
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
α-Amido sulphones as useful intermediates in the preparation of C-chiral α-aminophosphonates and α-aminophosphonic acids†
Joseph D.
Gbubele
 a,
Tomasz
Misiaszek
a,
Miłosz
Siczek
b and
Tomasz K.
Olszewski
*a
a,
Tomasz
Misiaszek
a,
Miłosz
Siczek
b and
Tomasz K.
Olszewski
*a
aDepartment of Physical and Quantum Chemistry, Faculty of Chemistry, Wrocław University of Science and Technology, ul. Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: tomasz.olszewski@pwr.edu.pl
bDepartment of Chemistry, University of Wrocław, F. Joliot-Curie 14, Wrocław, Poland
First published on 12th July 2023
Abstract
α-Amido sulphones have been used as useful starting materials in the preparation of C-chiral α-aminophosphonates and α-aminophosphonic acids. The developed methodology is based on a one-pot, base-catalysed in situ generation of an imine intermediate followed by addition of a phosphorus nucleophile. The presented protocol is simple and effective and can be applied to a variety of structurally diverse α-amido sulphones and phosphorus nucleophiles, leading to the desired pure products after simple crystallization in very good yields. Importantly, the use of H-phosphonate bearing a chiral auxiliary allows the reaction to be performed with high diastereoselectivity (a single diastereoisomer is generated and isolated) and the possibility of precise control of the configuration at the newly generated C-chiral centre.
Introduction
α-Amido sulfones are considered very useful and stable precursors of N-acylimines and therefore they have found many applications in modern organic synthesis, including asymmetric synthesis.1–3 Their utility stems from two aspects: the first one is their high stability and ease of preparation via simple acid-promoted three-component reactions of various aldehydes, sulfinates and carbamates,2 and the second one is their very simple transformation into reactive N-acylimine derivatives under either acidic or basic conditions (Scheme 1a). Selected protocols where α-amido sulfones have been used as substrates include reactions with organometallic reagents leading to the generation the N-acylimines required for the subsequent nucleophilic addition,4–7 reactions with allylating reagents leading to the synthesis of homoallylamino derivatives,8,9 reactions with cyanide ions providing α-amido nitriles,10–12 reactions with malonates and related derivatives,13–15 reactions with enolates from simple carbonyl derivatives,16–18 and finally reactions with nitronate anions leading to 2-nitroamine derivatives.19–21 | ||
| Scheme 1 Application of α-amido sulphones for the preparation of aminophosphonates. (a) Transformationioon of α-amido sulphones into reactive N-acylimines and N-acylium ions; (b) known protocols employing α-amido sulphones in the preparation of α-aminophopshonates; (c) known assymetric synthesis of α-aminophopshonates with the use of α-amido sulphones; (d) this work. | ||
Surprisingly, despite the extensive utility of α-amido sulfones in the preparation of structurally diverse compounds, their utility as imine precursors in the preparation of α-substituted aminophosphonates and aminophosphonic acids, important classes of compounds endowed with very interesting biological activities,22–26 including excellent enzyme inhibitors,27,28 drug candidates for antibiotics,29,30 and antibacterial and antifungal agents,31 is scarcely described in the literature.32
The only known applications include the reaction of α-amido sulfones with triethyl phosphite in the presence of indium(III) chloride33 or dialkyl trimethylsilyl phosphites in the presence of iron(III) chloride. Both reactions proceed through an N-acylium ion, as the reaction intermediate, and lead to α-aminophosphonic acid esters as racemic mixtures with acceptable yields (Scheme 1b).34 These methods, however, require the presence of metallic Lewis acid catalysts, malodorous or not easily available phosphorus nucleophiles, and dry conditions, under an inert atmosphere in a chlorinated solvent and the purification of the final products is tedious via column chromatography. In turn, the only known asymmetric preparation of optically active α-aminophosphonic acid derivatives with the use of α-amido sulfones is based on the reaction of dimethyl H-phosphonate under basic conditions in the presence of a chiral quaternary ammonium salt catalyst under phase-transfer catalytic conditions (Scheme 1c).35 This protocol is based on the formation of an N-acylimine and its further reaction with dimethyl H-phosphonate, leading to the desired optically active α-aminophosphonic acids and their methyl esters, albeit with moderate enantioselectivity.
Our interest in the preparation of new α-aminophosphonic acids and their esters36–40 prompted us to investigate the utility of α-amido sulphones as stable imine surrogates and practical intermediates in the preparation of these compounds. Herein we present the results of our research on the preparation of α-aminophosphonic acids and their esters not only as racemic mixtures but also in a highly diastereoselective fashion using a one-pot, effective and simple methodology based on the in situ generation of N-acylimines and their reaction with phosphorus nucleophiles (Scheme 1d).
