
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic vs. uncatalyzed [2 + 2] photocycloadditions of quinones with alkynes†
Alexander A.
Fadeev
 and
Martin
Kotora
*
and
Martin
Kotora
*
Department of Organic Chemistry, Faculty of Science, Charles University, Albertov 6, 128 43 Praha 2, Czech Republic. E-mail: kotora@natur.cuni.cz; Fax: +420 221 951 053; Tel: +420 221 951 326
First published on 11th July 2023
Abstract
Photoreactions of quinones with alkynes under catalytic and non-catalytic conditions were studied. In contrast to recent reports, simple irradiation with blue light is sufficient to trigger [2 + 2] photocycloadditions, which afford either fused cyclobutenes or reactive para-quinone methides (p-QMs) depending on the quinone structure. Revision of the chemo- and regioselectivity of the uncatalyzed photoreactions provided useful insight into their overlooked relatedness to the recently developed catalytic protocols. Experimental evidence indicates that the reactivity of the photochemically generated p-QMs is sufficient to perform uncatalyzed reactions with nucleophiles, which can help to explain the existing transformations and develop new cascade transformations involving quinones and alkynes.
1. Introduction
Photocatalytic reactions enable access to unique intermediates and products, inaccessible by classical chemical catalysis. The synergy between photochemistry and catalysis has created vast synthetic opportunities by establishing new reactivity modes, such as transition metal photoredox catalysis,1 organic photoredox catalysis (photo-organocatalysis)2 and metallaphotoredox catalysis.3 Photocatalytic approaches have been proven to be more general than purely photochemical methods by not requiring light absorption by reactants. On the other hand, purely photochemical reactions are ideally green, accessible and inexpensive and still remain a valuable synthetic tool.4Quinones play an important role in chemistry and biology, largely due to their redox and photoredox properties. Since the discovery of [2 + 2] photodimerization of thymoquinone by Liebermann in 1877,5 the photochemistry of quinones has received considerable attention and continues to grow.6 Reactions of quinones with alkynes have attracted increased interest due to the accessibility of the starting materials and the structural diversity of the products, which was later broadened with the aid of catalysis.
Uncatalyzed photoreactions of quinones with alkynes have been reported since the 1960s, yielding either fused cyclobutenes or p-quinone methides (p-QMs) via π–π* or n–π* quinone triplet states, respectively (Scheme 1).7 The reaction outcome strongly depends on the electronic properties of the used quinone: electron-poor p-benzoquinones give p-QMs, while electronically richer methoxy-p-benzoquinones and naphthoquinones give cyclobutenes as main products. Notably, highly polar substrates reportedly react even in the absence of light.8 In recent years, such reactions have found application in the synthesis of push–pull chromophores and spiro-systems,8 caged compounds,9 and activators of mRNAs.10
 | ||
| Scheme 1 Possible products of uncatalyzed photoreactions of quinones with alkynes. | ||
Over the past decade, several catalytic photoreactions involving quinones and alkynes have been developed, enabling the synthesis of structurally diverse products: aryl ketones,11,12 indoles,13 naphthofurans14 and various carbonyl compounds with a sterically hindered quaternary stereocenter in the α-position.15 Given that these photocatalytic methods have already demonstrated high potential in diversity-oriented synthesis and target-oriented synthesis aimed at pharmaceuticals and their precursors, the development of purely photochemical versions of such processes may help to minimize the generated waste, synthesis costs, and the risks associated with product contamination by transition metals. To achieve this, some of the aforementioned catalytic transformations relied on uncatalyzed photointeraction between quinones and alkynes to generate p-QMs as intermediates,15 although the subsequent uncatalyzed reactions with nucleophiles were found problematic.15a,e,f However, other reports excluded12,13 or did not consider11,14 such an interaction as part of the primary or secondary reaction pathway. Nevertheless, establishing pathways and conditions for product formation without a catalyst is important not only for the mechanistic understanding of such processes, but also for developing their enantioselective variants.15b,c
Herein, we report the possibilities of uncatalyzed [2 + 2] photocycloadditions of quinones with alkynes, compare their chemo- and regioselectivity with the recent catalytic versions and revise the structures of three reported cycloaddition products. In addition, we establish the conditions for an efficient uncatalyzed addition of nucleophiles to the photochemically generated p-QMs, highlighting its reversibility and utility in the development of synthetically useful cascade transformations using easily accessible quinones and alkynes.
