. Zhou, S. Li, B. Huang, C. Li, Y. Zhao, J. Chen, A. Chen, Y. Xiao, L. Liu and J. Zhang, Org. Biomol. Chem., 2017, 15, 4941–4945 RSC.
- T. Zhou, S. Li, B. Huang, C. Li, Y. Zhao, J. Chen, A. Chen, Y. Xiao, L. Liu and J. Zhang, Org. Biomol. Chem., 2017, 15, 5097 RSC.
- L. Ge, X. Lu, C. Cheng, J. Chen, W. Cao, X. Wu and G. Zhao, J. Org. Chem., 2016, 81, 9315–9325 CrossRef CAS PubMed.
- J. P. Tan, P. Yu, J. H. Wu, Y. Chen, J. Pan, C. Jiang, X. Ren, H. S. Zhang and T. Wang, Org. Lett., 2019, 21, 7298–7302 CrossRef CAS PubMed.
- Y. Chen, Z. Yu, Z. Jiang, J. P. Tan, J. H. Wu, Y. Lan, X. Ren and T. Wang, ACS Catal., 2021, 11, 14168–14180 CrossRef CAS.
- J. P. Tan, Y. Chen, X. Ren, Y. Guo, B. Yi, H. Zhang, G. Gao and T. Wang, Org. Chem. Front., 2022, 9, 156–162 RSC.
- S. Kumar, N. K. Vaishanv and K. Mohanan, Asian J. Org. Chem., 2022, 11, 3–7 Search PubMed.
- Z. Wang, T. Wang, W. Yao and Y. Lu, Org. Lett., 2017, 19, 4126–4129 CrossRef CAS PubMed.
- F. Jiang, G. Z. Luo, Z. Q. Zhu, C. S. Wang, G. J. Mei and F. Shi, J. Org. Chem., 2018, 83, 10060–10069 CrossRef CAS PubMed.
- H. H. Zhang, C. S. Wang, C. Li, G. J. Mei, Y. Li and F. Shi, Angew. Chem., Int. Ed., 2017, 56, 116–121 CrossRef CAS PubMed.
- Y. Cheng, Z. Fang, W. Li and P. Li, Org. Chem. Front., 2018, 5, 2728–2733 RSC.
- T. Zhou, T. Xia, Z. Liu, L. Liu and J. Zhang, Adv. Synth. Catal., 2018, 360, 4475–4479 CrossRef CAS.
- G. Zhan, M. L. Shi, Q. He, W. Du and Y. C. Chen, Org. Lett., 2015, 17, 4750–4753 CrossRef CAS PubMed.
- X. Chen, T. Wang, Z. Lu and P. Li, Org. Lett., 2022, 24, 3102–3106 CrossRef CAS PubMed.
- K. Zielke and M. Waser, Org. Lett., 2018, 20, 768–771 CrossRef CAS PubMed.
- Y. C. Liou, P. Karanam, Y. J. Jang and W. Lin, Org. Lett., 2019, 21, 8008–8012 CrossRef CAS PubMed.
- B. Xiong, S. Xu, Y. Liu, K. W. Tang and W. Y. Wong, J. Org. Chem., 2021, 86, 1516–1527 CrossRef CAS PubMed.
- A. Ali, H. K. Harit, M. Devi, D. Ghosh and R. P. Singh, J. Org. Chem., 2022, 87, 16313–16327 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |