
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Open-chain thiamine analogues as potent inhibitors of thiamine pyrophosphate (TPP)-dependent enzymes†
Alex H. Y.
Chan‡
 ,
Terence C. S.
Ho‡
and
Finian J.
Leeper
*
,
Terence C. S.
Ho‡
and
Finian J.
Leeper
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: fjl1@cam.ac.uk
First published on 24th July 2023
Abstract
A common approach to studying thiamine pyrophosphate (TPP)-dependent enzymes is by chemical inhibition with thiamine/TPP analogues which feature a neutral aromatic ring in place of the positive thiazolium ring of TPP. These are potent inhibitors but their preparation generally involves multiple synthetic steps to construct the central ring. We report efficient syntheses of novel, open-chain thiamine analogues which potently inhibit TPP-dependent enzymes and are predicted to share the same binding mode as TPP. We also report some open-chain analogues that inhibit pyruvate dehydrogenase E1-subunit (PDH E1) and are predicted to occupy additional pockets in the enzyme other than the TPP-binding pockets. This opens up new possibilities for increasing the affinity and selectivity of the analogues for PDH, which is an established anti-cancer target.
Introduction
Thiamine pyrophosphate (TPP)-dependent enzymes encompass a diverse range of catalytic activities but they all require the coenzyme TPP as its ylide 1 to catalyse the cleavage and formation of bonds adjacent to the carbonyl group of the acyl-donor substrate. For example in pyruvate dehydrogenase complex E1-subunit (PDH E1) an acetyl group is transferred from a donor (pyruvate) to an acceptor (lipoamide) through a ping–pong mechanism via intermediates 2–4 (Fig. 1a).1 Using thiamine/TPP analogues as small-molecule inhibitors has been a well-established approach to studying and/or manipulating cellular pathways involving TPP-dependent enzymes.2–20 The structures of three such inhibitors 5–7 are shown in Fig. 1b.2,14,15 They all possess a neutral central ring to capture the strong stabilising interactions between the enzyme and the catalytically active high-energy TPP ylide 1.7 However the need to make the central ring often lengthens the synthesis considerably. For this reason, many previous studies have focussed on an easily synthesised triazole as the central ring.19,20 In this paper we question the need for a central ring by synthesising “open-chain” thiamine/TPP analogues.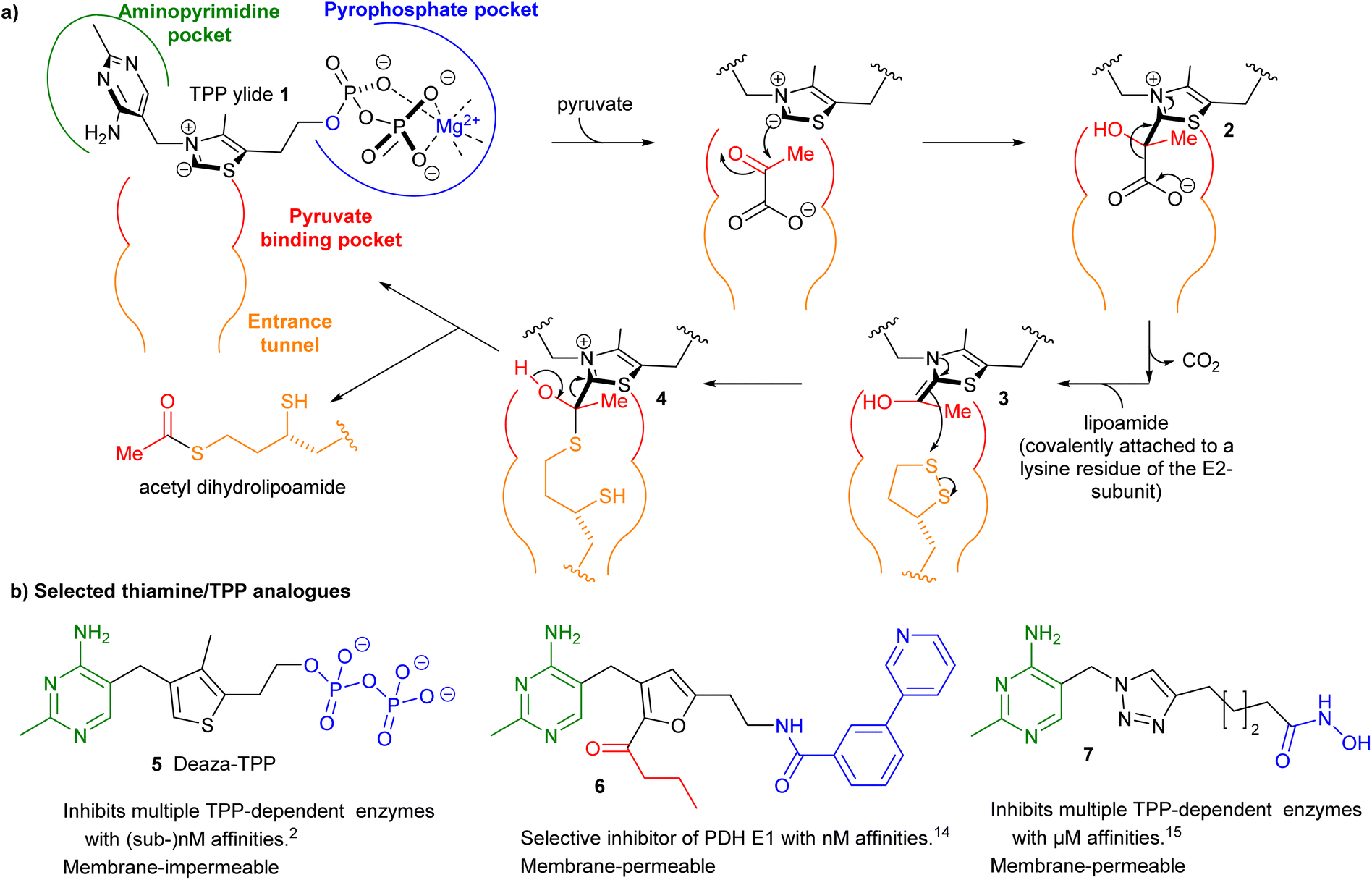 | ||
| Fig. 1 (a) Schematic depiction of the active site and mechanism of PDH E1. The relative position of the binding pockets in the active site is outlined. (b) Selected examples of thiamine/TPP analogues. The coloured substructures of the selected examples indicate the pockets that they occupy. | ||
Some open-chain thiamine analogues that inhibit TPP-dependent enzymes in cell-based studies have been reported,4 but they were operating as prodrugs: a ring-forming step (and enzymic pyrophosphorylation) was required for enzyme inhibition. He et al. have replaced the central ring with urea17 and N-acylhydrazone18 groups and these compounds showed potent inhibition of PDH E1 from Escherichia coli (and two other bacteria) but much less inhibition of mammalian PDH E1.18 Our focus in this paper is on inhibition of mammalian PDH E1 as recent papers have shown that certain cancers overexpress PDH E121–23 and a small-molecule inhibitor of PDH E1 suppresses development of one of these types of cancer in a mouse model.23 Devimistat (CPI-613), a lipoic acid derivative which targets PDH E1, shows strong antitumor activity against several cancers24 and has progressed into Phase III clinical trials.25,26 However, the inhibitors used in these studies were relatively unselective: fluoropyruvate23 inhibits a wide range of TPP-dependent enzymes, while devimistat24–26 targets all α-ketoacid dehydrogenases. There is, therefore, a need to develop selective inhibitors that are more easily synthesised than 6, for example.
We report herein open-chain thiamine analogues that bind in the TPP binding site and potently inhibit TPP-dependent enzymes. Some of our analogues are predicted by molecular docking to occupy alternative parts of the active site that are not involved in binding TPP. We believe that if TPP analogues could be designed that occupy all the available binding pockets, then they would be extremely potent and selective inhibitors of PDH E1.
Results and discussion
Our exploration of ring-opened TPP analogues was inspired by the conventional triazole-bearing compounds 12a–c. Bis-pyrimidines 12a–c were prepared via coupling of amine 9 with alkynoic acids 10a–c and then copper-catalysed alkyne–azide cycloaddition (CuAAC) between the resulting alkynes 11a–c and azide 812 (Scheme 1). Varying lengths of alkynes 10 were used to find the optimum linker length to allow one pyrimidine ring to bind in the aminopyrimidine pocket and one in the pyrophosphate pocket. Tested on porcine PDH E1, the longest bis-pyrimidine (12c) had the highest potency of the three (Table 1). In silico studies showed the expected binding mode of 12c in the TPP pocket (Fig. 2a and S2†) but also a second possible binding mode (with slightly lower docking score) in which the pyrimidine–CH2–amide motif occupies the binding region of TPP's pyrimidine–CH2–thiazolium (Fig. 2b and S2†). This alerted us to the possibility that open-chain analogues could be effective inhibitors. In silico docking studies also suggested that the aminopyrimidine ring in the aminopyrimidine pocket provided far more binding than the aminopyrimidine ring in the pyrophosphate pocket (Fig. S3†). In a previous paper14 we tested the effect of changing the substituents on the aminopyrimidine ring, but any change weakened the binding, so in the current study we kept the substituents unchanged. | ||
| Scheme 1 Synthesis of bis-pyrimidines 8a–c. Reagents and conditions: (i) NaN3, Na2SO3, H2O, 65 °C; (ii) H2(g), 10% Pd/C, MeOH, RT; (iii) 10a–c, DCC, DMAP, DMF, RT; (iv) CuSO4·5H2O, sodium ascorbate, t-BuOH, H2O, 40 °C. For all compounds: an = 1, bn = 2, cn = 3. | ||
 | ||
| Fig. 2 Schematic depiction of the two predicted binding modes of bis-pyrimidine 8c in the TPP pocket of PDH E1 (from Fig. S2†). Ar = 4-amino-2-methylpyrimidin-5-yl. The active site is viewed from the same angle as in Fig. 1a. | ||
| Compounds | Inhibitiona,b(%) [Compound]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[TPP] :[TPP] |
IC50a,c (μM) | vs. TPPd | ||
|---|---|---|---|---|---|
| 5:1 |
1:1 |
1:5 |
|||
|
a Data are the means of measurements in three technical replicates.
b Percentage inhibition determined for compounds at 50 μM with [TPP] = 10 μM (for 5:1); at 10 μM with [TPP] = 10 μM (for 1:1); at 10 μM with [TPP] = 50 μM (for 1:5).
c IC50 values (μM ± SEM) determined at [TPP] = 10 μM (refer to Fig. S1† for IC50 curves).
d Affinity of the compound relative to that of TPP (= [TPP]/IC50). ND, not determined.
|
|||||
| 12a | 53 ± 2 | 20 ± 2 | <15 | 42 ± 5 | 0.24 |
| 12b | 73 ± 3 | 32 ± 2 | <15 | 20 ± 4 | 0.50 |
| 12c | 82 ± 3 | 48 ± 2 | 18 ± 4 | 12 ± 1 | 0.83 |
| 14a | 27 ± 4 | ND | ND | ND | ND |
| 14b | 33 ± 3 | ND | ND | ND | ND |
| 14c | 28 ± 5 | ND | ND | ND | ND |
| 17a | 52 ± 2 | 18 ± 2 | ND | ND | ND |
| 17b | 61 ± 3 | 22 ± 3 | ND | ND | ND |
| 17c | 54 ± 4 | 19 ± 4 | ND | ND | ND |
| 18a | 44 ± 3 | <15 | <15 | ND | ND |
| 18b | 52 ± 2 | 20 ± 4 | <15 | ND | ND |
| 18c | 35 ± 4 | <15 | <15 | ND | ND |
| 21a | 60 ± 2 | 23 ± 2 | <15 | ND | ND |
| 21b | 72 ± 3 | 32 ± 2 | <15 | ND | ND |
| 21c | 80 ± 2 | 45 ± 3 | <15 | ND | ND |
| 22a | >90 | 67 ± 2 | 27 ± 3 | 4.9 ± 0.6 | 2.0 |
| 22b | >90 | 77 ± 3 | 35 ± 2 | 3.8 ± 0.5 | 2.6 |
| 22c | 80 ± 3 | 51 ± 4 | 20 ± 3 | 9.5 ± 0.7 | 1.1 |
| 23a | ND | 52 ± 3 | ND | ND | ND |
| 23b | ND | 60 ± 2 | ND | ND | ND |
To test the hypothesis, we replaced pyrimidine–CH2–triazole of bis-pyrimidines 12a–c with HOOC–CH2–triazole (14a–c) because a carboxylate group might interact ionically with the Mg2+ in the pyrophosphate pocket (Fig. 2b). Unfortunately, 14a–c, synthesised in one step from 11a–c and azidoacetic acid 13 (Scheme 2), were much weaker inhibitors than 12a–c (Table 1). Given that the second aminopyrimidine of 12c binds well in the pyrophosphate pocket, a benzyl group was introduced into 14a–c. Carboxylates 18a–c were synthesised from L-phenylalanine methyl ester 15via azide 16, then CuAAC with 11a–c and final hydrolysis of esters 17a–c (Scheme 2). This benzyl-substituted series (18a–c) was consistently more potent than 12a–c (Table 1). Computational docking suggested that this was because the phenyl ring occupies a relatively hydrophobic extension of the pyrophosphate pocket (Fig. 3). However, esters 17a–c had inhibitory potencies as high as carboxylates 18a–c (Table 1), suggesting that the ionic interactions between the carboxylate group and the Mg2+ ion did not compensate for the energy penalty of desolvation.
