
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion binding and transport with meso-alkyl substituted two-armed calix[4]pyrroles bearing urea and hydroxyl groups†
Małgorzata
Pamuła
 a,
Evgeny
Bulatov
a,
Luis
Martínez-Crespo
b,
Anniina
Kiesilä
a,
Julia
Naulapää
a,
Elina
Kalenius
a and
Kaisa
Helttunen
*a
a,
Evgeny
Bulatov
a,
Luis
Martínez-Crespo
b,
Anniina
Kiesilä
a,
Julia
Naulapää
a,
Elina
Kalenius
a and
Kaisa
Helttunen
*a
aUniversity of Jyvaskyla, Department of Chemistry, Nanoscience Center, P.O. Box 35, FI-40014 University of Jyvaskyla, Finland. E-mail: kaisa.j.helttunen@jyu.fi
bDepartment of Chemistry, Universitat de les Illes Balears, Cra. Valldemossa Km. 7.5, 07122 Palma de Mallorca, Spain
First published on 1st August 2023
Abstract
Calix[4]pyrroles bearing hydroxyl (1) or urea (3) groups attached to the meso-positions with propyl linkers were synthesized as cis- and trans-isomers. The anion binding properties of cis-1 and cis-3 were screened with ion-mobility mass spectrometry, where cis-1 formed complexes with Cl−, Br− and H2PO4−, whereas cis-3 formed complexes with most of the investigated anions, including Cl−, Br−, I−, NO3−, ClO4−, OTf−, SCN− and PF6−. The structures of the chloride complexes were further elucidated with density functional theory calculations and a crystal structure obtained for cis-1. In solution, chloride and dihydrogenphosphate anion binding with cis-1 and cis-3 were compared using 1H NMR titrations. To assess the suitability of two-armed calix[4]pyrroles as anion transporters, chloride transport studies of cis-1, cis-3 and trans-3 were performed using large unilamellar vesicles. The results revealed that cis-3 had the highest activity among the investigated calix[4]pyrroles, which was related to the improved affinity and isolation of chloride inside the binding cavity of cis-3 in comparison to cis-1. The results indicate that appending calix[4]pyrroles with two hydrogen bonding arms is a feasible strategy to obtain anion transporters and receptors with high anion affinity.
Introduction
Anion binding with synthetic receptors is a rapidly evolving field producing applications in anion sensing,1–5 extraction,6–9 and transport.10 Calix[4]pyrroles are a well-known class of neutral supramolecular receptors, where converging pyrrole rings provide hydrogen bond donors for anion binding.11,12 The anion binding properties can be tuned by extending the binding cavity with meso-substituents providing aromatic panels13–15 or more hydrogen bonding groups. Examples of the latter involve hosts bearing triazole,16 squaramide,17meso-acyl,18 and meso-pyrrole units.19 On the other hand, several non-planar strapped calix[4]pyrroles with enhanced anion binding affinities have been designed by connecting two meso-positions with an aliphatic linker equipped with amide, triazole or pyrrole groups.20,21 These hosts with increased number of hydrogen bond donors have typically improved affinity for chloride in comparison to the meso-octamethyl calix[4]pyrrole. However, a host design with flexible meso-alkyl linkers connected to additional hydrogen bond donors can be beneficial for binding larger polyatomic anions22 since it retains a relatively open calix[4]pyrrole cavity where the podand arms could wrap around an included guest. Aliphatic linkers in meso-substituents have proven efficient for inducing a large conformational change for a fluorescent chemosensor,22 providing perfluoroalkylated calix[4]pyrroles for fluoride extraction,6meso-tetrahexyl calix[4]pyrroles as chloride23 and cesium24 extractants, and acrylamide functionalized precursors for anion selective polymer membranes.25 However, for efficient anion binding the meso-alkyl arms should be on the same side of the binding cavity (cis),16,18,19 which leads in some cases to a challenging isomer separation.22For the last twenty years, increasing efforts have been directed towards development of anion receptors to be used as transmembrane transporters, due to their potential applications on the treatment of channelopathies (by replacing defective transmembrane proteins) and cancer (by inducing apoptosis in cells).26–29 Such transporters typically act via a mobile carrier mechanism, binding the anion on one side of the membrane to release it on the other side, the binding usually occurs via anion coordinating groups such as ureas, thioureas or pyrrole rings.30 In fact, several calix[4]pyrroles have been studied as anion transporters due to their binding properties. The simple octamethyl calix[4]pyrrole could transport chloride with moderate activity, but only as an ion pair with Cs+, which is a limitation for its use as a chloride transporter in biological systems.31 Further functionalization of the calix[4]pyrrole core led to compounds able to transport chloride more efficiently and coupled to either the transport of sodium (or potassium) in the same direction (Cl−/M+ symport) or the transport of nitrate in the opposite direction (Cl−/NO3− antiport).12,32 For example, the nitrate antiport function was achieved by fluorination of the β-pyrrole positions, which increased the hydrogen bond donor character of the pyrrole NHs.33 An alternative strategy to improve the binding and transport of chloride was to conveniently functionalize the meso-positions to afford strapped34–37 and aryl-extended calix[4]pyrroles.38,39 While the former contained well preorganized additional anion binding groups (such as amides and triazole rings) the latter had electron deficient aromatic panels able to form anion–π interactions. Moreover, similar calix[4]pyrrole-based receptors showed interesting properties on the transport of ion pairs40 and amino acids,41 providing additional evidence of the relevance of rational design to obtain efficient and selective transporters.
In this paper, we report the anion binding and transport properties of two-armed calix[4]pyrrole podands bearing two urea or hydroxyl groups connected with meso-alkyl linkers. The synthesis, separation of the cis and trans isomers and the intermolecular interactions in the crystal structures of the two-armed calix[4]pyrroles is also discussed.
