
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent α-amino acids via Heck–Matsuda reactions of phenylalanine-derived arenediazonium salts†
Rochelle
McGrory
,
Rebecca
Clarke
,
Olivia
Marshall
and
Andrew
Sutherland
 *
*
School of Chemistry, University of Glasgow, The Joseph Black Building, Glasgow, G12 8QQ, UK. E-mail: Andrew.Sutherland@glasgow.ac.uk
First published on 10th August 2023
Abstract
The Heck–Matsuda coupling reaction of arenediazonium salts derived from L-phenylalanine with various alkenes has been developed. A two-step process involving the preparation of a tetrafluoroborate diazonium salt from a 4-aminophenylalanine derivative, followed by a palladium(0)-catalysed Heck–Matsuda coupling reaction allowed access to a range of unnatural α-amino acids with cinnamate, vinylsulfone and stilbene side-chains. Analysis of the photophysical properties of these unnatural α-amino acids demonstrated that the (E)-stilbene analogues exhibited fluorescent properties with red-shifted absorption and emission spectra and larger quantum yields than L-phenylalanine.
Introduction
Unnatural α-amino acids are involved in a wide range of applications in medicine and across the life and physical sciences.1 As well as components of pharmaceutically active natural products and drugs, they are widely used as probes to study enzyme mechanism and function.2 There have also been significant recent efforts to develop fluorescent α-amino acids as imaging tools for chemical biology applications.3 In synthesis, unnatural α-amino acids are commonly used as catalysts, ligands and auxiliaries, as well as chiral building blocks for total synthesis.4Access to unnatural α-amino acids is often achieved by side-chain modification of proteinogenic analogues. Due to well-established aromatic chemistry, structural analogues of α-amino acids bearing arene side-chains are easily accessible.5 For example, L-phenylalanine-derived arenediazonium salts 1, which are readily prepared from 4-aminophenylalanine have been used for a variety of side-chain functionalisation reactions (Fig. 1a).6 These include the use of Baltz-Schiemann or Sandmeyer reactions for the preparation of halogenated derivatives such as 4-chlorophenylalanine 2.7 Arenediazonium salts of phenylalanine have been used to attach the amino acid to a solid support via a triazene linkage (e.g.3), for the synthesis of cyclic peptides.8 These intermediates have also been used for the synthesis of tetrazole analogues (4), which are inhibitors of DNA methyltransferase 1,9 while perfluoro-tert-butyl tyrosine 5, a probe for 19F NMR spectroscopy was prepared by a substitution reaction of an arenediazonium salt.10
 | ||
| Fig. 1 (a) Applications of phenylalanine-derived arenediazonium salts. (b) Synthesis of cinnamate-derived α-amino acids. (c) Synthesis of stilbene-derived α-amino acids. (d) Proposed work. | ||
We have a longstanding interest in the synthesis of unnatural α-amino acids and in particular, the development of new methodology for the preparation of fluorescent analogues.11 Recently, we reported the fluorescent properties of various benzotriazole-derived amino acids, that were prepared by the synthesis and cyclisation of tosylate arenediazonium salts.12 In these studies, the arenediazonium salts were prepared under mild conditions using a polymer-supported nitrite reagent and p-tosic acid. We were interested in the application of this type of methodology for the preparation of alkene-extended phenylalanine analogues as possible novel fluorophores. Phenylalanines with alkene side-chains have been prepared using various approaches. Cinnamate analogues are typically prepared by palladium(0)-catalysed Heck-type coupling of acrylates with halogen or triflate-substituted phenylalanines,13 while the Sengupta group described an example of methyl acrylate coupling with a phenylalanine-derived arenediazonium salt (Fig. 1b).14 Unnatural α-amino acids with stilbene side-chains have been prepared using a variety of approaches including the Wittig reaction,15 a Suzuki-Miyaura16 (Fig. 1c) or Negishi coupling reaction17 and, the iridium-catalysed reduction of alkyne analogues.18 Based on the diverse nature of these approaches, we were interested in the development of a single method that could be used for the synthesis of a variety of alkene-functionalised phenylalanine analogues. Herein, we report a two-step approach for the synthesis of unnatural α-amino acids with cinnamate, vinylsulfone and stilbene side-chains using the Heck–Matsuda coupling reaction of an arenediazonium salt intermediate (Fig. 1d). The photophysical properties of the (E)-stilbene analogues are also described, demonstrating stronger fluorescence and higher quantum yields than L-phenylalanine.
Results and discussion
The project began with a short synthesis of a suitably protected 4-aminophenylalanine derivative that could be used to explore diazotisation methods and subsequent Heck–Matsuda coupling reactions with alkenes.19 A two-step process was utilised for the protection of inexpensive and readily available L-3-nitrophenylalanine (6) (Scheme 1). The carboxylic acid was protected as methyl ester 7 in 88% yield using thionyl chloride and methanol, while the amino group was converted to benzyl carbamate 8 under standard conditions in 95% yield. Several methods were then examined for the chemoselective reduction of the nitro group. Although tin(II) chloride gave an 83% yield of L-4-aminophenylalanine 9, the transformation required a reaction time of 16 hours under reflux.9 Instead, a combination of zinc and acetic acid was found to be optimal and gave amine 9 in 94% yield, after a 4 hour reaction time under mild conditions. This efficient three-step route was found to be scalable, allowing the multigram synthesis of L-4-aminophenylalanine derivative 9. | ||
| Scheme 1 Three-step synthesis of L-4-aminophenylalanine derivative 9. | ||
Methods for diazonium salt formation of 4-aminophenylalanine 9 and subsequent Heck–Matsuda alkenylation were then explored. We previously reported the one-pot diazonium salt formation and Heck–Matsuda reaction of simple anilines using a polymer supported nitrite reagent and p-tosic acid for the diazotisation step.20 These mild conditions allowed fast (1.5 h) and effective olefination of a wide range of anilines. Application of this one-pot method using 4-aminophenylalanine 9, methyl acrylate and palladium acetate (10 mol%) gave methyl (E)-cinnamate 12a in 23% yield (Scheme 2). Although this procedure generated 12a cleanly via tosylate arenediazonium salt 10, a more efficient method was deemed necessary. The next attempt investigated synthesis of 12avia a more reactive tetrafluoroborate arenediazonium salt.21 Sengupta and Bhattacharyya demonstrated the one-pot olefination of an ethyl carbamate protected 4-aminophenylalanine derivative using tetrafluoroboric acid and sodium nitrite, followed by the addition of Pd(OAc)2 and methyl acrylate.14 Using a similar one-pot procedure with 4-aminophenylalanine 9, with initial formation of the tetrafluoroborate arenediazonium salt 11, followed by alkenylation with methyl acrylate and palladium acetate (1 mol%) gave cinnamate 12a in 19% yield. It was proposed that a more efficient overall synthesis of cinnamate 12a might be achieved by a two-pot process involving the preparation and isolation of tetrafluoroborate arenediazonium salt 11, followed by a separate Heck–Matsuda reaction. Treatment of 9 with tetrafluoroboric acid and sodium nitrite produced diazonium salt 11 as a red solid, which was isolated in 91% yield. Heck–Matsuda reaction of diazonium salt 11 with methyl acrylate and palladium acetate (1 mol%) at a reaction temperature of 50 °C and 0.5 h reaction time, gave cinnamate 12a in 71% yield (64% over the two steps).
 | ||
| Scheme 2 One- and two-pot synthesis of cinnamate 12a. | ||
The Heck–Matsuda reaction using tetrafluoroborate arenediazonium salt 11 was then examined for coupling with other alkenes (Scheme 3). The reactions were typically fast and showed completion after reaction times of 0.5–1 h. Only coupling with phenyl vinyl sulfone required a longer reaction time (5.5 h). The other variation required was catalyst loading. Cinnamate 12a was readily prepared in 71% yield using 1 mol% of Pd(OAc)2, while higher loadings of 5–10 mol% were required for the other alkenes. Overall, this allowed the synthesis of cinnamate 12a and vinylsulfone 12b, as well as a range of stilbenes (12c–12f) in yields of 44–80%. It should be noted that in all cases the products were isolated cleanly, with only the (E)-isomer observed by 1H NMR spectroscopy. The main limitation of this reaction was found with strongly electron-rich alkenes such as 4-methoxystyrene, which showed only trace conversion under standard conditions (50 °C, 1 h).
 | ||
Scheme 3 Scope of Heck–Matsuda reaction of arenediazonium salt 11 with various alkenes. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Using Pd(OAc)2 (1 mol%). bUsing Pd(OAc)2 (5 mol%). Using Pd(OAc)2 (1 mol%). bUsing Pd(OAc)2 (5 mol%). | ||
On synthesis of α-amino acids 12a–12f, the photophysical properties were measured. The UV/Visible absorption and photoluminescence spectra of the α-amino acids were recorded in methanol at a concentration of 2 or 5 μM. As expected, cinnamate 12a and vinylsulfone 12b showed weak fluorescence, due to the limited conjugation of the relatively small chromophores. In contrast, stilbene derived α-amino acids 12c–12f displayed interesting optical properties (Fig. 2 and Table 1).23 In comparison to parent amino acid, L-phenylalanine, the absorption spectra for 12c–12f (Fig. 2a) showed red-shifted bands with absorption maxima ranging from 295–317 nm and significantly larger molar extinction coefficients. Similarly, emission spectra (Fig. 2b) showed red-shifted bands from 349–357 nm and apart from 12f, larger quantum yields (5–7%). Amino acids 12c–12f also possessed good Stokes shifts, which is important for avoiding reabsorption. Overall, the bathochromic shift of absorption and emission bands for 12c–12f means these have the potential to be used as fluorophores in peptides and proteins without interference from fluorescent native α-amino acids (L-Phe, L-Tyr and L-Trp).
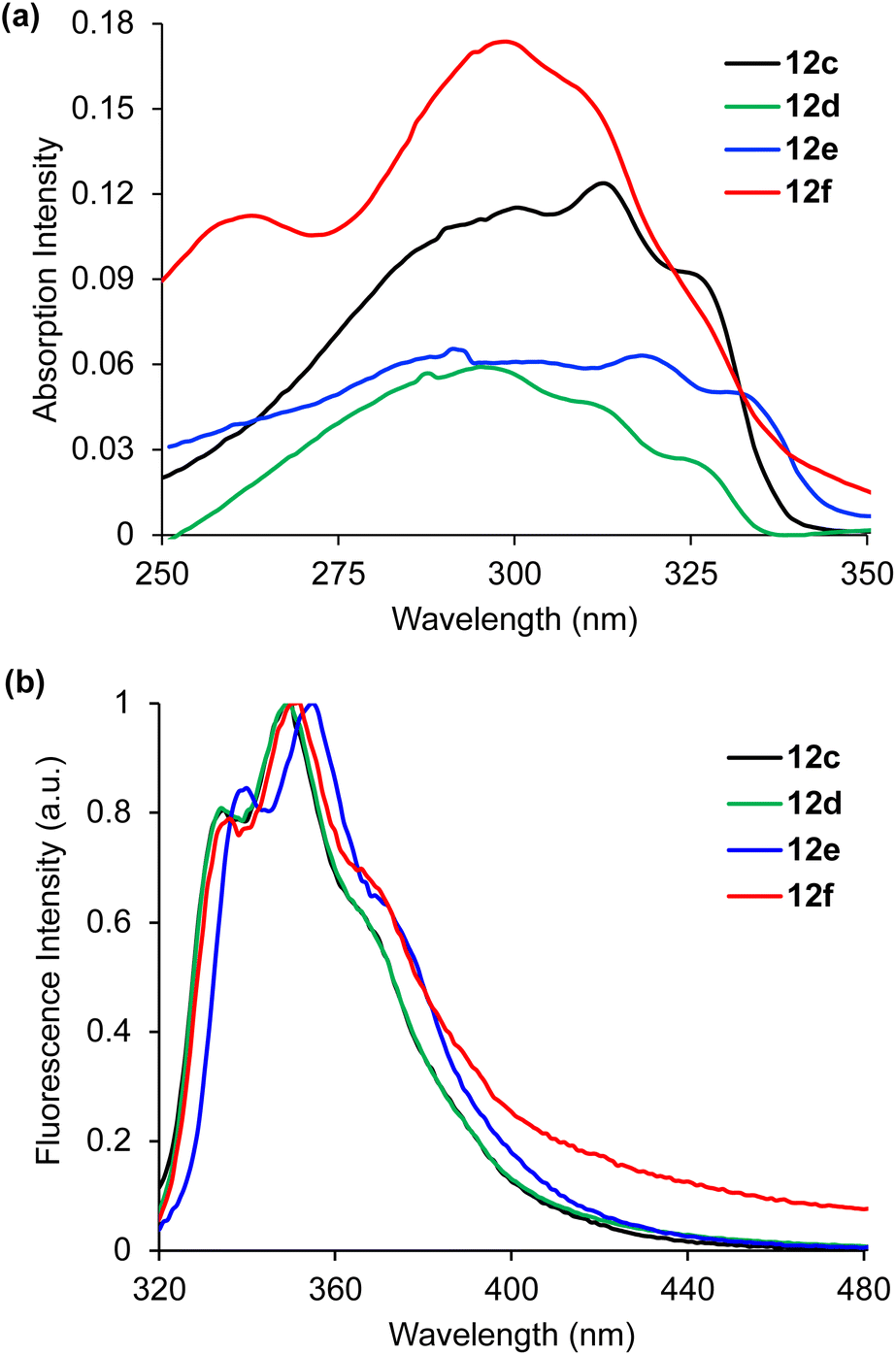 | ||
| Fig. 2 (a) Absorption spectra of amino acids 12c–12f recorded at 2 or 5 μM in MeOH. (b) Emission spectra of 12c–12f recorded at 2 or 5 μM in MeOH. | ||
a
| Amino acid | λ Abs (nm) | ε (cm−1 M−1) | λ Em (nm) | Stokes shift (cm−1) |
Φ
Fb |
|---|---|---|---|---|---|
| a All spectra were recorded at 2 or 5 μM in MeOH. b Quantum yields (ΦF) were determined in MeOH using anthracene and L-tryptophan as standards. | |||||
| L-Phe22 | 258 | 200 | 282 | 3299 | 0.024 |
| 12c | 314 | 20600 |
349 | 3194 | 0.05 |
| 12d | 295 | 20700 |
352 | 5489 | 0.07 |
| 12e | 317 | 22500 |
357 | 3535 | 0.06 |
| 12f | 300 | 24300 |
351 | 4843 | 0.01 |
The final stage of this project focused on demonstrating that the α-amino acids could be deprotected in the presence of the alkene moiety. It was also important to show that the parent α-amino acids retained the photophysical properties of the protected derivatives. Hence, as a proof-of-concept, stilbene α-amino acid 12c was selected (Scheme 4). Ester hydrolysis using caesium carbonate under mild conditions gave carboxylic acid 13 in 94% yield. The benzyl carbamate protecting group was then removed under acidic conditions and following recrystallisation, this gave stilbene α-amino acid 14 in 82% yield. Analysis of the photophysical data confirmed similar absorption and emission spectra, as well as quantum yield to that of 12c.23
 | ||
| Scheme 4 Two-step deprotection to stilbene-derived α-amino acid 14. | ||
Conclusions
In summary, a two-step process for the synthesis of alkene-extended analogues of L-phenylalanine has been developed using a Heck–Matsuda reaction. A readily accessible L-4-aminophenylalanine derivative was converted to a tetrafluoroborate arenediazonium salt and subsequent coupling with a range of alkenes allowed access to cinnamate, vinylsulfone and stilbene products. Analysis of the photophysical properties of these unnatural α-amino acids demonstrated that the stilbene analogues were fluorescent, exhibiting red-shifted absorption and emission, and for all but one example, higher quantum yields than the parent amino acid, L-phenylalanine. The bathochromic shift of the photophysical properties to wavelengths outwith that of fluorescent proteinogenic α-amino acids is significant and suggests application of these systems as potential peptidic probes. Current work is focused on investigating such applications.Experimental
All reagents and starting materials were obtained from commercial sources and used as received. All dry solvents were purified using a PureSolv 500 MD solvent purification system. Brine refers to a saturated solution of sodium chloride. Flash column chromatography was performed using Merck Millipore matrix silicagel 60 (40–63 μM). Merck aluminium-backed plates pre-coated with silica gel 60F254 were used for thin layer chromatography and were visualised with a UV lamp or by staining with KMnO4 or ninhydrin. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX 400 spectrometer or a Bruker DPX 500 spectrometer and data are reported as follows: chemical shift in ppm relative to tetramethylsilane (δH 0.00 and δC 0.00), or for 1H NMR, relative to residual chloroform (δH 7.26) or methanol (δH 3.31) as standard. For 13C NMR the chemical shifts are reported relative to the central resonance of CDCl3 (δC 77.2) or CD3OD (δC 49.0) as standard. Assignments are based on two-dimensional COSY, HSQC, HMBC and DEPT experiments. Infrared spectra were recorded on a Shimadzu IR Prestige-21 spectrometer or a Shimadzu FTIR-84005 spectrometer; wavenumbers are indicated in cm−1. Mass spectra were obtained either using a JEOL JMS-700 spectrometer for EI and CI, and Bruker Microtof-q or Agilent 6125B for ESI. Melting points were determined on a Reichert platform melting point apparatus or Stuart Scientific melting point apparatus. Optical rotations were determined as solutions irradiating with the sodium D line (λ = 589 nm) using an Autopol V polarimeter. [α]D values are given in units 10−1 deg cm−1 g−1. UV-Vis and fluorescence spectra were recorded on a fluorescence and absorbance spectrometer. Absorbance spectra were recorded with an integration time of 0.05 s and a band pass of 5 nm. Fluorescence spectra were recorded with excitation and emission band pass of 5 nm, an integration time of 2 s, and with detector accumulations set to 1. Quantum yields were determined using a comparative method against two standards.24 Anthracene (Φ = 0.27, in ethanol) and L-tryptophan (Φ = 0.14 in water) were used as standard references. The integrated fluorescence intensity of each compound was determined from the emission spectra given. Measurements were performed at a minimum of four different concentrations. Concentrations were chosen to ensure the absorption value was below 0.1 to avoid re-absorption effects. Integrated fluorescence intensity was plotted as a function of the measured absorbance and a linear fit was calculated. The resultant gradient was then used to calculate the quantum yield.Methyl (2S)-2-amino-3-(4′-nitrophenyl)propanoate (7)25
To a stirred solution of 4-nitro-L-phenylalanine (6) (7.00 g, 33.3 mmol) in methanol (140 mL) at 0 °C was added dropwise thionyl chloride (3.40 mL, 46.6 mmol). The reaction mixture was warmed to room temperature and then heated under reflux for 3 h. After cooling to room temperature, the reaction mixture was concentrated in vacuo. The reaction mixture was diluted in water (90 mL), basified to pH 8 using sodium bicarbonate and extracted with dichloromethane (3 × 90 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo to give methyl (2S)-2-amino-3-(4′-nitrophenyl)propanoate (7) (6.61 g, 88%) as a yellow oil. [α]20D +26.7 (c 0.1, EtOH), lit.25 [α]24D +34.2 (c 1.0, EtOH); δH (400 MHz, DMSO-d6) 2.91 (1H, dd, J 13.4, 7.8 Hz, 3-HH), 3.03 (1H dd, J 13.4, 5.9 Hz, 3-HH), 3.60 (3H, s, OCH3), 3.67 (1H, dd, J 7.8, 5.9 Hz, 2-H), 7.49 (2H, d, J 8.7 Hz, 2′-H and 6′-H), 8.14 (2H, d, J 8.7 Hz, 3′-H and 5′-H); δC (101 MHz, DMSO-d6) 39.9 (CH2), 51.6 (CH3), 55.2 (CH), 123.1 (2 × CH), 130.6 (2 × CH), 146.2 (C), 146.5 (C), 174.8 (C); m/z (ESI) 247 (MNa+. 100%).Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-nitrophenyl)propanoate (8)9
To a stirred solution of methyl (2S)-2-amino-3-(4′-nitrophenyl)propanoate (7) (4.19 g, 18.9 mmol) in water (50 mL) at 0 °C was added sodium bicarbonate (3.92 g, 46.7 mmol). A solution of benzyl chloroformate (3.16 mL, 22.4 mmol) in toluene (10 mL) was then added dropwise. The reaction mixture was warmed to room temperature and stirred for 18 h. The reaction mixture was diluted in water (100 mL) and extracted with ethyl acetate (3 × 100 mL). The organic layers were combined and washed with 1 M aqueous hydrochloric acid (200 mL), sodium bicarbonate (200 mL), water (200 mL) and brine (200 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 30% ethyl acetate in hexane gave methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-nitrophenyl)propanoate (8) (6.34 g, 95%) as an off-white solid. Spectroscopic data were consistent with the literature.9 Mp 58–59 °C; [α]23D +49.6 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.14 (1H, dd, J 13.8, 6.3 Hz, 3-HH), 3.30 (1H, dd, J 13.8, 5.7 Hz, 3-HH), 3.74 (3H, s, OCH3), 4.66–4.75 (1H, m, 2-H), 5.06 (1H, d, J 12.1 Hz, CHHPh), 5.12 (1H, d, J 12.1 Hz, CHHPh), 5.29 (1H, d, J 8.1 Hz, 2-NH), 7.20–7.40 (7H, m, 2′-H, 6′-H and Ph), 8.11 (2H, d, J 8.5 Hz, 3′-H and 5′-H); δC (101 MHz, CDCl3) 38.4 (CH2), 52.8 (CH3), 54.6 (CH), 67.3 (CH2), 123.9 (2 × CH), 128.4 (2 × CH), 128.5 (CH), 128.7 (2 × CH), 130.3 (2 × CH), 136.1 (C), 143.8 (C), 147.3 (C), 155.6 (C), 171.4 (C); m/z (ESI) 381 (MNa+. 100%).Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-aminophenyl)propanoate (9)9
To a stirred solution of methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-nitrophenyl)propanoate (8) (9.13 g, 25.5 mmol) in methanol (160 mL) was added zinc powder (16.7 g, 255 mmol) and acetic acid (14.6 mL, 255 mmol). The reaction mixture was stirred at room temperature for 4 h. The reaction mixture was filtered through a pad of Celite®, washed with methanol (100 mL) and concentrated in vacuo. The reaction mixture was diluted in ethyl acetate (300 mL) and was washed with water (5 × 250 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 40% ethyl acetate in hexane gave methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-aminophenyl)propanoate (9) (7.62 g, 94%) as a colourless oil. Spectroscopic data were consistent with the literature.9 [α]23D +48.8 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 2.97–3.02 (2H, m, 3-H2), 3.61 (2H, br s, 4′-NH2), 3.72 (3H, s, OCH3), 4.55–4.63 (1H, m, 2-H), 5.08 (d, 1H, J 12.7 Hz, OCHHPh), 5.11 (d, 1H, J 12.7 Hz, OCHHPh), 5.18 (1H, d, J 8.0 Hz, 2-NH), 6.57–6.62 (2H, m, 3′-H and 5′-H), 6.84–6.89 (2H, m, 2′-H and 6′-H), 7.28–7.40 (5H, m, Ph); δC (101 MHz, CDCl3) 37.5 (CH2), 52.4 (CH3), 55.1 (CH), 67.1 (CH2), 115.5 (2 × CH), 125.5 (C), 128.2 (2 × CH), 128.3 (CH), 128.7 (2 × CH), 130.3 (2 × CH), 136.5 (C), 145.6 (C), 155.8 (C), 172.3 (C); m/z (ESI) 351 (MNa+. 100%).Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11)
To a stirred solution of methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-aminophenyl)propanoate (9) (0.583 g, 1.78 mmol) in 48% aqueous fluoroboroic acid (0.902 mL) and water (0.950 mL) at 0 °C was added a solution of sodium nitrite (0.174 g, 2.49 mmol) in water (0.365 mL). The reaction mixture was stirred at 0 °C for 0.5 h which resulted in formation of a red precipitate. This was filtered and washed with cold water (5 mL). Purification by recrystallisation from acetone and diethyl ether gave methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11) (0.690 g, 91%) as a red solid. This was used immediately for subsequent reactions.Methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[(4′-methylcinnamate)phenyl]propanoate (12a)
Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11) (0.31 g, 0.73 mmol) was dissolved in methanol (1.5 mL). To this was added methyl acrylate (0.091 mL, 1.5 mmol) and palladium acetate (0.0020 g, 0.0089 mmol, 1 mol%). The reaction mixture was heated to 50 °C and stirred for 0.5 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by flash column chromatography, eluting with 40% ethyl acetate in hexane gave methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[(4′-methylcinnamate)phenyl]propanoate (12a) (0.21 g, 71%) as a colourless oil. νmax/cm−1 (neat) 3352 (NH), 2951 (CH), 1697 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1528, 1435, 1254, 1207, 1169; [α]19D +60.1 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.08 (1H, dd, J 13.9, 6.2 Hz, 3-HH), 3.18 (1H, dd, J 13.9, 5.6 Hz, 3-HH), 3.72 (3H, s, OCH3), 3.80 (3H, s, OCH3), 4.63–4.71 (1H, m, 2-H), 5.07 (d, 1H, J 12.4 Hz, OCHHPh), 5.11 (d, 1H, J 12.4 Hz, OCHHPh), 5.27 (1H, d, J 8.0 Hz, 2-NH), 6.40 (1H, d, J 16.0 Hz, 2′′-H), 7.11 (2H, d, J 8.1 Hz, 2′-H and 6′-H), 7.28–7.39 (5H, m, Ph), 7.42 (2H, d, J 8.1 Hz, 3′-H and 5′-H), 7.65 (1H, d, J 16.0 Hz, 1′′-H); δC (101 MHz, CDCl3) 38.2 (CH2), 51.8 (CH3), 52.6 (CH3), 54.8 (CH), 67.2 (CH2), 117.8 (CH), 128.2 (2 × CH), 128.37 (CH), 128.40 (2 × CH), 128.7 (2 × CH), 130.0 (2 × CH), 133.4 (C), 136.3 (C), 138.4 (C), 144.5 (CH), 155.7 (C), 167.5 (C), 171.9 (C); m/z (ESI) 398.1608 (MH+. C22H24NO6 requires 398.1598).