Results and discussion
The starting N-benzyloxycarbonylamino sulfones were conveniently prepared via a formic acid promoted three-component reaction of aldehydes, benzenesulfinic acid sodium salt and benzyl carbamate.41,42 α-Amido sulphones were conveniently obtained in pure form after simple crystallization. Subsequently, we selected α-amido sulfone 1a and diethyl H-phosphonate (2a) as model substrates to perform the hydrophosphonylation of N-benzyloxycarbonylamino sulfones under basic conditions (Table 1). During the optimization of the reaction conditions, we quickly realized that the generated N-acylimine 4a is highly unstable when removed from the reaction mixture and undergoes decomposition to the starting benzaldehyde (to circumvent this, strictly anhydrous conditions and a protective atmosphere of argon should be used). Since we were interested in developing a simple and robust protocol, we decided to generate the imine in situ and therefore all reactions were performed in one pot by mixing α-amido sulfone 1a, diethyl H-phosphonate 2a and an appropriate base and solvent under different conditions.|
|
||
|---|---|---|
| Entry | Conditions | Conv.b (%) |
| a General reaction conditions (unless otherwise stated): α-amido sulphone 1a (2.4 mmol), H-phosphonate 2a (2.4 mmol), and base (9.6 mmol, 4 equiv.). b Conversion based on the 31P NMR spectra of the crude reaction mixture. c Isolated yield in square brackets. d Reaction time: 8 h. | ||
| 1 | No base, r.t., 24 h, THF | 0 |
| 2 | K2CO3 (4 equiv.), r.t., 24 h, THF | 30% |
| 3 | K 2 CO 3 (4 equiv.), 66 °C, 4 h, THF | 99% [95%] |
| 4 | Pyridine (4 equiv.), 66 °C, 4 h, THF | 0 |
| 5 | Cs2CO3 (4 equiv.), 66 °C, 4 h, THF | 74 |
| 6 | Et3N (4 equiv.), 66 °C, 4 h, THF | 0 |
| 7 | NaOH (4 equiv.), 66 °C, 4 h, THF | 81 |
| 8 | Diisopropylamine (4 equiv.), 66 °C, 4 h, THF | 5 |
| 9 | L-Proline (4 equiv.), 66 °C, 4 h, THF | 0 |
| 10 | K2CO3 (4 equiv.), 64 °C, 4 h, MeOH | 0 |
| 11 | K2CO3 (4 equiv.), 77 °C, 4 h, ethyl acetate | 42 (56)d |
| 12 | K2CO3 (4 equiv.), 80 °C, 4 h, 2-methyl THF | 66 (89)d |
| 13 | K2CO3 (4 equiv.), 82 °C, 4 h, acetonitrile | 50 (94)d |
| 14 | K2CO3 (4 equiv.), 101 °C, 4 h, 1,4-dioxane | 54 (80)d |
| 15 | K2CO3 (2 equiv.), 66 °C, 4 h, THF | 60 |
| 16 | K2CO3 (6 equiv.), 66 °C, 4 h, THF | 97 |

The nature of the base was important, and we discovered that its presence is crucial for the reaction to occur (Table 1, entry 1). The best results were obtained with solid inorganic bases (Table 1, entries 2, 3, 5, and 7) rather than with organic bases (Table 1, entries 4, 6, 8, and 9). The best results were obtained with K2CO3 (4 equiv.) after 4 h in THF at 66 °C (Table 1, entry 3), and the desired product 3a was isolated in 95% yield after simple crystallization from diethyl ether. Using a greater amount of K2CO3 (6 equiv.) (Table 1, entry 16) did not improve the conversion and using a smaller amount of a base (2 equiv.) (Table 1, entry 15) lowered the reaction conversion. Furthermore, we also tested different solvents, and we selected THF for the examination of the substrate scope; however, quite good results were also obtained with acetonitrile (Table 1, entry 13), 2-methyl THF (Table 1, entry 12) and 1,4-dioxane (Table 1, entry 14), as representatives of “green solvents”, but the reactions required a longer time.
Having established the optimized reaction conditions, we decided to examine the substrate scope of the developed protocol (Table 2). It was satisfying to see that our methodology worked well when we replaced diethyl H-phosphonate with other structurally variable phosphonates such as n-butyl H-phosphonate, i-propyl H-phosphonate or bulkier benzyl H-phosphonate (compounds 3b–d, 3f–h, 3k, 3l, 3m and 3q). Likewise, the reaction worked very well with other substituted benzaldehyde derived α-amido sulphones, including those with electron withdrawing or electron donating groups, and also with heteroaromatic 2-pyridine derived α-amido sulphone 3n (Table 2). In all cases, the desired aromatic α-amino phosphonates were obtained in pure form in good to excellent isolated yields (74–95%) as white, bench-stable, non-hygroscopic solids, after simple crystallization from diethyl ether. Our methodology was also found to be suitable for the synthesis of aliphatic α-amino phosphonates 3o–r prepared from suitable aliphatic α-amido sulphones, although the yields of the final products were slightly lower (58–77%) than in the case of the aromatic derivatives. Additionally, the aliphatic α-amino phosphonates 3o–r were oils, but they were conveniently purified by simple filtration using a pad of silica gel.
|
|
||||
|---|---|---|---|---|
| Entry | α-Amido sulphone 1 | H-Phosphonate 2 | α-Aminophosphonate 3 | Yielda (%) |
a Yields are given for the purified, isolated products.
b General procedure for the hydrophosphonylation of α-amido sulphones 1: In a 50 mL flask, a suitable amount of H-phosphonate 2 (2.4 mmol, 1 equiv.) was added to a solution of α-amido sulphone 1 (2.4 mmol, 1 equiv.) and K2CO3 (9.6 mmol, 4 equiv.) in THF (15 mL). The mixture was heated at 66 °C for 4 h and then cooled and filtered. The solvent was removed under reduced pressure using a rotatory evaporator. The crude product was crystallized from diethyl ether in the case of solids and oily products were purified by column chromatography (eluent: hexane/ethyl acetate = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 3:1) to obtain pure products 3a–r. :1 to 3:1) to obtain pure products 3a–r.
|
||||
| 1 | 1a R1 = Ph | 2a R2 = Et | 3a R1 = Ph; R2 = Et | 95 |
| 2 | 1a R1 = Ph | 2b R2 = nBu | 3b R1 = Ph; R2 = nBu | 87 |
| 3 | 1a R1 = Ph | 2c R2 = iPr | 3c R1 = Ph; R2 = iPr | 94 |
| 4 | 1a R1 = Ph | 2d R2 = Bn | 3d R1 = Ph; R2 = Bn | 91 |
| 5 | 1b R1 = 4-Cl-Ph | 2a R2 = Et | 3e R1 = 4-Cl-Ph; R2 = Et | 95 |
| 6 | 1b R1 = 4-Cl-Ph | 2b R2 = nBu | 3f R1 = 4-Cl-Ph; R2 = nBu | 82 |
| 7 | 1b R1 = 4-Cl-Ph | 2c R2 = iPr | 3g R1 = 4-Cl-Ph; R2 = iPr | 95 |
| 8 | 1b R1 = 4-Cl-Ph | 2d R2 = Bn | 3h R1 = 4-Cl-Ph; R2 = Bn | 95 |
| 9 | 1c R1 = 4-F-Ph | 2a R2 = Et | 3i R1 = 4-F-Ph; R2 = Et | 82 |
| 10 | 1d R1 = 4-Me-Ph | 2a R2 = Et | 3j R1 = 4-Me-Ph; R2 = Et | 82 |
| 11 | 1d R1 = 4-Me-Ph | 2b R2 = nBu | 3k R1 = 4-Me-Ph; R2 = nBu | 74 |
| 12 | 1d R1 = 4-Me-Ph | 2c R2 = iPr | 3l R1 = 4-Me-Ph; R2 = iPr | 77 |
| 13 | 1d R1 = 4-Me-Ph | 2d R2 = Bn | 3m R1 = 4-Me-Ph; R2 = Bn | 91 |
| 14 | 1e R1 = 2-Pyridyl | 2a R2 = Et | 3n R1 = 2-Pyridyl; R2 = Et | 87 |
| 15 | 1f R1 = Et | 2a R2 = Et | 3o R1 = Et; R2 = Et | 62 |
| 16 | 1g R1 = iPr | 2a R2 = Et | 3p R1 = iPr; R2 = Et | 77 |
| 17 | 1g R1 = iPr | 2d R2 = Bn | 3q R1 = iPr; R2 = Bn | 62 |
| 18 | 1h R1 = iBu | 2a R2 = Et | 3r R1 = iBu; R2 = Et | 58 |

Subsequently we became interested in performing the hydrophosphonylation of α-amido sulphones in an asymmetric fashion. To do that, we used the H-phosphonate (R,R)-5, derived from (R,R)-TADDOL, as the chiral auxiliary and phosphorus nucleophile in the reaction with α-amido sulphones. Such a chiral H-phosphonate is a bench-stable reagent that can be conveniently prepared on a multigram-scale via the reaction of the easily available (R,R)-TADDOL and PCl3.43 In our preliminary attempts to perform the diastereoselective hydrophosphonylation of the model α-amido sulphone 1a with (R,R)-5, we applied the standard conditions used for the preparation of racemic α-amino phosphonates 3 with the hope that the presence of the chiral phosphorus nucleophile (R,R)-5 will favour the formation of the desired product 6 in a diastereoselective fashion (Table 3, entry 1). Unfortunately, the dr observed in product 6 was only moderate (dr 35:65) and hence we tried to verify the influence of the base on the diastereoselectivity of the reaction by testing different inorganic and organic bases under similar reaction conditions (Table 3, entries 2–6), but the results were again disappointing. We also tested the effect of the solvent in the case when Cs2CO3 was used as the base, but there was no improvement in the dr (Table 3, entries 7–9).
|
|
||
|---|---|---|
| Entry | Conditions | drb (%)/Conv.b [%] |
| a General reaction conditions (unless otherwise stated): α-amido sulphone 1a (2.4 mmol), H-phosphonate (R,R)-5 (2.4 mmol), and base (9,6 mmol, 4 equiv.). b Diastereoselectivity and conversion based on the 31P NMR spectra of the crude reaction mixture. — no reaction. c 6.5 g of metal oxide for 6 mmol of base. d The imine was first generated by reacting 1a with K2CO3 (2 equiv.) at 66 °C for 4 h in THF and after filtration, the filtrate containing the imine was transferred to a separate flask and cooled to −78 °C followed by the addition of an appropriate base and (R,R)-5. | ||
| 1 | K2CO3 (4 equiv.), 66 °C, 4 h, THF | 35:65 [80] |
| 2 | Cs2CO3 (4 equiv.), 66 °C, 4 h, THF | 33:67 [97] |
| 3 | NaOH (4 equiv.), 66 °C, 4 h, THF | 36:64 [87] |
| 4 | KOH (4 equiv.), 66 °C, 4 h, THF | 34:66 [90] |
| 5 | Pyridine (4 equiv.), 66 °C, 4 h, THF | — |
| 6 | Et3N (4 equiv.), 66 °C, 4 h, THF | — |
| 7 | Cs2CO3 (4 equiv.), 80 °C, 4 h, 2-methyl THF | 36:64 [89] |
| 8 | Cs2CO3 (4 equiv.), 110 °C, 4 h, toluene | 65:35 [97] |
| 9 | Cs2CO3 (4 equiv.), 82 °C, 4 h, acetonitrile | 45:55 [84] |
| 10 | KOH (2.5 equiv.), Fe2O3, r.t., 5 days, DCM | 60:40 [80]c |
| 11 | KOH (2.5 equiv.), Al2O3, r.t., 5 days, DCM | 25:75 [89]c |
| 12 | KOH (2.5 equiv.), ZnO, r.t., 5 days, DCM | 30:70 [76]c |
| 13 | KOH (2.5 equiv.), MgO, r.t., 5 days, DCM | 35:65 [74]c |
| 14 | K2CO3 (2.5 equiv.), Al2O3, r.t.,5 days, DCM | 22:78 [98]c |
| 15 | NaOH (2.5 equiv.), Al2O3, r.t.,5 days, DCM | 40:60 [98]c |
| 16 | n-BuLi (1 equiv.), 12 h, −78 °C, THF | —d |
| 17 | LDA (1 equiv.), 12 h, −78 °C, THF | —d |
| 18 | ZnEt2/(TMEDA) (1 equiv.), 12 h, −78 °C, THF | 100 [74]d |
| 19 | KOH (3 equiv.), −78 °C, 4 days, THF | 100 [95] |

We then decided to evaluate the possibility of performing the reaction under milder reaction conditions. To do so, we used the literature methodology for diastereoselective C–P bond formation based on the use of a weak inorganic base in combination with a solid metal oxide.44
Such heterogeneous reactions have been found to be performed under mild conditions as the metal oxides can activate P(O)H groups so that deprotonation of the P–H bond occurs in the presence of weaker bases. In our case, however, different combinations of simple bases (KOH, K2CO3 or NaOH) with different metal oxides (Fe2O3, Al2O3, ZnO and MgO) led to good conversions at room temperature, but the diastereoselectivity was only moderate (Table 3, entries 10–15).