2. Results and discussion
2.1. Photoreactions leading to p-QMs
Initially, the high synthetic potential of p-QMs16 prompted us to study the structure and stability of those that can be obtained from readily available quinones and alkynes. We believed that the electron-withdrawing effect of the exocyclic carbonyl group in the expected products would benefit their stability and avoid the introduction of bulky stabilizing groups (i.e. aryl and tert-butyl7,15), which may not be desired in further synthetic applications. First, a model reaction between p-benzoquinone and diphenylacetylene in acetonitrile under irradiation with blue light-emitting diodes (LEDs) showed that p-QM 1a can be obtained in 82% isolated yield (Scheme 2). The yield was further improved to 87% by using chloroform as the solvent. Reacting p-benzoquinone with 1-phenyl-1-propyne under these conditions selectively provided 1b in 85% yield. Electronically richer quinones exhibited higher reactivity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Thus, when phenylacetylene reacted with 1,4-naphthoquinone or with 2,6-dimethylbenzoquinone, p-QMs 1c and 1d were obtained in lower yields (32% and 40%) due to the concurrent formation of cyclobutenes 2c and 2d (12% and 29%). In fact, 1c was obtained as a mixture of stereoisomers Z-1c and E-1c in a 6/1 ratio. Pure Z-1c (Z/E > 20/1) was obtained by crystallization of the isomeric mixture from DCM/hexanes and its structure was confirmed by X-ray diffraction analysis (see the ESI, Section 3†). Prolonged exposure of photosensitive Z-1c to blue light caused its isomerization into E-1c and aromatization of the latter.7d,10a In contrast to electron-rich quinones, electron-poor tetrachloro-1,4-benzoquinone and 2,6-dibromo-1,4-benzoquinone reacted with diphenylacetylene chemoselectively to give p-QMs 1e and 1f as main products. Finally, several p-QMs were found to be unstable in the pure state but sufficiently stable in solution. For example, 1g was generated in solution from p-benzoquinone and 4-octyne in 70% yield. Unsymmetrical alkynes, such as 1-pentyne, phenylacetylene, and methyl phenylpropiolate, regioselectively reacted with p-benzoquinone to give respective solutions of labile p-QMs 1h, 1i and 1j in comparable yields.
C bonds. Thus, when phenylacetylene reacted with 1,4-naphthoquinone or with 2,6-dimethylbenzoquinone, p-QMs 1c and 1d were obtained in lower yields (32% and 40%) due to the concurrent formation of cyclobutenes 2c and 2d (12% and 29%). In fact, 1c was obtained as a mixture of stereoisomers Z-1c and E-1c in a 6/1 ratio. Pure Z-1c (Z/E > 20/1) was obtained by crystallization of the isomeric mixture from DCM/hexanes and its structure was confirmed by X-ray diffraction analysis (see the ESI, Section 3†). Prolonged exposure of photosensitive Z-1c to blue light caused its isomerization into E-1c and aromatization of the latter.7d,10a In contrast to electron-rich quinones, electron-poor tetrachloro-1,4-benzoquinone and 2,6-dibromo-1,4-benzoquinone reacted with diphenylacetylene chemoselectively to give p-QMs 1e and 1f as main products. Finally, several p-QMs were found to be unstable in the pure state but sufficiently stable in solution. For example, 1g was generated in solution from p-benzoquinone and 4-octyne in 70% yield. Unsymmetrical alkynes, such as 1-pentyne, phenylacetylene, and methyl phenylpropiolate, regioselectively reacted with p-benzoquinone to give respective solutions of labile p-QMs 1h, 1i and 1j in comparable yields.
 | ||
Scheme 2 Photoreactions leading to the predominant formation of p-QMs 1a–1j. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MeCN was used as the solvent, bafter subsequent purification by recrystallization, cyield in parentheses is based on the recovered diphenylacetylene, dNMR yields are given. MeCN was used as the solvent, bafter subsequent purification by recrystallization, cyield in parentheses is based on the recovered diphenylacetylene, dNMR yields are given. | ||
2.2. Photoreactions leading to cyclobutenes
The synthesis of cyclobutenes by [2 + 2] photocycloaddition of quinones with alkynes does not typically require any additional reagents,7,10,15 although trifluoroacetic acid and potassium persulfate were recently reported as necessary additives for certain transformations of this type.12 To evaluate the role of these reagents, we first attempted to perform [2 + 2] cycloadditions between such quinones and alkynes by the action of blue light alone (Scheme 3). All the reactions provided the cycloaddition products 2, indicating that stoichiometric amounts of trifluoroacetic acid and potassium persulfate are not essential for such photoreactions to proceed. On the other hand, we did not observe an increase in the reaction's efficiency when these reagents were employed according to Shah's method.12 To determine the reason for decreased yields in the catalyst-free photoreactions, we analyzed the reaction mixtures for possible byproducts. All the reactions proceeded with high conversion of quinones (>90%) and gave a similar outcome in acetonitrile and chloroform. 2-Methoxynaphthoquinone showed complete chemo- and regioselectivity in reactions with terminal alkynes, providing cyclobutenes 2b, 2e, 2f, and 2h–2k in 38–69% yields. A dimer of 2-methoxynaphthaquinone17 was formed in these reactions as the main byproduct (isolated in up to 22% yield when 2e was synthesized in MeCN). Conversely, menadione was not only less prone to dimerization, but also less chemo- and regioselective. Thus, products arising from Paternò–Büchi reactivity were detected in the crude mixtures, and cyclobutenes 2a and 2l–2n were formed as regioisomeric mixtures with the rr major/minor ≥ 5/1. Some other possible photochemical side reactions can be found in the literature.7e,18 | ||
| Scheme 3 Photoreactions affording fused cyclobutenes 2a–2n. aMeCN was used as the solvent, bbased on the 1H NMR analysis of the crude mixture, cunreacted diphenylacetylene was recovered (56%). | ||
According to the earlier reports, photocycloadditions of terminal alkynes to monosubstituted quinone double bonds proceed regioselectively to give predominantly syn-products.7c,e Shah, however, suggested an unusual reversal of regioselectivity in persulfate-mediated photoreactions of 2-methoxynaphthoquinone with cyclopropylacetylene and 1-ethynylcyclohexene based on the comparison of J-coupling in the 1H NMR spectra.12 When we conducted these photocycloadditions in a purely photochemical setup, the formed cyclobutenes 2j and 2k appeared spectrally identical to those reported by Shah et al. (the authors described cyclobutene hydrogens as singlets, yet we observed small J-coupling (<1 Hz)). To ascertain the structure, we obtained a single crystal of 2k, X-ray diffraction analysis of which disproved the proposed anti-arrangement in favor of the syn-arrangement (see the ESI, Section 3†). Compiled NMR data of compounds 2 showed that the reversal of syn-selectivity in the reactions with other terminal alkynes is also unlikely (see the ESI, Table S2†). Besides that, analysis of NMR data revealed that the spectra of cyclobutene 2d coincide with those of the recently reported 4-benzoyl-2,6-dimethylcyclohexa-2,5-dien-1-one11 – a product of a copper-catalyzed photointeraction between phenylacetylene and in situ generated 2,6-dimethylbenzoquinone. However, the structure of this reported product entails a rapid equilibrium shift to the fully aromatic tautomer. More likely, this compound possesses the structure of cyclobutene 2d and could be formed by the classical [2 + 2] photocycloaddition.
2.3. Product diversification using nucleophilic addition to p-QMs
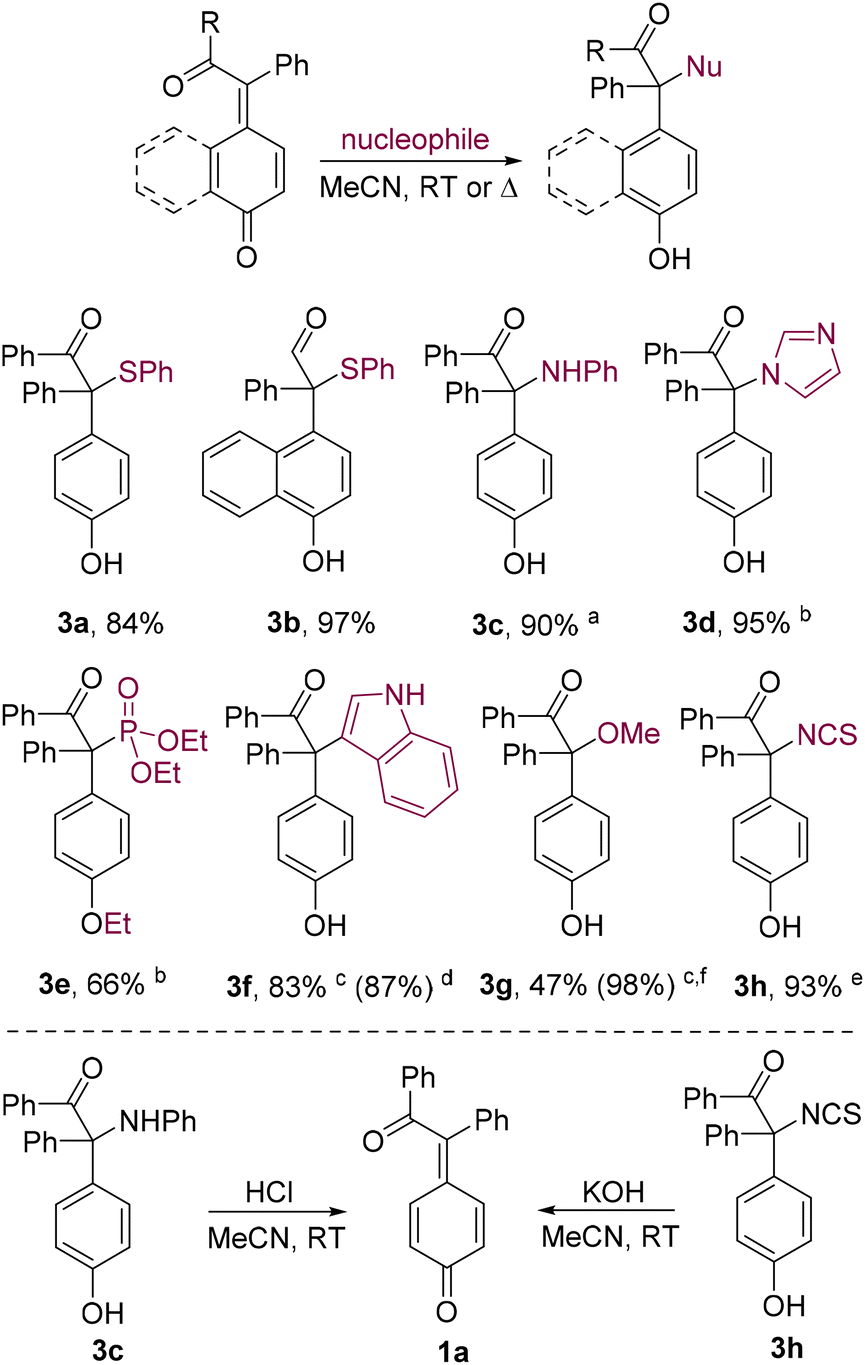
Recent studies have shown that [2 + 2] photocycloadditions of quinines with alkynes can work in tandem with catalytic 1,6-conjugate addition of nucleophiles to the in situ formed p-QMs.15 To address the less studied uncatalyzed addition, we first performed reactions of p-QM 1a with carbon and heteroatom nucleophiles under catalyst-free conditions (Scheme 4). Thus, 1,6-conjugate addition of thiophenol and aniline proceeded rapidly at room temperature even in the absence of Zn(OTf)2,15a providing adducts 3a, its benzo analogue 3b, and 3c in high yields (3c was found to be unstable and isolated as the O-acetate derivative 3c′). However, weaker nucleophiles reacted sluggishly at room temperature, and heating was found helpful. Thus, heating 1a with imidazole at 70 °C afforded the N-alkylation product 3d in nearly quantitative yield. Similarly, phosphonate 3e was prepared in 66% yield from 1a and triethyl phosphite in an Arbuzov-type reaction. The addition of indole became effective at 200 °C in a microwave reactor and furnished 3f in 83% yield (for comparison, catalysis with Zn(OTf)215a afforded 3f in a comparable yield of 87% at room temperature). Similarly, microwave-assisted addition of methanol to 1a provided 3g in 47% yield (98% based on recovered 1a). Treatment of 1a with potassium thiocyanate at room temperature led selectively to isothiocyanate 3h in 93% yield, although a sacrificial amount of trifluoroacetic acid was used to promote the reaction. However, this approach did not furnish the corresponding isocyanate when potassium cyanate was used instead. The lack of reactivity of potassium thiocyanate without the acid promoter is reflected in the reversibility of the addition: attempts to form the potassium phenoxide salt of 3h caused rapid elimination, reforming 1a. Likewise, the problem of isolating the pure aniline adduct 3c15a and its inability to form the hydrochloride salt can be attributed to the prominent elimination as well, which was confirmed by NMR monitoring of this process (see the ESI, Fig. S1†).
 | ||
| Scheme 4 Reversible conjugate addition of nucleophiles to 1a and 1c. aIsolated as the O-acetate derivative 3c′, bat 70 °C, cat 200 °C in a microwave reactor, dusing 5 mol% of Zn(OTf)2 as a catalyst, etrifluoroacetic acid was added to promote the reaction, fyield in parentheses is based on recovered 1a. | ||
Studying the nucleophilic addition of water to the photochemically generated p-QMs 1 revealed that the use of transition metal catalysis can be avoided not only for addition, but also for triggering the reorganization of the newly formed quaternary carbon center. First, the addition of water to p-QMs 1 can be easily achieved by switching from copper catalysis15d,e to either Brønsted-acid catalysis or simple heating, as exemplified by the formation of 4a from 1a (Scheme 5a). This reaction can also be performed in a one-pot fashion by generating p-QMs 1in situ from a quinone and an alkyne (Scheme 5b). Next, the thus obtained products from both terminal (4i) and internal (4a) alkynes undergo oxidation by p-benzoquinone under essentially the same conditions, forming p-hydroxybenzophenone in good yields and acylated hydroquinones as minor products (Scheme 5c). Notably, the formation of aryl ketones from 2,2-diaryl-2-hydroxyacetaldehydes was previously observed under different conditions.19 When p-benzoquinone was used in excess in the photoreaction with phenylacetylene, p-hydroxybenzophenone was formed directly (Scheme 5d), indicating that no additional reagents11,12 are principally necessary to perform such a transformation. The increased yield of the acid-catalyzed reaction (51%) compared to the catalyst-free reaction (27%) can be explained by a faster conversion of the labile p-QM intermediate 1i.
 | ||
| Scheme 5 Transformations based on nucleophilic addition of water to p-QMs 1a, 1g and 1i. | ||
Another example of a cascade reaction involving a photochemically formed p-QM is the acid-catalyzed interaction of Z-1c with 1-naphthol, which only afforded the unexpected rearranged compound 5 in 27% yield. Mechanistically, its formation could involve a 1,2-phenyl shift in the initial product of 1,6-conjugate addition followed by isomerization, as depicted in Scheme 6. Interestingly, compound 5 was also obtained by Shah and co-workers in a photochemical reaction between 1-naphthol and phenylacetylene mediated by thiophenol and trifluoroacetic acid.14 Possibly, the formation of 5 under Shah's conditions could represent a cascade process involving the oxidation of 1-naphthol into 1,4-naphthoquinone followed by the photoreaction with phenylacetylene to give 1c as an intermediate, which could then react with a second molecule of 1-naphthol to form 5.
 | ||
| Scheme 6 Unexpected outcome of the acid-catalyzed reaction of Z-1c with 1-naphthol and its plausible mechanism. | ||
In general, mechanistic pathways for the catalyst-free light-triggered reactions involving quinones and alkynes can be outlined as shown in Scheme 7. First, the irradiation of quinone BQ in the presence of alkyne A can either afford the [2 + 2] carbocycloaddition product 2 or proceed as [2 + 2] Paternò–Büchi cycloaddition followed by electrocyclic ring opening of the spirocyclic oxetene intermediate to form p-QM 1.7 The ion-radical pair [BQ˙−A˙+] was found to play a pivotal role in the Paternò–Büchi photocycloaddition process and may be involved in the concurrent formation of cyclobutenes 2 as well. Next, the photochemically generated electrophilic p-QMs 1 can undergo reversible addition of nucleophiles present in the reaction mixture. The products of the addition of water as a nucleophile can be oxidized by the photoexcited quinone to the final product – p-hydroxyaryl ketone. The loss of the acyl group during this step is reflected in the formation of the acylated byproducts (Scheme 5c), possibly by the photo-Friedel–Crafts reaction pathway.20 Finally, the oxidation of the formed hydroquinone HQ into the starting quinone BQ by a sacrificial oxidant can close the cycle. A distinctive feature of the overall process is that visible light alone allows a quinone to play the role of a starting material in the beginning and an oxidant in the end, yet allowing the control of reaction chemoselectivity by the quinone substitution character and the ratio of the reactants.