O), 1528, 1435, 1254, 1207, 1169; [α]19D +60.1 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.08 (1H, dd, J 13.9, 6.2 Hz, 3-HH), 3.18 (1H, dd, J 13.9, 5.6 Hz, 3-HH), 3.72 (3H, s, OCH3), 3.80 (3H, s, OCH3), 4.63–4.71 (1H, m, 2-H), 5.07 (d, 1H, J 12.4 Hz, OCHHPh), 5.11 (d, 1H, J 12.4 Hz, OCHHPh), 5.27 (1H, d, J 8.0 Hz, 2-NH), 6.40 (1H, d, J 16.0 Hz, 2′′-H), 7.11 (2H, d, J 8.1 Hz, 2′-H and 6′-H), 7.28–7.39 (5H, m, Ph), 7.42 (2H, d, J 8.1 Hz, 3′-H and 5′-H), 7.65 (1H, d, J 16.0 Hz, 1′′-H); δC (101 MHz, CDCl3) 38.2 (CH2), 51.8 (CH3), 52.6 (CH3), 54.8 (CH), 67.2 (CH2), 117.8 (CH), 128.2 (2 × CH), 128.37 (CH), 128.40 (2 × CH), 128.7 (2 × CH), 130.0 (2 × CH), 133.4 (C), 136.3 (C), 138.4 (C), 144.5 (CH), 155.7 (C), 167.5 (C), 171.9 (C); m/z (ESI) 398.1608 (MH+. C22H24NO6 requires 398.1598).
Methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(2′′-phenylsulfonylvinyl)phenyl]propanoate (12b)
Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11) (0.230 g, 0.539 mmol) was dissolved in methanol (1.5 mL). To this was added phenyl vinyl sulfone (0.181 g, 1.08 mmol) and palladium acetate (0.0120 g, 0.054 mmol, 10 mol%). The reaction mixture was heated to 50 °C and stirred for 5.5 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by flash column chromatography, eluting with 50% ethyl acetate in hexane gave methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(2′′-phenylsulfonylvinyl)phenyl]propanoate (12b) (0.114 g, 44%) as a yellow solid. Mp 55–60 °C; νmax/cm−1 (neat) 3314 (NH), 2585 (CH), 2180, 2025, 1717 (CO), 1520, 1308, 1146, 752; [α]21D +58.4 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.06 (1H, dd, J 13.8, 6.2 Hz, 3-HH), 3.15 (1H, dd, J 13.8, 5.7 Hz, 3-HH), 3.71 (3H, s, OCH3), 4.61–4.71 (1H, m, 2-H), 5.05 (d, 1H, J 12.0 Hz, OCHHPh), 5.10 (d, 1H, J 12.0 Hz, OCHHPh), 5.22 (1H, d, J 8.2 Hz, 2-NH), 6.82 (1H, d, J 15.4 Hz, 2′′-H), 7.12 (2H, d, J 8.0 Hz, 2′-H and 6′-H), 7.27–7.41 (7H, m, Ph, 3′-H and 5′-H), 7.51–7.67 (4H, m, 1′′-H, 3′′′-H, 4′′′-H and 5′′′-H), 7.92–7.98 (2H, m, 2′′′-H and 6′′′-H); δC (101 MHz, CDCl3) 38.4 (CH2), 52.6 (CH3), 54.7 (CH), 67.2 (CH2), 127.3 (CH), 127.8 (2 × CH), 128.3 (2 × CH), 128.4 (CH), 128.7 (2 × CH), 128.9 (2 × CH), 129.5 (2 × CH), 130.2 (2 × CH), 131.4 (C), 133.5 (CH), 136.3 (C), 139.6 (C), 140.9 (C), 142.1 (CH), 155.6 (C), 171.7 (C); m/z (ESI) 480.1486 (MH+. C26H26NO6S requires 480.1475).
Methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[(4′-(phenylethenyl)phenyl]propanoate (12c)
Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11) (0.178 g, 0.417 mmol) was dissolved in methanol (1.5 mL). To this was added styrene (0.0950 mL, 0.834 mmol) and palladium acetate (0.00900 g, 0.0401 mmol, 10 mol%). The reaction mixture was heated to 50 °C and stirred for 1 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by flash column chromatography, eluting with 40% diethyl ether in hexane gave methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[(4′-phenylethenyl)phenyl]propanoate (12c) (0.123 g, 71%) as a white solid. Mp 123–128 °C; νmax/cm−1 (neat) 3356 (NH), 2955 (CH), 2920 (CH), 2025, 1717 (CO), 1528, 1435, 1285, 1261, 1231, 1215, 1038, 756; [α]18D +46.4 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.09 (1H, dd, J 13.9, 6.0 Hz, 3-HH), 3.15 (1H, dd, J 13.9, 5.7 Hz, 3-HH), 3.74 (3H, s, OCH3), 4.64–4.72 (1H, m, 2-H), 5.08 (d, 1H, J 12.8 Hz, OCHHPh), 5.12 (d, 1H, J 12.8 Hz, OCHHPh), 5.22 (1H, d, J 8.3 Hz, 2-NH), 7.05–7.11 (4H, m, 2′-H, 6′-H, 1′′-H and 2′′-H), 7.23–7.40 (8H, m, Ph, 3′′′-H, 4′′′-H and 5′′′-H), 7.42 (2H, d, J 8.1 Hz, 3′-H and 5′-H), 7.48–7.53 (2H, m, 2′′′-H and 6′′′-H); δC (101 MHz, CDCl3) 38.2 (CH2), 52.5 (CH3), 54.9 (CH), 67.2 (CH2), 126.7 (2 × CH), 126.9 (2 × CH), 127.8 (CH), 128.3 (CH), 128.35 (2 × CH), 128.37 (C), 128.7 (2 × CH), 128.8 (2 × CH), 128.9 (2 × CH), 129.8 (2 × CH), 135.3 (C), 136.5 (C), 137.5 (C), 155.8 (C), 172.1 (C); m/z (ESI) 416.1843 (MH+. C26H26NO4 requires 416.1856).
Methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(4′′′-fluorophenylethenyl)phenyl]propanoate (12d)
Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11) (0.225 g, 0.526 mmol) was dissolved in methanol (1.5 mL). To this was added 4-fluorostyrene (0.126 mL, 1.05 mmol) and palladium acetate (0.00600 g, 0.0267 mmol, 5 mol%). The reaction mixture was heated to 50 °C and stirred for 1 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by flash column chromatography, eluting with 40% diethyl ether in hexane gave methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(4′′′-fluorophenylethenyl)phenyl]propanoate (12d) (0.150 g, 66%) as a white solid. Mp 100–105 °C; νmax/cm−1 (neat) 3372 (NH), 2955 (CH), 1705 (CO), 1512, 1227, 1038, 833; [α]20D +5.3 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.08 (1H, dd, J 14.0, 6.1 Hz, 3-HH), 3.15 (1H, dd, J 14.0, 5.7 Hz, 3-HH), 3.73 (3H, s, OCH3), 4.63–4.72 (1H, m, 2-H), 5.09 (d, 1H, J 12.0 Hz, OCHHPh), 5.13 (d, 1H, J 12.0 Hz, OCHHPh), 5.23 (1H, d, J 8.3 Hz, 2-NH), 6.93–7.12 (6H, m, 2′-H, 6′-H, 1′′-H, 2′′-H, 2′′′-H and 6′′′-H), 7.28–7.50 (9H, m, Ph and 3′-H, 5′-H, 3′′′-H and 5′′′-H); δC (101 MHz, CDCl3) 38.2 (CH2), 52.5 (CH3), 54.9 (CH), 67.2 (CH2), 115.8 (2 × CH, 2JCF 21.8 Hz), 126.8 (2 × CH), 127.6 (CH), 128.11 (2 × CH, 3JCF 8.1 Hz), 128.13 (CH), 128.3 (2 × CH), 128.4 (CH), 128.7 (2 × CH), 129.8 (2 × CH), 133.6 (C, 4JCF 3.6 Hz), 135.3 (C), 136.3 (C), 136.4 (C), 155.8 (C), 162.5 (C, 1JCF 247.1 Hz), 172.1 (C); m/z (ESI) 456.1580 (MNa+. C26H24FNNaO4 requires 456.1582).
Methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(4′′′-chlorophenylethenyl)phenyl]propanoate (12e)
Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11) (0.212 g, 0.496 mmol) was dissolved in methanol (1.5 mL). To this was added 4-chlorostyrene (0.119 mL, 0.993 mmol) and palladium acetate (0.0110 g, 0.450 mmol, 10 mol%). The reaction mixture was heated to 50 °C and stirred for 1 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by flash column chromatography, eluting with 40% diethyl ether in hexane followed by 100% diethyl ether gave methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(4′′′-chlorophenylethenyl)phenyl]propanoate (12e) (0.179 g, 80%) as a white solid. Mp 110–116 °C; νmax/cm−1 (neat) 3369 (NH), 2957 (CH), 1721 (CO), 1707 (CO), 1523, 1226, 1038, 821; [α]25D +61.9 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.08 (1H, dd, J 13.9, 6.1 Hz, 3-HH), 3.16 (1H, dd, J 13.9, 5.7 Hz 3-HH), 3.73 (3H, s, OCH3), 4.64–4.72 (1H, m, 2-H), 5.09 (d, 1H, J 12.0 Hz, OCHHPh), 5.13 (d, 1H, J 12.0 Hz, OCHHPh), 5.22 (1H, d, J 8.3 Hz, 2-NH), 7.00–7.04 (2H, m, 1′′-H and 2′′-H), 7.06–7.12 (2H, m, 2′-H and 6′-H), 7.29–7.46 (11H, m, Ph, 3′-H, 5′-H, 2′′′-H, 3′′′-H, 5′′′-H and 6′′′-H); δC (101 MHz, CDCl3) 38.2 (CH2), 52.5 (CH3), 54.9 (CH), 67.2 (CH2), 126.9 (2 × CH), 127.5 (CH), 127.8 (2 × CH), 128.3 (CH), 128.4 (2 × CH), 128.7 (2 × CH), 128.96 (CH), 129.00 (2 × CH), 129.8 (2 × CH), 133.4 (C), 135.6 (C), 135.9 (C), 136.1 (C), 136.4 (C), 155.7 (C), 172.0 (C); m/z (ESI) 472.1289 (MNa+. C26H2435ClNNaO4 requires 472.1286).
Methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(3′′′-formylphenylethenyl)phenyl]propanoate (12f)
Methyl (2S)-2-(benzyloxycarbonylamino)-3-(4′-diazophenyl)propanoate tetrafluoroborate (11) (0.204 g, 0.478 mmol) was dissolved in methanol (1.5 mL). To this was added 3-vinylbenzaldehyde (0.121 mL, 0.955 mmol) and palladium acetate (0.0110 g, 0.450 mmol, 10 mol%). The reaction mixture was heated to 50 °C and stirred for 1 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by flash column chromatography, eluting with 40% diethyl ether in hexane gave methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[4′-(3′′′-formylphenylethenyl)phenyl]propanoate (12f) (0.105 g, 50%) as a white solid. Mp 110–114 °C; νmax/cm−1 (neat) 3322 (NH), 2955 (CH), 1736 (CO), 1694 (CO), 1528, 1296, 1250, 1022, 733; [α]20D +4.2 (c 0.1, CHCl3); δH (400 MHz, CDCl3) 3.09 (1H, dd, J 13.9, 6.1 Hz, 3-HH), 3.17 (1H, dd, J 13.9, 5.8 Hz, 3-HH), 3.74 (3H, s, OCH3), 4.64–4.73 (1H, m, 2-H), 5.09 (d, 1H, J 12.0 Hz, OCHHPh), 5.13 (d, 1H, J 12.0 Hz, OCHHPh), 5.24 (1H, d, J 8.2 Hz, 2-NH), 7.08–7.15 (3H, m, 2′-H, 6′-H and 2′′-H), 7.18 (1H, d, J 16.0 Hz, 1′′-H), 7.29–7.39 (5H, m, Ph), 7.44 (2H, d, J 8.1 Hz, 3′-H and 5′-H), 7.53 (1H, t, J 7.6 Hz, 5′′′-H), 7.72–7.80 (2H, m, 4′′′-H and 6′′′-H), 8.02 (1H, s, 2′′′-H), 10.06 (1H, s, 3′′′-CHO); δC (101 MHz, CDCl3) 38.1 (CH2), 52.5 (CH3), 54.9 (CH), 67.1 (CH2), 127.0 (2 × CH), 127.1 (CH), 127.2 (CH), 128.2 (2 × CH), 128.3 (CH), 128.6 (2 × CH), 129.0 (CH), 129.5 (CH), 129.8 (2 × CH), 130.1 (CH), 132.4 (CH), 135.7 (C), 135.9 (C), 136.3 (C), 136.9 (C), 138.4 (C), 155.7 (C), 172.0 (C), 192.4 (CH); m/z (ESI) 466.1626 (MNa+. C27H25NNaO5 requires 466.1625).
(2S,1′′E)-2-(Benzyloxycarbonylamino)-3-[(4′-phenylethenyl)phenyl]propanoic acid (13)
To a stirred solution of methyl (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[(4′-phenylethenyl)phenyl]propanoate (12c) (0.087 g, 0.21 mmol) in methanol (3 mL), dioxane (1.75 mL) and water (1.75 mL) was added caesium carbonate (0.089 g, 0.27 mmol). The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was concentrated in vacuo, diluted in water (5 mL) and acidified to pH 1 using 1 M aqueous hydrochloric acid. The reaction mixture was extracted with dichloromethane (3 × 10 mL) and ethyl acetate (3 × 10 mL). The organic layers were combined, dried (MgSO4), filtered and concentrated in vacuo to give (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[(4′-(phenylethenyl)phenyl]propanoic acid (13) (0.079 g, 94%) as a white solid. Mp 160–164 °C; νmax/cm−1 (neat) 3333 (NH), 3144, 2928 (CH), 2357, 1694 (CO), 1524, 1447, 1223, 814; [α]25D +28.0 (c 0.1, MeOH); δH (400 MHz, CD3OD) 2.92 (1H, dd, J 13.9, 9.5 Hz, 3-HH), 3.19 (1H, dd, J 13.9, 4.8 Hz, 3-HH), 4.35–4.49 (1H, m, 2-H), 4.98 (d, 1H, J 12.0 Hz, OCHHPh), 5.05 (d, 1H, J 12.0 Hz, OCHHPh), 7.13 (2H, s, 1′′-H and 2′′-H), 7.15–7.37 (10H, m, Ph, 2′-H, 6′-H, 3′′′-H, 4′′′-H and 5′′′-H), 7.44 (2H, d, J 7.7 Hz, 3′-H and 5′-H), 7.53 (2H, d, J 7.7 Hz, 2′′′-H and 6′′′-H); δC (101 MHz, CD3OD) 38.4 (CH2), 56.7 (CH), 67.5 (CH2), 127.5 (2 × CH), 127.6 (2 × CH), 128.5 (CH), 128.6 (2 × CH), 128.9 (CH), 129.41 (2 × CH), 129.43 (2 × CH), 129.7 (2 × CH), 130.7 (2 × CH), 137.4 (C), 138.1 (C), 138.2 (C), 138.9 (C), 158.4 (C), 175.1 (C); m/z (ESI) 402.1683 (MH+. C25H24NO4 requires 402.1705).
(2S,1′′E)-2-Amino-3-[(4′-(phenylethenyl)phenyl]propanoic acid hydrochloride (14)
A solution of (2S,1′′E)-2-(benzyloxycarbonylamino)-3-[(4′-(phenylethenyl)phenyl]propanoic acid (13) (0.025 g, 0.062 mmol) in 4 M hydrochloric acid in dioxane (2 mL) was heated under reflux for 4 h. To this was added 6 M aqueous hydrochloric acid (2 mL) and the reaction mixture was heated under reflux for 18 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Purification by recrystallisation from methanol and diethyl ether gave (2S,1′′E)-2-amino-3-[(4′-(phenylethenyl)phenyl]propanoic acid hydrochloride (14) (0.014 g, 82%) as a white solid. Mp 224–230 °C; νmax/cm−1 (neat) 3364 (NH), 2913 (CH), 1736 (CO), 1489, 1219, 826; [α]21D −6.0 (c 0.1, MeOH); δH (400 MHz, CD3OD) 3.16 (1H, dd, J 14.5, 7.6 Hz, 3-HH), 3.27–3.36 (1H, m, 3-HH), 4.22–4.27 (1H, m, 2-H), 7.20 (2H, s, 1′′-H and 2′′-H), 7.26 (1H, t, J 7.4 Hz, 4′′′-H), 7.32 (2H, d, J 8.0 Hz, 2′-H and 6′-H), 7.36 (2H, t, J 7.4 Hz, 3′′′-H and 5′′′-H), 7.56 (2H, d, J 7.4 Hz, 2′′′-H and 6′′′-H), 7.59 (2H, d, J 8.0 Hz, 3′-H and 5′-H); δC (101 MHz, CD3OD) 35.8 (CH2), 53.8 (CH), 126.2 (2 × CH), 126.8 (2 × CH), 127.4 (CH), 127.5 (CH), 128.3 (2 × CH), 128.8 (CH), 129.4 (2 × CH), 133.5 (C), 137.15 (C), 137.25 (C), 170.0 (C); m/z (ESI) 268.1325 (MH+. C17H18NO2 requires 268.1332).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the University of Glasgow (Ph.D. studentship to RM) and the Engineering and Physical Sciences Research Council (EP/S029168/1) is gratefully acknowledged.References
- For reviews see: (a) I. Wagner and H. Musso, Angew. Chem., Int. Ed. Engl., 1983, 22, 816 CrossRef; (b) J. P. Greenstein and M. Winitz, Chemistry of the Amino Acids, ed. R. E. Krieger, FL, 1984, vol. 1–3 Search PubMed; (c) Chemistry and Biochemistry of the Amino Acids, ed. G. C. Barrett, Chapman and Hall, London, 1985 Search PubMed; (d) M. J. O'Donnell, α-Amino Acid Synthesis, Tetrahedron, 1988, 44, 5253 CrossRef; (e) R. M. Williams, Synthesis of Optically Active α-Amino Acids, Pergamon Press, Oxford, 1989, vol. 7 Search PubMed.
- (a) Y. Ohfune, Acc. Chem. Res., 1992, 25, 360 CrossRef CAS; (b) A. Sutherland and C. L. Willis, Nat. Prod. Rep., 2000, 17, 621 RSC; (c) D. A. Dougherty, Curr. Opin. Chem. Biol., 2000, 4, 645 CrossRef CAS PubMed; (d) Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 5127 CrossRef CAS; (e) H. Vogt and S. Bräse, Org. Biomol. Chem., 2007, 5, 406 RSC; (f) R. Smits, C. D. Cadicamo, K. Burger and B. Koksch, Chem. Soc. Rev., 2008, 37, 1727 RSC; (g) M. Mortensen, R. Husmann, E. Veri and C. Bolm, Chem. Soc. Rev., 2009, 38, 1002 RSC; (h) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764 CrossRef CAS PubMed.