Next, we turned our attention towards the modification of the reaction conditions, and more specifically the reaction temperature, and we decided to lower the temperature as much as possible in order to favour the diastereoselective formation of one of the diastereoisomers (Table 3, entries 16–19). The preliminary attempts that we have performed required the generation of the imine from α-amido sulphone 1a (K2CO3 [2 equiv.], 66 °C, 4 h, THF) and after the filtration of the base, the filtrate containing the crude imine was transferred to a separate flask and cooled to −78 °C, followed by the addition of a base and (R,R)-5. Such a reaction when performed with n-Buli or LDA (Table 3, entries 16 and 17) did not produce the desired product; however, the use of ZnEt2/(TMEDA)45,46 resulted in the formation of a single diastereoisomer of 6a, albeit with a low conversion (74%). Inspired by this result, we decided to perform the reaction directly at −78 °C but with the use of KOH (3 equiv.) as a simple, solid inorganic base (useful also in the synthesis of racemic α-aminophosphonates, Table 1) and α-amido sulfone 1a in the presence of (R,R)-5 (Table 3, entry 19). To our great satisfaction, during this reaction, the imine was generated in situ and reacted directly with (R,R)-5, producing the desired α-aminophosphonate 6a as a single diastereoisomer after 4 days at −78 °C. Subsequently, the optimized reaction conditions were used in order to verify the substrate scope of this reaction (Table 4). We were pleased to see that aromatic α-amido sulphones 1a–f reacted very well under such reaction conditions, producing the desired α-aminophosphonates 6a–f as single diastereoisomers in good, isolated yields (85–95%). Importantly, neither the presence of electron-donating (6d) or electron-withdrawing (6b and 6c) groups nor the place of substitution on the aromatic ring (6e) or the size of the aromatic substituent (6f) affected the diastereoselectivity of the reaction, and in each case, a single diastereoisomer of the desired α-aminophosphonates 6a–f was formed. In the case of aliphatic α-amido sulfone 1k and 1h, presumably due to their lower reactivity, the yields were lower (65–75%) and the dr was up to 9:1. The absolute configuration of the newly formed asymmetric carbon in α-aminophosphonates (R,R,R)-6a–h was found to be (R), as determined by X-ray analysis of the crystals of compound (R,R,R)-6a (Fig. 1).47
 | ||
| Fig. 1 Determination of the absolute configuration in compounds (R,R,R)-6a and (S,S,S)-6d by X-ray analysis. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | α-Amido sulphone 1 | H-Phosphonate 5 | α-Aminophosphonate 6 | Diastereomeric ratio (dr) | Yielda (%) |
|
a Yields are given for the purified, isolated products.
b General procedure for the asymmetric hydrophosphonylation of α-amido sulphones 1: In a 50 mL flask, a suitable amount of TADDOL derived H-phosphonate 5 (1 equiv.) was added to a solution of the appropriate α-amido sulphone 1 (1 equiv.) in THF (15 mL). After the resulting mixture was cooled to −78 °C, finely ground KOH (3 equiv.) was added to one portion to the mixture. The reaction mixture was stirred vigorously at the same temperature without any precaution to exclude air or moisture. After 4 days, saturated aq. NH4Cl (ca. 15 mL) was added, and then the mixture was allowed to warm to room temperature. The organic layer was separated, and the aqueous layer was extracted three times with toluene (3 × 15 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered and the filtrate was concentrated to give the crude product. The concentrated crude product was then purified by column chromatography using CH2Cl2 (100%) to CH2Cl2/MeOH 97:3 to afford the desired pure products 6a–h.
|
|||||
| 1 | 1a R1 = Ph | (R,R)-5 | (R,R,R)-6a R1 = Ph | Single dia. | 85 |
| 2 | 1c R1 = 4-F-Ph | (R,R)-5 | (R,R,R)-6b R1 = 4-F-Ph | Single dia. | 93 |
| 3 | 1b R1 = 4-Cl-Ph | (R,R)-5 | (R,R,R)-6c R1 = 4-Cl-Ph | Single dia. | 95 |
| 4 | 1d R1 = 4-Me-Ph | (R,R)-5 | (R,R,R)-6d R1 = 4-Me-Ph | Single dia. | 91 |
| 5 | 1i R1 = 2,4-MeO-Ph | (R,R)-5 | (R,R,R)-6e R1 = 2,4-MeO-Ph | Single dia. | 89 |
| 6 | 1j R1 = 2-naphthyl | (R,R)-5 | (R,R,R)-6f R1 = 2-naphthyl | Single dia. | 90 |
| 7 | 1k R1 = Me | (R,R)-5 | 6g R1 = Me | 9:1 |
65 |
| 8 | 1h R1 = iBu | (R,R)-5 | 6h R1 = R1 = iBu | 9:1 |
75 |
| 9 | 1a R1 = Ph | (S,S)-5 | (S,S,S)-6a R1 = Ph | Single dia. | 88 |
| 10 | 1c R1 = 4-F-Ph | (S,S)-5 | (S,S,S)-6b R1 = 4-F-Ph | Single dia. | 91 |
| 11 | 1d R1 = 4-Me-Ph | (S,S)-5 | (S,S,S)-6d R1 = 4-Me-Ph | Single dia. | 93 |
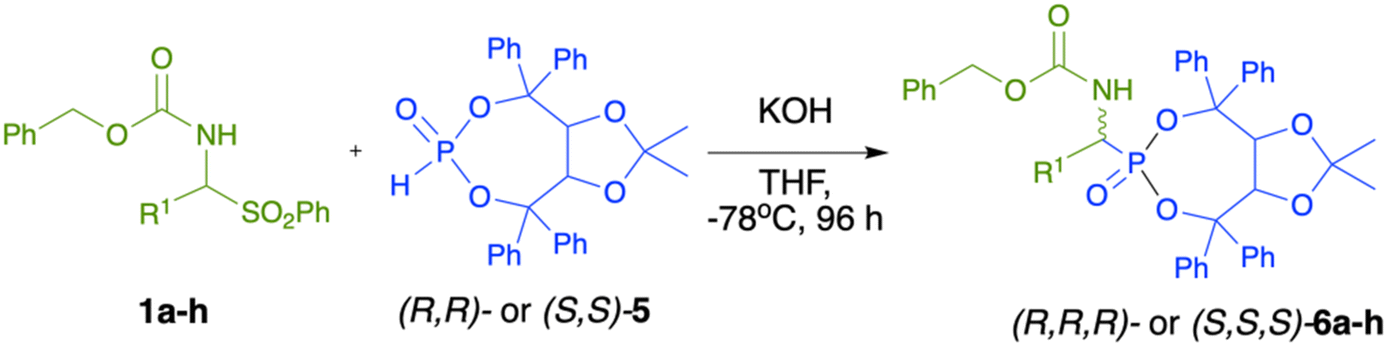
Interestingly, when we used the opposite enantiomer of the chiral phosphorus auxiliary (R,R)-5, namely (S,S)-5, under similar reaction conditions, the absolute configuration at the newly generated asymmetric α-carbon atom changed to (S). This was unambiguously confirmed by the X-ray analysis of the crystals of compound (S,S,S)-6d (Fig. 1).47 Additionally, circular dichroism (CD) spectroscopy measurements performed for the solutions of compounds (R,R,R)-6d and (S,S,S)-6d revealed that the two compounds are enantiomers as their CD spectra were mirror images of each other.47 The possibility of selective preparation of both enantiomers of α-aminophosphonates by simple use of either of the two easily available enantiomers of the chiral auxiliary (R,R)-5 or (S,S)-5 represents a real advantage of the developed methodology and clearly shows the influence of the chiral auxiliary on the diastereoselectivity of the hydrophosphonylation of α-amido sulphones.