Results and discussion
Synthesis
Dipyrromethane 4 was synthesized according to a literature protocol42 as a precursor for two-armed calix[4]pyrroles. Two-armed meso-hydroxypropyl calix[4]pyrrole 1 has previously been prepared by a reductive cleavage of a bis-ester strapped host (Scheme S-1†) via three steps from 4 with an overall yield of 8.6%.22,42 Direct acid catalyzed condensation of dipyrromethane 4 using three equivalents of trifluoroacetic acid in acetone produced a mixture of cis-1 and trans-1 (Scheme 1). A fraction of pure trans-1 was isolated by precipitation at 10% yield, and cis-1 was separated from rest of the trans-1 by silica column chromatography at 12–19% yield. 1H NMR spectrum of cis-1 in acetone-d6 displays a multiplet resonance at 5.75 ppm for eight β-pyrrolic protons, a broad singlet at 7.98 ppm for NH protons, and a triplet at 3.36 ppm for OH protons. In addition, the 1H NMR spectrum shows three separate multiplets for the CH2 protons of the meso-arms. Singlets at 1.48, 1.45 and 1.43 ppm arise from the three non-equivalent meso-methyl groups of the cis isomer assuming that the conformations are averaged in the NMR timescale.15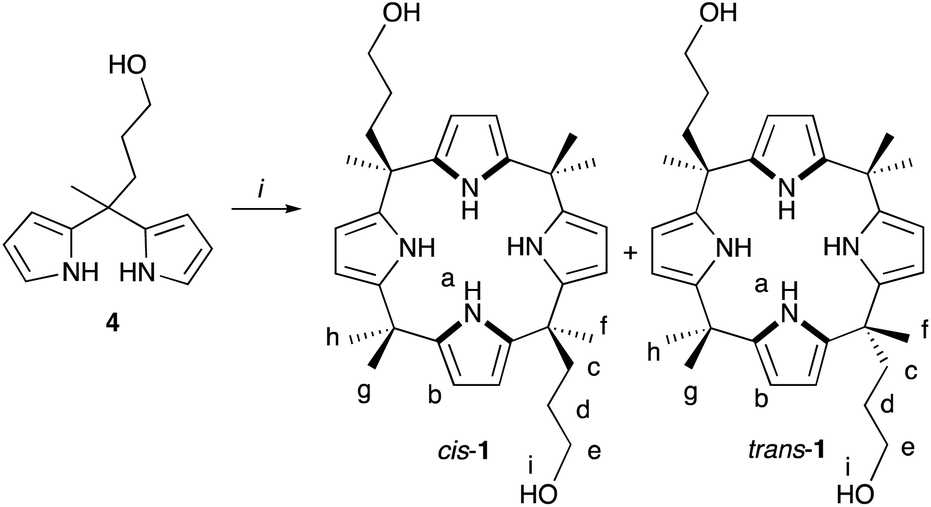 | ||
| Scheme 1 Synthesis of two-armed calix[4]pyrrole 1. Conditions: (i) trifluoroacetic acid in acetone. The letters indicate the NMR assignment. | ||
In order to increase the strength and number of hydrogen bond donors in the meso-groups, two-armed calix[4]pyrrole 3 with urea substituents was synthesized from dipyrromethane 4 (Scheme 2). First, the hydroxyl group in 4 was converted to a phthalimide group using previously reported Mitsunobu reaction.43 The resulting dipyrromethane 5 was reacted with trifluoroacetic acid in acetone yielding 2 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis and trans isomers at 40% yield. Several methods were attempted for the separation of the isomers of 2, including preparative TLC, flash column chromatography and normal phase HPLC, but without success. However, small difference in solubility in acetonitrile allowed the separation of cis-2 and trans-2 on a milligram scale to perform NMR analysis of ca. 90% pure isomers (Fig. S-12–22†) and crystal structure determination (see section 8.1 of the ESI†). Next, hydrazinolysis of the phthalimide groups was performed for the isomer mixture of 2 to yield the corresponding free amines. Ing-Manske reaction in previously reported reaction conditions25,43 for similar compounds did not proceed, most likely due to low solubility of 2 in ethanol, but successful conversion was obtained in a mixture of dichloromethane and ethanol. The deprotected intermediate was immediately reacted with p-nitrophenyl isocyanate to yield bis-urea functionalized calix[4]pyrrole 3. Fortunately, column chromatography of the crude provided pure trans-3 in 15% yield and pure cis-3 in 12% yield. 1H NMR spectra of cis-3 and trans-3 in acetone-d6 showed the expected number of meso-methyl proton signals for each isomer. The urea protons resonated as a singlet at 8.5 (j) and as a triplet at 6.0 ppm (i), and pyrrolic NH protons at 8.0 ppm for both isomers. 1H,1H-ROESY experiments for cis-3 (Fig. S-28 and 29†) and trans-3 (Fig. S-35 and 36†) in acetone-d6 revealed in both cases through-space correlations between pyrrole NH and all three methyl groups (f, g, h), as well as the pyrrole CH (b) and all three methyl groups and meso-CH2 groups (c) of the arms. Such correlations coincide with the 1,3-alternate conformation of the calix[4]pyrrole ring where the adjacent pyrrole NH groups point up and down, respectively, and may indicate the presence of several different conformations averaged in the NMR timescale.
:1 mixture of cis and trans isomers at 40% yield. Several methods were attempted for the separation of the isomers of 2, including preparative TLC, flash column chromatography and normal phase HPLC, but without success. However, small difference in solubility in acetonitrile allowed the separation of cis-2 and trans-2 on a milligram scale to perform NMR analysis of ca. 90% pure isomers (Fig. S-12–22†) and crystal structure determination (see section 8.1 of the ESI†). Next, hydrazinolysis of the phthalimide groups was performed for the isomer mixture of 2 to yield the corresponding free amines. Ing-Manske reaction in previously reported reaction conditions25,43 for similar compounds did not proceed, most likely due to low solubility of 2 in ethanol, but successful conversion was obtained in a mixture of dichloromethane and ethanol. The deprotected intermediate was immediately reacted with p-nitrophenyl isocyanate to yield bis-urea functionalized calix[4]pyrrole 3. Fortunately, column chromatography of the crude provided pure trans-3 in 15% yield and pure cis-3 in 12% yield. 1H NMR spectra of cis-3 and trans-3 in acetone-d6 showed the expected number of meso-methyl proton signals for each isomer. The urea protons resonated as a singlet at 8.5 (j) and as a triplet at 6.0 ppm (i), and pyrrolic NH protons at 8.0 ppm for both isomers. 1H,1H-ROESY experiments for cis-3 (Fig. S-28 and 29†) and trans-3 (Fig. S-35 and 36†) in acetone-d6 revealed in both cases through-space correlations between pyrrole NH and all three methyl groups (f, g, h), as well as the pyrrole CH (b) and all three methyl groups and meso-CH2 groups (c) of the arms. Such correlations coincide with the 1,3-alternate conformation of the calix[4]pyrrole ring where the adjacent pyrrole NH groups point up and down, respectively, and may indicate the presence of several different conformations averaged in the NMR timescale.
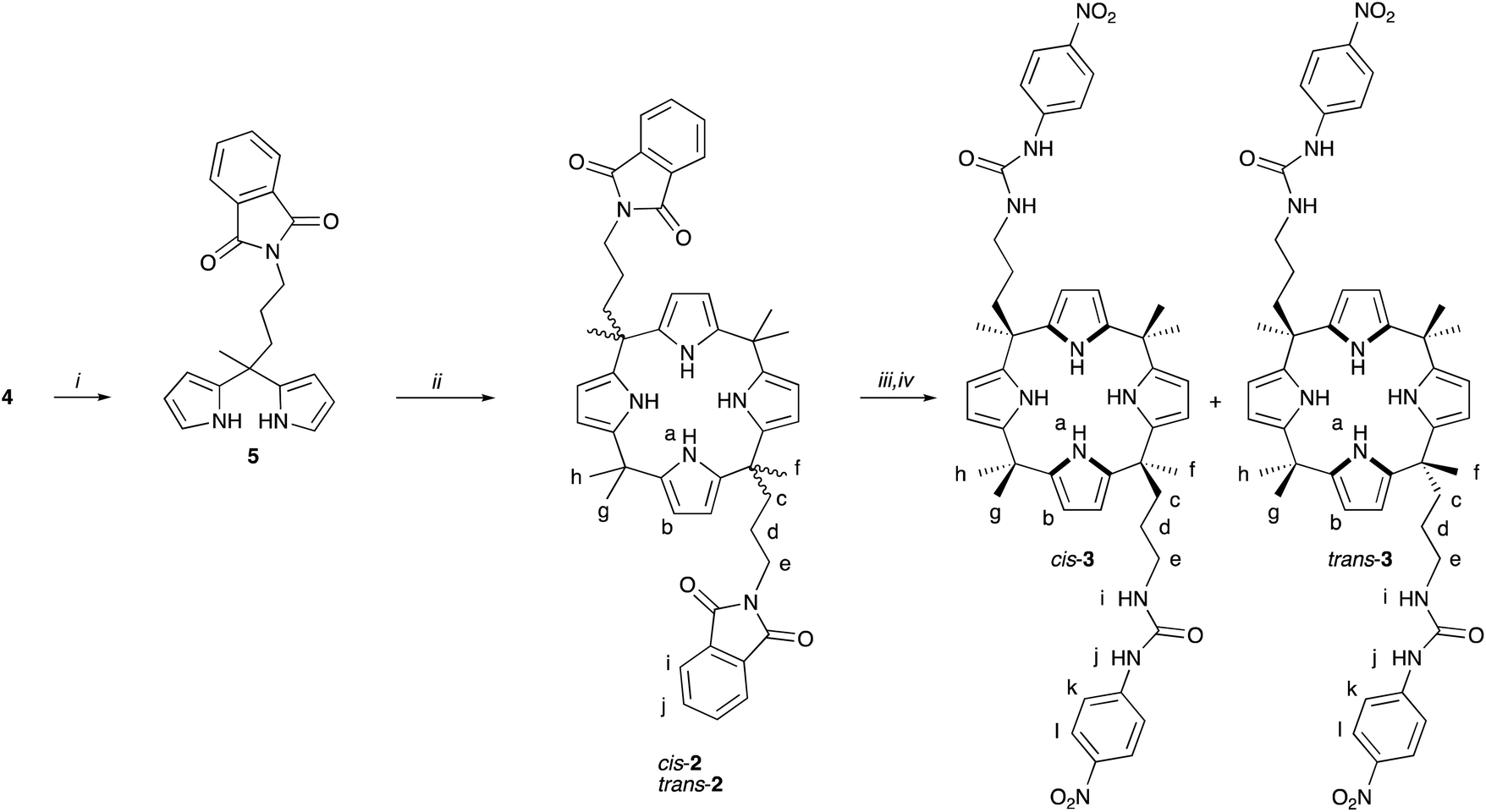 | ||
| Scheme 2 Synthesis of two-armed calix[4]pyrroles 2 and 3. Conditions: (i) PPh3, diisopropyl azodicarboxylate and phthalimide in dichloromethane, (ii) trifluoroacetic acid in acetone, (iii) hydrazine in dichloromethane/ethanol, and (iv) p-nitrophenyl isocyanate in tetrahydrofuran. The letters indicate the NMR assignment. | ||
X-ray crystallography
Single crystals of trans-1 and cis-1 were obtained by vapour diffusion of acetone into THF solutions of the pure isomers (Fig. 1). A solvent-free form of trans-1 was crystallized, whereas cis-1 was crystallized as a THF solvate cis-1·0.25 THF. Both crystal structures display the host in 1,3-alternate conformation and the hydroxypropyl arms are oriented axially or equatorially in the trans-1 and cis-1, respectively (Fig. 1a and c). Intermolecular self-inclusion of both podand arms into the cavities of two neighboring molecules is mediated by hydrogen bonding of the OH group with two pyrrole NH groups. In case of trans-1, calix[4]pyrroles pack into a hydrogen bonded chain (Fig. 1b), whereas cis-1 forms an assembly of a 3D hydrogen bonded network with a helical architecture and channels partially filled with THF molecules (Fig. 1d). | ||
| Fig. 1 (a) Crystal structure of trans-1 (anisotropic displacement parameters are drawn at 50% probability level), (b) hydrogen-bonded chain in the crystal structure of trans-1 (neighbouring molecules are presented in different colours and CH hydrogen atoms are omitted for clarity), (c) crystal structure of cis-1·0.25 THF (solvent molecule is omitted), (d) hydrogen-bonded helix in the crystal structure of cis-1·0.25 THF (neighbouring molecules are presented in different colours and the solvent molecules and CH hydrogen atoms are omitted for clarity). | ||
The complex cis-1·TMACl·0.5 EtOH was crystallized by slow evaporation of ethanol solution of cis-1 with excess of tetramethylammonium chloride. The crystal structure reveals that upon interaction with the guest anion, the host adopts cone conformation of the calix[4]pyrrole core and axial orientation of the hydroxypropyl arms (Fig. 2a). The anion is hydrogen bonded by the four pyrrole NH groups (N⋯Cl distance 3.33–3.38 Å). In addition, one of the hydroxypropyl arms interacts with the anion in half of the molecules within the crystal (O⋯Cl distance 3.41 Å), and half of the molecules are hydrogen bonding with the neighboring host forming a dimeric arrangement of complexes. The other arm does not hydrogen bond with the guest (O⋯Cl distance 3.90 Å), and instead, is either hydrogen bonding to the neighboring host or participating in the hydrogen bond network connecting the host–guest complex dimers and ethanol molecules into hydrogen bonded chains (Fig. 2b). Such an arrangement of the hydrogen bonds suggests that the OH hydrogen bonding with the anion is not strongly favoured, as only 25% of the OH groups of the host participate in the OH⋯Cl− interactions. The TMA cations reside in the shallow electron rich cavities of the calix[4]pyrroles on one side and in proximity to the chloride anions on the other side (Fig. S-60†) closing the sides of the dimeric assemblies. Thus, the mode of interaction of cis-1 with TMACl where the cation is located in the shallow cavity and the anion is bound by the macrocycle in cone conformation is characteristic to most calix[4]pyrroles in the solid state, whereas meso-p-hydroxyphenyl calix[4]pyrrole has displayed exo-cavity chloride complexation by hydroxyl groups.44
 | ||
| Fig. 2 (a) Crystal structure of cis-1·TMACl·0.5 EtOH (anisotropic displacement parameters are drawn at 50% probability level), (b) hydrogen bonded chain (neighbouring cis-1 molecules are presented in different colours and CH hydrogen atoms are omitted for clarity), (c) DFT-optimized structure of the Cl−⊂cis-1 (B3LYP/6-311++G(d,p)). | ||
ESI-MS experiments
Anion binding properties of cis-1 and cis-3 were screened with ion-mobility mass spectrometry (IM-MS, Fig. 3) and using tetrabutylammonium (TBA) salts of halides F−, Cl−, Br− and I−, oxoanions NO3−, H2PO4−, HSO4−, ClO4− and OTf−, and SCN−, BF4−, and PF6−. The studies were focused to the cis-isomers of 1 and 3 because these isomers are expected to give higher anion affinity than their corresponding trans-isomers.16,18 In case of cis-1, in addition to the deprotonated host [1-H]−, 1:1 anion complexes with Cl−, Br− and H2PO4− were observed. All anion complexes gave one peak in their IM-MS arrival time distributions indicating single conformer formation.