 | ||
| Scheme 7 Summarized mechanistic pathways for the uncatalyzed light-triggered reactions involving a quinone (BQ), a hydroquinone (HQ) and an alkyne (A). | ||
3. Conclusions
In conclusion, we reinvestigated the photochemical interaction between quinones and alkynes in both non-catalytic and catalytic protocols. We revised its chemo- and regioselectivity and showed that (a) such [2 + 2] photocycloadditions can be performed without any additional reagents, (b) the subsequent 1,6-conjugate addition of nucleophiles to the photochemically generated p-QMs is reversible and can be efficiently achieved even without catalysis, and (c) the formation of p-hydroxyaryl ketones from quinones and alkynes can also proceed in an uncatalyzed fashion when an excess of quinone is used. Overall, uncatalyzed light-mediated reactions between quinones and alkynes offer milder and greener alternatives to their catalytic versions, yet allowing the preparation of diverse products from readily available starting materials. At the same time, we believe that understanding the uncatalyzed photoreactivity may help to develop novel catalytic transformations with even broader synthetic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. F. and M. K. gratefully acknowledge the financial support from the “Grant Schemes at CU” (reg. no. CZ.02.2.69/0.0/0.0/19_073/0016935), the Czech Science Foundation (grant no. 21-39639L), and the Charles University Research Centre Program (grant no. UNCE/SCI/014). The authors would like to thank Dr Ivana Císařová for single crystal X-ray diffraction analysis.References
- (a) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (c) M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS.
- (a) M. Oelgemöller, Chem. Rev., 2016, 116, 9664–9682 CrossRef PubMed; (b) M. D. Kärkäs, J. A. Porco, Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (c) M. Tavakolian and M. Hosseini-Sarvari, ACS Sustainable Chem. Eng., 2021, 9, 4296–4323 CrossRef CAS; (d) S. Protti, D. Ravelli and M. Fagnoni, Trends Chem., 2022, 4, 305–317 CrossRef CAS.
- C. Liebermann, Ber. Dtsch. Chem. Ges., 1877, 10, 2177–2179 CrossRef.
- (a) J. M. Bruce, Photochemistry of Quinones, John Wiley & Sons Ltd, 1974 Search PubMed; (b) K. Maruyama and A. Osuka, Recent advances in the photochemistry of quinones, John Wiley & Sons Ltd, 1988 Search PubMed; (c) Y. Ando and K. Suzuki, Chem. – Eur. J., 2018, 24, 15955–15964 CrossRef CAS PubMed.
- (a) H. E. Zimmerman and L. Craft, Tetrahedron Lett., 1964, 5, 2131–2136 CrossRef; (b) D. Bryce-Smith, G. I. Fray and A. Gilbert, Tetrahedron Lett., 1964, 5, 2137–2139 CrossRef; (c) S. P. Pappas and B. C. Pappas, Tetrahedron Lett., 1967, 8, 1597–1600 CrossRef; (d) S. P. Pappas and N. A. Portnoy, J. Org. Chem., 1968, 33, 2200–2203 CrossRef CAS; (e) S. Farid, W. Kothe and G. Pfundt, Tetrahedron Lett., 1968, 9, 4147–4150 CrossRef; (f) E. Bosch, S. M. Hubig and J. K. Kochi, J. Am. Chem. Soc., 1998, 120, 386–395 CrossRef CAS.
- (a) S. Kato, M. T. R. Beels, P. La Porta, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht, I. Biaggio and F. Diederich, Angew. Chem., Int. Ed., 2010, 49, 6207–6211 CrossRef CAS PubMed; (b) C. Dengiz, O. Dumele, S. Kato, M. Zalibera, P. Cias, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht, G. Gescheidt and F. Diederich, Chem. – Eur. J., 2014, 20, 1279–1286 CrossRef CAS PubMed; (c) C. Dengiz and T. M. Swager, Synlett, 2017, 28, 1427–1431 CrossRef CAS; (d) T. Shoji, J. Higashi, S. Ito, M. Yasunami and N. Morita, Heterocycles, 2011, 83, 2271–2274 CrossRef CAS; (e) B. A. Trofimov, L. N. Sobenina, Z. V. Stepanova, I. A. Ushakov, L. M. Sinegovskaya, T. I. Vakul'skaya and A. I. Mikhaleva, Synthesis, 2010, 470–476 CrossRef CAS; (f) C. Dengiz, C. Prange, P. Gawel, N. Trapp, L. Ruhlmann, C. Boudon and F. Diederich, Tetrahedron, 2016, 72, 1213–1224 CrossRef CAS.