Finally, to demonstrate the synthetic utility of the obtained α-aminophosphonates, we have shown that the model compounds 3a and 3n and the pure diastereoisomers of (R,R,R)-6a,b can be selectively and easily deprotected (Scheme 2).
 | ||
| Scheme 2 Further transformations of the obtained α-aminophosphonates. | ||
The acid hydrolysis of 3a afforded free α-aminophosphonic acid 7a (89% yield). In turn, hydrogenation of acid labile heteroaromatic 3n or benzaldehyde derived 3a α-aminophosphonates in the presence of Pd/C led to α-aminophosphonate 8n (92% yield) or 8a (89% yield), phosphonic analogue of 2-phenylglycine, with a free amino group suitable for subsequent incorporation into the peptide sequence.
In the case of pure diastereoisomers of (R,R,R)-6a,b, acid hydrolysis led to pure enantiomers of free α-aminophosphonic acids (R)-7a (90% yield) and (R)-7b (87% yield) without racemisation.
Conclusions
We have demonstrated that α-amido sulphones are easily available, bench-stable surrogates of imines and useful starting materials for the preparation of structurally diverse α-aminophosphonates and α-aminophosphonic acids. The developed methodology is based on the in situ generation of an imine during a three-component reaction of α-amido sulphones, H-phosphonates and a base under suitable conditions. The in situ generated imine undergoes hydrophosphonylation with various H-phosphonates in just 4 h in boiling THF, leading to α-aminophosphonates in high yields. In turn, when hydrophosphonylation is performed at −78 °C in the presence of a chiral H-phosphonate derived from easily available TADDOL, the desired α-aminophosphonates are obtained in a diastereoselective fashion (dr up to 100%). The asymmetric synthesis can be performed with precise control of the chirality at the newly generated α-carbon atom and selectively either (R)- or (S)-diastereoisomers can be produced. The developed methodology is operationally simple, and the products are obtained, in a majority of cases, in pure form after simple crystallization. The obtained α-aminophosphonates can be easily deprotected leading to α-aminophosphonic acids (also in enantiomerically pure form) that can find further application as useful building blocks in organic synthesis or medicinal chemistry.Experimental
Materials and methods
All the substrates and solvents were of analytical grade purchased from Polish suppliers (Sigma-Aldrich and POCh) and used without further purification. Unless otherwise specified solvents were removed using a rotary evaporator. The 1H, 13C and 31P NMR spectra were collected on a Jeol 400yh instrument (400 MHz for 1H NMR, 162 MHz for 31P NMR and 101 MHz for 13C NMR) and were processed using dedicated software (Delta 5.0.5). Samples of the product were diluted with CDCl3, DMSO-d6 or D2O with reference to the respective residual 1H or 13C signals of the solvents. Coupling constant are reported in hertz (Hz). Multiplicities are reported with the abbreviations: s (singlet), brs (broad singlet), d (doublet), t (triplet), and m (multiplet) and the reported J values are those observed from the splitting patterns in the spectrum and may not reflect the true coupling constant values. Analytical thin layer chromatography was performed on SIL G/UV254 plates and visualization was accomplished using UV light (254 nm). Column chromatography (FC) was performed using Sigma-Aldrich® silica gel high purity-grade (SiO2 70–230 mesh). The optical rotations were measured on a Bellingham + Stanley ADP440 + polarimeter and [α]TD values are given in deg cm3 g−1 dm−1; concentrations, c, are listed in 0.05 g/5 mL−1. Mass spectra were recorded using a Waters LCT Premier XE mass spectrometer (electrospray ionization, ESI) (Waters, Milford, MA, USA) and melting points were determined using SRS melting point apparatus OptiMelt MPA 100 (Stanford Research System, Sunnyvale, CA, USA) and are reported at the Faculty of Chemistry, Wroclaw University of Science and Technology. CCDC 2248672 for (R,R,R)-6a and 2248673† for (S,S,S)-6d contain the supplementary crystallographic data for this paper.:1 to 3:1) to obtain the pure products 3a–r.
:3 to afford the desired pure products 6a–h.
