
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of 3-hydroxy- and 3-amino-3-alkynyl-2-oxindoles by the dimethylzinc-mediated addition of terminal alkynes to isatins and isatin-derived ketimines†
Elena
Prieto
 ,
Jorge D.
Martín
,
Javier
Nieto
* and
Celia
Andrés
*
,
Jorge D.
Martín
,
Javier
Nieto
* and
Celia
Andrés
*
Instituto CINQUIMA and Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Valladolid, Paseo de Belén, 7, 47011 Valladolid, Spain. E-mail: franciscojavier.nieto@uva.es
First published on 3rd August 2023
Abstract
A common protocol for enantioselective alkynylation of isatins and isatin-derived ketimines using terminal alkynes and Me2Zn in the presence of a catalytic amount of a chiral perhydro-1,3-benzoxazine with moderate to excellent enantioselectivity under mild reaction conditions is described. The additions to ketimines present a novel approach to chiral amines being derivatives of oxindoles. The reaction is broad in scope with respect to aryl- and alkyl-substituted terminal alkynes and isatin derivatives. In isatins, the alkynylation occurs at the Si face of the carbonyl group, whereas in the ketimine derivatives it occurs at the Re face of the imine.
Introduction
The 3-heteroatom-substituted oxindoles are an important class of compounds whose structure is present in a wide variety of natural and synthetic products that exhibit a wide range of interesting biological activities and pharmaceutical properties.1 Studies of the structure–biological activity relationship have shown that their biological activities are significantly affected both by the configuration of the stereocentre in C-3 and by its substitution pattern.2 Therefore, in recent years, the asymmetric synthesis of chiral 3-hydroxy and 3-aminooxindoles has become a hot topic in organic synthesis.3,4Among the 2-oxindole derivatives, we are interested in the 3-hydroxy- and 3-amino-3-alkynyl-2-oxindole derivatives that can be prepared by alkynylation of isatins and isatin-derived ketimines, since some of these have been found to exhibit significant pharmacological activities (Fig. 1). For example, 3-(hex-1-yn-1-yl)-3-hydroxy-1-methylindolin-2-one (I) shows excellent biological activity in Echinococcus multilocularis metacestodes, which can cause severe liver problems,5 3-(cyclopropylethynyl)-3-hydroxy-1,5-dimethylindolin-2-one (II) is more active than efavirenz against HIV-1 replication6 and 5-chloro-3-ethenyl-3-hydroxy-1-(prop-2-yn-1-yl)indolin-2-one (III) is a good inhibitor of the enzyme Akt kinase with potential anticancer activity.7 In addition, 3-alkynyl-3-hydroxy-2-oxindoles and 3-alkynyl-3-amino-2-oxindoles are commonly used as versatile synthons in a wide variety of synthetic applications,8 notably the preparation of biologically active spirooxindoles9 and other heterocyclic derivatives (Fig. 2).10
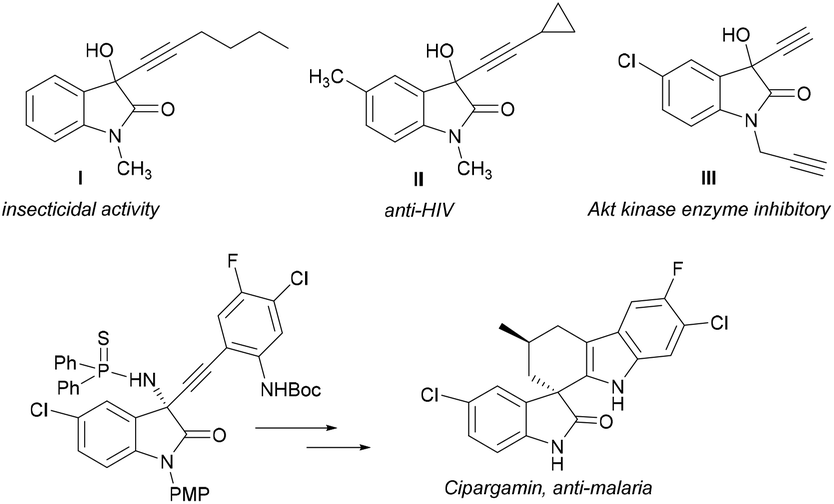 | ||
| Fig. 1 Examples of bioactive 3-alkynyl-3-hydroxy-2-oxindoles and preparation of the antimalarial agent cipargamin from a chiral 3-alkynyl-3-amino-2-oxindole derivative. | ||
 | ||
| Fig. 2 Some representative examples of the preparation of spirooxindoles and other heterocyclic derivatives from chiral 3-alkynyl-3-hydroxy-2-oxindoles. | ||
Although there are different examples of alkynylations of isatins leading to racemic mixtures of products,11 the asymmetric version of this reaction has only been studied recently. Different Cu(I) complexes have emerged as efficient catalysts for this transformation, as reported by Liu,12 Guo13 and Zhou14 who employed Cu(I)/chiral guanidine, Cu(I)/chiral phosphine and Cu(I)/chiral aminophenol systems respectively. A Cu(II)/bisoxazoline complex has also been shown to be effective in the catalytic enantioselective addition of ynamides to isatins.15 Alternatively, Maruoka has developed a competitive Ag(I)/chiral phase transfer catalytic system for the alkynylation of trityl-protected isatin derivatives,16 and the Zhang group has developed the alkynylation of N-p-methoxybenzyl-protected isatins using a bifunctional Ag(I)/amidophosphine-urea catalyst.17 In addition, Zn(II)/chiral hydroxyl oxazoline ligand and Co(II)/bisoxazolinephosphine catalytic systems have been successfully employed by Chen18 and Li,19 respectively. Zhao recently also developed a novel Zn/Co bimetallic catalysis system in the presence of a chiral bisphosphine with excellent results.20
Enantioselective alkynylation of isatin-derived ketimines has been much less explored, and to the best of our knowledge, there are only two examples of the catalytic enantioselective addition of terminal alkynes to isatin-derived ketimines. The Shibasaki group developed the addition of alkynylzinc derivatives to thiophosphinoyl ketimines catalysed by a mesitylcopper/bisphosphine complex21 and applied this protocol to the enantioselective synthesis of the antimalarial agent cipargamin (Fig. 1),9g and the Liu group used a chiral Cu(I)/guaninide complex in the alkynylation of isatin-derived N-Boc ketimines.22
Despite much progress made to date, the development of new mild, and efficient catalytic systems for the enantioselective alkynylation of isatins and isatin-derived ketimines with a broad scope for both alkynes and isatins remains challenging and interesting, and to the best of our knowledge, a common method for both is not known.
In previous works, we have shown that conformationally restricted chiral perhydro-1,3-benzoxazines behave as excellent ligands in the enantioselective addition of organozinc derivatives to aldehydes and activated ketones, including the enantioselective alkynylation of α-keto esters and 1,2-diketones with alkynyl zinc derivatives,23 and in this way, we envisioned their utilization as ligands for the enantioselective addition of alkynyl zinc reagents to isatin derivatives. These chiral perhydro-1,3-benzoxazines can be easily prepared in a few steps from (−)-8-aminomenthol.23a,d,24
Given the difference in structure and reactivity of substrates such as α-keto esters, α-diketones, isatins and isatin-derived ketimines, it is not usual to find the same chiral ligand that allows the alkynylation of all of them with a high enantioselectivity.
Results and discussion
Enantioselective alkynylation of isatins
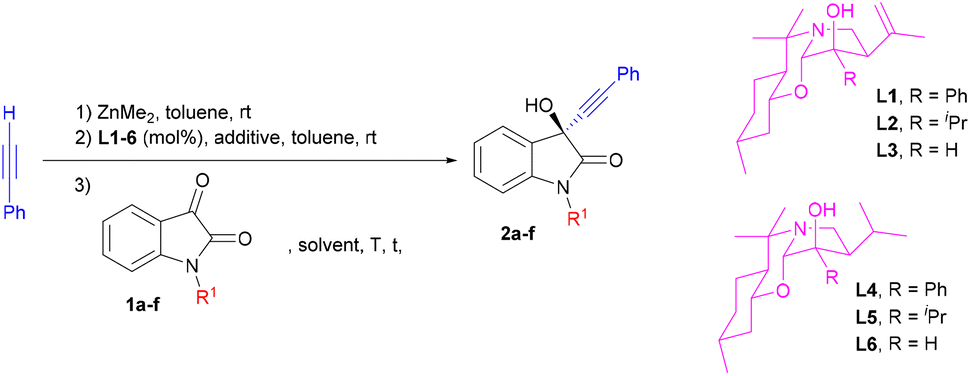
Initially, asymmetric alkynylation of isatins with the in situ generated zinc acetylide from phenylacetylene and Me2Zn was chosen as a reaction model to evaluate both the catalytic activity of a range of chiral perhydro-1,3-benzoxazines (L1–6) and the optimal substituent on the nitrogen of the isatin (Table 1).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Isatin, R | Ligand (mol %) | Additive (mol %) | Solvent | T (ºC) | t (h) | Yieldb (%) | erc |
| a Reaction conditions: 0.1 mmol of 1a–f, 0.4 mmol of dimethylzinc and 0.4 mmol of phenylacetylene. b Yield of isolated product after purification by flash column chromatography. c Enantiomeric ratio determined by HPLC on a chiral stationary phase. d Only 0.2 mmol of dimethylzinc and 0.25 mmol of phenylacetylene were used. | ||||||||
| 1 | 1a, CH3 | L1 (20) | — | Toluene | 0 | 20 | 2a (36) | 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24 :24 |
| 2 | 1a, CH3 | L2 (20) | — | Toluene | 0 | 20 | 2a (79) | 84:16 |
| 3 | 1a, CH3 | L3 (20) | — | Toluene | 0 | 20 | 2a (36) | 72:28 |
| 4 | 1a, CH3 | L4 (20) | — | Toluene | 0 | 20 | 2a (55) | 77:23 |
| 5 | 1a, CH3 | L5 (20) | — | Toluene | 0 | 20 | 2a (40) | 73:27 |
| 6 | 1a, CH3 | L6 (20) | — | Toluene | 0 | 20 | 2a (38) | 72:28 |
| 7 | 1a, CH3 | L2 (20) | — | Toluene | −20 | 20 | 2a (72) | 91:9 |
| 8 | 1a, CH3 | L2 (20) | — | Toluene | −30 | 35 | 2a (60) | 90:10 |
| 9 | 1a, CH3 | L2 (20) | B(OEt)3 (10) | Toluene | −20 | 12 | 2a (78) | 86:14 |
| 10 | 1a, CH3 | L2 (20) | DiMPEG (10) | Toluene | −20 | 20 | 2a (48) | 87:13 |
| 11 | 1a, CH3 | L2 (20) | iPrOH (10) | Toluene | −20 | 20 | 2a (52) | 85:15 |
| 12 | 1a, CH3 | L2 (20) | Ti(OiPr)4 (10) | Toluene | −20 | 20 | 2a (64) | 84:16 |
| 13 | 1a, CH3 | L2 (20) | B(OEt)3 (10) | Toluene | −40 | 45 | 2a (70) | 86:14 |
| 14 | 1b, C6H5–CH2 | L2 (20) | — | Toluene | −20 | 20 | 2b (68) | 92:8 |
| 15 | 1c, 2,6-Cl2–C6H3–CH2 | L2 (20) | — | Toluene | −20 | 20 | 2c (72) | 93:7 |
| 16 | 1d, C(C6H5)3 | L2 (20) | — | Toluene | −20 | 24 | 2d (67) | 92:8 |
| 17 | 1e, Ac | L2 (20) | — | Toluene | −20 | 24 | 2e (68) | 93:7 |
| 18 | 1f, Ts | L2 (20) | — | Toluene | −20 | 3 | 2f (52) | 83:17 |
| 19 | 1c, 2,6-Cl2–C6H3–CH2 | L2 (20) | — | CH2Cl2:toluene 3:1 |
−20 | 22 | 2c (78) | 96:4 |
| 20d | 1c, 2,6-Cl2–C6H3–CH2 | L2 (20) | — | CH2Cl2:toluene 3:1 |
−20 | 22 | 2c (50) | 88:12 |
| 21 | 1c, 2,6-Cl2–C6H3–CH2 | L2 (10) | — | CH2Cl2:toluene 3:1 |
−20 | 22 | 2c (60) | 90:10 |
Alkynylation of N-methyl isatin 1a in toluene at 0 °C in the presence of ligands L1–6 (20 mol%) afforded 2a in moderate enantioselectivity. The ligand L2 led to the product in a good yield and with the best enantioselectivity (Table 1, entry 2). Then, we chose ligand L2 to continue with the optimization of the reaction conditions, modifying the solvent, temperature, and N-protecting group at the isatin and using catalytic amounts of additives. On lowering the reaction temperature to −20 °C, the enantioselectivity improved to a good 91:9 er with a slightly decreased yield (entry 7), however, at −30 °C the reaction slows down and the enantioselectivity does not improve (entry 8). We also tested some previously reported additives,25 such as Lewis acids, isopropanol and DiMPEG (entries 9–13), but they did not improve the enantioselectivity. With −20 °C as a suitable temperature, we tested different N-protecting groups (entries 14–18). The best result was found when N-2,6-dichlorobenzyl isatin 1c was used, achieving the product in 72% with a 93:7 er (entry 14). No decrease in the enantiocontrol was observed when an electron-withdrawing acetyl group was used, and the product 2e was obtained in a similar yield (entry 17), however, the N-tosyl isatin 1f led to a moderate enantioselectivity and yield due to the formation of several by-products after only 3 h at −20 °C (entry 18). When the reaction of phenylacetylene with isatin 1c was carried out using a 3:1 dichloromethane–toluene mixture as solvent, a significant improvement in both yield and enantioselectivity was observed (78%, er = 96:4, entry 19).
Finally, a considerable decrease in the chemical yield and enantioselectivity was observed when only 2 equivalents of the alkynylzinc reagent were employed or the ligand loading was reduced to 10 mol% (entries 20 and 21).
The absolute configuration of the newly formed stereogenic centre on 2a, 2b and 2d was established as R. This configuration was assigned by comparing the sign of the optical rotation with literature data for the same compounds12–14,16,18 and has been extended to all the other 3-hydroxy-3-alkynyloxindoles synthesized based on mechanistic analogy. On the other hand, the absolute configuration of the alkynylation product of 1e was confirmed to be R by X-ray diffraction analysis of a single crystal of 2e.
With the optimized conditions in hand (entry 19 in Table 1), we proceeded to explore the scope of the reaction of N-2,6-dichlorobenzyl isatin 1c with different terminal alkynes, as illustrated in Table 2.
| a Reaction conditions. (1) 0.4 mmol of alkyne, 0.4 mmol of dimethylzinc, toluene, rt; (2) 0.02 mmol of L2 toluene, rt; (3) 0.1 mmol of 1c, dichloromethane, −20 °C, 22 h. Yield of isolated products after purification by flash column chromatography. Enantiomeric ratio determined by HPLC on a chiral stationary phase. |
|---|

|
To our satisfaction, a wide range of phenylacetylene derivatives bearing different electron-withdrawing and electron-donating substituents on the ortho, meta or para positions of the phenyl group were tolerated, giving the 3-alkynyl-3-hydroxy-2-oxindoles 3a–j in a moderate to good yield and excellent enantioselectivity. When the reaction was carried out in the presence of aliphatic alkynes a decrease in yield and enantioselectivity was observed, particularly with the bulky tert-butyl derivative, which afforded the alcohol 3k in a poor yield of 38% with moderate enantioselectivity (er = 92:8). An exception was the 4-bromobutyne which provided propargyl alcohol 3n in a moderate chemical yield, but an excellent enantioselectivity (er = 99:1). Trimethylsilylacetylene also provided the corresponding tertiary alcohol 3o with a good enantioselectivity of 95:5 er.
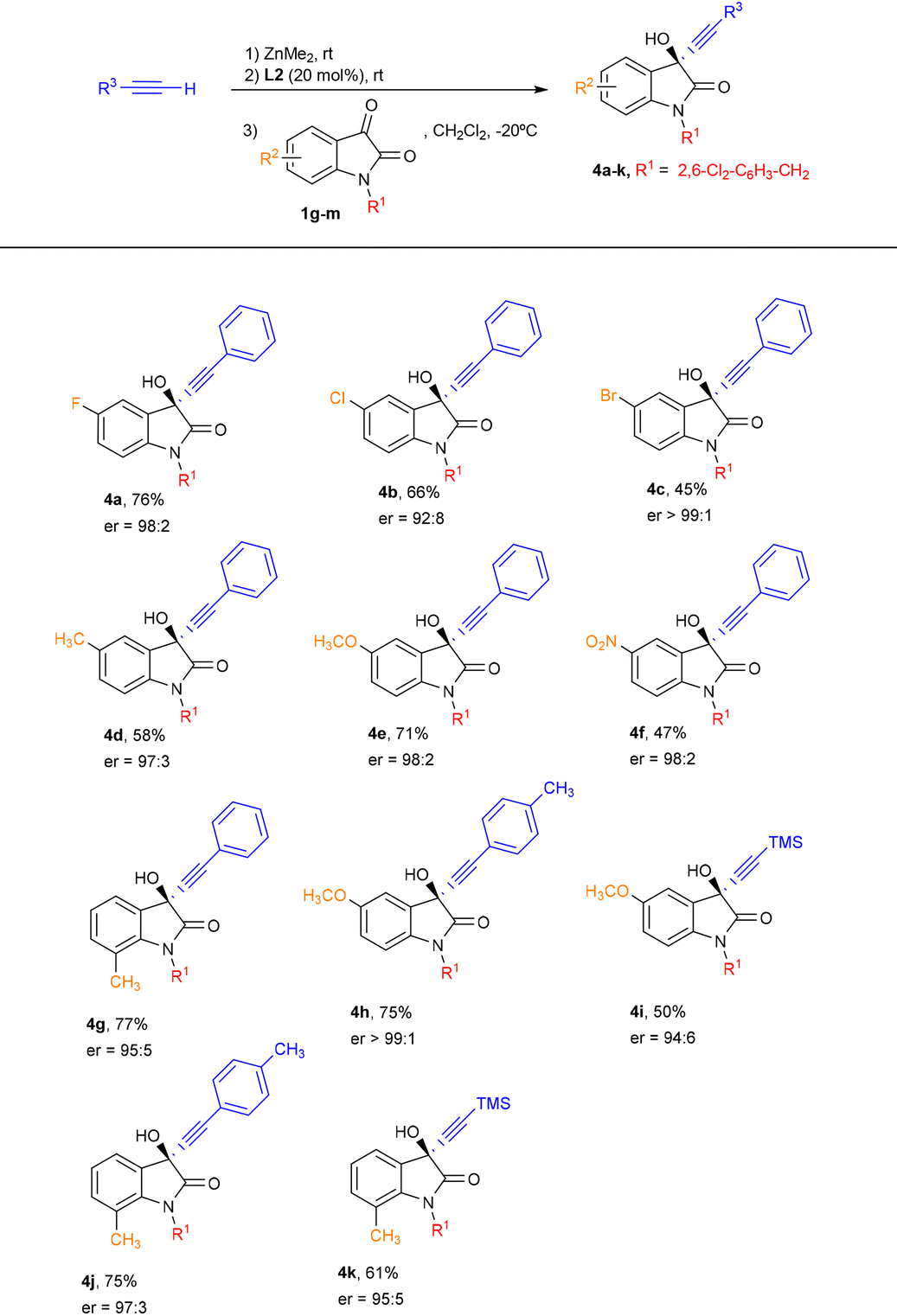
We next explored the scope of the reaction of phenylacetylene with isatins bearing different electron-withdrawing and electron-donating groups in the aromatic ring (Table 3). Pleasingly, substituents at position 5 did not have a remarkable influence in the enantiocontrol and the products 4a–f were obtained in a moderate to good yield and excellent enantioselectivity. In addition, when 7-methyl-substituted isatin was employed, the propargylic alcohol 4g was obtained in a competitive 77% and 95:5 er. Furthermore, good yields and excellent enantioselectivities were observed when p-tolylacetylene was reacted with 5-methoxy and 7-methyl-substituted isatins (products 4h and 4j, respectively), although a slight decrease in both yield and enantiocontrol was noted when using trimethylsilylacetylene with these isatins (products 4i and 4k).
| a Reaction conditions. (1) 0.4 mmol of alkyne, 0.4 mmol of dimethylzinc, toluene, rt; (2) 0.02 mmol of L2 toluene, rt; (3) 0.1 mmol of 1g–m, dichloromethane, −20 °C, 22 h. Yield of isolated products after purification by flash column chromatography. Enantiomeric ratio determined by HPLC on a chiral stationary phase. |
|---|
|
Finally, to show the potential of our catalytic system we tested our ligand in the enantioselective synthesis of two pharmacologically active5,6 compounds, 5a and 5b, derived from N-methylisatin (Scheme 1). Both chiral propargylic alcohols were obtained in a good yield under our optimized reaction conditions. Cyclopropinyl derivative 5a was achieved with a high enantioselectivity, which was only moderate for compound 5b.
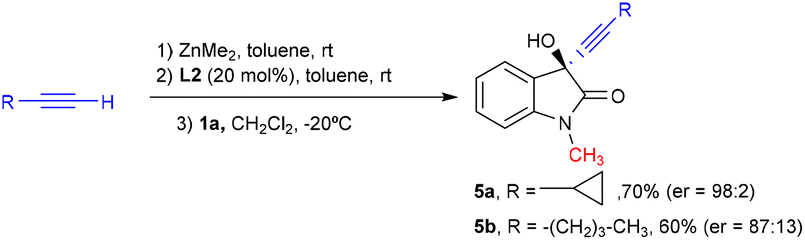 | ||
| Scheme 1 Enantioselective synthesis of the pharmacologically interesting derivatives 5a–b. | ||
Enantioselective alkynylation of isatin-derived ketimines
The alkynylation of the isatin-derived N-phenyl ketimine 6a with phenylacetylene was selected as the model reaction to optimize the reaction conditions. We first tested ligands L1, L2, L4 and L5 (20 mol%) using a mixture CH2Cl2:toluene 2:1 as the solvent at room temperature, and the results are collected in Table 4.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ketimine | R1 | R2 | Ligand (mol %) | Yieldb (%) | erc |
|
a Unless specified, all the reactions were performed with phenylacetylene (0.4 mmol), dimethylzinc (0.4 mmol), L (0.02 mmol, 20 mol%) and 6a–m (0.1 mmol) in a mixture of CH2Cl2:toluene 2:1 (1.5 mL) at rt for 18 h.
b Yield of isolated product after purification by flash column chromatography.
c Enantiomeric ratio determined by HPLC on a chiral stationary phase.
d A mixture of CH2Cl2:toluene 1:2 was used as the solvent.
e Performed at 0 °C for 60 h.
f Only 0.2 mmol of dimethylzinc and 0.2 mmol of phenylacetylene were employed.
|
||||||
| 1 | 6a | 2,6-Cl2–C6H3–CH2 | Ph | L1 (20) | 7a (89) | 75:25 |
| 2 | 6a | 2,6-Cl2–C6H3–CH2 | Ph | L2 (20) | 7a (92) | 99:1 |
| 3 | 6a | 2,6-Cl2–C6H3–CH2 | Ph | L4 (20) | 7a (92) | 74:26 |
| 4 | 6a | 2,6-Cl2–C6H3–CH2 | Ph | L5 (20) | 7a (95) | 79:21 |
| 5d | 6a | 2,6-Cl2–C6H3–CH2 | Ph | L2 (20) | 7a (84) | 90:10 |
| 6e | 6a | 2,6-Cl2–C6H3–CH2 | Ph | L2 (20) | 7a (12) | 98:2 |
| 7 | 6b | 2,6-Cl2–C6H3–CH2 | 4-CF3–C6H4 | L2 (20) | 7b (95) | >99:1 |
| 8 | 6c | 2,6-Cl2–C6H3–CH2 | 4-OCH3–C6H4 | L2 (20) | 7c (71) | >99:1 |
| 9 | 6d | 2,6-Cl2–C6H3–CH2 | 2-OCH3–C6H4 | L2 (20) | 7d (85) | 65:35 |
| 10 | 6e | 2,6-Cl2–C6H3–CH2 | 2-CH3–C6H4 | L2 (20) | 7e (38) | 95:5 |
| 11 | 6f | 2,6-Cl2–C6H3–CH2 | 2,4,6-(CH3)3–C6H2 | L2 (20) | — | — |
| 12 | 6g | 2,6-Cl2–C6H3–CH2 | 3,5-Cl2–C6H3 | L2 (20) | 7g (98) | >99:1 |
| 13 | 6h | Bn | 3,5-Cl2–C6H3 | L2 (20) | 7h (93) | 94:6 |
| 14 | 6i | CH3 | 3,5-Cl2–C6H3 | L2 (20) | 7i (90) | 96:4 |
| 15 | 6j | MOM | 3,5-Cl2–C6H3 | L2 (20) | 7j (93) | 95:5 |
| 16 | 6k | CPh3 | 3,5-Cl2–C6H3 | L2 (20) | 7k (95) | 87:13 |
| 17 | 6l | Ac | 3,5-Cl2–C6H3 | L2 (20) | — | — |
| 18 | 6m | H | 3,5-Cl2–C6H3 | L2 (20) | — | — |
| 19 | 6g | 2,6-Cl2–C6H3–CH2 | 3,5-Cl2–C6H3 | L2 (10) | 7g (65) | 95:5 |
| 20f | 6g | 2,6-Cl2–C6H3–CH2 | 3,5-Cl2–C6H3 | L2 (20) | 7g (36) | — |

In the presence of ligands L1, L4 and L5, the reaction afforded amine 7a in a high yield with moderate enantioselectivity (Table 4, entries 1, 3 and 4). Pleasingly, the product was achieved in a 92% yield and excellent enantioselectivity (99:1 er) when ligand L2 was employed (Table 4, entry 2).
The reaction seemed to be sensitive to solvent conditions, as shown when the reaction was performed in a 1:2 CH2Cl2:toluene mixture instead of a 2:1 one, both yield and enantiocontrol decreased (Table 4, entry 5). The reaction was also carried out using L2 at 0 °C, but under these conditions product 7a was obtained in a low yield after 60 hours (Table 4, entry 6). Thus, ligand L2 in a mixture of toluene:CH2Cl2 1:2 at room temperature (Table 4, entry 2) was selected as the optimized condition for this catalytic system.
With the optimized condition in hand, we studied the effect of different aryl substituents R2 at the imine position, as well as the influence of nitrogen protecting groups R1 at the isatin moiety. Reactions of ketimines 6b and 6g bearing electron withdrawing substituents at the aromatic ring of the imine position proceeded with a full conversion to the desired product and excellent enantioselectivity (Table 4, entries 7 and 12). No detrimental effect on the enantiocontrol was observed when the p-anisidine derivative 6c was tested although the chemical yield decreased to 71% (Table 4, entry 8). The reaction seemed to be sensitive to the steric effect of substituents in the ortho position of the aromatic ring, as shown in entries 9–11 in Table 4. Product 7e bearing an o-toluidine moiety was achieved with a high enantioselectivity but only in a 38% yield and the use of the more sterically hindered mesityl group resulted in no conversion of the starting ketimine 6f (Table 4, entries 10 and 11, respectively). In contrast, o-anisidine 6d afforded the product 7d in good yield, but the enantiomeric ratio was low, probably due to a different coordination between the zinc alkoxide catalyst and the 2-methoxyphenylimine derivative (Table 4, entry 9).
3,5-Dichloroaniline was chosen as the imine substituent in order to proceed with the evaluation of different protecting groups on the nitrogen of the isatin moiety, as collected in entries 13–18 in Table 4. Reactions between phenylacetylene and benzyl-, methyl- and methoxymethyl-protected substrates 6h, 6i and 6j proceeded with a high chemical yield and good enantioselectivity (Table 4, entries 13–15). The use of the high sterically hindered trityl derivative 6k did not affect product yield, but the enantioselectivity decreased slightly to er 87:13 (Table 4, entry 16). No product was obtained when the reaction was carried out using the unprotected imine 6m, recovering the starting material after 18 hours of stirring at rt (Table 4, entry 18), nor with the acetyl derivative 6l, which decomposed into several by-products (Table 4, entry 17).
Finally, when we tested the reaction between imine 6g and phenylacetylene with a lower ligand charge (10 mol%) the catalytic system proved to be effective, but we observed a decrease in both product yield and enantioselectivity, obtaining 7g in a 65% yield and 95:5 er (Table 4, entry 19). Unfortunately, using 2 equiv. of phenylacetylene and dimethylzinc instead of 4 equiv. resulted in a much slower reaction and poor chemical yield (Table 4, entry 20).
Since the 3,5-dichloroaniline moiety constitutes an important structural motif in many biologically active compounds,26 1-(2,6-dichlorobenzyl)-3-((3,5-dichlorophenyl)imino)indolin-2-one 6g was selected to continue studying the scope of the reaction with terminal alkynes (Table 5).
| a Reaction conditions: (1) 0.4 mmol of alkyne, 0.4 mmol of dimethylzinc, toluene rt; (2) ligand L2 0.02 mmol (20 mol%), toluene, rt; (3) ketimine 1g 0.1 mmol, CH2Cl2, rt 18 h. Isolated yields are given. The er values were determined by HPLC analysis on a chiral stationary phase. |
|---|

|
To our delight, our catalytic system again tolerated a wide range of terminal alkynes, including phenylacetylene derivatives bearing electron-donating and electron-withdrawing substituents at different positions of the aromatic ring, and aliphatic alkynes. All the products 8a–o were obtained with a high chemical yield and enantioselectivity, including propargylamines 8i, 8k, 8l and 8n derived from functionalized alkynes. Only the bulky 3,3-dimethylbut-1-yne led to propargylamine 8o with a lower enantioselectivity.