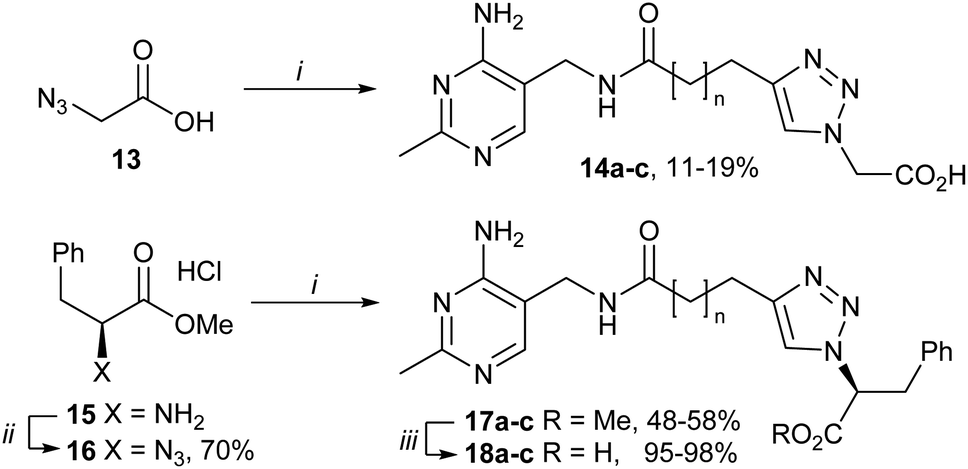 | ||
| Scheme 2 Synthesis of carboxylates 14a–c and 18a–c. Reagents and conditions: (i) 11a–c, CuSO4·5H2O, sodium ascorbate, DMF, 40 °C; (ii) triflyl azide, CuSO4·5H2O, DIPEA, MeOH, DCM, RT; (iii) KOH, THF, H2O, RT. For all compounds: an = 1, bn = 2, cn = 3. | ||
 | ||
| Fig. 3 Schematic depiction of the predicted binding modes of carboxylate 18b in the TPP pocket of PDH E1 (from Fig. S4†). The active site is viewed from the same angle as in Fig. 1a. | ||
To address this issue, the anionic carboxylate was replaced by an uncharged metal binding group (MBG), hydroxamate.27 Hydroxamates 22a–c were prepared from L-phenylalanine via azides 19 and 20, then CuAAC with alkynes 11a–c and final debenzylation (Scheme 3). These hydroxamates 22a–c were much stronger inhibitors than the bis-pyrimidines 12a–c or the carboxylates 18a–c, with affinity 22b > 22a > 22c (Table 1).