 | ||
| Fig. 3 (−)ESI-MS spectrum measured from 10 μM solution of cis-3 and TBACl (1:5 ratio) in acetonitrile. Inset on the left shows the theoretical (red) and experimental (black) isotopic distribution for [3 + Cl]− and the inset on the right shows its IM-MS arrival time distribution. | ||
The experimental collision cross section values, DTCCSN2, for the anion complexes were very similar (between 230.2 and 234.2 Å2), but clearly larger compared to that of the deprotonated host [1-H]− (221.8 Å2). Whereas cis-1 formed more selectively complexes with anions, the cis-3 showed higher and broader tendency for anion complexation and formed anion adducts with Cl−, Br−, I−, NO3−, ClO4−, OTf−, SCN− and PF6−. The ESI-MS spectrum with TBACl is shown as a representative example in Fig. 3. Interestingly, the abundancy of Cl−⊂cis-3 was six-fold higher than Cl−⊂cis-1 suggesting higher affinity of cis-3 for chloride.
As with cis-1, also in the case of cis-3, the anion complexes showed very similar DTCCSN2 values, indicating similar structure and binding site for all anions (see Table S1† for comparison). The only exception is the complex with triflate [3 + OTf]−, for which DTCCSN2 was 5 to 10 Å2 larger compared to other anion complexes. In general, halide complexes resulted in smaller DTCCSN2 values and higher abundancies than the oxoanions. The experimental DTCCSN2 derived from the IM-MS experiments were compared with theoretical values obtained for DFT energy minimized structures (see DFT section) of deprotonated cis-1, cis-3, and the Cl− complexes of cis-1 and cis-3, where the bound host adopted cone conformation. The difference between the theoretical and experimental values was ∼3% in case of [1-H]− and Cl−⊂cis-1 and ∼7% with [3-H]− and Cl−⊂cis-3 indicating a good agreement between the gas phase theoretical and experimental structures.
NMR binding studies
Anion binding with cis-1 was investigated in DMSO-d6 with 1H NMR spectroscopy using TBA salts. Previous binding studies of the host with fluoride and acetate anions have shown 1:1 stoichiometry and slow complexation/decomplexation kinetics on the NMR timescale.22 The addition of incremental amounts of chloride induced gradual shifting of the proton signals indicating that the free and bound receptor are displaying fast binding kinetics (Fig. 4). The pyrrole NH protons appearing originally at 9.19 ppm first broadened and disappeared until with excess of chloride emerged at 10.8 ppm. The OH protons resonating at 4.35 were shifted slightly upfield to 4.27 ppm suggesting that they do not interact with the anion in solution, where hydrogen bonding to the solvent molecules can dominate due to their abundance. The chemical shift changes of the β-pyrrolic protons (0.23 ppm upfield) and meso-CH2 protons of the arms (0.33 ppm downfield) upon addition of 10 eq. of chloride were fitted to a theoretical 1:1 binding isotherm and the fit returned a binding constant of 1.0·103 M−1 (Table 1). Furthermore, a meso-methyl group (g) assigned to be on the same side of the calix[4]pyrrole plane as the hydroxypropyl arms shifted 0.24 ppm downfield. These spectral features indicated that the anion is bound into the cavity of the calix[4]pyrrole in agreement with the XRD and IM-MS studies.
 | ||
| Fig. 4 1H NMR titration of cis-1 (1.1 mM) with TBACl in DMSO-d6 indicating complexed species with primed letters (an asterisk denotes the NMR solvent). | ||
:1 complexes
| Host | Guest | Solvent | NMR kinetics | K 11 (×10−4 M−1) |
|---|---|---|---|---|
|
a Fitted to a 1:1 binding model with HypNMR 2008.
b Fitted to a 1:2 binding model.
c Higher order complexes formed with excess guest.
|
||||
| cis-1 | Cl− | DMSO-d6 | Fast | 0.1a |
| cis-1 | H2PO4− | DMSO-d6 | Slow | >1 |
| cis-3 | Cl− | Acetone-d6 | Fast | >1b |
| cis-3 | H2PO4− | Acetone-d6 | Slow/intermediate | >1c |
| cis-3 | H2PO4− | DMSO-d6 | Slow/intermediate | >1c |
Next, cis-3 was titrated with TBACl in acetone-d6 which induced significant downfield shifts of pyrrole and urea NH protons (Fig. 5). The pyrrole NH initially broadened and at 1 eq. of added guest appeared at 10.3 ppm and gradually shifted more downfield with excess guest. Even though the titration was carried out until 19 eq. of chloride, complete saturation of the chemical shift changes was not observed. The distal urea NH (j, alpha to the nitrophenyl ring) also experienced a significant 2.8 ppm downfield shift, whereas the proximal urea NH (i) shifted slightly less, 1.5 ppm downfield. In addition, the CH2 proton alpha to the urea nitrogen (e) experienced initial downfield shift until at 1.6 eq. the signal started to shift slightly upfield (Fig. S-40†). Also, nitrophenyl protons experienced similar non-monotonic chemical shift changes suggesting that with excess of chloride, higher order complexes are formed. A theoretical 1:2 binding isotherm gave a relatively good fit for the data, from which the binding constant for the 1:1 complex was K11 > 104 M−1.
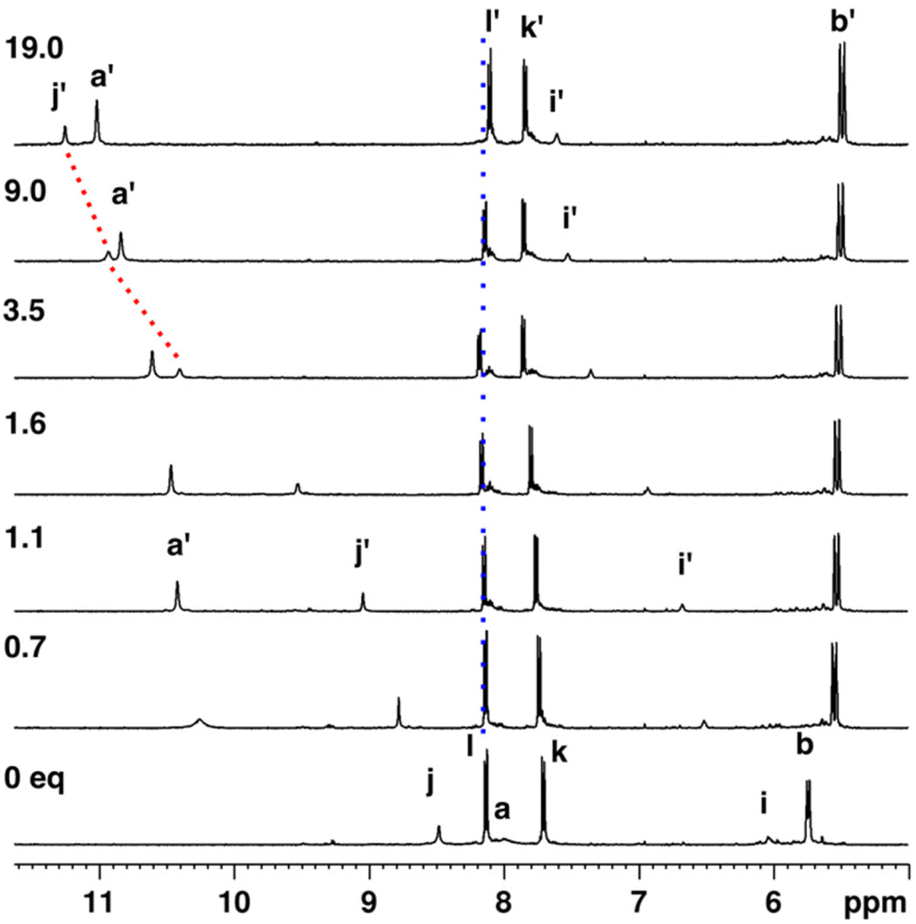 | ||
| Fig. 5 1H NMR titration of cis-3 (1.0 mM) with TBACl in acetone-d6 indicating complexed species with primed letters. | ||
To investigate the structure of the 1:1 chloride complex in solution, 1H,1H-ROESY was recorded after addition of 1 equivalent of the guest. The ROESY spectrum showed close proximity between pyrrole NH (a′) and one meso-methyl group (g′) on the same side of the pyrrole ring plane as the arms, and between pyrrole NH and arm protons (c′, Fig. 6). In addition, pyrrole CH protons (b) showed ROE correlation only with two meso-methyl groups (f′, h′) that are directed below the pyrrole ring plane. This indicates that binding of chloride stabilizes the cone conformation in solution.
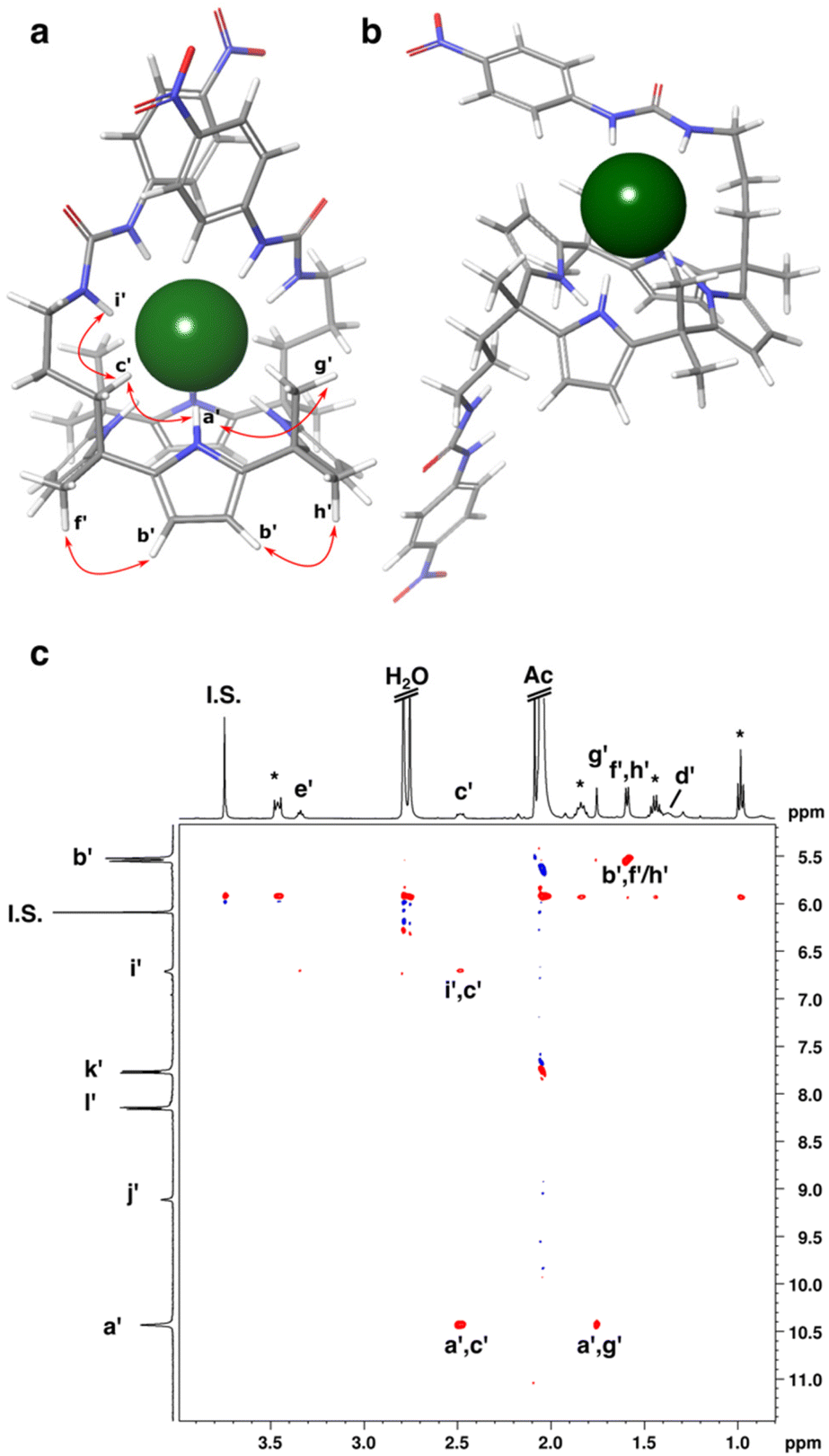 | ||
| Fig. 6 Energy minimized structures (B3LYP/6-311++G(d,p)) of (a) Cl−⊂cis-3 showing ROE correlations with red arrows, (b) Cl−⊂trans-3. (c) 1H,1H-ROESY of cis-3 (1.0 mM) with 1 eq. of TBACl in acetone-d6 (the asterisks denote the TBA signals). | ||
The complexation of oxoanion H2PO4− by cis-1 and cis-3 was also studied by NMR titrations. In case of cis-1 in DMSO-d6 (Fig. S-39†), complexation induced the emergence of a new set of signals indicating slow binding kinetics on the NMR timescale. The chemical shift changes were similar to the Cl− complex. For cis-3, titrations were carried out in acetone-d6 (Fig. S-42 and 43†) and in DMSO-d6 (Fig. S-44 and 45†). In both experiments the pyrrole NH and CH protons (a, b) experienced significant broadening and slow or intermediate exchange kinetics with their bound counterparts, whereas the urea NH protons broadened to the baseline. At approximately 1 eq. of added guest the bound pyrrole CH protons sharpened and no signals of the free host were visible, from which a binding constant of K11 > 104 M−1 was estimated. With excess of guest the signals kept shifting suggesting that higher order complexes may form.
ITC
The affinities of cis-1 and cis-3 for chloride in a polar organic solvent were compared using isothermal titration calorimetry (Table 2). A mixture of 3% DMSO in acetonitrile was chosen as a solvent to ensure the solubility of the hosts and for comparison with the previous literature data for cis-1.22 The K11 for cis-1 was 9.5·104, which showed almost two orders of magnitude increase in comparison to the NMR titration in DMSO-d6. This most likely reflects the better solvation of the receptor in DMSO than in acetonitrile. On the other hand, the value is slightly lower than previously obtained with ITC (1.7·105 M−1) using higher host concentration (∼1 mM vs. ∼20 μM). In similar conditions, cis-3 gave 1.3 kcal mol−1 more negative enthalpy and slightly higher binding constant than cis-1 as expected due to formation of more hydrogen bonds between the guest and the urea groups. It is of interest to note that the affinity for chloride with two-armed calix[4]pyrroles can be improved by adding urea groups. Previous studies have shown that the rather flexible design of cis-1 provides a modest improvement for chloride binding, whereas binding of polyatomic oxoanion acetate is increased by an order of magnitude in comparison to meso-octamethyl calix[4]pyrrole.22:1 complex) in acetonitrile with 3% DMSO at 25 °C (errors 15–20%)
| Host | ΔG (kcal mol−1) | ΔH (kcal mol−1) | TΔS (kcal mol−1) | K 11 (×10−4 M−1) |
|---|---|---|---|---|
| cis-1 | −6.8 | −5.8 | 1.0 | 9.5 |
| cis-3 | −7.2 | −7.1 | 0.1 | 17.0 |
DFT calculations
DFT calculations at B3LYP/6-311++G(d,p) level using dispersion correction were performed to obtain energy minimized structures of free cis-1 and cis-3, as well as their chloride complexes Cl−⊂cis-1 and Cl−⊂cis-3 in the gas phase. The obtained structures were used to calculate the theoretical TMLJCCSN2 values for comparison with the experimental values from the IM-MS measurements (Table S1†). The initial structures used for the geometry optimizations were created from crystallographic data of cis-1·TMACl·0.5 EtOH. Upon geometry optimization of the complexes, both receptors retained their cone conformation with pyrrolic NH protons experiencing most favorable interactions with the anions. In both cases, calix[4]pyrrole arms adopt axial orientation engaging each of the arms in hydrogen bonding interactions with the anion. According to the optimized structure of the Cl−⊂cis-1 complex, inclusion of the anion within the calix[4]pyrrole binding site forms hydrogen bonds with N⋯Cl distance in the range of 3.40–3.41 Å and the two hydroxyl groups point towards the anion with O⋯Cl distances of 3.40 Å (Fig. 2c). This indicates that in the gas phase, unlike in the crystal structure and in solution, both OH groups form hydrogen bonds with the anion due to the absence of competing hydrogen bond acceptors.In the Cl−⊂cis-3 complex, the presence of chloride anion in the cavity entails an orientation of the arms where they bend towards each other with urea groups in anti–anti conformation (Fig. 6a). In such an assembly, each urea group is engaged in two hydrogen bonds with average N⋯Cl distance of 3.37 Å, creating a favorable arrangement for inclusion of a small spherical anion such as chloride buried in the binding site. Additionally, the anion receives four hydrogen bonds from pyrrolic NH groups with distances in the range of 3.41–3.44 Å. The complex is further stabilized by parallel-displaced π⋯π stacking interactions between aromatic rings of the nitrobenzene moieties with 3.42 Å between a centroid defined by the carbon atoms of one phenyl ring and a mean plane of the second phenyl ring carbons.
In addition, DFT calculation was carried out for the Cl−⊂trans-3 complex. In that case, calix[4]pyrrole core is arranged in a partial cone conformation decreasing the number of pyrrolic hydrogen bonds to three. Since one of the arms in Cl−⊂trans-3 complex points away from the anion, only one urea group can hydrogen bond with the anionic guest leaving the anion more exposed than in Cl−⊂cis-3 (Fig. 6b). Thus, cis-3 provides better binding site for chloride with more hydrogen bonds to stabilize the host–guest complex.
Anion transport
Given the anion binding properties shown by compounds cis-1 and cis-3, they were good candidates to mediate transmembrane anion transport as mobile carriers able to stabilize the anion in the lipophilic interior of the membrane as 1:1 transporter–anion complexes (the formation of 1:2 or other polyanionic complexes is not considered, given the higher resistance that those species would offer to diffuse through the membrane). Thus, we studied the anion transport properties of those compounds using the well-established lucigenin assay (Fig. 7). Large unilamellar vesicles (LUVs) of POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and cholesterol (7:3 ratio) with an average diameter of ∼160 nm (Fig. S-53†) were prepared with NaNO3 (225 mM) and the chloride sensitive dye lucigenin (0.8 mM) encapsulated and suspended in NaNO3 (225 mM). Initially, the transporter was added to a sample of vesicles as an external stock solution in an organic solvent (acetone or methanol) and the mixture was equilibrated by stirring it at 25 °C inside a fluorometer. The transport was initiated by an extravesicular pulse of NaCl (25 mM) and the kinetics of chloride influx was monitored via the quenching of the lucigenin fluorescence over time. Ideally, the transport of Cl− into the vesicles is compensated by transport of NO3− out of the vesicles, a process known as Cl−/NO3− antiport.
 | ||
| Fig. 7 Chloride transport by calix[4]pyrroles cis-1, cis-3 and trans-3 (at the transporter:lipid ratio indicated) into LUVs of POPC:cholesterol (70:30) suspended in 225 mM NaNO3 solution (interior and exterior), monitored by the quenching of the fluorescence of encapsulated lucigenin (0.8 mM) upon addition of 25 mM NaCl. The blank curve was recorded in absence of transporter. (a) Comparison between the different receptors. (b) Concentration dependence of the transport activity of cis-3. | ||
While compound cis-1 showed no significant activity, compound cis-3 showed efficient transport activity at a concentration of 1 transporter per 200 lipids (Fig. 7a). We also tested isomer trans-3, which appeared to be approximately 10 times less active than cis-3 (cis-3 at 1:2000 shows similar activity to that of trans-3 at 1:200) (Fig. 7a). This was interpreted as a more favourable geometry of the cis-3 allowing all the anion binding groups to interact with one chloride anion, which is not the case for the trans isomer (see Fig. 6a and b). From the optimized geometries for the two isomers of 3, a higher affinity for chloride would be expected for the cis isomer. However, taking into account rather similar affinities observed for cis-1 and cis-3, the presumed affinity differences alone cannot explain the observed anion transport efficiencies. On the other hand, optimal binding geometry can play an additional role on the anion transport not related to the binding affinity. It has been previously observed that the presence of multiple anion binding groups in a molecule, in case they all cannot participate on the coordination of a single anion, can have a detrimental effect on the transmembrane transport ability of the molecule, probably due to the formation of additional interactions within the membrane environment.45 The strong geometry dependence observed for the isomers of 3 supported the hypothesis of these compounds working as mobile carriers, rather than forming self-assembled channels.
To further study transporter cis-3 we performed dose–response experiments. As expected, the transport rate decreased with the concentration of transporter (Fig. 7b), but clear transport activity was still observed at a concentration of 1 transporter per 5000 lipids. The initial rates calculated for the transport curves showed a linear dependence of the concentration of transporter (Fig. S-55 and section 6.3 of the ESI†), in agreement with a mobile carrier mechanism that relies on the formation of a 1:1 transporter–anion complex. Moreover, when the NO3− inside and outside the vesicles was substituted by SO42−, an anion much more difficult to transport due to its high hydration energy, the transport efficiency of compounds cis-3 and trans-3 decreased significantly (Fig. S-54†). This confirmed that Cl−/NO3− antiport is the predominant mechanism instead of Na+/Cl− symport, and ruled out the formation of non-specific pores in the membrane upon addition of the transporters as the cause of the fluorescence response.
Experimental
See ESI† for experimental details.Conclusions
We have successfully prepared and studied two-armed meso-substituted calix[4]pyrrole podands bearing hydroxyl and urea groups. Isomer separation was successful for the more polar meso-substituents in 1 and 3, whereas it worked for the phthalimide-derivative 2 only in milligram scale to afford crystal structures. Crystal structures of pure isomers of 1 and 2 showed always 1,3-alternate conformation for the macrocycle, whereas the orientation of the arms varied depending on the packing of the hosts.IM-MS analysis indicated that cis-1 and cis-3 formed anion complexes with a single binding geometry in cone conformation. 1H NMR titrations permitted to identify stable complexes formed by compounds cis-1 and cis-3 with Cl− and H2PO4−. The NMR titrations revealed that in DMSO solution the binding of dihydrogenphosphate by cis-1 is stronger than that of chloride, and in both cases the interaction with the pyrrole NHs was the main cause of the complex formation. Moreover, the crystal structure of cis-1 with TMACl shows the formation of the hydrogen bonds between the four pyrrole NHs of the calix[4]pyrrole core, in a cone conformation, and the chloride anion. The NMR titrations suggested little contribution of the hydroxyl groups to the coordination of the anion, in line with the crystal structure where only one of the two hydroxyl groups was interacting with the anion. On the other hand, titrations of cis-3 in acetone indicated clear implication of both the pyrrole NHs and the urea NHs on the coordination of the anions, which also resulted in higher order complexation with excess guest. The higher number of hydrogen bond donor groups involved in the complexation of chloride by cis-3 compared to cis-1 was reflected in significantly higher abundancy of chloride complexes of cis-3 than cis-1 in ESI-MS, and slightly higher binding enthalpy and binding constant observed in the ITC titrations. Furthermore, the effect of the additional urea groups in cis-3 on transport activity is drastic. While cis-1 is inactive, cis-3 can perform efficient transport via the formation of a 1:1 complex (mobile carrier mechanism). In line with the DFT model of complex Cl−⊂cis-3, an efficient coordination by the eight acidic NHs of the molecule and a good shielding of the anion are probably the main reasons for the transport activity observed. Moreover, comparison to the trans-3 isomer revealed that the geometry of cis-3 plays an important role on its anion transport activity, probably because this geometry permits the interaction of the anion with all the acidic NHs. Our study shows that the design of partially flexible calix[4]pyrroles bearing hydrogen bond donors in the meso-positions may prove a feasible strategy to increase binding and transport affinity or selectivity to larger anions such as H2PO4− and nucleotides.
Author contributions
Conceptualization – KH; Formal analysis – MP, EB, LM-C, AK, KH; Funding acquisition – LM-C, KH; Investigation – MP, EB, LM-C, AK, JN, KH; Project administration – KH; Supervision – EB, EK, KH; Validation – MP, EB, AK; Visualization – MP, EB, LM-C, AK, KH; Writing – original draft – MP, EB, LM-C, AK, EK, KH; Writing – review and editing – LM-C, EK, KH.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Prof. Pablo Ballester for hosting KH for a research stage and the Research Support Area of ICIQ for data acquisition. Dr Laura Laverdure is sincerely thanked for consulting the computational part of this work. Academy of Finland is acknowledged for financial support (projects 309910, 314287, and 335685). LM-C acknowledges the Ministerio de Universidades (Spanish government) for the María Zambrano grant awarded under the Recovery, Transformation and Resilience Plan (NextGenerationEU).References
- A. F. Sierra, D. Hernández-Alonso, M. A. Romero, J. A. González-Delgado, U. Pischel and P. Ballester, J. Am. Chem. Soc., 2020, 142, 4276–4284 CrossRef CAS PubMed.
- M. Pushina, P. Koutnik, R. Nishiyabu, T. Minami, P. Savechenkov and P. Anzenbacher Jr., Chem. – Eur. J., 2018, 24, 4879–4884 CrossRef CAS PubMed.
- H. Wang, L. O. Jones, T. Zhao, I. Hwang, V. M. Lynch, N. M. Khashab, G. C. Schatz, Z. A. Page and J. L. Sessler, Chem. Sci., 2023, 14, 4120–4125 RSC.
- S. Amharar and A. Aydogan, Dyes Pigm., 2022, 197, 109918 CrossRef CAS.
- E. Baysak, S. Yuvayapan, A. Aydogan and G. Hizal, Sens. Actuators, B, 2018, 258, 484–491 CrossRef CAS.
- S. Maji and D. Mandal, Chem. – Asian J., 2017, 12, 2369–2373 CrossRef CAS PubMed.