- For recent reviews, see: (a) A. T. Krueger and B. Imperiali, ChemBioChem, 2013, 14, 788 CrossRef CAS PubMed; (b) R. Kubota and I. Hamachi, Chem. Soc. Rev., 2015, 44, 4454 RSC; (c) A. H. Harkiss and A. Sutherland, Org. Biomol. Chem., 2016, 14, 8911 RSC; (d) Z. Cheng, E. Kuru, A. Sachdeva and M. Vendrell, Nat. Rev., 2020, 4, 275 CAS.
- For example, see: (a) C. J. Easton, Chem. Rev., 1997, 97, 53 CrossRef CAS PubMed; (b) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef PubMed; (c) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed.
- S. Sengupta and S. Chandrasekaran, Org. Biomol. Chem., 2019, 17, 8308 RSC.
- For examples, see: (a) Y. Wang and Q. Lin, Org. Lett., 2009, 11, 3570 CrossRef CAS PubMed; (b) X.-Y. Liu, S.-J. Zhai, F.-F. Feng, F.-G. Zhang and J.-A. Ma, ChemCatChem, 2020, 12, 5623 CrossRef CAS; (c) S.-J. Zhai, D. Cahard, F.-G. Zhang and J.-A. Ma, Chin. Chem. Lett., 2022, 33, 863–866 CrossRef CAS; (d) O. Nwajiobi, A. K. Verma and M. Raj, J. Am. Chem. Soc., 2022, 144, 4633 CrossRef CAS PubMed.
- R. A. Houghten and H. Rapoport, J. Med. Chem., 1974, 17, 556 CrossRef CAS PubMed.
- C. Torres-García, D. Pulido, F. Albericio, M. Royo and E. Nicolás, J. Org. Chem., 2014, 79, 11409 CrossRef PubMed.
- B. Zhu, J. Ge and S. Q. Yao, Bioorg. Med. Chem., 2015, 23, 2917 CrossRef CAS PubMed.
- C. M. Tressler and N. J. Zondlo, Org. Lett., 2016, 18, 6240 CrossRef CAS PubMed.
- (a) L. S. Fowler, D. Ellis and A. Sutherland, Org. Biomol. Chem., 2009, 7, 4309 RSC; (b) L. Gilfillan, R. Artschwager, A. H. Harkiss, R. M. J. Liskamp and A. Sutherland, Org. Biomol. Chem., 2015, 13, 4514 RSC; (c) A. H. Harkiss, J. D. Bell, A. Knuhtsen, A. G. Jamieson and A. Sutherland, J. Org. Chem., 2019, 84, 2879 CrossRef CAS PubMed; (d) J. D. Bell, A. H. Harkiss, D. Nobis, E. Malcolm, A. Knuhsten, C. R. Wellaway, A. G. Jamieson, S. W. Magennis and A. Sutherland, Chem. Commun., 2020, 56, 1887 RSC.
- (a) R. J. Faggyas, N. L. Sloan, N. Buijs and A. Sutherland, Eur. J. Org. Chem., 2019, 5344 CrossRef CAS; (b) J. D. Bell, T. E. F. Morgan, N. Buijs, A. H. Harkiss, C. R. Wellaway and A. Sutherland, J. Org. Chem., 2019, 84, 10436 CrossRef CAS PubMed; (c) L. M. Riley, T. N. Mclay and A. Sutherland, J. Org. Chem., 2023, 88, 2453 CrossRef CAS PubMed.
- For example, see: (a) J. W. Tilley, R. Sarabu, R. Wagner and K. Mulkerins, J. Org. Chem., 1990, 55, 906 CrossRef CAS; (b) H. Fretz, Tetrahedron Lett., 1996, 37, 8475 CrossRef CAS; (c) M. K. Gurjar, T. P. Khaladkar, R. G. Borhade and A. Murugan, Tetrahedron Lett., 2003, 44, 5183 CrossRef CAS.
- S. Sengupta and S. Bhattacharyya, Tetrahedron Lett., 1995, 36, 4475 CrossRef CAS.
- (a) G. Jones and S. Wright, J. Chem. Soc. C, 1971, 141 RSC; (b) L. Chen, J. W. Tilley, R. W. Guthrie, F. Mennona, T.-N. Huang, G. Kaplan, R. Trilles, D. Miklowski, N. Hruby, W. Schwinge, B. Wolitzky and K. Rowan, Bioorg. Med. Chem. Lett., 2000, 10, 729 CrossRef CAS PubMed.
- (a) M. J. Burk, J. R. Lee and J. P. Martinez, J. Am. Chem. Soc., 1994, 116, 10847 CrossRef CAS; (b) J. X. Qiao, K. J. Fraunhoffer, Y. Hsiao, Y.-X. Li, C. Wang, T. C. Wang and M. A. Poss, J. Org. Chem., 2016, 81, 9499 CrossRef CAS PubMed.
- M. Matsushita, K. Yoshida, N. Yamamoto, P. Wirsching, R. A. Lerner and K. D. Janda, Angew. Chem., Int. Ed., 2003, 42, 5984 CrossRef CAS PubMed.
- (a) Y. Wang, Z. Huang and Z. Huang, Nat. Catal., 2019, 2, 529 CrossRef CAS; (b) Z. Huang, Y. Wang, X. Leng and Z. Huang, J. Am. Chem. Soc., 2021, 143, 4824 CrossRef CAS PubMed.
- (a) K. Kikukawa and T. Matsuda, Chem. Lett., 1977, 159 CrossRef CAS; (b) K. Kikukawa, K. Nagira and T. Matsuda, Bull. Chem. Soc. Jpn., 1977, 50, 2207 CrossRef CAS; (c) K. Kikukawa, K. Nagira, N. Terao, F. Wada and T. Matsuda, Bull. Chem. Soc. Jpn., 1979, 52, 2609 CrossRef CAS.
- R. J. Faggyas, M. Grace, L. Williams and A. Sutherland, J. Org. Chem., 2018, 83, 12595 CrossRef CAS PubMed.
- Tosylate arenediazonium salts have been shown to be relatively stable: V. D. Filimonov, M. Trusova, P. Postnikov, E. A. Krasnokutskaya, Y. M. Lee, H. Y. Hwang, H. Kim and K.-W. Chi, Org. Lett., 2008, 10, 3961 CrossRef CAS PubMed.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579 CrossRef CAS PubMed.
- The individual absorption and fluorescence spectra for α-amino acids 12c–f and 14 are presented in the ESI.†.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067 RSC.
- S. Gerisch, H.-D. Jakubke and H.-J. Kreuzfeld, Tetrahedron: Asymmetry, 1995, 6, 3039 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and emission spectra, 1H and 13C NMR spectra for all compounds. See DOI: https://doi.org/10.1039/d3ob01096a |
| This journal is © The Royal Society of Chemistry 2023 |