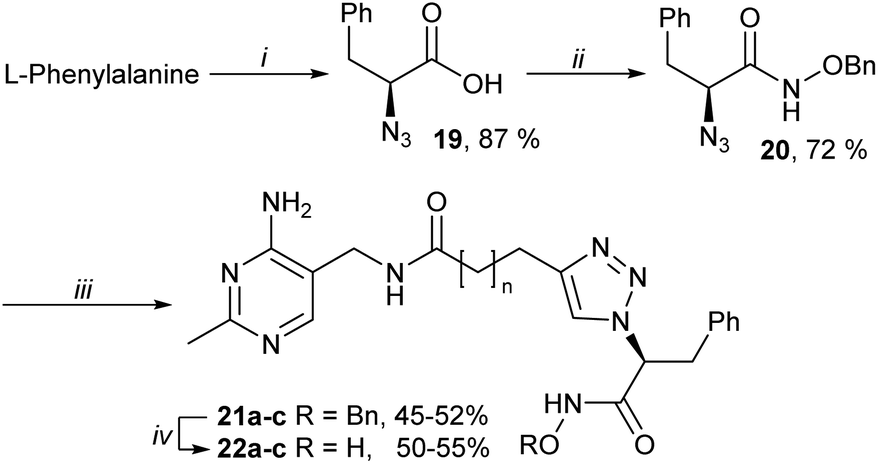 | ||
| Scheme 3 Synthesis of hydroxamates 22a–c. Reagents and conditions: (i) triflyl azide, CuSO4·5H2O, K2CO3, MeOH, H2O, RT; (ii) CDI, NH2OBn·HCl, THF, DMF, RT; (iii) 11a–c, CuSO4·5H2O, sodium ascorbate, t-BuOH, H2O, 40 °C; (iv) BCl3, DCM, RT. For all compounds: an = 1, bn = 2, cn = 3. | ||
Insights into the binding mode of 22a–c were obtained through in silico studies. As expected, the ionic interactions of 18a–c have been replaced by a bidentate interaction between the hydroxamate MBG and the Mg2+ but the phenyl group, instead of being positioned in the pyrophosphate pocket, as with 18a–c (Fig. 3), was predicted to flip back towards the amide linkage, occupying a region close to where the 4-Me group of TPP normally resides, with the amide linkage protruding into the pyruvate binding pocket (as depicted in Fig. 4a). This supported the pharmacophore of thiamine/TPP analogues that the binding region of the central neutral ring (mimicking the ylide of TPP) highly favours non-polar groups.1–3,7,16,28
 | ||
| Fig. 4 Schematic depictions of the predicted binding modes of (a) hydroxamate 22b and (b) O-benzylhydroxamate 21c to PDH E1 (from Fig. S5 & S6). The active site is viewed from the same angle as in Fig. 1a. | ||
The TPP-competitive nature of 22a–c was confirmed as the observed potency decreased with increasing [TPP] (Table 1). A full IC50 determination was conducted at 10 μM TPP for 12a–c and 22a–c (Fig. S1†) and showed that 22a and 22b bound 2.0 and 2.6 times tighter than TPP. Given the reported KM of TPP = 50 nM,29 the calculated KI values of 17a and 17b for PDH E1 (using [TPP]/IC50 = KM(TPP)/KI) were in the low-nanomolar range (17a: 25 nM; 17b: 19 nM).
As a control, O-benzyl hydroxamates 21a–c were tested and found to be weaker inhibitors than 17a–c. However, given that the O-benzyl group should greatly diminish the metal-binding capability, the residual activities of 21a–c were surprisingly high. Therefore, they were subjected to computational docking. Interestingly, completely new binding modes were predicted: the tricyclic terminus was positioned into the substrate binding pockets, instead of the pyrophosphate pocket (Fig. 4b). This novel binding mode is presumably possible because these open-chain analogues have greater conformational flexibility than their ring-bearing counterparts. It also provided a possible explanation for the relative affinities among the series 21a–c: the longer homologues (21b and 21c) positioned the C-benzyl into a side-pocket off the lipoamide-binding entrance tunnel (Fig. 4b), while the shortest 21a could not reach this pocket and positioned the C-benzyl group in the pyruvate/lipoamide-binding pockets. In all three compounds the O-benzyl group occupied the entrance tunnel.
Hydroxamates 23a,b, enantiomers of 22a,b, were synthesised from D-phenylalanine (as in Scheme 3) and they were only slightly weaker inhibitors than 22a,b (Table 1). In silico studies predicted that the enantiomers shared similar binding modes, with the hydroxamate as the MBG and with the phenyl ring flipping towards the amide linkage (not shown). The relatively low sensitivity of binding to chirality can be attributed to the conformational flexibility of these open-chain analogues.