:1; [α]20D = −163.7 (c = 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 7.60–7.46 (m, 4H), 7.45–7.13 (m, 21H), 5.54 (d, J = 7.9 Hz, 1H), 5.35–4.93 (m, 4H), 4.34–4.18 (m, 1H), 1.38 (dd, J = 17.7, 7.3 Hz, 3H), 0.77 (s, 3H), 0.58 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 155.64 (d, 1JCO = 6.4 Hz), *155.41 (d, 1JCO = 8.8 Hz), 144.44, 143.73, 143.38, 139.55, 136.24, 129.75, 128.84, 128.76, 128.64, 128.58, 128.39, 128.36, 128.28, 128.22, 128.12, 127.92, 127.83, 127.39, 127.33, 127.20, 127.08, 126.87, 126.65, *114.4, 114.12, 90.87 (d, 2JCP = 13.5 Hz), 86.87 (d, 2JCP = 8.8 Hz), 79.68, 78.99, 67.19, *67.02, 45.04 (d, 1JCP = 167.66 Hz), *44.44 (d, 1JCP = 166.65 Hz), 26.94 (d, 2JCP = 6.3 Hz), *26.66 (d, 2JCP = 7.9 Hz), 16.20, 15.71; 31P NMR (162 MHz, CDCl3) δ: 19.38, *18.88; HRMS (ESI) calculated for C41H40NO7PNa [M + Na]+: 712.2440 found 721.2435.
:9; [α]20D = −153.8 (c = 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 7.61–7.48 (m, 4H), 7.42–7.06 (m, 21H), 5.54 (d, J = 7.9 Hz, 1H), 5.27–5.07 (m, 2H), 5.01 (d, J = 10.5 Hz, 1H), 4.94 (d, J = 12.1 Hz, 1H), 4.30–4.16 (m, 1H), 1.77–1.56 (m, 2H), 0.90 (d, J = 6.6 Hz, 3H), 0.80 (d, J = 6.4 Hz, 3H), 0.74 (s, 3H), 0.60 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 156.03 (d, 1JCO = 4.3 Hz), *155.73 (d, 1JCO = 5.1 Hz), 144.69 (d, 3JCP = 7.2 Hz), *144.52 (d, 3JCP = 5.8 Hz), 143.78, 139.56, 136.26, 129.82, 128.94, 128.73, 128.65, 128.60, 128.54, 128.44, 128.39, 128.35, 128.26, 128.17, 128.06, 127.77, 127.36, 127.32, 127.23, 126.95, 126.67, *114.33, 114.16, 91.10 (d, 2JCP = 13.8 Hz), *90.61 (d, 2JCP = 14.1 Hz), *87.07 (d, 2JCP = 9.1 Hz), 86.80 (d, 2JCP = 8.9 Hz), *80.21, 79.79, *79.49, 78.93, 67.23, *67.06, 47.97 (d, 1JCP = 165.64 Hz, *47.31 (d, 1JCP = 165.64 Hz), *38.36 (d, 2JCP = 4.1 Hz), 37.82 (d, 2JCP = 3.4 Hz), 26.94, *26.72, 24.63, *24.46, *24.32, *23.52, 23.48, *21.27, 21.18; 31P NMR (162 MHz, CDCl3) δ: *19.21, 18.90; HRMS (ESI) calculated for C44H46NO7PNa [M + Na]+: 754.2910 found 754.2913.
Conflicts of interest
There are no conflicts to declare.References
- E. Marcantoni, A. Palmieri and M. Petrini, Recent synthetic applications of α-amido sulfones as precursors of N-acylimino derivatives, Org. Chem. Front., 2019, 6, 2142–2182 RSC.
- M. Petrini, α-Amido Sulfones as Stable Precursors of Reactive N-Acylimino Derivatives, Chem. Rev., 2005, 105, 3949–3977 CrossRef CAS PubMed.
- B. Yin, Y. Zhang and Li.-W. Xu, Recent Applications of α-Amido Sulfones as in situ Equivalents of Activated Imines for Asymmetric Catalytic Nucleophilic Addition Reactions, Synthesis, 2010,(21), 3583–3595 CrossRef CAS.
- M. G. Moloney, T. Panchal and R. Pike, Trans-2,5-Disubstituted pyrrolidines: rapid stereocontrolled access from sulfones, Org. Biomol. Chem., 2006, 4, 3894–3897 RSC.
- X. Guinchard and J.-N. Denis, Reactions of In Situ Generated N-Boc Nitrones with Aromatic and Heteroaromatic Grignard Reagents: Application to the Synthesis of Zileuton, J. Org. Chem., 2008, 73, 2028–2031 CrossRef CAS PubMed.
- H. Nakagawa, J. C. Rech, R. W. Sindelar and J. A. Ellmann, Catalytic Enantioselective Addition of Arylboronic Acids to N-Boc Imines Generated in Situ, Org. Lett., 2007, 9, 5155–5157 CrossRef CAS PubMed.
- Z. Liu and M. Shi, Catalytic asymmetric addition of arylboronic acids to N-Boc imines generated in situ using C2-symmetric cationic N-heterocyclic carbenes (NHCs) Pd2+ diaquo complexes, Tetrahedron, 2011, 66, 2619–2623 CrossRef.
- M. Lombardo, E. Mosconi, F. Pasi, M. Petrini and C. Trombini, An Efficient Diastereoselective Route to Differentially Protected anti-4-Amino-1-alken-3-ols, J. Org. Chem., 2007, 72, 1834–1837 CrossRef CAS PubMed.
- Z.-F. Xie, Z. Chai, G. Zhao and J.-D. Wang, A Convenient Route to Synthesize N-Protected α,α-Difluorohomoallylic Amines by gem-Difluoroallylation of α-Amido Sulfones, Synthesis, 2008, 3805–3809 CAS.
- H. Yan, J. S. Oh, J.-W. Lee and C. E. Song, Scalable organocatalytic asymmetric Strecker reactions catalysed by a chiral cyanide generator, Nat. Commun., 2012, 3, 1212 CrossRef PubMed.
- T. Ooi, Y. Uematsu, J. Fujimoto, K. Fukumoto and K. Maruoka, Advantage of in situ generation of N-arylsulfonyl imines from α-amide sulfones in the phase-transfer-catalyzed asymmetric Strecker reaction, Tetrahedron Lett., 2007, 48, 1337–1340 CrossRef CAS.
- T. Ooi, Y. Uematsu and K. Maruoka, Asymmetric Strecker Reaction of Aldimines Using Aqueous Potassium Cyanide by Phase-Transfer Catalysis of Chiral Quaternary Ammonium Salts with a Tetranaphthyl Backbone, J. Am. Chem. Soc., 2006, 128, 2548–2549 CrossRef CAS PubMed.
- S. Lou, P. Dai and S. E. Schaus, Asymmetric Mannich Reaction of Dicarbonyl Compounds with α-Amido Sulfones Catalyzed by Cinchona Alkaloids and Synthesis of Chiral Dihydropyrimidones, J. Org. Chem., 2007, 72, 9998–10008 CrossRef CAS PubMed.
- H. Zhu, X. Jiang, X. Li, C. Hou, Y. Jiang, K. Hou, R. Wang and Y. Li, Highly Enantioselective Synthesis of N-Protected β-Amino Malonates Catalyzed by Magnetically Separable Heterogeneous Rosin-Derived Amino Thiourea Catalysts: A Stereocontrolled Approach to β-Amino Acids, ChemCatChem, 2013, 5, 2187–2190 CrossRef CAS.
- D. Li, Y. Tan, P. Peng, S. Li, N. Zhang, Y. Liu and H. Yan, Asymmetric Mannich Reaction and Construction of Axially Chiral Sulfone-Containing Styrenes in One Pot from α-Amido Sulfones Based on the Waste–Reuse Strategy, Org. Lett., 2018, 20, 4959–4963 CrossRef CAS PubMed.
- Y. Hayashi, D. Sakamoto, H. Shomura and D. Hashizume, Asymmetric Mannich Reaction of α-Keto Imines Catalyzed by Diarylprolinol Silyl Ether, Chem. – Eur. J., 2013, 19, 7678–7681 CrossRef CAS PubMed.
- H. Wu, H. An, S. Mo and T. Kodadek, Asymmetric synthesis of vinylogous β-amino acids and their incorporation into mixed backbone oligomers, Org. Biomol. Chem., 2017, 15, 3255–3264 RSC.
- X.-S. Hua, Y. Dub, J.-S. Yu, F.-M. Liao, P.-G. Ding and J. Zhou, A Highly Efficient Gold(I)-Catalyzed Mukaiyama–Mannich Reaction of α-Amino Sulfones with Fluorinated Silyl Enol Ethers To Give β-Amino α-Fluorinated Ketones, Synlett, 2017, 28, 2194–2198 CrossRef.
- Y. Wei, W. He, Y. Liu, P. Liu and S. Zhang, Highly Enantioselective Nitro-Mannich Reaction Catalyzed by Cinchona Alkaloids and N-Benzotriazole Derived Ammonium Salts, Org. Lett., 2012, 14, 704–707 CrossRef CAS PubMed.
- K. M. Johnson, M. S. Rattley, F. Sladojevich, D. M. Barber, M. G. Nuñez, A. M. Goldys and D. J. Dixon, A New Family of Cinchona-Derived Bifunctional Asymmetric Phase-Transfer Catalysts: Application to the Enantio- and Diastereoselective Nitro-Mannich Reaction of Amidosulfones, Org. Lett., 2012, 14, 2492–2495 CrossRef CAS PubMed.
- B. Wang, Y. Liu, C. Sun, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Asymmetric Phase-Transfer Catalysts Bearing Multiple Hydrogen-Bonding Donors: Highly Efficient Catalysts for Enantio- and Diastereoselective Nitro-Mannich Reaction of Amidosulfones, Org. Lett., 2014, 16, 6432–6435 CrossRef CAS PubMed.
- M. M. Abdou, Synopsis of recent synthetic methods and biological applications of phosphinic acid derivatives, Tetrahedron, 2020, 76, 131251 CrossRef CAS.
- J. B. Rodriguez and C. Gallo-Rodriguez, The Role of the Phosphorus Atom in Drug Design, ChemMedChem, 2019, 14, 190 CAS.
- G. P. Horsman and D. L. Zechel, Phosphonate Biochemistry, Chem. Rev., 2017, 117, 5704 CrossRef CAS PubMed.
- Ch. M. Sevrain, M. Berchel, H. Couthon and P.-A. Jaffres, Phosphonic acid: preparation and applications, Beilstein J. Org. Chem., 2017, 13, 2186 CrossRef CAS PubMed.
- A. Mucha, P. Kafarski and L. Berlicki, Remarkable Potential of the α -Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry, J. Med. Chem., 2011, 54(17), 5955–5980 CrossRef CAS PubMed.
- D. V. Patel, K. Rielly-Gauvin, D. E. Ryono, C. A. Free, W. L. Rogers, S. A. Smith, J. M. DeForrest, R. S. Oehl and E. W. Petrillo, α-Hydroxy Phosphinyl-Based Inhibitors of Human Renin, J. Med. Chem., 1995, 38, 4557–4569 CrossRef CAS PubMed.
- P. P. Giannousis and P. A. Bartlett, Phosphorus amino acid analogs as inhibitors of leucine aminopeptidase, J. Med. Chem., 1987, 30, 1603 CrossRef CAS PubMed.
- K.-K. A. Wang, T. L. Ng, P. Wang, Z. Huang, E. P. Balskus and W. A. van der Donk, Glutamic acid is a carrier for hydrazine during the biosyntheses of fosfazinomycin and kinamycin, Nat. Commun., 2018, 9, 1 CrossRef PubMed.
- J. Wang, M. F. Ansari, J.-M. Lin and C.-He Zho, Design and Synthesis of Sulfanilamide Aminophosphonates as Novel Antibacterial Agents towards Escherichia coli, Chin. J. Chem., 2021, 39, 2251–2263 CrossRef CAS.
- E. C. L. Marrs, L. Varadi, A. F. Bedernjak, K. M. Day, M. Gray, A. L. Jones, S. P. Cummings, R. J. Anderson and J. D. Perry, Phosphonopeptides Revisited, in an Era of Increasing Antimicrobial Resistance, Molecules, 2020, 25, 1445 CrossRef CAS PubMed.
- F. Orsini, G. Sello and M. Sisti, Aminophosphonic acids and derivatives. Synthesis and biological applications, Curr. Med. Chem., 2010, 17, 264–289 CrossRef CAS PubMed.
- B. Das, K. Damodar and N. Bhunia, A Simple and Efficient Access to α-Amino Phosphonates from N-Benzyloxycarbonylamino Sulfones Using Indium(III) Chloride, J. Org. Chem., 2009, 74, 5607–5609 CrossRef CAS PubMed.
- V. Boyapati and B. Das, Improved facile synthesis of α-amino phosphonates by the reaction of α-amido sulfones with dialkyl trimethyl silyl phosphites catalyzed by Fe(III) chloride, Synth. Commun., 2017, 47, 449–456 CrossRef.
- F. Fini, G. Micheletti, L. Bernardi, D. Peterson, M. Fochi and A. Ricci, An easy entry to optically active α-amino phosphonic acid derivatives using phase-transfer catalysis (PTC), Chem. Commun., 2008, 4345–4347 RSC.
- J. D. Gbubele and T. K. Olszewski, Asymmetric synthesis of organophosphorus compounds using H–P reagents derived from chiral alcohols, Org. Biomol. Chem., 2021, 19, 2823–2846 RSC.
- A. Brol and T. K. Olszewski, Synthesis and stability of 1-aminoalkylphosphonic acid quaternary ammonium salts, Org. Biomol. Chem., 2021, 19, 6422–6430 RSC.
- A. Brol and T. K. Olszewski, Deamination of 1-Aminoalkylphosphonic Acids: Reaction Intermediates and Selectivity, Molecules, 2022, 27, 8849 CrossRef CAS PubMed.
- E. Proniewicz and T. K. Olszewski, SERS/TERS Characterization of New Potential Therapeutics: The Influence of Positional Isomerism, Interface Type, Oxidation State of Copper, and Incubation Time on Adsorption on the Surface of Copper(I) and (II) Oxide Nanoparticles, J. Med. Chem., 2022, 65(5), 4387–4400 CrossRef CAS PubMed.
- P. Cybulska, Y.-M. Legrand, A. Babst-Kostecka, S. Diliberto, A. Leśniewicz, E. Oliviero, V. Bert, C. Boulanger, C. Grison and T. K. Olszewski, Green and Effective Preparation of α-Hydroxyphosphonates by Ecocatalysis, Molecules, 2022, 27, 3075 CrossRef CAS PubMed.
- A. Louise Tillman, J. Ye and D. J. Dixon, Direct enantio- and diastereoselective Mannich reactions of malonate and β-keto esters with N-Boc and N-Cbz aldimines catalysed by a bifunctional cinchonine derivative, Chem. Commun., 2006, 1191–1193 RSC.
- We have deliberately selected benzyl carbamate as this group can be conveniently removed by hydrogenation under neutral conditions, without affecting the phosphonate moiety and the obtained free amine could be further attached to the peptide fragment or further modified.
- For a recent example of the synthesis of TADDOL H-phosphonate 5, see: (a) Y.-X. Wang, S.-L. Qi, Yu.-X. Luan, X.-W. Han, S. Wang and H. Chen, Mengchun Ye Enantioselective Ni–Al Bimetallic Catalyzed exo-Selective C– H Cyclization of Imidazoles with Alkenes, J. Am. Chem. Soc., 2018, 140(16), 5360–5364 CrossRef CAS PubMed. For the preparation of P-stereogenic phosphonates, use of P-chiral H-phosphinates can be recommended, see: (b) T. Chen and Li.-B. Han, Optically active H-phosphinates and their stereospecific transformations into optically active P-stereogenic organophosphoryl compounds, Synlett, 2015, 26, 1153–1163 CrossRef CAS; (c) Q. Li, T. Chen, Q. Xu and L.-B. Han, Rhodium- and Iridium-catalyzed asymmetric addition of optically pure P-chiral H-phosphinates to aldehydes leading to optically active α-hydroxyphosphinates, Chem. – Eur. J., 2016, 22, 6213–6217 CrossRef CAS PubMed; (d) J. Yang, T. Chen, Y. Zhou, S.-F. Yin and Li.-B. Han, Mechanistic studies on the palladium-catalyzed cross dehydrogenative coupling of P(O)–H compounds with terminal alkynes: Stereochemistry and reactive intermediates, Organometallics, 2015, 34(20), 5095–5098 CrossRef CAS; (e) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, Bo Qu, Y. Xu, Z. Li, J. T. Reeves, J.-N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, Efficient asymmetric synthesis of P-chiral phosphine oxides via properly designed and activated benzoxazaphosphinine-2-oxide agents, J. Am. Chem. Soc., 2013, 135(7), 2474–2477 CrossRef CAS PubMed.
- L. Tedeschi and D. Enders, Asymmetric Synthesis of β-Phosphono Malonates via Fe2O3-Mediated Phospha-Michael Addition to Knoevenagel Acceptors, Org. Lett., 2001, 3, 3515–3517 CrossRef CAS PubMed.
- F. Palacios, T. K. Olszewski and J. Vicario, Diastereoselective hydrophosphonylation of imines using (R,R)-TADDOL phosphite. Asymmetric synthesis of α-aminophosphonic acid derivatives, Org. Biomol. Chem., 2010, 8, 4255–4258 RSC.
- D. Enders, L. Tedeschi and J. W. Bats, Asymmetric Synthesis of α-Substituted β-Nitrophosphonic Acids by Phospha-Analogous Michael Addition to Aromatic Nitroalkenes, Angew. Chem., Int. Ed., 2000, 39, 4605 CrossRef CAS PubMed.
- See the ESI† for more details.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2248672 and 2248673. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00924f |
| This journal is © The Royal Society of Chemistry 2023 |