We next explored the scope of the reaction of terminal alkynes with isatin-derived ketimines bearing electron-donating and electron-withdrawing substituents at the aromatic ring (Table 6). Again, the enantiocontrol seemed not to be influenced by the electronic effect of these substituents and the 3-amino-3-alkynyl-oxindoles 9a–m were obtained in an excellent chemical yield and high enantioselectivity, with the exception of the nitro derivative 9f, which was obtained with a moderate enantioselectivity (89:11 er).
| Reaction conditions: (1) 0.4 mmol of alkyne, 0.4 mmol of dimethylzinc, toluene rt; (2) ligand L2 0.02 mmol (20 mol%), toluene, rt; (3) ketimine 5n–t 0.1 mmol, CH2Cl2, rt, 18 h. Isolated yields are given. The er values were determined by HPLC analysis on a chiral stationary phase. |
|---|

|
To evaluate the synthetic potential of the catalyst, we tested the scalability of this method by carrying out the reaction on a gram scale. Under the optimized reaction conditions, 1.13 g (2.5 mmol) of 6g reacted with phenylacetylene, giving a lower but still acceptable yield (1.18 g, 85% yield) of the desired product 6g with an enantioselectivity of 98:2 er.
The absolute configuration of the newly formed stereogenic centre on 8c was established by X-ray diffraction analysis and has been extended to all the other 3-amino-3-alkynyl-oxindoles 7a–k, 8a–o, and 9a–m based on mechanistic analogy.
Interestingly, a change in facial selectivity is observed in the alkynylation of the isatin-derived ketimines compared to that of isatins. In the isatins, the alkynylzinc derivative attacks the Si face of the carbonyl group, whereas in the isatin-derived ketimines the attack occurs on the Re face of the imine (Fig. 3).
 | ||
| Fig. 3 Opposite facial selectivity in the enantioselective alkynylation of isatins and isatin-derived ketimines in the presence of L2. | ||
A detailed mechanistic discussion is difficult at present due to the different coordination possibilities of the starting substrates in a bimetallic catalyst system, such as the one proposed by Noyori for the alkylation of carbonyl compounds with organozinc reagents in the presence of 1,2-aminoalcohols as chiral ligands.27 In addition, the isatin imines exist as an equilibrium mixture of E/Z isomers,28 further complicating the coordination possibilities. Further work to uncover the reaction mechanism and the application of these enantioselective reactions in other asymmetric transformations is in progress in our laboratories.
Conclusions
In summary, we have developed a common and efficient method for the preparation of chiral 3-hydroxy- and 3-amino-3-alkynyl-oxindoles via the Me2Zn-mediated addition of terminal alkynes to isatins and isatin-derived ketimines using the same chiral ligand L2. A variety of isatins and ketimine derivatives bearing electron-withdrawing or electron-donating substituents on the aromatic ring and aromatic and aliphatic terminal alkynes were investigated, which gave rise to 1,2-addition products in excellent yields and high enantioselectivities. The process could be scaled up maintaining the same efficiency.Experimental section
The experimental and analytical procedures are described in detail in the ESI.†Author contributions
E. P. developed the catalytic enantioselective alkynylations of isatins and isatin-derived ketimines, conducted most of the experiments, analysed the results and wrote the original draft, J. D. M. performed part of the experiments of the enantioselective alkynylation of isatins. J. N. conceived, designed, and supervised the project, and prepared the manuscript. C. A. conceived, designed and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Junta de Castilla y León (projects FEDER-VA115P17, and VA149G18) for financial support. The authors also thank Dr José M. Martín-Álvarez for his assistance in the determination of the X-ray structures.Notes and references
- (a) S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20–38 Search PubMed; (b) H.-p. Zhang, Y. Kamano, Y. Ichihara, H. Kizu, K. Komiyama, H. Itokawa and G. R. Pettit, Tetrahedron, 1995, 51, 5523–5528 Search PubMed; (c) S. Hibino and T. Choshi, Nat. Prod. Rep., 2001, 18, 66–87 Search PubMed; (d) H. V. Erkizan, Y. Kong, M. Merchant, S. Schlottmann, J. S. Barber-Rotenberg, L. Yuan, O. D. Abaan, T.-h. Chou, S. Dakshanamurthy, M. L. Brown, A. Üren and J. A. Toretsky, Nat. Med., 2009, 15, 750–756 Search PubMed; (e) M. Schönberger, C. Leggett, S. W. Kim and J. M. Hooker, Bioorg. Med. Chem. Lett., 2010, 20, 3103–3106 Search PubMed; (f) T. Emura, T. Esaki, K. Tachibana and M. Shimizu, J. Org. Chem., 2006, 71, 8559–8564 Search PubMed; (g) M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175–1180 Search PubMed; (h) Y. Khetmalis, M. Shivani, S. Murugesan and K. V. G. C. Sekhar, Biomed. Pharmacother., 2021, 141, 111842 Search PubMed; (i) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 Search PubMed.
- (a) P. Hewawasam, M. Erway, S. L. Moon, J. Knipe, H. Weiner, C. G. Boissard, D. J. Post-Munson, Q. Gao, S. Huang, V. K. Gribkoff and N. A. Meanwell, J. Med. Chem., 2002, 45, 1487–1499 Search PubMed; (b) T. Tokunaga, W. E. Hume, T. Umezome, K. Okazaki, Y. Ueki, K. Kumagai, S. Hourai, J. Nagamine, H. Seki, M. Taiji, H. Noguchi and R. Nagata, J. Med. Chem., 2001, 44, 4641–4649 Search PubMed; (c) J. S. Barber-Rotenberg, S. P. Selvanathan, Y. Kong, H. V. Erkizan, T. M. Snyder, S. P. Hong, C. L. Kobs, N. L. South, S. Summer, P. J. Monroe, M. Chruszcz, V. Dobrev, P. N. Tosso, L. J. Scher, W. Minor, M. L. Brown, S. J. Metallo, A. Üren and J. A. Toretsky, Oncotarget, 2012, 3, 172–182 Search PubMed; (d) J. Totobenazara, P. Bacalhau, A. A. San Juan, C. S. Marques, L. Fernandes, A. Goth, A. T. Caldeira, R. Martins and A. J. Burke, ChemistrySelect, 2016, 1, 3580–3588 Search PubMed.
- (a) S. Mohammadi, R. Heiran, R. P. Herrera and E. Marqués-López, ChemCatChem, 2013, 5, 2131–2148 Search PubMed; (b) P. Brandao and A. J. Burke, Tetrahedron, 2018, 74, 4927–4957 Search PubMed; (c) Z.-Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443–1454 Search PubMed; (d) A. Kumara and S. S. Chimni, RSC Adv., 2012, 2, 9748–9762 Search PubMed; (e) B. Yu, H. Xing, D.-Q. Yu and H.-M. Liu, Beilstein J. Org. Chem., 2016, 12, 1000–1039 Search PubMed; (f) G. Zhu, Y. Li, G. Bao, W. Sun, L. Huang, L. Hong and R. Wang, ACS Catal., 2018, 8, 1810–1816 Search PubMed.
- (a) J. Kaur and S. S. Chimni, Org. Biomol. Chem., 2018, 16, 3328–3347 Search PubMed; (b) J. Kaur, S. S. Chimni, S. Mahajana and A. Kumar, RSC Adv., 2015, 5, 52481–52496 Search PubMed; (c) P. Chauhan and S. S. Chimni, Tetrahedron: Asymmetry, 2013, 24, 343–356 Search PubMed; (d) J.-S. Yua, F. Zhoua, Y.-L. Liua and J. Zhou, Synlett, 2015, 2491–2504 Search PubMed; (e) S. Hajra, S. S. Roy, A. Biswas and S. A. Saleh, J. Org. Chem., 2018, 83, 3633–3644 Search PubMed.
- M. E. Sarciron, P. Audin, I. Delabre, C. Gabrion, A. F. Petavy and J. Paris, J. Pharm. Sci., 1993, 82, 605–609 Search PubMed.
- (a) N. Boechat, W. B. Kover, V. Bongertz, M. M. Bastos, N. C. Romeiro, M. L. G. Azevedo and W. Wollinger, Med. Chem., 2007, 3, 533–542 Search PubMed; (b) S. Chander, C.-R. Tang, A. Penta, P. Wang, D. P. Bhagwat, N. Vanthuyne, M. Albalat, P. Patel, S. Sankpal, Y.-T. Zheng and M. Sankaranarayanan, Bioorg. Chem., 2018, 79, 212–222 Search PubMed.
- P. S. Prathima, R. Bikshapathi, Y. Poornachandra, V. Himabindu, G. J. Kumar, N. Jagadeesh, C. G. Kumar and V. J. Rao, Anti-Cancer Agents Med. Chem., 2017, 17, 1963–1970 Search PubMed.
- (a) N. Kumarswamyreddy, M. Prakash, S. Jayakumar and V. Kesavan, RSC Adv., 2015, 5, 54316–54320 Search PubMed; (b) K. Laxmikeshav, A. P. Sakla, S. Rasane, S. E. John and N. Shankaraiah, ChemistrySelect, 2020, 5, 7004–7012 Search PubMed; (c) S. Yaragorla, A. Pareek and R. Dada, Adv. Synth. Catal., 2017, 359, 3068–3075 Search PubMed; (d) S. Yaragorla, R. Dada and A. Pareek, ChemistrySelect, 2018, 3, 495–499 Search PubMed; (e) S. Yaragorla, R. Dada, P. Rajesh and M. Sharma, ACS Omega, 2018, 3, 2934–2946 Search PubMed; (f) Y.-C. Lee, S. Patil, C. Golz, C. Strohmann, S. Ziegler, K. Kumar and H. A. Waldmann, Nat. Commun., 2017, 14043 Search PubMed; (g) D. Xie, X. Xu, S. Long, X.-Y. Tang and L. Wang, J. Org. Chem., 2022, 87, 7852–7863 Search PubMed; (h) T. Khan, P. Rajesh, D. Arun and S. Yaragorla, Org. Biomol. Chem., 2021, 19, 10201–10209 Search PubMed.
- (a) N. Kumarswamyreddy and V. Kesavan, Eur. J. Org. Chem., 2016, 5301–5308 Search PubMed; (b) H. J. Roh, S. Y. Kim, B. K. Min and J. N. Kim, Tetrahedron Lett., 2017, 58, 21–24 Search PubMed; (c) I. R. Siddiqui, Rahila, S. Shamim, P. Rai, Shireen, M. A. Waseem and A. A. H. Abumhdi, Tetrahedron Lett., 2013, 54, 6991–6994 Search PubMed; (d) H. J. Roh, G. Kim, S. Cho, J. Y. Ryu, J. Lee and J. N. Kim, Tetrahedron Lett., 2018, 59, 1484–1488 Search PubMed; (e) H. J. Roh, D. Y. Seo, J. Y. Ryu, J. Lee and J. N. Kim, Tetrahedron Lett., 2017, 58, 4094–4098 Search PubMed; (f) Y.-W. Xu, L. Li and X.-P. Hu, Org. Lett., 2020, 22, 9534–9538 Search PubMed; (g) H. Takada, N. Kumagai and M. Shibasaki, Org. Lett., 2015, 17, 4762–4765 Search PubMed.
- (a) B. Kalvacherla, S. Batthula, S. Balasubramanian and R. K. Palakodety, Org. Lett., 2018, 20, 3824–3828 Search PubMed; (b) S. Yaragorla, R. Dada, G. Singh, A. Pareek, M. Rana and A. K. Sharma, ChemistrySelect, 2016, 1, 6902–6906 Search PubMed; (c) H. J. Roh, J. W. Lim, J. Y. Ryu, J. Lee and J. N. Kim, Tetrahedron Lett., 2016, 57, 4280–4283 Search PubMed.
- (a) M. Chouhan, K. R. Senwar, K. Kumar, R. Sharma and V. A. Nair, Synthesis, 2014, 195–202 Search PubMed; (b) Z.-y. Wu, Z.-w. Zhang, L.-l. Ding, M. Xiang and S.-y. Luo, Tetrahedron Lett., 2020, 61, 15257711 Search PubMed; (c) G. Chen, Y. Wang, S. Gao, H.-P. He, S.-L. Li, J.-X. Zhang, J. Ding and X.-J. Hao, J. Heterocycl. Chem., 2009, 46, 217–220 Search PubMed; (d) V. K. Sing, A. Upadhyay and R. K. P. Singh, Tetrahedron Lett., 2017, 58, 156–158 Search PubMed; (e) F. Lazreg, M. Lesieur, A. J. Samson and C. S. J. Cazin, ChemCatChem, 2016, 8, 209–213 Search PubMed; (f) B. Paplal, K. Sathish, S. Nagaraju and D. Kashinath, Catal. Commun., 2020, 135, 105874 Search PubMed; (g) X.-P. Fu, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, Green Chem., 2011, 13, 549–553 Search PubMed; (h) K. Dhara, A. Kapat, T. Ghosh and J. Dash, Synthesis, 2016, 4260–4268 Search PubMed.
- Q. Chen, Y. Tang, T. Huang, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 5286–5289 Search PubMed.
- N. Xu, D.-W. Gu, J. Zi, X.-Y. Wu and X.-X. Guo, Org. Lett., 2016, 18, 2439–2442 Search PubMed.
- H. He, Z. Yang, Y. Chai, R. Wu, P. Cheng, J. Zhou and H. Zhou, Org. Lett., 2021, 23, 5739–5743 Search PubMed.
- M. Moskowitz and C. Wolf, Angew. Chem., Int. Ed., 2019, 58, 3402–3406 Search PubMed.
- S. Paria, H.-J. Lee and K. Maruoka, ACS Catal., 2019, 9, 2395–2399 Search PubMed.
- D. Jiang, P. Tang, Q. Tan, Z. Yang, L. He and M. Zhang, Chem. – Eur. J., 2020, 26, 15830–15834 Search PubMed.
- L. Chen, G. Huang, M. Liu, Z. Huang and F.-E. Chen, Adv. Synth. Catal., 2018, 360, 3497–3501 Search PubMed.
- J.-F. Chen and C. Li, Org. Lett., 2020, 22, 4686–4691 Search PubMed.
- R.-Z. Huang, Z.-C. Ma, Y. Huang and Y. Zhao, J. Org. Chem., 2023, 88, 7755–7763 Search PubMed.
- L. Yin, Y. Otsuka, H. Takada, S. Mouri, R. Yazaki, N. Kumagai and M. Shibasaki, Org. Lett., 2013, 15, 698–701 Search PubMed.
- Q. Chen, L. Xie, Z. Li, Y. Tang, P. Zhao, L. Lin, X. Feng and X. Liu, Chem. Commun., 2018, 54, 678–681 Search PubMed.
- (a) C. Andrés, R. Infante and J. Nieto, Tetrahedron: Asymmetry, 2010, 21, 2230–2237 Search PubMed; (b) R. Infante, J. Nieto and C. Andrés, Org. Biomol. Chem., 2011, 9, 6691–6699 Search PubMed; (c) R. Infante, J. Nieto and C. Andrés, Synthesis, 2012, 1343–1348 Search PubMed; (d) R. Infante, J. Nieto and C. Andrés, Chem. – Eur. J., 2012, 18, 4375–4379 Search PubMed; (e) R. Infante, A. Gago, J. Nieto and C. Andrés, Adv. Synth. Catal., 2012, 354, 2797–2804 Search PubMed; (f) R. Infante, J. M. Martín-Álvarez, C. Andrés and J. Nieto, Org. Lett., 2017, 19, 1516–1519 Search PubMed; (g) E. Prieto, R. Infante, J. Nieto and C. Andrés, Org. Biomol. Chem., 2021, 19, 3859–3867 Search PubMed.
- C. Andrés, I. González, J. Nieto and C. D. Rosón, Tetrahedron, 2009, 65, 9728–9736 Search PubMed.
- (a) E. M. Volgl, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 1570–1577 Search PubMed; (b) J. Rudolph, N. Hermanns and C. Bolm, J. Org. Chem., 2004, 69, 3997–4000 Search PubMed; (c) J. Rudolph, M. Lormann, C. Bolm and S. Dahmen, Adv. Synth. Catal., 2005, 347, 1361–1368 Search PubMed.
- (a) J. Y. Lee, Y. S. Shin, S. Jeon, S. I. Lee, S. Noh, J.-E. Cho, M. S. Jang, S. Kim, J. H. Song, H. R. Kim and C. M. Park, Bioorg. Med. Chem. Lett., 2021, 39, 127885 Search PubMed; (b) Y. Zhang, K. T. Zhao, S. G. Fox, J. Kim, D. R. Kirsch, R. J. Ferrante, R. I. Morimoto and R. B. Silverman, J. Med. Chem., 2015, 58, 5942–5949 Search PubMed; (c) J.-P. Wu, J. Emeigh, D. A. Gao, D. R. Goldberg, D. Kuzmich, C. Miao, I. Potocki, K. C. Qian, R. J. Sorcek, D. D. Jeanfavre, K. Kishimoto, E. A. Mainolfi, G. Nabozny, Jr., C. Peng, P. Reilly, R. Rothlein, R. H. Sellati, J. R. Woska, Jr., S. Chen, J. A. Gunn, D. O'Brien, S. H. Norris and T. A. Kelly, J. Med. Chem., 2004, 47, 5356–5366 Search PubMed; (d) A. C. Pappas and D. J. Fisher, Pestic. Sci., 1979, 10, 239–246 Search PubMed.
- (a) M. Yamakawa and R. Noyori, J. Am. Chem. Soc., 1995, 117, 6327–6335 Search PubMed; (b) M. Yamakawa and R. Noyori, Organometallics, 1999, 18, 128–133 Search PubMed.
- Q.-X. Guo, Y.-W. Liu, X.-C. Li, L.-Z. Zhong and Y.-G. Peng, J. Org. Chem., 2012, 77, 3589–3594 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, NMR spectra data (1H and 13C) and chiral HPLC chromatograms for new synthesized compounds. CCDC 1872814 and 2097363. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01023f |
| This journal is © The Royal Society of Chemistry 2023 |