Hydroxamate 22b is a drug-like molecule:30 possessing the neutral hydroxamate as the MBG (unlike TPP's polyanionic pyrophosphate moiety31), molecular weight = 438 g mol−1, hydrogen bond (HB) donors = 5, HB acceptors = 8, and calculated logP = 0.6 (calculated using MarvinSketch 21.1). 22b is also an efficient ligand for PDH E1, with ligand efficiency of 0.34 kcal mol−1 per heavy atom.32 The PDH E1 used throughout the study was commercially available porcine PDH E1, which is a widely accepted alternative to human PDH E1 as they share almost identical sequences (>95%).14 Taken together, hydroxamate 22b is expected to be a membrane-permeable, potent inhibitor of human PDH E1. In recent years, evidence has suggested that small molecule inhibitors of PDH E1 may be effective against cancers that over-express the PDH complex (PDHc).21–24 Thus, we hope to test 22b on cancer cell lines and to compare its effects with other cellular probes for PDHc.13–15,23
It was also of interest to evaluate the activity of 22b on other TPP-dependent enzymes, such as pyruvate decarboxylase (PDC), pyruvate oxidase (PO) and 2-oxoglutarate dehydrogenase (OGDH) E1. Hydroxamate 22b displayed modest inhibition of bacterial OGDH E1 and PDC, but little or no activity on bacterial PO or yeast PDC (Table S1†). With bacterial OGDH E1 and PDC, the level of inhibition by 22b reduced with increasing concentrations of TPP (Table S1†) suggesting a TPP-competitive mode of action.
Comparing the two hydroxamates 22b and 7, 22b is the better inhibitor of PDH E1 (KI = 19 nM vs. 40 nM (ref. 15)), which shows that replacing the central ring of thiamine/TPP analogues 5–7 with an open-chain amide has not lost too much of the binding energy and that is more than compensated by the favourable binding of the C-benzyl group. Furan 6 is 4-5-times tighter binding than 22b (KI = 4.2 nM)14 but that was the result of extensive optimisation of the amide moiety that occupies the pyrophosphate pocket and of the acyl group attached to the furan (at the equivalent position to C-2 of TPP), whereas no optimisation of 22b has been undertaken.
One thing that is clear from our docking studies is that there are further binding sites that can be exploited to improve the affinity of inhibitors other than the TPP-binding sites. We believe 21c is a relatively good inhibitor because it binds in the pyruvate and lipoamide binding sites and the side-pocket (Fig. 4b), whereas 22b does not take advantage of these sites but instead has a MBG to bind to the Mg2+ in the pyrophosphate pocket (Fig. 4a). This ability to use different binding pockets to get the best binding is only possible because of the flexibility of the open chain analogues. If an inhibitor could be developed that bound well in all the available pockets then it would be very potent indeed. The various pockets in PDH E1 were visualised using the program Caver Analyser 1.033 and Fig. 5 shows the two active site cavities of this α2β2 tetramer. This clearly shows the binding pockets already mentioned, binding the aminopyrimidine, central ring, pyrophosphate, pyruvate and lipoamide/entrance tunnel, plus the side pocket to the left of the entrance tunnel in this view. This side-tunnel seems to be considerably larger than required to accommodate the C-benzyl of 21c (as shown in Fig. 5b). In addition, there seems to be a further side-pocket to the right of the entrance tunnel, which might also be profitably used in the design of inhibitors.
 | ||
| Fig. 5 Active site cavities of human PDH E1 (PDB: 6CFO). The protein is an α2β2 tetramer and each chain, in cartoon view, is coloured differently (β chains have the lighter colours). The active sites are viewed from the same angle as in Fig. 1a. (a) The cavity is in gold and bound in the cavity is the adduct between TPP and inhibitor acetyl phosphinate (red dots, part of chain C in 6CFO); the Mg2+ ion is a cyan dot. (b) The second cavity (which contained the ligand in chain A in 6CFO) is in blue and bound in the cavity is the docked structure of 21c (yellow dots). The Mg2+ ion is a cyan dot. Images calculated and displayed by Caver Analyser 1.0.33 | ||
Conclusions
We have demonstrated that the synthesis of open-chain thiamine analogues is much easier than the synthesis of analogues with neutral central rings such as deazaTPP 5 and furan 6. Furthermore, the flexibility of the open chain allows the pendent groups to explore all the available binding sites, instead of just the one site dictated by the conformationally restricted ring. Hydroxamate 22b is a potent, drug-like inhibitor of TPP-dependent enzymes, particularly mammalian PDH E1, with tighter binding (to PDH E1) than that of TPP. It should be possible to modify 22b to add groups that bind in the other available pockets identified here and so arrive at an even more potent inhibitor. This may help uncover the roles of PDH E1 in cancer development and lead to a treatment for cancers that rely on PDHc for their growth.Conflicts of interest
All the authors declare there is no conflict of interest.Acknowledgements
A.H.Y.C. and T.C.S.H. are supported by K.M. Medhealth.References
- R. A. W. Frank, F. J. Leeper and B. F. Luisi, Cell. Mol. Life Sci., 2007, 64, 892–905 CrossRef CAS PubMed.