- Y.-C. He, Y.-M. Yan, H.-B. Tong, Z.-X. Ren, J.-H. Wang, Y.-B. Zhang, J.-B. Chao and M.-L. Wang, Chem. Commun., 2020, 56, 9364–9367 RSC.
- H. Wang, L. O. Jones, I. Hwang, M. J. Allen, D. Tao, V. M. Lynch, B. D. Freeman, N. M. Khashab, G. C. Schatz, Z. A. Page and J. L. Sessler, J. Am. Chem. Soc., 2021, 143, 20403–20410 CrossRef CAS PubMed.
- Y.-C. He, H.-B. Tong, C.-G. Xu, Z.-X. Ren, Y.-Z. Wang, Y.-M. Yan and M.-L. Wang, Org. Lett., 2023, 25, 1737–1741 CrossRef CAS PubMed.
- L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Chem, 2022, 8, 46–118 CAS.
- P. A. Gale, J. L. Sessler and V. Král, Chem. Commun., 1998, 1–8 RSC.
- D. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 532–546 RSC.
- L. Bonomo, E. Solari, G. Toraman, R. Scopelliti, C. Floriani and M. Latronico, Chem. Commun., 1999, 140, 2413–2414 RSC.
- P. Anzenbacher Jr., K. Jursíková, V. M. Lynch, P. A. Gale and J. L. Sessler, J. Am. Chem. Soc., 1999, 121, 11020–11021 CrossRef.
- G. Bruno, G. Cafeo, F. H. Kohnke and F. Nicolò, Tetrahedron, 2007, 63, 10003–10010 CrossRef CAS.
- H. Kim, K.-I. Hong, J. H. Lee, P. Kang, M.-G. Choi and W.-D. Jang, Chem. Commun., 2018, 54, 10863–10865 RSC.
- Y.-C. He, Z.-X. Ren, X.-F. Zhao, Y.-B. Zhang, J.-H. Wang, J.-B. Chao and M.-L. Wang, Tetrahedron, 2019, 75, 130491 CrossRef.
- S. P. Mahanta, B. S. Kumar and P. K. Panda, Chem. Commun., 2011, 47, 4496–4498 RSC.
- K.-C. Chang, T. Minami, P. Koutnik, P. Y. Savechenkov, Y. Liu and P. Anzenbacher Jr., J. Am. Chem. Soc., 2014, 136, 1520–1525 CrossRef CAS PubMed.
- C.-H. Lee, H. Miyaji, D.-W. Yoon and J. L. Sessler, Chem. Commun., 2008, 7345, 24–34 RSC.
- S. Peng, Q. He, G. I. Vargas-Zúñiga, L. Qin, I. Hwang, S. K. Kim, N. J. Heo, C.-H. Lee, R. Dutta and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 865–907 RSC.
- J. Yoo, E. Jeoung and C. H. Lee, Supramol. Chem., 2009, 21, 164–172 CrossRef CAS.
- N. J. Williams, V. S. Bryanstev, R. Custelcean, C. A. Seipp and B. A. Moyer, Supramol. Chem., 2016, 28, 176–187 CrossRef CAS.
- N. J. Williams, S. Roy, C. O. Reynolds, R. Custelcean, V. S. Bryantsev and B. A. Moyer, Chem. Commun., 2019, 55, 3590–3593 RSC.
- G. V. Zyryanov, T. H. Kinstle and P. Anzenbacher Jr., Synlett, 2008, 1171–1174 CAS.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497 RSC.
- J. T. Davis, P. A. Gale and R. Quesada, Chem. Soc. Rev., 2020, 49, 6056–6086 RSC.
- L. E. Bickerton, T. G. Johnson, A. Kerckhoffs and M. J. Langton, Chem. Sci., 2021, 12, 11252–11274 RSC.
- L. Martínez-Crespo and H. Valkenier, ChemPlusChem, 2022, 87, e202200266 CrossRef PubMed.
- C. C. Tong, R. Quesada, J. L. Sessler and P. A. Gale, Chem. Commun., 2008, 6321–6323 RSC.
- S. H. Park, S. H. Park, E. N. W. Howe, J. Y. Hyun, L. J. Chen, I. Hwang, G. Vargas-Zuñiga, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Chem, 2019, 5, 2079–2098 CAS.
- P. A. Gale, C. C. Tong, C. J. E. Haynes, O. Adeosun, D. E. Gross, E. Karnas, E. M. Sedenberg, R. Quesada and J. L. Sessler, J. Am. Chem. Soc., 2010, 132, 3240–3241 CrossRef CAS PubMed.
- M. G. Fisher, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light, F. P. Schmidtchen and C. C. Tong, Chem. Commun., 2009, 3017–3019 RSC.
- M. Yano, C. C. Tong, M. E. Light, F. P. Schmidtchen and P. A. Gale, Org. Biomol. Chem., 2010, 8, 4356–4363 RSC.
- S.-K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS PubMed.
- I.-W. Park, J. Yoo, B. Kim, S. Adhikari, S. K. Kim, Y. Yeon, C. J. E. Haynes, J. L. Sutton, C. C. Tong, V. M. Lynch, J. L. Sessler, P. A. Gale and C.-H. Lee, Chem. – Eur. J., 2012, 18, 2514–2523 CrossRef CAS PubMed.
- L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324–8330 CrossRef CAS PubMed.
- L. Martínez-Crespo, J. L. Sun-Wang, P. Ferreira, C. F. M. Mirabella, G. Aragay and P. Ballester, Chem. – Eur. J., 2019, 25, 4775–4781 CrossRef PubMed.
- S. H. Park, I. Hwang, D. A. McNaughton, A. J. Kinross, E. N. W. Howe, Q. He, S. Xiong, M. D. Kilde, V. M. Lynch, P. A. Gale, J. L. Sessler and I. Shin, Chem, 2021, 7, 3325–3339 CAS.
- L. Martínez-Crespo, J. L. Sun-Wang, A. F. Sierra, G. Aragay, E. Errasti-Murugarren, P. Bartoccioni, M. Palacín and P. Ballester, Chem, 2020, 1–17 Search PubMed.
- D.-W. Yoon, H. Hwang and C.-H. Lee, Angew. Chem., Int. Ed., 2002, 41, 1757–1759 CrossRef CAS PubMed.
- C.-H. Lee, J.-S. Lee, H.-K. Na, D.-W. Yoon, H. Miyaji, W.-S. Cho and J. L. Sessler, J. Org. Chem., 2005, 70, 2067–2074 CrossRef CAS PubMed.
- M. Pamuła, E. Bulatov and K. Helttunen, J. Mol. Struct., 2023, 1273, 134268 CrossRef.
- L. Martínez-Crespo, L. Halgreen, M. Soares, I. Marques, V. Félix and H. Valkenier, Org. Biomol. Chem., 2021, 19, 8324–8337 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, DFT coordinates. CCDC 2248771–2248776. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00919j |
| This journal is © The Royal Society of Chemistry 2023 |