- J. Álvarez-García, V. Rubio-Pisabarro, C. Silva-López and M. M. Cid, Org. Lett., 2020, 22, 4527–4531 CrossRef PubMed.
- (a) X. Chen, C. Huang, W. Zhang, Y. Wu, X. Chen, C. Zhang and Y. Zhang, Chem. Commun., 2012, 48, 6432–6434 RSC; (b) S.-B. Tan, C. Huang, X. Chen, Y. Wu, M. Zhou, C. Zhang and Y. Zhang, Bioorg. Med. Chem., 2013, 21, 6124–6131 CrossRef CAS PubMed.
- A. Sagadevan, V. P. Charpe, A. Ragupathi and K. C. Hwang, J. Am. Chem. Soc., 2017, 139, 2896–2899 CrossRef CAS PubMed.
- S. Sultan, M. Bhat, M. A. Rizvi and B. A. Shah, J. Org. Chem., 2019, 84, 8948–8958 CrossRef CAS PubMed.
- A. Sagadevan, A. Ragupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896–13901 CrossRef CAS PubMed.
- N. Chalotra, I. H. Shah, S. Raheem, M. A. Rizvi and B. A. Shah, J. Org. Chem., 2021, 86, 16770–16784 CrossRef CAS PubMed.
- (a) A. Sharma, V. Dixit, S. Kumar and N. Jain, Org. Lett., 2021, 23, 3409–3414 CrossRef CAS PubMed; (b) Z.-W. Qiu, L. Long, Z.-Q. Zhu, H.-F. Liu, H.-P. Pan, A.-J. Ma, J.-B. Peng, Y.-H. Wang, H. Gao and X.-Z. Zhang, ACS Catal., 2022, 12, 13282–13291 CrossRef CAS; (c) L. Dai, J. Guo, Q. Huang and Y. Lu, Sci. Adv., 2022, 8, eadd2574 CrossRef CAS PubMed; (d) J. Kumar, A. Ahmed, S. Kumar, S. Raheem, M. A. Rizvi and B. A. Shah, New J. Chem., 2022, 46, 10967–10973 RSC; (e) V. P. Charpe, A. Ragupathi, A. Sagadevan, Y.-S. Ho, M.-J. Cheng and K. C. Hwang, Chem. – Eur. J., 2023, e202300110 CrossRef CAS PubMed; (f) V. Dixit, A. Sharma, A. Jangid and N. Jain, Adv. Synth. Catal., 2023, 365, 892–899 CrossRef CAS.
- For recent reviews, see: (a) A. Parra and M. Tortosa, ChemCatChem, 2015, 7, 1524–1526 CrossRef CAS; (b) C. G. S. Lima, F. P. Pauli, D. C. S. Costa, A. S. de Souza, L. S. M. Forezi, V. F. Ferreira and F. de C. da Silva, Eur. J. Org. Chem., 2020, 2650–2692 CrossRef CAS; (c) J.-Y. Wang, W.-J. Hao, S.-J. Tu and B. Jiang, Org. Chem. Front., 2020, 7, 1743–1778 RSC; (d) G. Singh, R. Pandey, Y. A. Pankhade, S. Fatma and R. V. Anand, Chem. Rec., 2021, 21, 4150–4173 CrossRef CAS PubMed; (e) Y. Hussain, Tamanna, M. Sharma, A. Kumar and P. Chauhan, Org. Chem. Front., 2022, 9, 572–592 RSC.
- J. V. Ellis and J. E. Jones, J. Org. Chem., 1975, 40, 485–488 CrossRef CAS.
- (a) F. A. L. Anet and D. P. Mullis, Tetrahedron Lett., 1969, 10, 737–740 CrossRef; (b) W. Kothe, Tetrahedron Lett., 1969, 10, 5201–5204 CrossRef.
- (a) S. M. McElvain, S. B. Mirviss and C. L. Stevens, J. Am. Chem. Soc., 1951, 73, 3807–3811 CrossRef CAS; (b) L. Syper, Tetrahedron, 1987, 43, 2853–2871 CrossRef CAS; (c) T. Kataoka and T. Iwama, Tetrahedron Lett., 1995, 36, 245–248 CrossRef CAS.
- For a recent example, see: L. J. Mitchell, W. Lewis and C. J. Moody, Green Chem., 2013, 15, 2830–2842 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2240461 and 2240462. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00636k |
| This journal is © The Royal Society of Chemistry 2023 |