- S. Mann, C. Perez Melero, D. Hawksley and F. J. Leeper, Org. Biomol. Chem., 2004, 2, 1732 RSC.
- K. M. Erixon, C. L. Dabalos and F. J. Leeper, Org. Biomol. Chem., 2008, 6, 3561 RSC.
- Y. Le Huerou, I. Gunawardana, A. A. Thomas, S. A. Boyd, J. de Meese, W. deWolf, S. S. Gonzales, M. Han, L. Hayter, T. Kaplan, C. Lemieux, P. Lee, J. Pheneger, G. Poch, T. T. Romoff, F. Sullivan, S. Weiler, S. K. Wright and J. Lin, Bioorg. Med. Chem. Lett., 2008, 18, 505–508 CrossRef CAS PubMed.
- A. A. Thomas, J. De Meese, Y. Le Huerou, S. A. Boyd, T. T. Romoff, S. S. Gonzales, I. Gunawardana, T. Kaplan, F. Sullivan, K. Condroski, J. P. Lyssikatos, T. D. Aicher, J. Ballard, B. Bernat, W. DeWolf, M. Han, C. Lemieux, D. Smith, S. Weiler, S. K. Wright, G. Vigers and B. Brandhuber, Bioorg. Med. Chem. Lett., 2008, 18, 509–512 CrossRef CAS PubMed.
- A. A. Thomas, Y. Le Huerou, J. De Meese, I. Gunawardana, T. Kaplan, T. T. Romoff, S. S. Gonzales, K. Condroski, S. A. Boyd, J. Ballard, B. Bernat, W. DeWolf, M. Han, P. Lee, C. Lemieux, R. Pedersen, J. Pheneger, G. Poch, D. Smith, F. Sullivan, S. Weiler, S. K. Wright, J. Lin, B. Brandhuber and G. Vigers, Bioorg. Med. Chem. Lett., 2008, 18, 2206–2210 CrossRef CAS PubMed.
- X. Pei, K. M. Erixon, B. F. Luisi and F. J. Leeper, Biochemistry, 2010, 49, 1727–1736 CrossRef CAS PubMed.
- X. W. A. Chan, C. Wrenger, K. Stahl, B. Bergmann, M. Winterberg, I. B. Müller and K. J. Saliba, Nat. Commun., 2013, 4, 2060 CrossRef PubMed.
- A. Iqbal, E.-H. Sahraoui and F. J. Leeper, Beilstein J. Org. Chem., 2014, 10, 2580–2585 CrossRef PubMed.
- F. Rabe von Pappenheim, M. Aldeghi, B. Shome, T. Begley, B. L. de Groot and K. Tittmann, Nat. Chem. Biol., 2020, 16, 1237–1245 CrossRef CAS PubMed.
- E. Grabowska, M. Czerniecka, U. Czyżewska, A. Zambrzycka, Z. Łotowski and A. Tylicki, J. Enzyme Inhib. Med. Chem., 2021, 36, 122–129 CrossRef CAS PubMed.
- A. H. Y. Chan, I. Fathoni, T. Ho, K. J. Saliba and F. J. Leeper, RSC Med. Chem., 2022, 13, 817–821 RSC.
- A. H. Y. Chan, T. C. S. Ho, K. Agyei-Owusu and F. J. Leeper, Org. Biomol. Chem., 2022, 20, 8855–8858 RSC.
- A. H. Y. Chan, T. C. S. Ho, D. R. Parle and F. J. Leeper, Org. Biomol. Chem., 2023, 21, 1755–1763 RSC.
- A. H. Y. Chan, T. C. S. Ho, I. Fathoni, R. Pope, K. J. Saliba and F. J. Leeper, ACS Med. Chem. Lett., 2023, 14, 621–628 CrossRef CAS PubMed.
- A. H. Y. Chan, T. C. S. Ho, R. Irfan, R. A. A. Hamid, E. S. Rudge, A. Iqbal, A. Turner, A. K. H. Hirsch and F. J. Leeper, Bioorg. Chem., 2023, 138, 106602 CrossRef CAS PubMed.
