Polynitro-functionalized 4-phenyl-1H-pyrazoles as heat-resistant explosives†‡
Received
15th June 2023
, Accepted 20th July 2023
First published on 20th July 2023
Abstract
A new class of heat-resistant explosives was synthesized by coupling N-methyl-3,5-dinitropyrazole with polynitrobenzene moieties through carbon–carbon bonds. Simple Pd(0)-based Suzuki cross-coupling reactions between N-methyl-4-bromo-3,5-dinitropyrazole and 4-chloro/3-hydroxy-phenylboronic acid followed by nitration, amination and oxidation lead to the formation of C–C connected penta-nitro energetic derivatives 6 and 10. Various other energetic derivatives, such as amino (5), azido (7), nitramino (8) and energetic salts (11–14), were also explored to fine-tune their properties. All the compounds were thoroughly characterized using IR, NMR [1H, 13C{1H}], differential scanning calorimetry (DSC), elemental analysis, and HRMS studies. Compounds 5, 10 and 13 were further characterized through 15N NMR, and the crystal structures of 6 and 14 were confirmed through single-crystal X-ray diffraction studies. The physicochemical and energetic properties of all the energetic compounds were explored. Most of the synthesized compounds demonstrated high thermal stability (decomposition temperature Tdec > 250 °C), among which compounds 5 and 6 showed excellent thermal stability, having decomposition temperatures above 300 °C. The excellent thermal stability, acceptable sensitivity and good energetic properties of compounds 5, 6, 10 and 13 make them promising heat-resistant explosives. Furthermore, these compounds were found to be more thermally stable than the known N-methyl-3,5-dinitropyrazole-based and C–N coupled 3,4,5-trinitrobenzene-azole-based energetic compounds.
Introduction
With the advancement of our civilization, the demand for more natural resources (coal, gas, oil, etc.) by humankind, as well as interest in the quest for space exploration, has been increasing rapidly. To meet these demands, new heat-resistant explosives must be developed with excellent thermal stability (decomposition temperature Tdec > 250 °C), along with improved energetic performance and sensitivity.1–5 Traditional heat-resistant explosives such as hexanitrostilbene (HNS) have excellent thermal stability (Tdec > 318 °C), but are less powerful and are highly sensitive to impact (impact sensitivity IS = 5 J).6 There are a few other polynitrobenzene-based heat-resistant explosives like 2,4,6-triamino-1,3,5-trinitrobenzene (TATB, Tdec = 350 °C), 2,6-bis(picrylamino)-3,5-dinitropyridine (PYX, Tdec = 325 °C), hexanitrobiphenyl (HNBP, Tdec > 250 °C), etc.7–10 However, it is observed that due to high carbon content, most of these polynitrobenzene-based energetic compounds exhibit poor energetic performance.11,12 Recently, there has been a surge in the number of new energetic compounds based on nitrogen and oxygen-rich heterocyclic ring systems like azoles, oxadiazoles, pyrazine, tetrazene, etc.13–22 Energetic materials with such nitrogen-based heterocyclic rings as their backbone are highly endothermic and considered environmentally friendly as they release non-toxic dinitrogen (N2) gas after their decomposition. Therefore, recently, several heat-resistant explosives based on polynitrobenzene moieties connected to nitrogen-rich heterocyclic rings have been reported.23,24 Such hybrid systems have relatively high densities and high heats of formation, leading to improved detonation and sensitivity properties.
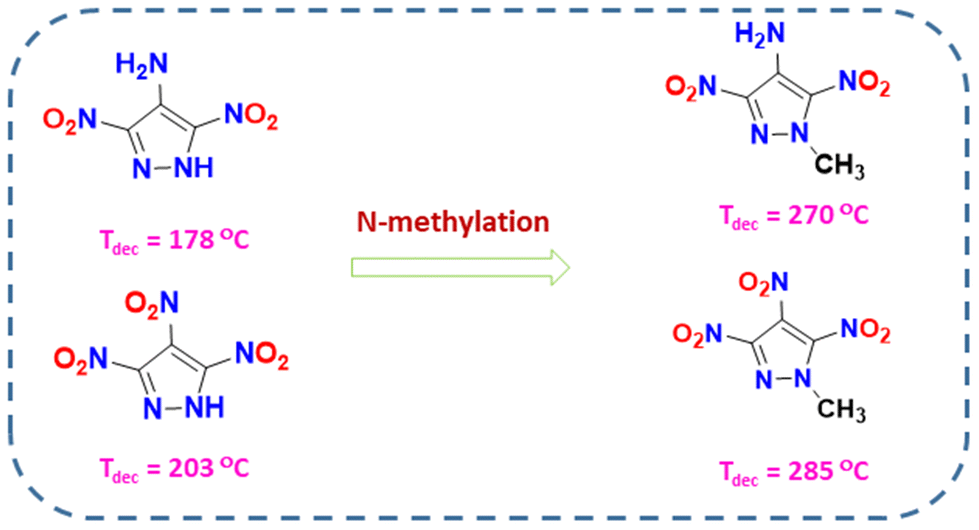
In the last decade, most advanced energetic materials have been based on five-membered nitrogen-rich heterocyclic systems (azoles).25–31 Among azoles, pyrazole-based energetic compounds are particularly in demand due to their stability and easy derivatization at the three carbon atoms in the ring.32–36 4-Substituted-3,5-dinitropyrazoles with various substituents at the 4th position (e.g., –NH2, –NO2, –N3, –NNO2, etc.) are the most common and belong to different classes of energetic materials.37–39 Energy and stability properties of these compounds can be fine-tuned by changing the explosophores at the 4th position and by various N-functionalization strategies. Shreeve and coworkers have investigated a series of 4-substituted-3,5-dinitropyrazoles where N-methylation and N-amination efficiently enhanced the overall stability (thermal and insensitivity). N-methylation specifically facilitates significant improvements in thermal stability (Fig. 1).37 Therefore, the N-methylated 3,5-dinitropyrazole skeleton might be a desired framework for achieving increased thermal stability and improved energetic performance.
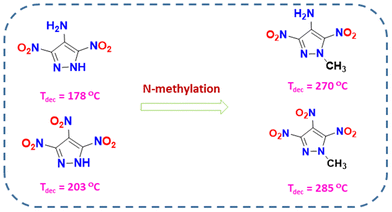 |
| | Fig. 1 Effect of N-methylation on 4-substituted-3,5-dinitropyrazole derivatives. | |
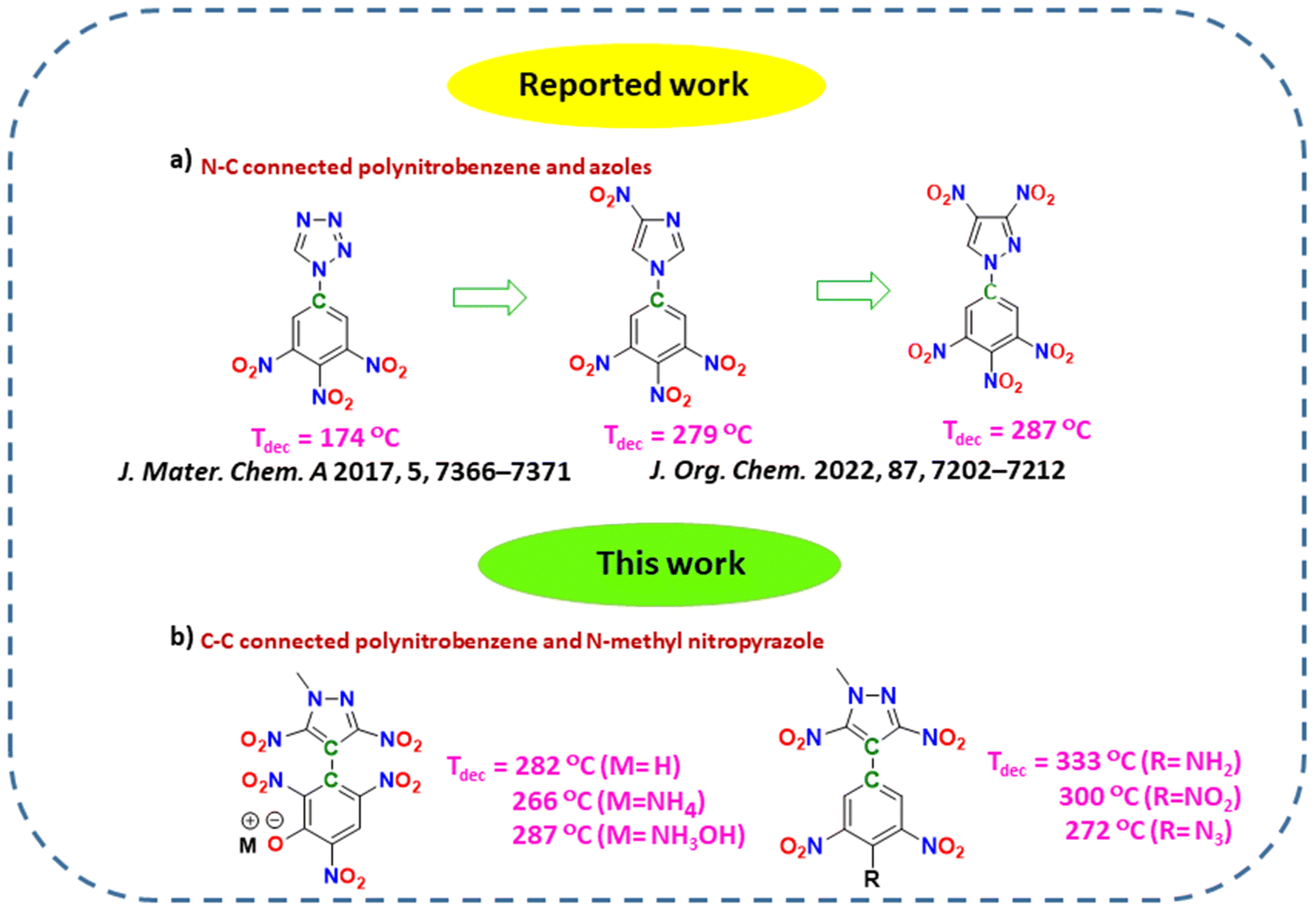
In recent years, compounds having the 3,4,5-trinitrobenzene moiety and different azoles connected via C–N bonds have been reported, and it was observed that as we move from tetrazole to pyrazole, there is a significant improvement in thermal stability (Fig. 2).23,24 Our group has recently reported various heat-resistant energetic compounds based on dinitropyrazole, and we have shown that C–C connected compounds are more thermally stable than C–N connected compounds.40 Keeping all these factors in mind, in this work, we have decided to connect a N-methyl-3,5-dinitropyrazole moiety with various polynitrobenzene moieties via C–C bonds. Other explosophores like –OH, –NH2, –N3, and –NHNO2 were also introduced into the polynitrobenzene skeletons to modify the properties further. Additionally, a hydroxy moiety in 2,4,6-trinitrophenol in 10 allowed the formation of various energetic salts with decreased mechanical sensitivity. Most of the trinitrobenzene–dinitropyrazole-based compounds reported in this work were found to have excellent thermal stability, good physical stability and detonation properties. Compounds 5 (Tdec = 333 °C) and 6 (Tdec = 300 °C) showed overall properties that were better than those of the commonly used heat-resistant explosive, HNS. Furthermore, these compounds were found to be more thermally stable among the known N-methyl-3,5-dinitropyrazole-based and C–N coupled 3,4,5-trinitrobenzene-azole-based energetic compounds.
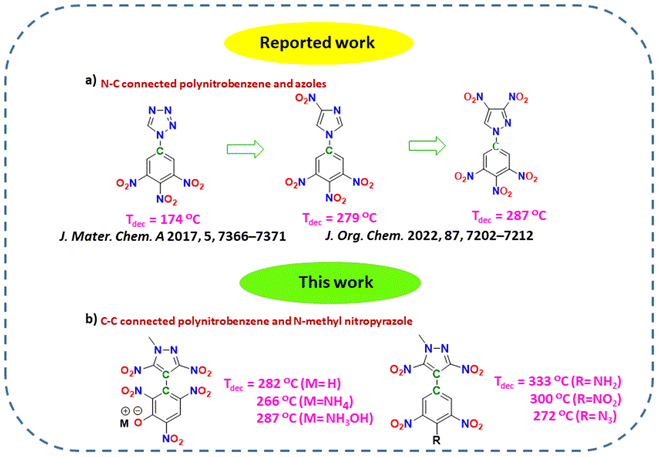 |
| | Fig. 2 (a) Comparison of thermal stability of N–C connected polynitrobenzene and azole-based compounds. (b) Structures of thermally stable C–C connected polynitrobenzene and N-methyl-3,5-dinitropyrazole-based compounds. | |
Results and discussion
Synthesis
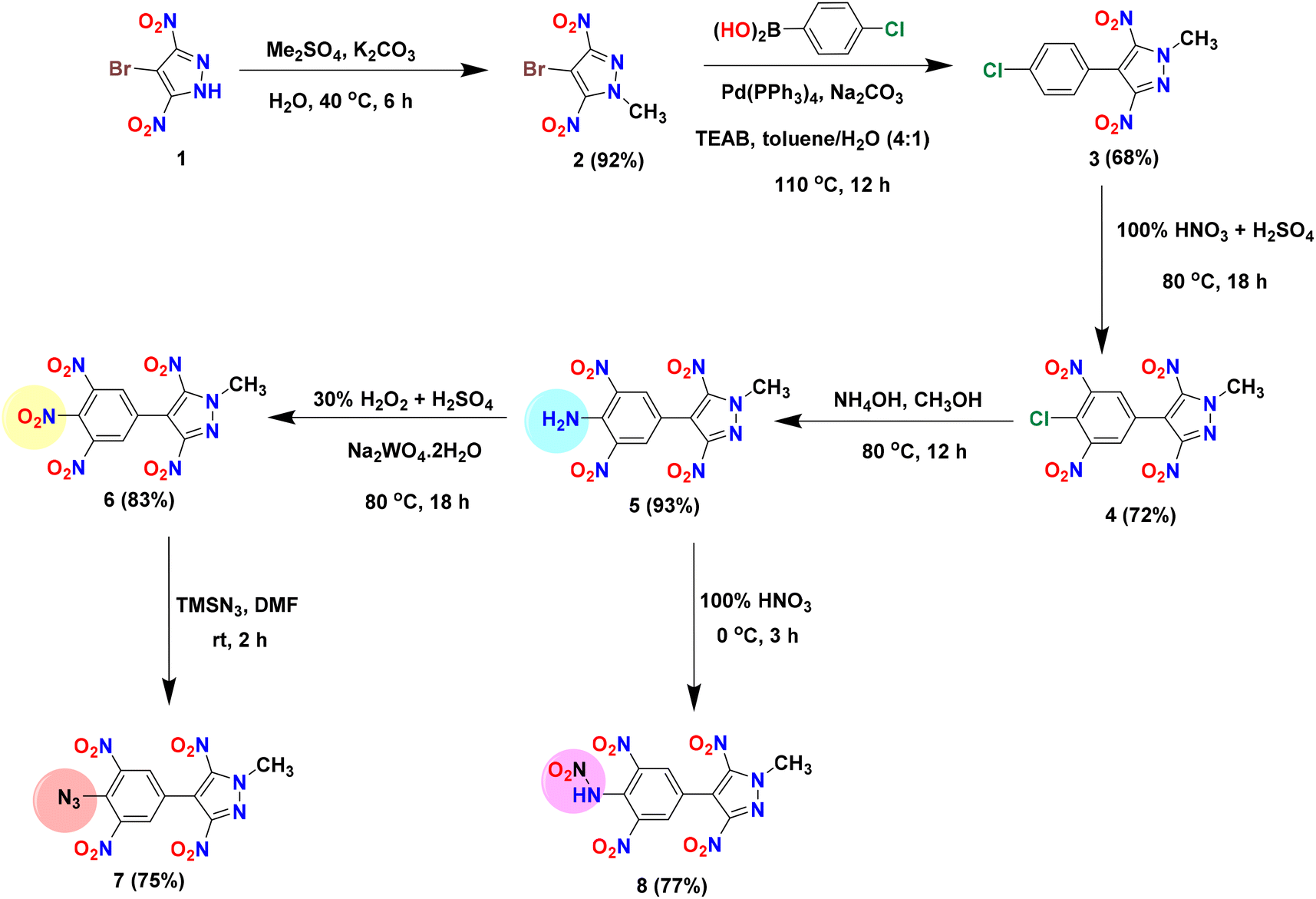
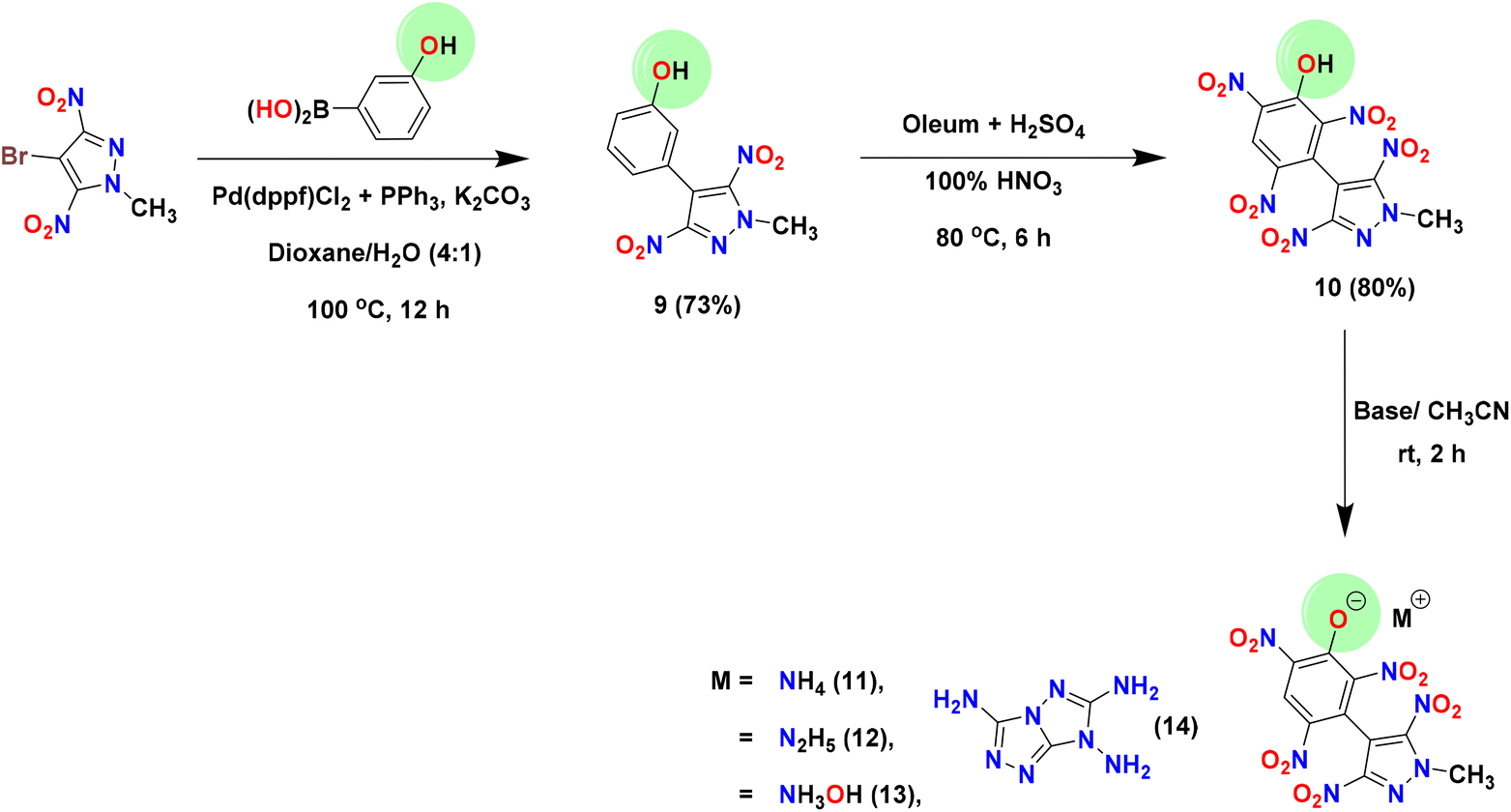
The precursor, 4-bromo-3,5-dinitropyrazole (1), was prepared according to literature procedures.41 Dimethyl sulfate was used to methylate the acidic NH site in 1, affording the electrophilic coupling partner 4-bromo-1-methyl-3,5-dinitro-1H-pyrazole (2) in 92% yield. Compound 2 was used as an organohalide for the Pd(0)-catalyzed Suzuki–Miyaura cross-coupling reaction with 4-chlorophenylboronic acid and 3-hydroxyphenylboronic acid to afford C–C coupled products 3 and 9 in 68% and 73% yields, respectively. The optimized yields for 3 and 9 were observed with Pd(PPh3)4 and Pd(dppf)Cl2/PPh3 [dppf = 1,1′-bis(diphenylphosphino)ferrocene] as catalysts, respectively. To convert the 4-chlorophenyl moiety of 3 to a 3,4,5-trinitrophenyl moiety (compound 6), the first nitration of 3 was carried out using a nitrating mixture (98% H2SO4 + 100% HNO3) to give 4 in 72% yield. Subsequently, amination (using 28% aq. ammonia) followed by oxidation with 30% H2O2 in the presence of sodium tungstate resulted in compounds 5 (93%) and 6 (83%), respectively. The azido derivative (compound 7) was obtained by the reaction of 6 with trimethylsilyl azide (TMSN3) in DMF, whereas the nitramino derivative (compound 8) was prepared by the reaction of 5 with 100% HNO3 (Scheme 1). In contrast to compound 3, the 3-hydroxyphenyl derivative (compound 9) was converted to the 2,4,6-trinitrophenol derivative (compound 10) in a single step (in 80% yield) using oleum and a nitrating mixture. Since compound 10 has an acidic phenolic OH, further energetic salts 11–14 were prepared by reacting 10 with one equivalent of various nitrogen-rich bases in an acetonitrile solution (Scheme 2).
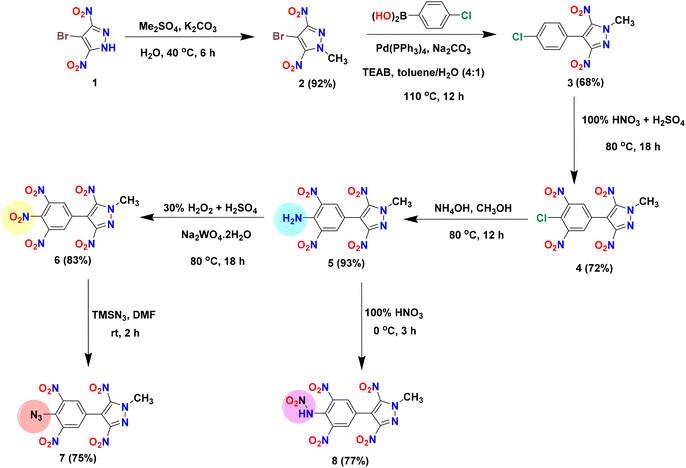 |
| | Scheme 1 Synthesis of compounds 2–8. | |
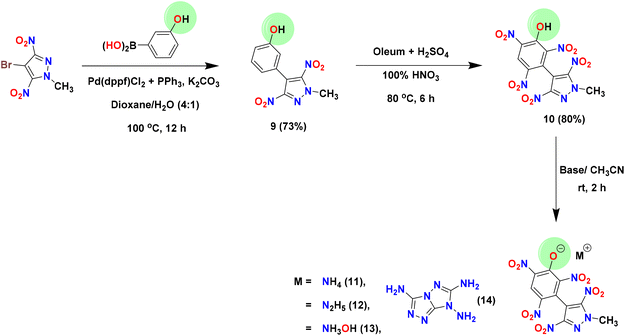 |
| | Scheme 2 Synthesis of compounds 9–14. | |
Spectral analysis of compounds 3–14
All the compounds prepared in this study were thoroughly characterized by NMR [1H, 13C{1H}, and, in a few cases, 15N NMR] spectra, IR, HRMS and elemental analysis studies. Signals corresponding to the methyl group in 3–14 were observed in the range of 4.28–4.38 ppm in the 1H NMR spectra, and 41.19–43.71 ppm in the 13C{1H} NMR spectra. In the 1H NMR spectra, the peak corresponding to the NH2 protons in 5 was observed at 8.59 ppm, whereas the peak due to the OH group in 9 was observed at 9.63 ppm. The presence of a labile proton (OH) in 10 was confirmed by a D2O exchange experiment, where the additional peak due to HOD was observed at 3.86 ppm (ESI Fig. 19‡). In the 1H NMR spectra of the neutral pentanitro derivatives, the signal corresponding to two phenyl protons in 6 (9.13 ppm) was found to be slightly deshielded compared to that of the phenyl proton of 10 (8.88 ppm). Signals corresponding to the phenyl proton in salts 11–14 were found to be slightly deshielded (8.90–8.99 ppm) compared to that of neutral compound 10 (8.88 ppm). In the 13C{1H} NMR spectra, peaks due to aromatic carbons in 3–14 were seen in the 107.74–161.22 ppm range.
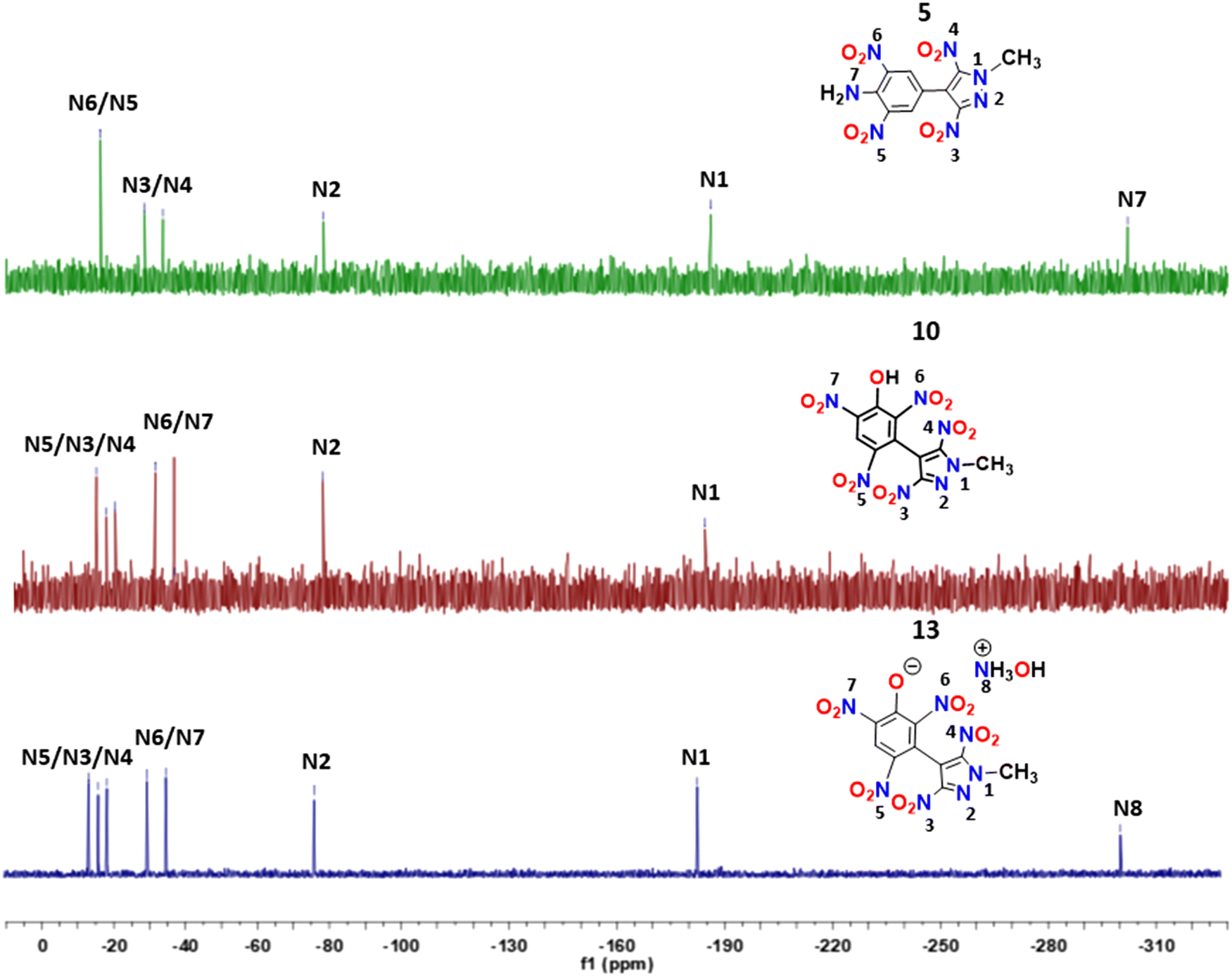
The 15N NMR spectra of compounds 5, 10 and 13 were recorded in DMSO-d6, and the chemical shift measurements were done by taking nitromethane as an external standard. The signals corresponding to chemically non-equivalent nitrogens were identified by comparing them with the analogous compounds previously reported in the literature.11–15 The methyl-substituted nitrogen atoms (N1) of the pyrazole ring in these compounds were found to be shielded (−183.80 to −186.23 ppm) compared to the other ring nitrogen (N2) of the pyrazole ring (−76.72 to −78.41 ppm). In compound 5, the signal corresponding to the nitro groups (N5/N6) present in the benzene moiety was found to be downfield shifted at −16.33 ppm compared to the signals (−28.62 and −33.71 ppm) of the nitro groups (N3/N4) present on the pyrazole moiety. In compounds 10 and 13, signals corresponding to the pyrazole nitro groups were observed in the range of −16.45 to −18.91 ppm, slightly deshielded compared to pyrazole nitro groups of 5. The 15N NMR signals due to nitro groups attached to the benzene ring in compounds 10 and 13 resonated in the range of −35.32 to −13.54 ppm. The resonance corresponding to NH2 (N7) in 5 and hydroxylammonium (N8) moieties in 13 were found to be the most shielded at −302.10 ppm and −301.99 ppm, respectively (Fig. 3).
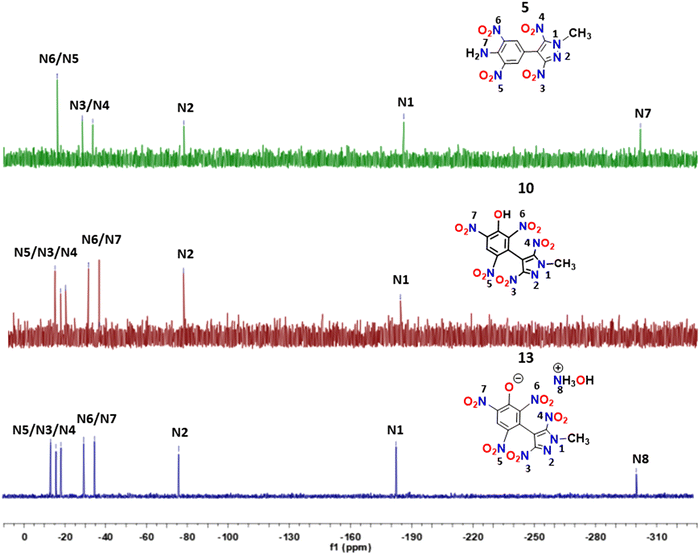 |
| | Fig. 3
15N NMR spectra of compounds 5, 10 and 13. | |
Crystal structure
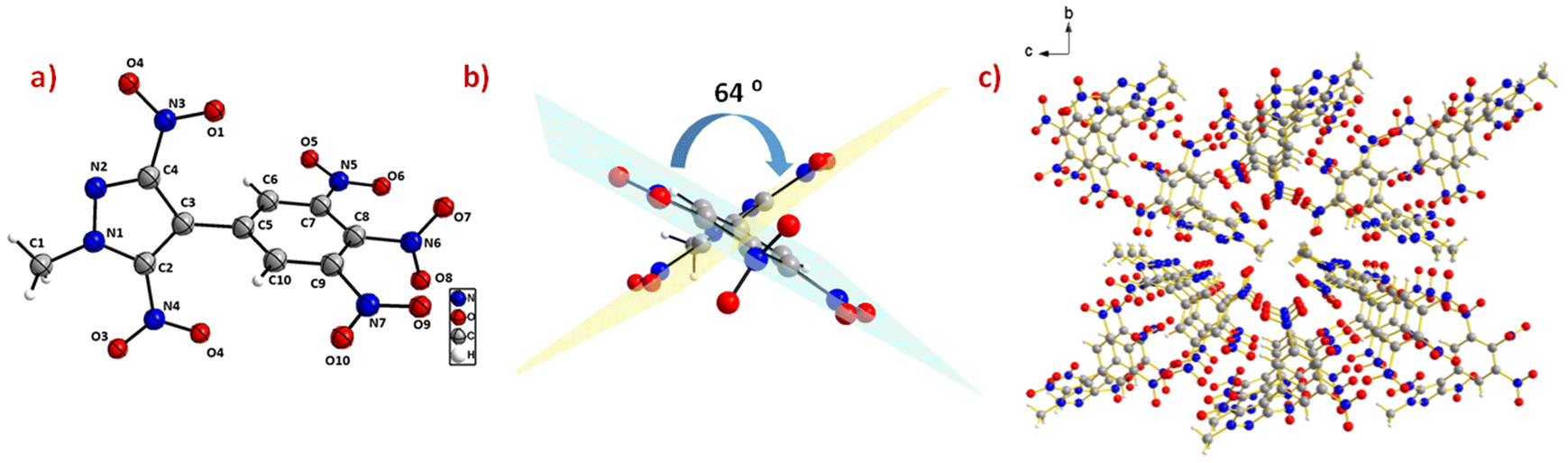
Suitable single crystals of compounds 6 and 14 were grown by slow evaporation of their saturated solutions in acetonitrile/water and water, respectively. The crystal structures and packing diagrams of 6 and 14 are depicted in Fig. 4 and 5. Detailed information about the crystallographic data of these crystals is provided in the ESI.‡ Compound 6 crystallizes in the monoclinic space group P21/n with a crystal density of 1.72 gcm−3. The pyrazole and benzene rings are found to be non-coplanar with a dihedral angle of 64.3(8)°. In the benzene moiety, the nitro groups connected to the carbons C(7) and C(9) are in the plane of the benzene, whereas the nitro group connected to the C(8) atom is almost perpendicular to the plane of the benzene ring, with an angle of 83.1(3)°. The C–C bond length between the two rings, C(3)–C(5), is 1.48(3) Å. In the phenyl ring, all C–C bond lengths lie in the range of 1.38–1.39 Å, whereas the C–C bond lengths in the pyrazole ring lie between 1.38–1.40 Å (Fig. 4).
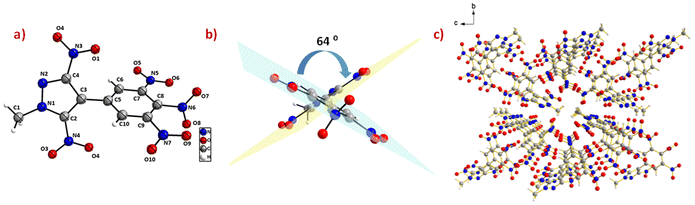 |
| | Fig. 4 (a) Thermal ellipsoid plot (50%) and labelling scheme for 6. (b) Dihedral angle between the two rings. (c) Ball and stick packing diagram view. | |
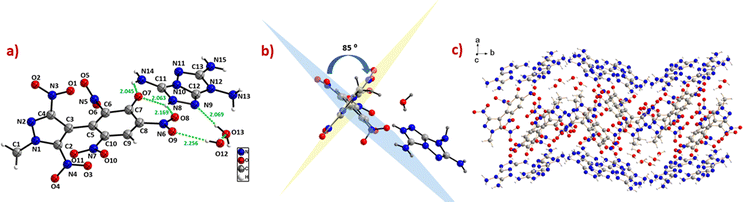 |
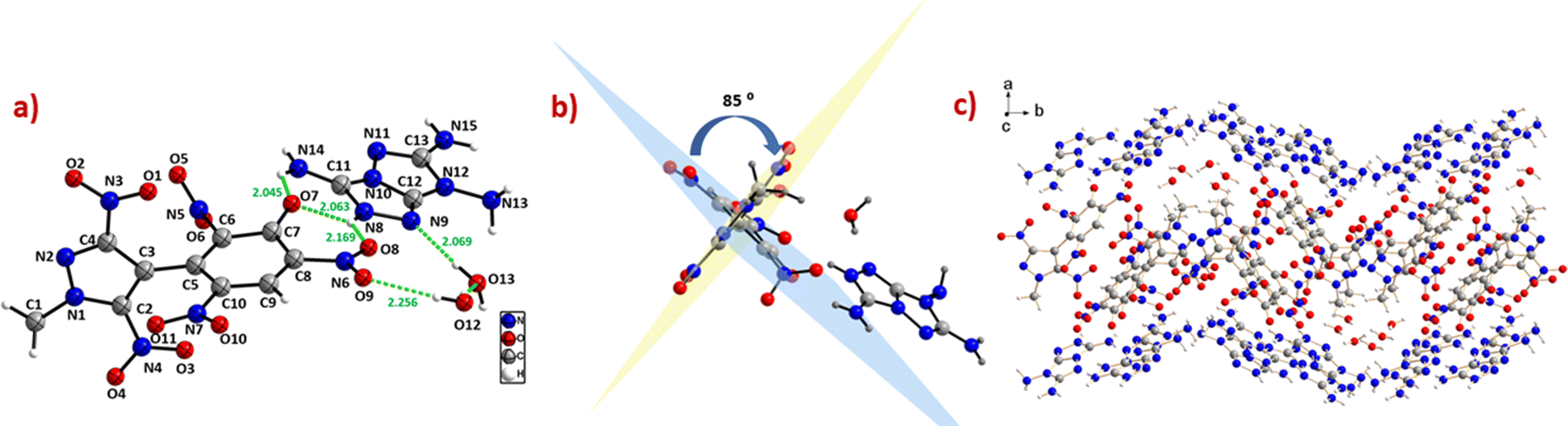
| | Fig. 5 (a) Thermal ellipsoid plot (50%) and labelling scheme for 14. (b) Dihedral angle between the two rings. (c) Ball and stick packing diagram view. | |
Compound 14 belongs to the monoclinic space group P121/c1 with a crystal density of 1.75 g cm−3. Compound 14 crystallizes as a dihydrate, where the water molecules contributed to the formation of hydrogen bonds [O(12)–H(12)B⋯O(9), 2.256 Å, and O(13)–H(13)A⋯N(9), 2.069 Å, as depicted in Fig. 5a]. Additionally, the extensive hydrogen bonding interactions between the 7H-[1,2,4]triazolo[4,3-b][1,2,4]triazole-3,6,7-triamine (TATOT) cation and anion (N(14)–H(14)B⋯O(7), 2.045 Å; N(8)–H(8)⋯O(7), 2.063 Å; N(8)–H(8)⋯O(8), 2.169 Å) could be seen in the single crystal of compound 14. The dihedral angle between the mean planes of the phenyl and pyrazole rings is 84.7(9)°, indicating that both rings are almost perpendicular. In the phenyl moiety, the nitro groups attached to C(8) and C(10) are almost in the plane of the benzene ring, whereas the nitro group connected to C(6) is twisted out of the plane, with an angle of 68.1(1)°. The C–C bond length between the two rings in 14 is 1.49(3)° Å, comparable to 6. In the phenyl ring, the C–C bond lengths lie between 1.38–1.44 Å, whereas in the pyrazole ring, the C–C bond lengths lie between 1.37–1.40 Å. Multiple inter- and intramolecular hydrogen bonding and mixed stacking interactions were observed in the crystal packing of 14 (Fig. 5).
Hirshfeld surface analysis and NCI analysis
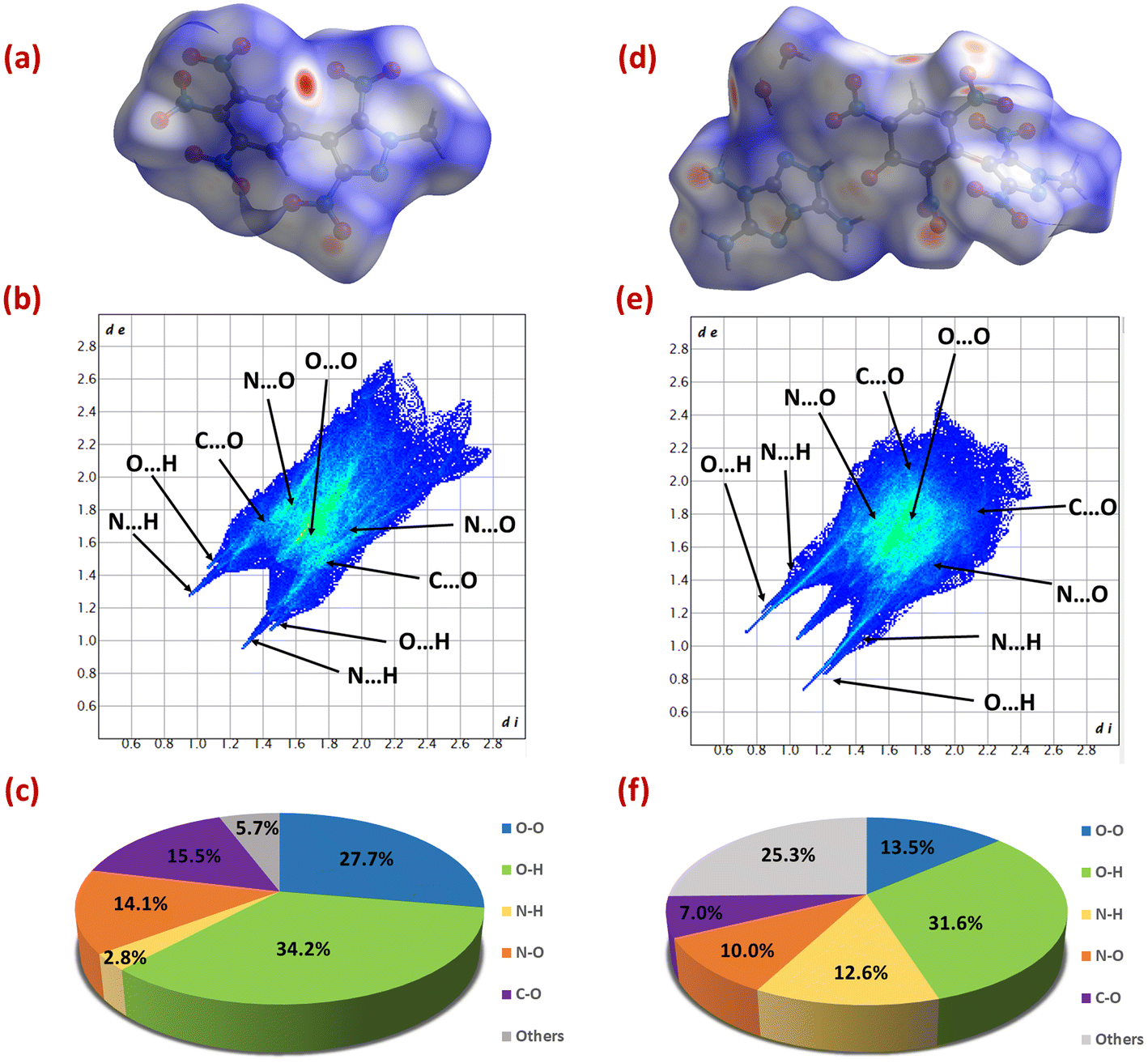
To explore additional information about structure–property relationships, Hirshfeld surface analysis and 2D fingerprint plots were investigated for 6 and 14 using CrystalExplorer 17.5 software.42 The red and blue regions on the Hirshfeld surface represent high and low close-contact interatomic interactions, respectively. In compounds 6 and 14, the red dots are mainly due to hydrogen bonding interactions (O⋯H and N⋯H), contributing 37% and 44.2% of the total interactions, respectively. These hydrogen bonding interactions can be seen in the form of spikes on the 2D fingerprint plots of 6 and 14. In the case of 6, a high abundance of O⋯O interactions were found, and they contributed 27.7% of the total interactions. The O⋯O interactions are a significant indicator of increased sensitivity in explosives. As this proportion increases, more nitro groups are exposed to the surface, increasing the chances of explosion.43,44 Therefore, despite having a high percentage of hydrogen bonding interactions, 6 (IS: 16 J, FS: 240 N) has a moderate sensitivity value. Whereas the high percentage of hydrogen bonding interactions in 14 supports its excellent physical stability (Fig. 6).
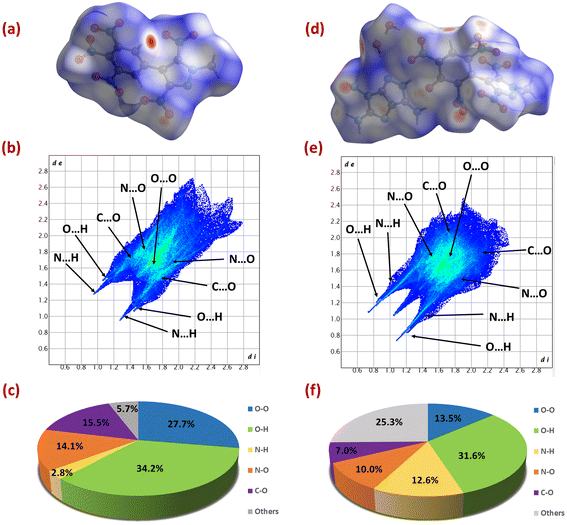 |
| | Fig. 6 Hirshfeld surfaces (a and d), fingerprint plots (b and e) and populations (c and f) of the close contacts of 6 and 14, respectively. | |
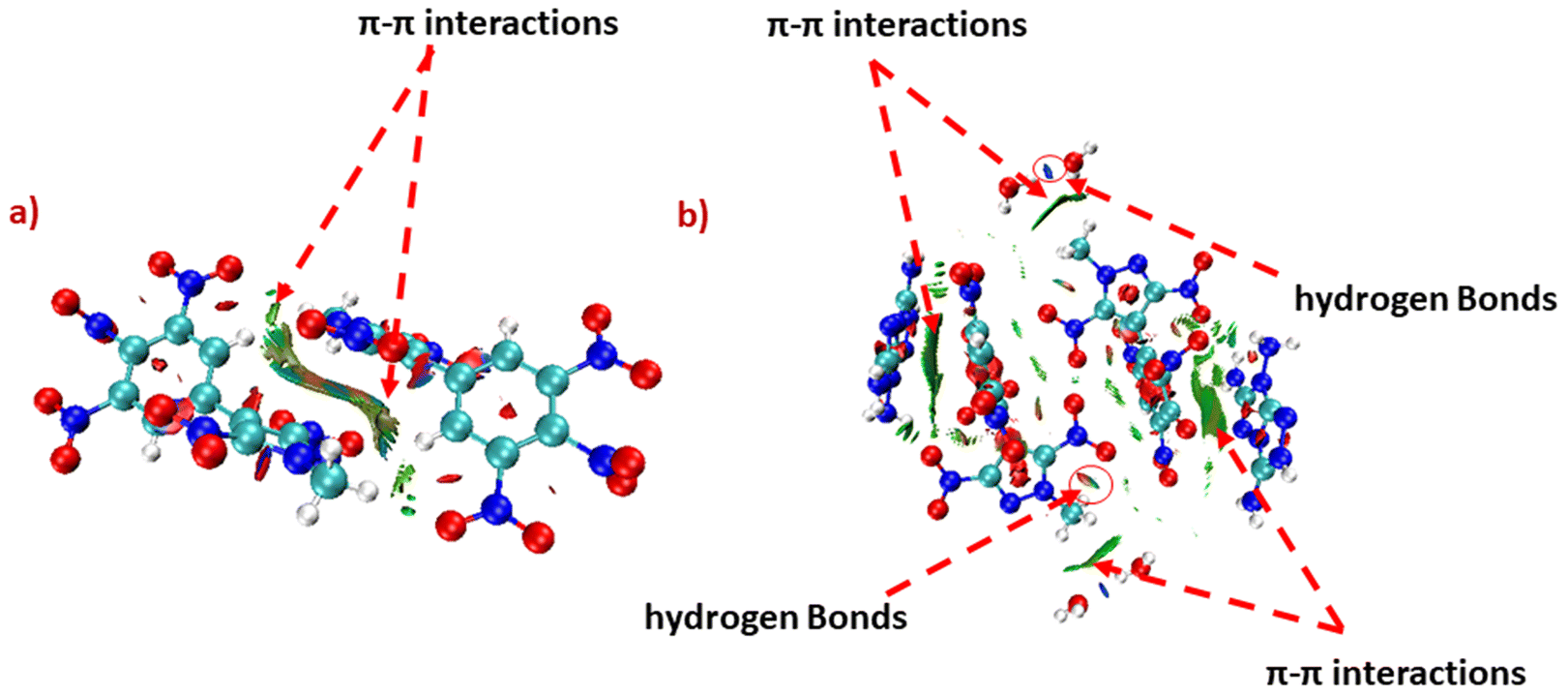
To correlate physical stability to the intramolecular interactions of energetic compounds, the non-covalent interaction analysis for compounds 6 and 14 was studied using Multiwfn software, and plots were visualized with the help of VMD software.45 The large green isosurface area between the two molecules of compounds 6 and 14 indicates strong π–π interactions. Apart from that, the green and blue ellipses can be seen in 14, which indicates hydrogen bonding. However, these hydrogen bonding interactions are not visible in 6. The presence of strong π–π interactions provides excellent thermal stability to 6 (Tdec = 300 °C). Whereas, due to the presence of strong π–π interactions as well as intramolecular hydrogen bonding, 14 exhibits insensitivity (IS > 40 J, FS > 360 N) towards external stimuli, as well as an acceptable thermal stability (Tdec = 235 °C) (Fig. 7).
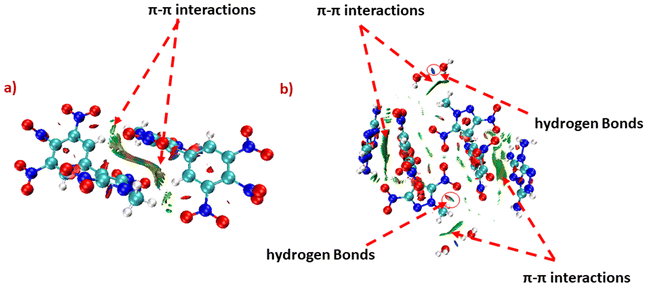 |
| | Fig. 7 Noncovalent interaction (NCI) analysis for compounds 6 (a) and 14 (b). | |
Electrostatic potential (ESP) and bond dissociation energy (BDE) studies
To explain the effect of different explosophores on the mechanical sensitivity of neutral compounds 5–8 and 10, their electrostatic potential (ESP) was studied based on an optimized crystal structure derived from the Gaussian 09 program and Multiwfn software. The red and blue areas on the surface of the ESP maps correspond to local maxima and minima ESP values, respectively (Fig. 8). According to the principle, compounds with higher negative charge accumulation result in lower mechanical sensitivity. From the ESP-mapped diagrams (Fig. 8a–d) of compounds 5–8, the order of negative charge accumulation near the phenyl C–NO2 bonds followed the sequence of 7 < 8 < 6 < 5, indicating that the azido derivative (compound 7) is the most sensitive. In contrast, the amino derivative (compound 5) is the least sensitive, which is consistent with the experimental results. A very significant blue area (electronegative region) and large negative charge accumulations can be seen in the case of 10 (IS: 32 J, FS: 360 N), which suggests its lower mechanical sensitivity. Furthermore, to support the thermal stability of synthesized compounds 5–8, we have computed the bond dissociation energy (BDE) of the longest C–NO2 bonds. The higher BDE value indicates more energy required for the bond rupture, which reveals better bond strength. Previous reports state that the BDE of the weakest bond in an energetic material should be higher than 80–120 kJ mol−1 to have better thermostability and be less prone to pyrolysis initiation.21–23 The bond lengths of the longest C–NO2 bond in compounds 5–8 are 1.4547, 1.4823, 1.4734, and 1.4757 Å, respectively; their corresponding BDEs are 293.39, 219.69, 255.55, and 234.07 kJ mol−1. The BDE of the N–NO2 bond in compound 8 is 140.53 kJ mol−1. All the compounds reveal BDEs higher than 120 kJ mol−1, indicating their higher stability, which allows them to withstand thermal initiation and supports the DSC data.
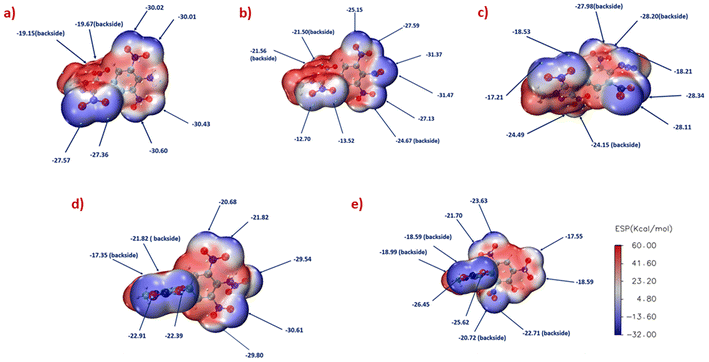 |
| | Fig. 8 ESP-mapped molecular vdW surfaces of compounds 5(a), 6(b), 7(c), 8(d) and 10(e). | |
Thermal stability and sensitivity
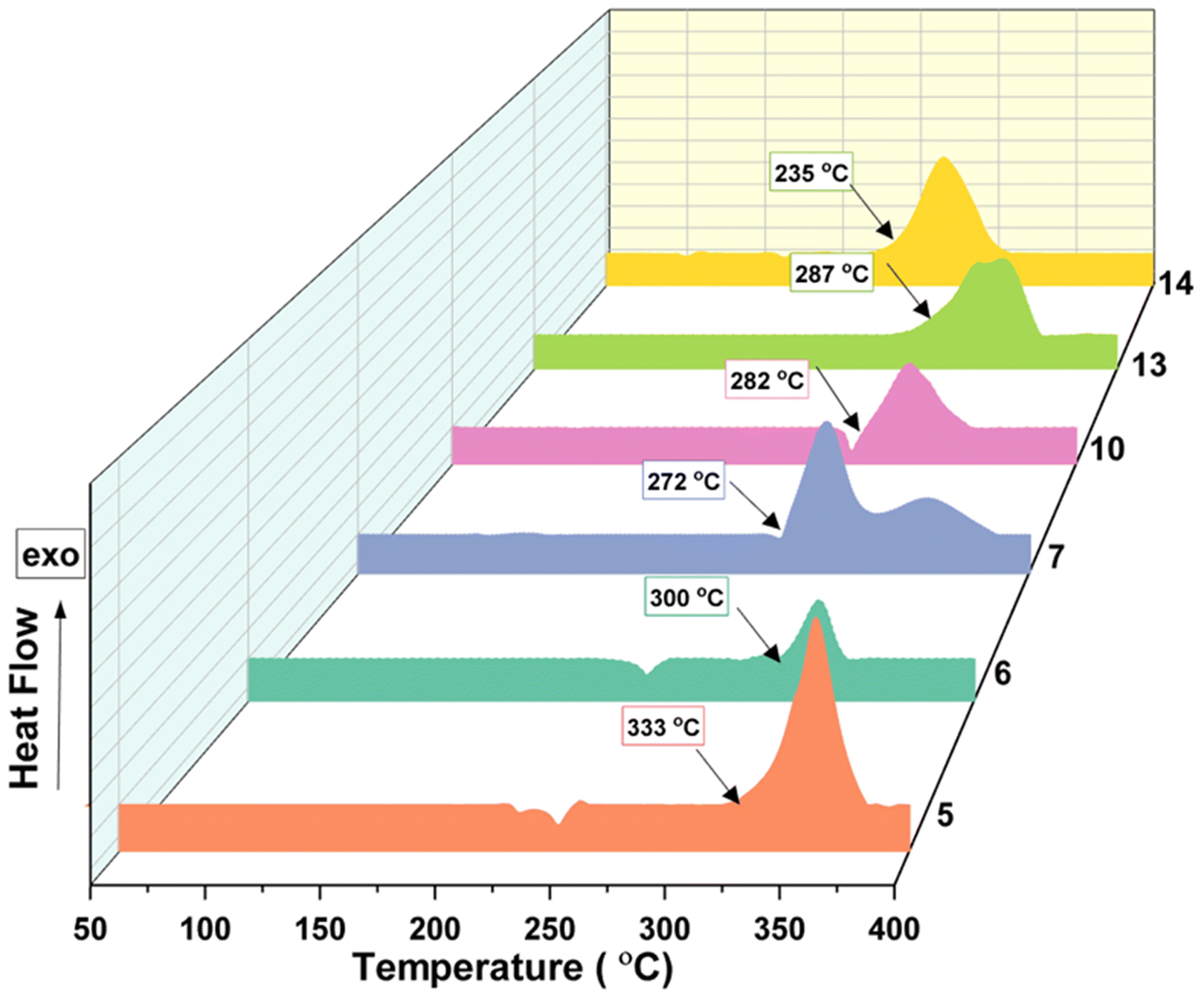
The thermal behaviour of compounds 5–8 and 10–14 was evaluated using differential scanning calorimetry (DSC) at the heating rate of 5 °C min−1. Compounds 5 (Tdec = 333 °C) and 6 (Tdec = 300 °C) exhibit excellent thermal stability comparable to that of the traditionally used heat-resistant explosive, HNS (Tdec = 318 °C). The azido derivative 7 (Tdec = 272 °C) also showed a good decomposition temperature, whereas the nitramine derivative 8 (Tdec = 193 °C) showed relatively lower thermal stability. The neutral compound 10 (Tdec = 282 °C), along with its salts 11 (Tdec = 266 °C) and 13 (Tdec = 287 °C), exhibits a decomposition temperature that is higher than RDX (Tdec = 207 °C) and approaches the value of TNT (Tdec = 295 °C). Compared to other salts, the decomposition temperature of the hydrazinium salt, 12 (Tdec = 172 °C) was relatively low, whereas the TATOT salt, 14 (Tdec = 235 °C) exhibits acceptable thermal stability (Fig. 9).
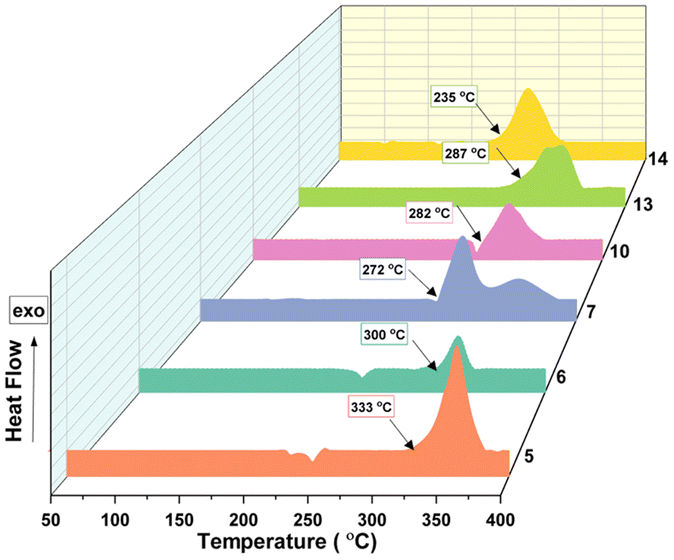 |
| | Fig. 9 DSC plots of compounds 5–14 at a heating rate of 5 °C min−1. | |
The physical stability of compounds 5–8 and 10–14 towards impact and friction was measured using a standard BAM fall hammer and a BAM friction tester, respectively. Compounds 5 and energetic salts 11–14 were found to be insensitive toward impact and friction (IS > 40 J, FS > 360 N). Compounds 6 (IS: 16 J, FS: 240 N), 8 (IS: 10 J, FS: 120 N) and 10 (IS: 32 J, FS > 360 N) showed moderate sensitivity, whereas the azido derivative 7 (IS: 5 J, FS: 60 N) was found to be the most sensitive.
Physicochemical and energetic performance
Table 1 summarizes the physicochemical properties and energetic performance of compounds 5–14. The densities of all the compounds were measured at room temperature (25 °C) using a helium gas pycnometer, and the values ranged from 1.68 (5)–1.78 (10) g cm−3. The heats of formation for all the energetic compounds were calculated using Gaussian 09 program suite. Compounds 5–8 have high positive ΔHf values that vary between 122.5 and 534.0 kJ mol−1, and 7 has the highest ΔHf because of the azido functionality. Due to a higher oxygen content in compounds 10–13, they exhibit negative heats of formation (ΔHf) ranging from −244.4 kJ mol−1 to −76.0 kJ mol−1, whereas 7H-[1,2,4]triazolo[4,3-b][1,2,4]triazole-3,6,7-triamine (TATOT) salt 14 (ΔHf = 255.8 kJ mol−1) showed a positive heat of formation. The detonation performance of all the compounds was calculated using EXPLO5 (version 6.06).46 These properties were calculated based on their measured density and heat of formation values. The value of detonation velocities and pressures range from 7059–7692 m s−1 and 19.6–24.7 GPa, respectively. All of compounds 5–14 are found to be denser and more energetic than TNT (Dv = 6824 m s−1 and P = 19.4 GPa). Also, apart from 5, all the other compounds are more energetic than HNS (Dv = 7164 m s−1 and P = 21.5 GPa).47
Table 1 Physicochemical and energetic properties of compounds 5–8 and 10–14
| Compd. |
ρ
(g cm−3) |
D
v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (m s−1) b (m s−1) |
P
(GPa) |
ΔHfd (kJ mol−1) |
T
me (°C) |
T
decf (°C) |
ISg (J) |
FSh (N) |
|
Density measured using a gas pycnometer at 25 °C.
Calculated detonation velocity.
Calculated detonation pressure.
Heat of formation.
Melting point (peak).
Temperature of decomposition (onset).
Impact sensitivity.
Friction sensitivity.
|
|
5
|
1.68 |
7059 |
19.6 |
122.5 |
246 |
333 |
>40 |
>360 |
|
6
|
1.74 |
7607 |
24.2 |
152.6 |
242 |
300 |
16 |
240 |
|
7
|
1.72 |
7552 |
23.2 |
534.0 |
— |
272 |
5 |
60 |
|
8
|
1.75 |
7692 |
24.4 |
196.3 |
— |
193 |
10 |
120 |
|
10
|
1.78 |
7687 |
24.7 |
−76.0 |
275 |
282 |
32 |
>360 |
|
11
|
1.74 |
7476 |
22.7 |
−244.4 |
— |
266 |
>40 |
>360 |
|
12
|
1.73 |
7603 |
23.5 |
−96.4 |
107 |
172 |
>40 |
>360 |
|
13
|
1.75 |
7690 |
24.7 |
−196.9 |
— |
287 |
>40 |
>360 |
|
14
|
1.72 |
7436 |
21.4 |
255.8 |
— |
235 |
>40 |
>360 |
| TNT |
1.65 |
6824 |
19.4 |
−59.4 |
|
295 |
15 |
>353 |
| RDX |
1.80 |
8795 |
34.9 |
92.6 |
|
204 |
7.5 |
120 |
| HNS47 |
1.75 |
7164 |
21.6 |
78.2 |
|
318 |
5 |
240 |
Conclusions
A family of new thermally stable energetic materials based on N-methyl-3,5-dinitropyrazole and polynitrobenzene moieties linked with C–C bonds was synthesized. The physicochemical and energetic properties of all the energetic compounds were explored. To study the effect of various explosophores, functional groups such as –NH2/–N3/–NHNO2/–OH were introduced to the polynitrobenzene skeletons. Most of the compounds synthesized in this study are more energetic, less sensitive and thermally stable than traditional explosives TNT and HNS. Compounds 5 (Tdec = 333 °C) and 6 (Tdec = 300 °C) showed excellent thermal stability, having decomposition temperatures comparable to that of well-known heat-resistant explosive HNS (Tdec = 318 °C). Also, these compounds are found to be more thermally stable among the known N-methyl-3,5-dinitropyrazole-based and C–N coupled 3,4,5-trinitrobenzene-azole-based energetic compounds. The structure–property relationship was studied using X-ray crystallographic studies, Hirshfeld surface and NCI analyses, and electrostatic potential and bond dissociation energy studies. Compounds 6, 10 and 13 exhibit excellent physical and detonation properties suggesting they could be potential replacements for the conventional heat-resistant explosive HNS.
Experimental section
Caution!
Although no accidents were observed during the synthesis, handling, and characterization, all the compounds reported in this work are potentially explosive materials and may explode unpredictably under certain conditions. All the compounds must be synthesized only on a small scale (<100 mg). Any mechanical actions involving grinding or scratching must be avoided. In addition, all the manipulations must be strictly carried out in a fume hood behind a polycarbonate safety shield. Eye protection, a face shield, and leather gloves must be worn while handling these compounds.
General methods
All reagents were purchased from Aldrich, TCI or GLR Innovations in analytical grade and were used as supplied, if not stated otherwise. 1H, 13C NMR and 15N spectra were recorded using a 500 MHz (JEOL ECZ500R/S1) NMR spectrometer operating at 500, 125 and 50.69 MHz, respectively. Chemical shifts in the 1H and 13C NMR spectra are reported relative to the external standard of Me4Si, while chemical shifts in the 15N NMR spectra are reported relative to the external standard of ammonia. The melting and decomposition temperatures were obtained on a differential scanning calorimeter (S11 6300 EXSTAR) at a scan rate of 5 °C min−1. IR spectra were recorded using KBr pellets for solids on a PerkinElmer FT-IR spectrometer. Densities were measured at room temperature by employing Anton Paar Ultrapyc 5000 gas pycnometer. Quadrupole-time-of-flight high-resolution mass spectra (QTOF–HRMS) were obtained in ESI mode. Elemental analyses were carried out on an elementar model Vario-EI-III. The impact and friction sensitivity measurements were made using a standard BAM fall hammer (OZM) and a BAM friction tester (FST ProEX).
Suitable crystals of 6 and 14 were obtained by slow evaporation of their saturated solutions in acetonitrile/water and water. The single-crystal diffraction studies were carried out on a Bruker SMART APEX CCD diffractometer with a Mo Kα (λ = 0.710 73 Å) sealed tube. All crystal structures were solved by direct methods. The program SAINT (version 6.22) was used for the integration of the intensity of reflections and scaling. The program SADABS was used for absorption correction. The crystal structures were solved and refined using the SHELXTL (version 6.12) package.48 All hydrogen atoms were included in idealized positions, and a riding model was used. Non-hydrogen atoms were refined with anisotropic displacement parameters.
The heats of formation for energetic compounds 5–8 and 10–14 were obtained using isodesmic reactions (ESI‡). The geometric optimization and frequency analyses of the structures were performed at the B3PW91/6-31G(d,p) level without any symmetry restriction as implemented in the Gaussian 09 program.49,50 All the optimized derivatives were confirmed to be true local energy minima on the potential energy surface without any imaginary frequencies. All calculated gas-phase enthalpies for covalent materials were converted to solid-phase values by subtracting the empirical heats of sublimation obtained based on the molecular surface properties.51 For salts 11–14, the solid-phase heats of formation were calculated based on the Born–Haber energy cycle.52,53
4-Bromo-1-methyl-3,5-dinitro-1H-pyrazole (2).
A solution of potassium carbonate (20.90 g, 152 mmol) was prepared in 120 mL water. After stirring for 10 minutes, 4-bromo-3,5-dinitro-1H-pyrazole (1) (6.00 g, 25 mmol) was added. The reaction mixture was stirred for 30 minutes, followed by the addition of dimethyl sulphate (15.90 g, 126 mmol). The stirring was continued for 6 h at 40 °C. The reaction mixture was then cooled and poured into water. The precipitate formed was collected by filtration, washed with water, and dried under vacuum to obtain pure compound 2 as a white solid. Yield: 5.80 g, 92%. 1H NMR (DMSO-d6, ppm): 4.28 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 149.2, 144.1, 90.2, 43.1 ppm. IR (ν, cm−1): 3448, 2857, 1568, 1497, 1429, 1389, 1330, 1301, 1036, 876, 748. Elemental analysis for C4H3BrN4O4 (249.93): calcd: C 19.14, H 1.20, N 22.32%; found: C 19.16, H 1.31, N 22.26%.
4-(4-Chlorophenyl)-1-methyl-3,5-dinitro-1H-pyrazole (3).
A mixture of 2 (3.00 g, 11.90 mmol), 4-chlorophenylboronic acid (2.79 g, 17.80 mmol), Na2CO3 (3.15 g, 29.70 mmol) and TEAB (0.24, 1.19 mmol) was suspended in toluene (60 mL) and H2O (15 mL), and carefully degassed for 15–20 minutes. The catalyst, Pd(PPh3)4 (0.37 g, 0.03 mmol), was then added and the reaction mixture was stirred further for 20 minutes under a nitrogen atmosphere. The reaction mixture was then sealed and refluxed for 12 hours. After cooling down, extraction was done with ethyl acetate (3 × 20 mL) and washed with brine. The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column, eluting with hexane/EtOAc (20:1) to afford 3 as a light-yellow solid. Yield: 2.30 g, 68%. 1H NMR (DMSO-d6, ppm): 7.54 (d, 2H, CH), 7.50 (d, 2H, CH), 4.31 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 148.5, 142.7, 133.2, 130.9, 127.5, 125.4, 114.2, 41.9 ppm. IR (ν, cm−1): 3438, 2992, 2937, 1617, 1533, 1334, 1117, 766, 716. Elemental analysis for C10H7ClN4O4 (282.02): calcd: C 42.50, H 2.50, N 19.82%; found: C 42.41, H 2.58, N 19.89%.
4-(4-Chloro-3,5-dinitrophenyl)-1-methyl-3,5-dinitro-1H-pyrazole (4).
Compound 3 (3.70 g, 12.30 mmol) was added to concentrated sulfuric acid (25 mL) and stirred until a clear solution was obtained. It was then cooled down to 0 °C and 100% nitric acid (6 mL) was added dropwise. The reaction mixture was stirred at 80 °C for 12 h. After cooling, the mixture was poured into crushed ice (300 g) and the precipitate obtained was filtered, washed with water (10 mL), and dried in air to yield 4 as a white solid. Yield: 3.51 g, 72%. Tm (5 °C min−1): 191 °C (onset). 1H NMR (DMSO-d6, ppm): 8.70 (s, 2H, CH), 4.34 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 149.0, 148.4, 130.3, 129.4, 119.3, 114.5, 110.9, 42.8 ppm. IR (ν, cm−1): 3449, 3084, 1631, 1555, 1442, 1354, 919, 734. Elemental analysis for C10H5ClN6O8 (371.99): calcd: C 32.23, H 1.35, N 22.55%; found: C 32.14, H 1.42, N 22.48%.
4-(1-Methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,6-dinitroaniline (5).
An aqueous solution of ammonia (5 mL) was added to a solution of compound 4 (1.5 g, 4.02 mmol) in methanol (30 mL). The resulting solution was refluxed for 12 h in a sealed tube. Upon completion, the reaction mixture was cooled to RT. The product was precipitated during cooling. The solid was filtered and washed with methanol (10 mL) and dried in air to afford 5 as a yellow solid. Yield: 1.32 g, 93%. Tm (5 °C min−1): 246 °C (onset), Tdec (5 °C min−1): 333 °C (onset). 1H NMR (DMSO-d6, ppm): 8.74 (s, 2H, CH), 8.59 (s, 2H, NH2), 4.33 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 149.4, 143.7, 140.8, 135.5, 134.5, 112.5, 112.2, 42.7 ppm. 15N NMR (DMSO-d6): −16.3, −28.6, −33.7, −78.4, −186.2, −302.1. IR (ν, cm−1): 3779, 3472, 3065, 1637, 1548, 1511, 1328, 1248, 1091, 955. Elemental analysis for C10H7N7O8 (353.04): calcd: C 34.01, H 2.00, N 27.76%; found: C 34.08, H 2.06, N 27.71%. HRMS (ESI) m/z [M − H]− calcd for C10H6N7O8: 352.0278; found: 352.0279.
1-Methyl-3,5-dinitro-4-(3,4,5-trinitrophenyl)-1H-pyrazole (6).
Compound 5 (0.30 g, 0.84 mmol) was dissolved in concentrated sulfuric acid (20 mL), and Na2WO4·2H2O (0.20 g, 0.62 mmol) was added in small portions to the reaction mixture at 0 °C. Hydrogen peroxide (30%, 3 mL) was then added dropwise, and the resulting mixture was stirred at RT for 18 h and then poured into ice-cold water. The precipitate obtained was filtered and washed with cold water to afford 6 as a pale yellow solid. Yield: 0.27 g, 83%. Tm (5 °C min−1): 242 °C (onset), Tdec (5 °C min−1): 300 °C (onset). 1H NMR (DMSO-d6, ppm): 9.13 (s, 2H, CH), 4.37 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 149.3, 146.8, 143.6, 139.5, 139.9, 131.4, 117.2, 112.2, 42.8 ppm. IR (ν, cm−1): 3451, 2978, 1573, 1496, 1437, 1327, 1118, 1027, 859, 745. Elemental analysis for C10H5N7O10 (383.01): calcd: C 31.34, H 1.32, N 25.59%; found: C 31.21, H 1.39, N 25.51%. HRMS (ESI) m/z [M]− calcd for C10H5N7O10: 383.0098; found: 383.0096.
4-(4-Azido-3,5-dinitrophenyl)-1-methyl-3,5-dinitro-1H-pyrazole (7).
Trimethylsilyl azide (0.06 g, 0.52 mmol) was added to a solution of 6 (0.10 g, 0.26 mmol) in dimethyl formamide (3 mL). The reaction mixture was stirred at ambient temperature for 2 h. The reaction mixture was poured into ice. The yellow-colored precipitate was collected by suction filtration, washed with cold water, and dried under vacuum to afford 7. Yield: 0.04 g, 75%. Tdec (5 °C min−1): 272 °C (onset). 1H NMR (DMSO-d6, ppm): 8.63 (s, 2H, CH), 4.34 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 149.1, 143.7, 130.7, 128.3, 125.1, 114.6, 111.4, 42.9 ppm. IR (ν, cm−1): 3444, 2139, 1632, 1552, 1332, 1106, 920, 717. Elemental analysis for C10H5N9O8 (379.03): calcd: C 31.67, H 1.33, N 33.24%; found: C 31.68, H 1.42, N 33.28%. HRMS (ESI) m/z [M − H]− calcd for C10H4N9O8: 378.0183; found: 378.0196.
N-(4-(1-Methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,6-dinitrophenyl)nitramide (8).
Compound 5 (0.15 g, 0.42 mmol) was added to concentrated sulfuric acid (25 mL), and stirred until a clear solution was obtained. It was then cooled down to 0 °C, and 100% nitric acid (1 mL) was added dropwise. The reaction mixture was stirred at room temperature for 3 h and then poured into cold water (30 mL). The dark-yellow precipitate obtained was filtered, washed with water (5 mL), and dried in air to yield 8. Yield: 0.12 g, 77%. Tdec (5 °C min−1): 193 °C (onset). 1H NMR (DMSO-d6, ppm): 9.15 (s, 2H, CH), 4.38 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 149.5, 147.0, 143.8, 140.2, 131.7, 117.7, 112.5, 43.1 ppm. IR (ν, cm−1): 3449, 2844, 1632, 1457, 1320, 1310, 1113, 607. Elemental analysis for C10H6N8O10 (398.02): calcd: C 30.16, H 1.52, N 28.14%.; found: C 30.12, H 1.55, N 28.06%. HRMS (ESI) m/z [M − NO2 − H]− calcd for C10H7N7O8: 353.0356; found: 353.0125.
3-(1-Methyl-3,5-dinitro-1H-pyrazol-4-yl)phenol (9).
A mixture of 2 (2 g, 7.96 mmol), (3-hydroxyphenyl)boronic acid (1.3 g, 9.55 mmol), and K2CO3 (2.75 g, 19.90 mmol) was taken into a sealed tube and suspended in dioxane (40 ml) and water (10 ml) solution (4:1). After that, the solution mixture was carefully degassed for 15–20 minutes under a nitrogen atmosphere. Then, catalyst Pd(dppf)Cl2 (0.40 g, 0.55 mmol) and PPh3 (0.20 g, 0.79 mmol) were added and the reaction mixture was further stirred under a N2 atmosphere for 15 minutes. Then, the reaction mixture was quickly sealed and heated to 100 °C for 12 h. After cooling down the solution, water was added (20 ml), and the mixture was extracted with ethyl acetate (3 × 40 mL), and washed with brine. The collected organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The obtained crude mixture was purified by chromatography on a silica-gel column, eluting with hexane/EtOAc (10:1) to afford 9 as a yellow solid. Yield: 1.53 g, 73%. 1H NMR (DMSO-d6, ppm): 9.63 (s, 1H, OH), 7.25 (m, 1H, CH), 6.86 (m, 1H, CH), 6.80 (m, 2H, CH), 4.29 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 157.0, 149.3, 143.2, 129.4, 128.0, 120.1, 116.4, 116.0, 116.0, 42.5 ppm. IR (ν, cm−1): 3477, 2926, 1584, 1534, 1413, 1325, 1293, 1159, 901, 860. Elemental analysis for C10H8N4O5 (264.05): calcd: C 45.46, H 3.05, N 21.21%; found: C 45.34, 3.11, 21.22%.
3-(1-Methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,4,6-trinitrophenol (10).
Compound 9 (1.2 g, 4.53 mmol) was added to 98% sulfuric acid (6 ml), and then the slurry temperature was maintained at 0 °C. Then, 20% oleum (8 ml) was added slowly with stirring, keeping the temperature of the mixture below 30 °C. The mixture was stirred at room temperature for 15 minutes, and then heated to 90 °C for 3 h. After cooling down, the reaction mixture was again kept in an ice bath to maintain the temperature at 0 °C. To this solution, successive addition of 98% sulfuric acid (4 ml) and 100% HNO3 (4 ml) was done slowly. The mixture was stirred at room temperature for 15 minutes, and then heated to 90 °C for 3 h. After cooling down the solution, the mixture was poured into crushed ice. The precipitate was filtered, and thoroughly washed with water to obtain compound 10 as a white solid. Yield: 1.45 g, 80%. Tm (5 °C min−1): 275 °C (onset), Tdec (5 °C min−1): 282 °C (onset). 1H NMR (DMSO-d6, ppm): 8.88 (s, 1H, CH), 4.36 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 159.3, 148.7, 146.9, 142.8, 137.9, 126.4, 123.2, 120.5, 107.8, 42.7 ppm. 15N NMR (DMSO-d6): −13.5, −16.4, −18.8, −30.0, −35.3, −76.9, −183.7. IR (ν, cm−1): 3436, 3124, 2921, 1636, 1595, 1548, 1448, 1333, 1278, 1176, 1098. Elemental analysis for C10H5N7O11 (399.00): calcd: C 30.09, H 1.26, N 24.56%; found: C 30.03, H 1.28, N 24.44%. HRMS (ESI) m/z [M − H]− calcd for C10H5N10O11: 399.9969; found: 399.9965.
General procedure for the synthesis of the energetic salts 11–14
3-(1-Methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,4,6-trinitrophenol (10) (0.15 g, 0.37 mmol) was suspended in 5 mL acetonitrile. A solution of 28% aqueous ammonia (0.012 g, 0.37 mmol), hydrazine monohydrate (0.017, 0.37 mmol), 50% aqueous hydroxylamine (0.012 g, 0.37 mmol) or 7H-[1,2,4]triazolo[4,3-b][1,2,4]triazole-3,6,7-triamine (0.038 g, 0.37 mmol) was added. The reaction mixture was stirred for 2 h at room temperature. The solvent was evaporated under reduced pressure and dried over vacuum to obtain salts 11–14.
Ammonium 3-(1-methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,4,6-trinitrophenolate (11).
Yield: 0.14 g, 93% (yellow solid). Tdec (5 °C min−1): 266 °C (onset) 1H NMR (CD3CN, ppm): 8.98 (s, 1H, CH), 6.33 (s, 4H, NH4+), 4.28 (s, 3H, CH3). 13C{1H} NMR (CD3CN, ppm): δ 161.0, 150.2, 148.3, 143.8, 138.5, 127.7, 126.4, 122.3, 109.1, 43.7 ppm. IR (ν, cm−1): 3499, 2931, 1608, 1530, 1434, 1338, 1296, 1245, 1158, 708. Elemental analysis for C10H8N8O11 (416.03): calcd: C 28.86, H 1.94, N 26.92%; found: C 28.79, H 1.99, N 26.79%.
Hydrazinium 3-(1-methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,4,6-trinitrophenolate (12).
Yield: 0.15 g, 93% (orange solid). Tm (5 °C min−1): 107 °C (onset), Tdec (5 °C min−1): 172 °C (onset). 1H NMR (CD3CN, ppm): 8.99 (s, 1H, CH), 4.92 (s, 5H, N2H5+), 4.28 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 161.2, 150.4, 148.5, 143.9, 138.7, 127.8, 126.8, 122.4, 109.1, 43.7 ppm. IR (ν, cm−1): 3445, 2921, 1603, 1525, 1457, 1328, 1296, 1094, 961, 708. Elemental analysis for C10H9N9O11 (431.04): calcd: C 27.85, H 2.10, N 29.23%; found: C 27.74, H 2.14, N 29.33%.
Hydroxylammonium 3-(1-methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,4,6-trinitrophenolate (13).
Yield: 0.13 g, 81% (light yellow solid). Tdec (5 °C min−1): 287 °C (onset). 1H NMR (DMSO-d6, ppm): 8.90 (s, 1H, CH), 4.36 (s, 3H, CH3). 13C{1H} NMR (DMSO-d6, ppm): δ 159.4, 148.8, 147.0, 142.8, 138.0, 126.5, 123.4, 120.7, 107.8, 42.8 ppm. 15N NMR (DMSO-d6): −13.7, −16.5, −18.9, −30.1, −35.3, −76.7, −183.8, −302.0. IR (ν, cm−1): 3440, 3219, 2935, 2710, 1599, 1530, 1466, 1328, 1296, 1158, 956, 708. Elemental analysis for C10H8N8O12 (432.02): calcd: C 27.79, H 1.87, N 25.93%; found: C 27.81, H 1.94, N 25.87%.
3,6,7-Triamino-7H-[1,2,4]triazolo[4,3-b][1,2,4]triazol-2-ium 3-(1-methyl-3,5-dinitro-1H-pyrazol-4-yl)-2,4,6-trinitrophenolate (14).
Yield: 0.17 g, 85% (fluorescent yellow). Tdec (5 °C min−1): 235 °C (onset). 1H NMR (DMSO-d6, ppm): 8.88 (s, 1H, CH), 8.21 (s, 2H, NH2), 7.24 (s, 2H, NH2), 4.36 (s, 3H, CH3), 4.02 (s, 2H, NH2). 13C{1H} NMR (DMSO-d6, ppm): δ 160.2, 159.3, 148.7, 147.4, 146.9, 142.7, 141.1, 137.9, 126.5, 123.2, 120.6, 107.7, 42.7 ppm. IR (ν, cm−1): 3499, 3352, 3229, 1704, 1645, 1566, 1438, 1333, 1258, 1153, 708. Elemental analysis for C13H11N15O11 (553.07): calcd: C 28.22, H 2.00, N 37.97%; found: C 28.31, H 2.11, N 37.88%.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support from the Science and Engineering Research Board (SERB/CRG/2021/004447). D. K. acknowledges the Department of Science and Technology (DST), India, for financial support under the DST INSPIRE faculty award scheme [DST/INSPIRE/04/2017/001464]. We thank DST-FIST [SR/FST/CS-II/2018/72(C)] and IITR for funding the 500 MHz NMR and single-crystal X-ray diffraction facilities at IIT Roorkee. K. P. and P. B. acknowledge DST-Inspire and MHRD for research fellowships, respectively.
References
-
T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, Berlin, 2019 Search PubMed.
-
M. Gozin and L. L. Fershtat, Nitrogen-Rich Energetic Materials, Wiley-VCH, 2022 Search PubMed.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2017, 23, 1743–1747 CrossRef CAS PubMed.
- D. Kumar and A. J. Elias, Resonance, 2019, 24, 1253–1271 CrossRef.
- Y. Tang, D. Kumar and J. M. Shreeve, J. Am. Chem. Soc., 2017, 139, 13684–13687 CrossRef CAS PubMed.
-
D. S. Viswanath, T. K. Ghosh and V. M. Boddu, Emerging Energetic Materials: Synthesis, Physicochemical, and Detonation Properties, Springer, 2018 Search PubMed.
- Y. Wang, Y. Liu, S. Song, Z. Yang, X. Qi, K. Wang, Y. Liu, Q. Zhang and Y. Tian, Nat. Commun., 2018, 9, 2444 CrossRef PubMed.
- P. A. Pagoria, Propellants, Explos., Pyrotech., 2016, 41, 452–469 CrossRef CAS.
- T. M. Klapötke and T. G. Witkowski, ChemPlusChem, 2016, 81, 357–360 CrossRef PubMed.
- J. P. Agrawal, Mehilal, U. S. Prasada and R. N. Survea, New J. Chem., 2000, 24, 583–585 RSC.
- H. Li, L. Zhang, N. Petruik, K. Wang, Q. Ma, D. Shem-Tov, F. Zhao and M. Gozin, ACS Cent. Sci., 2020, 6, 54–75 CrossRef CAS PubMed.
- W. Ma, Z.-Q. Zhang, Q. Ma, J. Tang, W. Yang, H. Yang, G. Cheng and G.-J. Fan, Cryst. Growth Des., 2023, 23, 1127–1132 CrossRef CAS.
- J. Singh, R. J. Staples and J. M. Shreeve, J. Mater. Chem. A, 2023, 11, 12896–12901 RSC.
- J. Singh, R. J. Staples and J. M. Shreeve, Org. Lett., 2022, 24, 8832–8836 CrossRef CAS PubMed.
- D. Kumar, C. He, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2016, 4, 9220–9228 RSC.
- W. Hu, J. Tang, X. Ju, Z. Yi, H. Yang, C. Xiao and G. Cheng, ACS Cent. Sci., 2023, 9, 742–747 CrossRef CAS PubMed.
- L. Pei, Q. Lai, P. Yin and S. Pang, Cryst. Growth Des., 2023, 23, 3595–3602 CrossRef CAS.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2017, 5, 16767–16775 RSC.
- Q. Sun, X. Li, Q. Lin and M. Lu, Org. Biomol. Chem., 2018, 16, 8034–8037 RSC.
- A. K. Yadav, M. K. Jujam, V. D. Ghule and S. Dharavath, Chem. Commun., 2023, 59, 4324–4327 RSC.
- S. Banik, P. Kumar, V. D. Ghule, S. Khanna, D. Allimuthu and S. Dharavath, J. Mater. Chem. A, 2022, 10, 22803–22811 RSC.
- G. Zhao, P. Yin, D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2019, 141, 19581–19584 CrossRef CAS PubMed.
- N. Kommu, M. Balaraju, V. D. Ghule and A. K. Sahoo, J. Mater. Chem. A, 2017, 5, 7366–7371 RSC.
- S. Vangara, N. Kommu, V. Thaltiri, M. Balaraju and A. K. Sahoo, J. Org. Chem., 2022, 87, 7202–7212 CrossRef CAS PubMed.
- J. Zhang, Y.-F. Ling, G.-X. Wang, L. Zhang and J. Luo, Org. Biomol. Chem., 2018, 16, 4784–4788 RSC.
- D. Kumar, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2016, 4, 9931–9940 RSC.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2017, 23, 7876–7881 CrossRef CAS PubMed.
- V. A. Kuehl, A. H. Cleverland, C. J. Snyder and D. E. Chavez, ACS Omega, 2023, 8, 18408–18413 CrossRef CAS PubMed.
- J. Cai, J. Zhang, J. Xiong, R. Li, T. Fei, Q. Lai, P. Yin and S. Pang, Chem. Eng. J., 2023, 467, 143527 CrossRef CAS.
- S. Seth and C. Pathak, Cryst. Growth Des., 2023, 23, 4669–4679 CrossRef CAS.
- G. Zhao, D. Kumar, P. Yin, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, Org. Lett., 2019, 21, 1073–1077 CrossRef CAS PubMed.
- J. Singh, R. J. Staples and J. M. Shreeve, ACS Appl. Mater. Interfaces, 2021, 13, 61357–61364 CrossRef CAS PubMed.
- Y. Tang, W. Huang, A. K. Chinnam, J. Singh, R. J. Staples and J. M. Shreeve, Inorg. Chem., 2021, 60, 8339–8345 CrossRef CAS PubMed.
- G. Herve, C. Roussel and H. Graindorge, Angew. Chem., Int. Ed., 2010, 49, 3177–3181 CrossRef CAS PubMed.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2017, 5, 10437–10441 RSC.
- P. Zhang, D. Kumar, L. Zhang, D. Shem-Tov, N. Petrutik, A. K. Chinnam, C. Yao, S. Pang and M. Gozin, Molecules, 2019, 24, 4324 CrossRef PubMed.
- C. He, J. Zhang, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 2863–2868 RSC.
- P. Yin, J. Zhang, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2014, 20, 16529–16536 CrossRef CAS PubMed.
- D. Fischer, J. L. Gottfried, T. M. Klapotke, K. Karaghiosoff, J. Stierstorfer and T. G. Witkowski, Angew. Chem., Int. Ed., 2016, 55, 16132–16135 CrossRef CAS PubMed.
- K. Pandey, P. Bhatia, P. Dolui, V. D. Ghule and D. Kumar, Asian J. Org. Chem., 2022, 11, 202–210 CrossRef.
- M. Tomanová, L. Jedinák, J. Kosar, L. Kvapil, P. Hradil and P. Cankař, Org. Biomol. Chem., 2017, 15, 10200–10211 RSC.
-
M. J. turner, J. J. Mckinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
- M. Reichel, D. Dosch, T. Klapötke and K. Karaghiosoff, J. Am. Chem. Soc., 2019, 141, 19911–19916 CrossRef CAS PubMed.
- S. Li, R. Bu, R. Gou and C. Zhang, Cryst. Growth Des., 2021, 21, 6619–6634 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
-
M. Sucéska, EXPLO5 6.01, Brodarski Institute, Zagreb, Croatia, 2013 Search PubMed.
- T. Yan, J. Ma, H. Yang and G. Cheng, Chem. Eng. J., 2022, 429, 132416 CrossRef CAS.
-
SMART: Bruker Molecular Analysis Research Tools, version 5.618, Bruker Analytical X-ray Systems, Madison, WI, 2000 Search PubMed.
-
R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem. A, 2006, 110, 1005–1013 CrossRef CAS PubMed.
- H. D. B. Jenkins, D. Tudela and L. Glasser, Inorg. Chem., 2002, 41, 2364–2367 CrossRef CAS PubMed.
- V. D. Ghule, J. Phys. Chem. C, 2013, 117, 16840–16849 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Jean'ne M. Shreeve on the occasion of her 90th Birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2178043 and 2270163. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00949a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Prachi
Bhatia
a,
Prachi
Bhatia