- J.-B. He, L.-L. Feng, J. Li, R.-J. Tao, Y.-L. Ren, J. Wan and H.-W. He, Bioorg. Med. Chem., 2014, 22, 89–94 CrossRef CAS PubMed.
- H. He, H. Xia, Q. Xia, Y. Ren and H. He, Bioorg. Med. Chem., 2017, 25, 5652–5661 CrossRef CAS PubMed.
- Y. Zhou, J. Feng, L. Feng, D. Xie, H. Peng, M. Cai and H. He, J. Agric. Food Chem., 2019, 67, 12538–12546 CrossRef CAS PubMed.
- J. Feng, H. He, Y. Zhou, X. Guo, H. Liu, M. Cai, F. Wang, L. Feng and H. He, Bioorg. Med. Chem., 2019, 27, 2413–2420 CrossRef CAS PubMed.
- S. M. Davidson, T. Papagiannakopoulos, B. A. Olenchock, J. E. Heyman, M. A. Keibler, A. Luengo, M. R. Bauer, A. K. Jha, J. P. O'Brien, K. A. Pierce, D. Y. Gui, L. B. Sullivan, T. M. Wasylenko, L. Subbaraj, C. R. Chin, G. Stephanopolous, B. T. Mott, T. Jacks, C. B. Clish and M. G. Vander Heiden, Cell Metab., 2016, 23, 517–528 CrossRef CAS PubMed.
- C. T. Hensley, B. Faubert, Q. Yuan, N. Lev-Cohain, E. Jin, J. Kim, L. Jiang, B. Ko, R. Skelton, L. Loudat, M. Wodzak, C. Klimko, E. McMillan, Y. Butt, M. Ni, D. Oliver, J. Torrealba, C. R. Malloy, K. Kernstine, R. E. Lenkinski and R. J. DeBerardinis, Cell, 2016, 164, 681–694 CrossRef CAS PubMed.
- J. Chen, I. Guccini, D. Di Mitri, D. Brina, A. Revandkar, M. Sarti, E. Pasquini, A. Alajati, S. Pinton, M. Losa, G. Civenni, C. V. Catapano, J. Sgrignani, A. Cavalli, R. D'Antuono, J. M. Asara, A. Morandi, P. Chiarugi, S. Crotti, M. Agostini, M. Montopoli, I. Masgras, A. Rasola, R. Garcia-Escudero, N. Delaleu, A. Rinaldi, F. Bertoni, J. de Bono, A. Carracedo and A. Alimonti, Nat. Genet., 2018, 50, 219–228 CrossRef CAS PubMed.
- Z. Zachar, J. Marecek, C. Maturo, S. Gupta, S. D. Stuart, K. Howell, A. Schauble, J. Lem, A. Piramzadian, S. Karnik, K. Lee, R. Rodriguez, R. Shorr and P. M. Bingham, J. Mol. Med., 2011, 89, 1137–1148 CrossRef CAS PubMed.
- P. A. Philip, M. E. Buyse, A. T. Alistar, C. M. S. P. R. Lima, S. Luther, T. S. Pardee and E. Van Cutsem, Future Oncol., 2019, 15, 3189–3196 CrossRef CAS PubMed.
- T. S. Pardee, S. Luther, M. Buyse, B. L. Powell and J. Cortes, Future Oncol., 2019, 15, 3197–3208 CrossRef CAS PubMed.
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed.
- S. Lüdtke, P. Neumann, K. M. Erixon, F. Leeper, R. Kluger, R. Ficner and K. Tittmann, Nat. Chem., 2013, 5, 762–767 CrossRef PubMed.
- D. A. Walsh, R. H. Cooper, R. M. Denton, B. J. Bridges and P. J. Randle, Biochem. J., 1976, 157, 41–67 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- E. S. Rudge, A. H. Y. Chan and F. J. Leeper, RSC Med. Chem., 2022, 13, 375–391 RSC.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- A. Pavelka, E. Sebestova, B. Kozlikova, J. Brezovsky, J. Sochor and J. Damborsky, IEEE/ACM Trans. Comput. Biol. Bioinf., 2016, 13, 505–517 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods and results for enzyme assays and computational docking; synthetic methods, compound characterisation and NMR spectra. See DOI: https://doi.org/10.1039/d3ob00884c |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |