
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemically enabled oxidative aromatization of pyrazolines†
Silja
Hofmann‡
a,
Martin
Linden‡
a,
Julian
Neuner
a,
Felix N.
Weber
a and
Siegfried R.
Waldvogel
 *ab
*ab
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, Mainz, 55128, Germany. E-mail: waldvogel@uni-mainz.de; Web: https://www.aksw.uni-mainz.de/prof-dr-s-r-waldvogel/
bInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS FMS), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 17th May 2023
Abstract
Pyrazoles are a very important structural motif widely found in pharmaceuticals and agrochemicals. An electrochemically enabled approach for the sustainable synthesis of pyrazoles via oxidative aromatization of pyrazolines is presented. Inexpensive sodium chloride is employed in a dual role as a redox mediator and supporting electrolyte in a biphasic system (aqueous/organic). The method is applicable to a broad scope and can be conducted in the simplest electrolysis set-up using carbon-based electrodes. Hence, the method allows for simple work-up strategies such as extraction and crystallization, which enables application of this green synthetic route on a technically relevant scale. This is underlined by demonstration of a multi-gram scale electrolysis without loss in yield.
Introduction
Pyrazoles represent an important and common structural motif in modern pharmaceuticals due to their biological potency.1 Depending on their substitution pattern and the functional groups attached to the pyrazole moiety, they have already found application in treatments for various diseases and have potential for further use.2 One prominent example is sildenafil (3), a phosphodiesterase inhibitor for treatment of erectile dysfunction (Chart 1). Furthermore, pyrazoles exhibit metabolic,3 cardiac,4 and gastrointestinal effects,5 antibacterial,6 antipsychotic,7 anti-inflammatory,8 antinociceptive9 properties and are promising candidates for the development of new anticancer drugs.10 Pyrazoles are widely found in active compounds in agrochemicals, e.g. fungicides (pyraclostrobin, 2)11 or herbicides (benzofenap, 1).12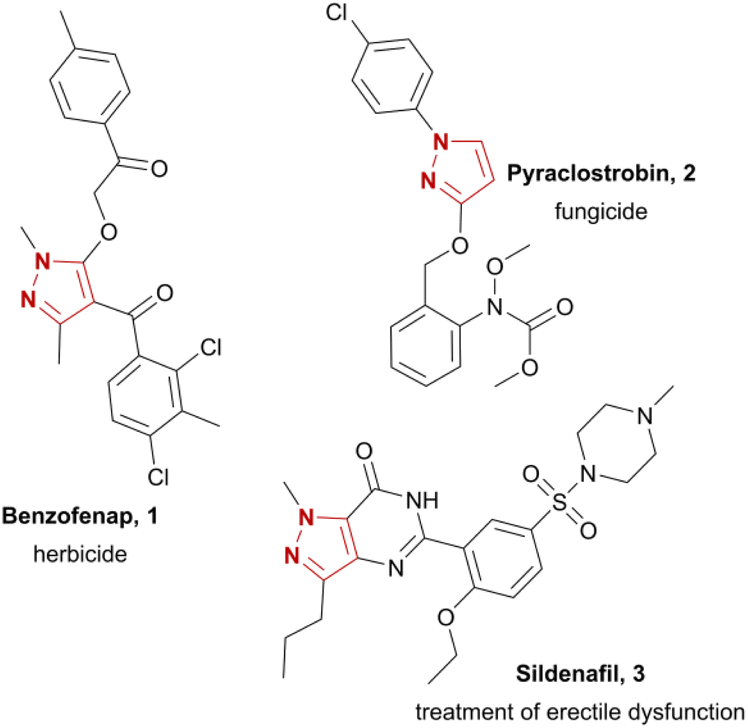 | ||
| Chart 1 Representative examples of pharmaceutically and agrochemically important pyrazoles. | ||
Common synthetic strategies are summarized in Scheme 1. These involve condensations of 1,3-diketones13 or ynones14 with hydrazines. However, regioisomeric mixtures of the desired pyrazole are frequently obtained and the syntheses of these starting materials can be capricious. Hydroamination reactions followed by C–N couplings can be conducted in flow with inexpensive copper catalysts but are severely limited in scope.15 Direct access to pyrazoles is possible by cyclization of readily available hydrazones with dipolarophiles.16 Nonetheless, these methods largely require expensive transition metal catalysts17 or additional oxidizers, e.g. oxygen18 or peroxides,19 or rely on costly nitro-olefins.20 There are synthetic strategies which feature a two-step approach via initial formation of a pyrazoline from readily available starting materials and subsequent oxidation. For the latter step, oxidizing agents such as oxygen,21 peroxides,22 or halogens23,24 in over-stoichiometric quantities are necessary, which typically necessitates a transition metal catalyst.25 To name one example, the synthesis of pyraclostrobin (2) involves the oxidative aromatization of a pyrazolidinone as a key step. This reaction conventionally requires copper-, cobalt- or iron catalysts in combination with oxygen.26 As modern chemistry moves towards greener methodologies, current synthetic strategies must fulfil the requirements of resource efficiency and sustainability as well as enhanced safety of the process. Electrochemistry aligns with these principles of green chemistry, as hazardous and often expensive redox reagents can be substituted with electric current.27 This also facilitates inherently safe processes, allowing for precise control over the reaction as the electron transfer is initially constrained to the electrode surface.28 In addition, the generation of reagent waste can be minimized as solvents and supporting electrolytes can be recycled.29 The down-stream processing is often more costly than the electrochemical conversion and decisive for translation into large scale application.30 The use of widely available and robust carbon-based electrode materials like boron-doped diamond (BDD) ensures low-maintenance set-ups.31 In summary, the reaction can become almost waste- and pollutant-free if electricity from renewable resources is employed.
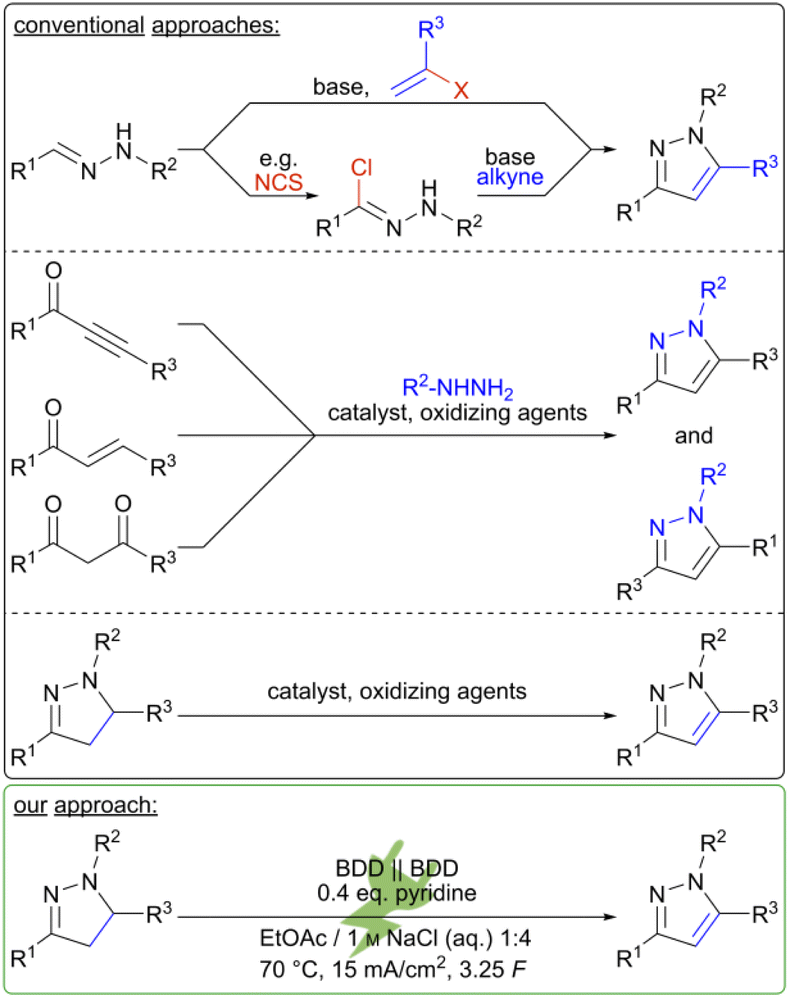 | ||
| Scheme 1 Conventional and electrochemical approaches for the synthesis of pyrazoles. | ||
Electrochemical reactions of pyrazoles have been of scientific interest for decades;32 in particular, the functionalization of pyrazoles at position 4 have been studied. Besides electrochemical chlorination,33,34 bromination34,35 or even iodination reactions,36 a methodology for an electrochemical selenylation has also been established.37 Additionally, electrochemical N-arylation reactions of unsubstituted or 4-nitro-substituted pyrazoles have been studied.38 Furthermore, the iodide-mediated synthesis of highly sulfonylated pyrazoles from sulfonylhydrazines in an undivided electrolysis cell has been reported.39 An intramolecular approach to the pyrazole moiety uses hypervalent iodine-mediated C–N-coupling of α,β-unsaturated hydrazones granting access to 1,3,5-substituted pyrazoles. However, this requires electron-withdrawing substituents on the nitrogen.40 Electrosynthesis to pyrazole derivatives including N–N bond formation usually does not provide the hetero-aromatic system.41 Remarkably, the oxidation of pyrazolines to their corresponding pyrazoles is less common. Potentiostatic oxidation of both 3-aminopyrazoline42 and 1,3,5-triphenylpyrazoline43 led to dimerization as well as the formation of the corresponding pyrazoles. An electrochemical iodide-mediated approach for the synthesis of pyrazolines and pyrazoles from readily available hydrazones gave access to a library of pyrazolines in up to excellent yields.44 Application of a redox mediator enhances selectivity and suppresses side reactions, e.g. polymerizations or over-oxidations.45 However, using alkynes as dipolarophiles yielded the corresponding pyrazoles in rather poor yields. We have developed an electrochemical method for the oxidation of pyrazolines to their corresponding pyrazoles using inexpensive sodium chloride in a double role as supporting electrolyte and mediator. BDD was chosen as electrode material in an environmentally friendly solvent system of water and ethyl acetate, allowing for facile work-up.46 The synthesis was performed under the simplest conditions: an undivided beaker-type electrolysis cell using a constant current set up. These are all pre-requisites for operation in multi-gram scale.
Results and discussion
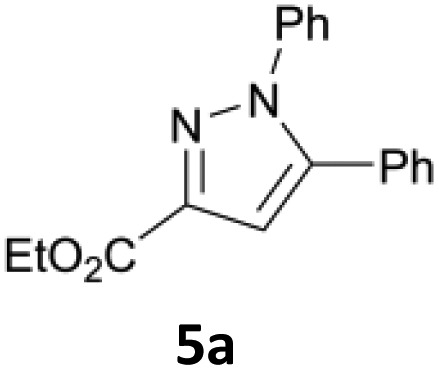
Initially, the electrochemical oxidation of pyrazolines was investigated using ethyl 1H-4,5-dihydro-1,5-diphenylpyrazole-3-carboxylate 4a as test substrate. First, the influence of halide, organic solvent, electrode material, and temperature were tested (Table 1). Initial reactions were performed using conditions based on previous work on the mediated electrochemical synthesis of pyrazolines.44 Here, graphite electrodes and a biphasic solvent system (ratio of the layers organic/aqueous 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), with a sodium halide concentration of 1 M with respect to the aqueous phase was used along with high stirring speed (1000 rpm). The concentration of the test substrate 4a was set to 0.34 mmol (concentrations of pyrazolines are given relative to the organic layer). At 25 °C, among all tested halides, only sodium chloride yielded the desired pyrazole 5a in 26%. The yield for 5a increased with elevated temperature, and hit a maximum yield of 59% at 70 °C. Within the tested electrode materials (graphite (Cgr), glassy carbon (Cgl), BDD, and various dimension stable anodes (DSA)), BDD gave 5a in highest yield of 62%. Alteration of the counter electrode to stainless steel did not improve product formation.47 Further reaction optimization with regards to temperature, current density, and amount of charge was conducted using a Design of Experiments (DoE) approach. This enables fast and precise investigation of the reaction process, while interactions between the factors and outliers are considered for the statistical analysis of the results.48 A fractional factorial design was chosen (24–1 resolution of III with central point and rotatable axis points, each data point run in triplicate). Target optimization led to the following reaction conditions: BDD electrodes, EtOAc/1 M NaCl (aq.) 1:4 (v/v), 70 °C, 15 mA cm−2 current density, and an applied charge of 2.5 F, which yielded 5a in 73% (calibrated GC yield). Next, the addition of tetraalkylammonium chlorides as co-supporting electrolytes to the electrolysis mixture (maintaining a total chloride concentration of 1 M) was investigated. Employing tetrabutylammonium chloride in the electrolysis showed almost no beneficial effect. Use of ammonium salts with shorter alkyl chains (Me or Et) proved to be detrimental (see Table S8, ESI†).
:4), with a sodium halide concentration of 1 M with respect to the aqueous phase was used along with high stirring speed (1000 rpm). The concentration of the test substrate 4a was set to 0.34 mmol (concentrations of pyrazolines are given relative to the organic layer). At 25 °C, among all tested halides, only sodium chloride yielded the desired pyrazole 5a in 26%. The yield for 5a increased with elevated temperature, and hit a maximum yield of 59% at 70 °C. Within the tested electrode materials (graphite (Cgr), glassy carbon (Cgl), BDD, and various dimension stable anodes (DSA)), BDD gave 5a in highest yield of 62%. Alteration of the counter electrode to stainless steel did not improve product formation.47 Further reaction optimization with regards to temperature, current density, and amount of charge was conducted using a Design of Experiments (DoE) approach. This enables fast and precise investigation of the reaction process, while interactions between the factors and outliers are considered for the statistical analysis of the results.48 A fractional factorial design was chosen (24–1 resolution of III with central point and rotatable axis points, each data point run in triplicate). Target optimization led to the following reaction conditions: BDD electrodes, EtOAc/1 M NaCl (aq.) 1:4 (v/v), 70 °C, 15 mA cm−2 current density, and an applied charge of 2.5 F, which yielded 5a in 73% (calibrated GC yield). Next, the addition of tetraalkylammonium chlorides as co-supporting electrolytes to the electrolysis mixture (maintaining a total chloride concentration of 1 M) was investigated. Employing tetrabutylammonium chloride in the electrolysis showed almost no beneficial effect. Use of ammonium salts with shorter alkyl chains (Me or Et) proved to be detrimental (see Table S8, ESI†).
|
|
||
|---|---|---|
| Deviation from standard conditions | T/°C | Yielda |
| Screening reactions performed in 5 mL Teflon cells. n(Pyrazoline) = 0.34 mmol.a Yield determined using GC analysis with 1,3,5-trimethoxybenzene as internal standard after external calibration. | ||
| None | 25 | 26% |
| 1 M NaBr | 25 | 0% |
| 1 M NaI | 25 | 0% |
| None | 65 | 38% |
| 2.5 M NaCl | 65 | 34% |
| None | 70 | 59% |
| BDD || BDD | 70 | 62% |
| Cgl || Cgl | 70 | 0% |
| DSA (IrO2 on Ta) || Cgr | 70 | 43% |
| DSA (Ru–Ir mixed oxide on Ta) || Cgr | 70 | 57% |
| DSA (Ru–Ir mixed oxide on Ta) || VA1.4571 | 70 | 49% |
| DSA (Ru–Ir mixed oxide on Ti) || Cgr | 70 | 55% |
Previous work has shown the importance of finely dispersed emulsions for efficient conversion in biphasic systems;44 therefore, the set-up was changed to 25 mL jacketed beaker-type cells with more powerful cross-shaped stirring bars to allow for more efficient mixing. Within the first scale-up experiment (1.7 mmol pyrazoline 4a) it was discovered full conversion of the substrate was possible at 3.25 F instead of 2.5 F, providing 5a in 69% yield. Therefore, the optimum amount of charge was adjusted for further experiments to reflect this discovery. To yield the corresponding pyrazole, elimination of hydrogen halide must take place. Thus, small amounts of different bases were used within the reaction.
Of the tested bases sodium carbonate, triethylamine, and pyridine, addition of 0.4 eq. pyridine allowed for an increase in yield of 5a to 75% (see Table S9, ESI†). Lastly, the influence of substrate concentration on the reaction was tested. Fortunately, the reaction proceeded smoothly in the concentration range of 0.34 M (75% 5a) and 0.75 M (77% 5a), while lower concentrations of 0.25 M proved detrimental, yielding 55%. Employing even higher concentrations of pyrazoline (1 M 4a) also resulted in a drop in yield, likely due to poor solubility of the starting material in the organic layer. The scope of the reaction was investigated, and different pyrazoles could be obtained in moderate to excellent yields. The results are summarized in Scheme 2. In particular, it was found that pyrazolines bearing an ester group in the 3-position were oxidized smoothly, yielding pyrazoles 5a–5h in 50–93% yield, including 4,5-disubstituted pyrazole 5f (89%). In contrast, 4,5-disubstituted pyrazole 5j′ was only obtained in poor yield of 15%, likely due to enhanced ring strain of the condensed norbornene. Furthermore, the influence of different substituents in the positions 1, 3, and 5 was investigated. The N-methyl analogue 5e′ was obtained in 80% yield, and even the N-(2-hydroxethyl)-substituted pyrazoline was converted successfully yielding 36% of the corresponding pyrazole 5f′. In addition, the tolerance of aliphatic and styryl substituents in position 3 was demonstrated in yields up to 43% (5g′–5i′). Interestingly, the cyclopropyl-substituted pyrazole 5h′ (41%) was obtained in a yield comparable to the methyl-substituted analogue 5g′ (43%) with no side products or products indicating opening of the cyclopropyl ring.
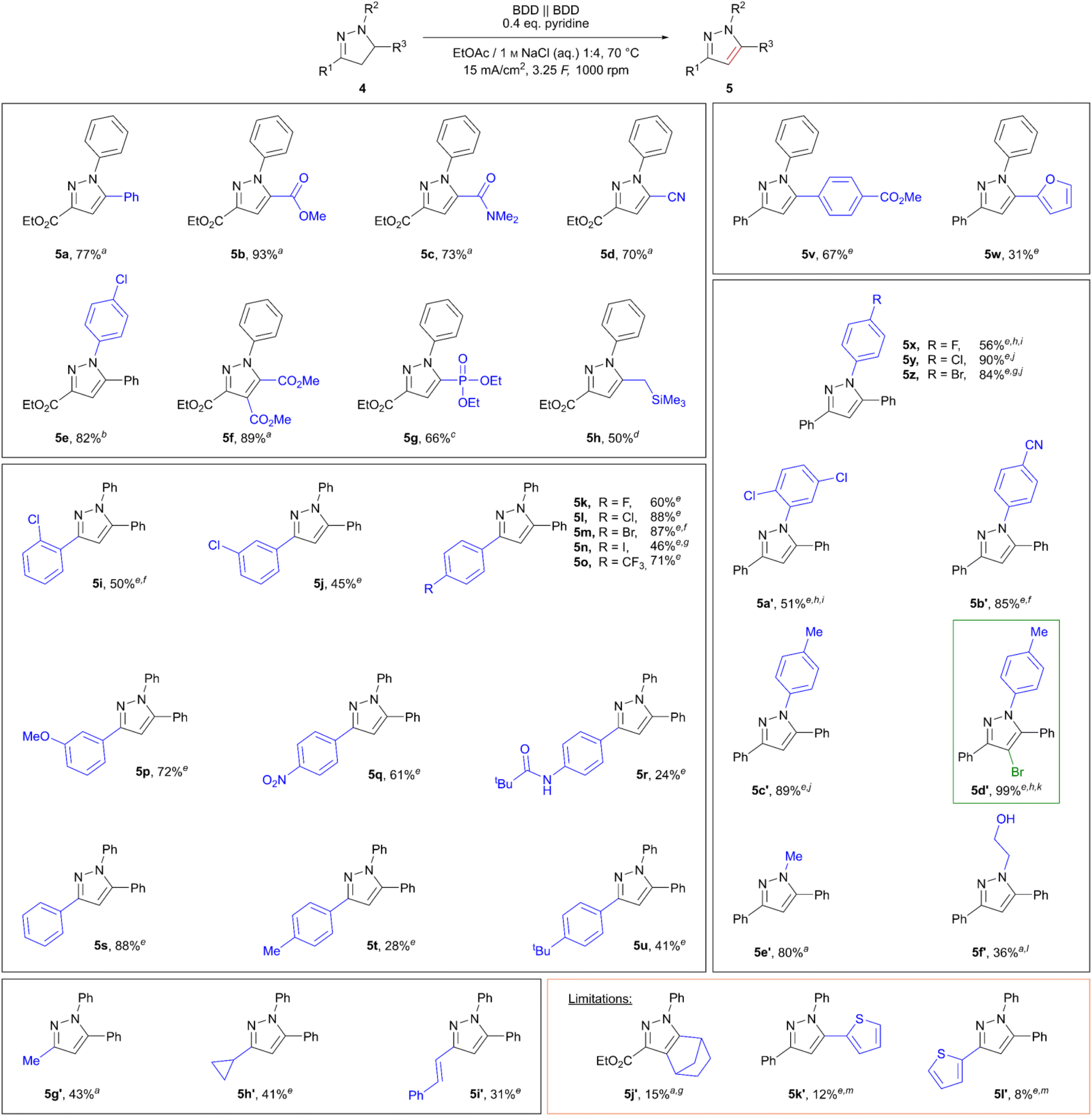 | ||
| Scheme 2 Optimized reaction conditions and scope of the oxidative aromatization of pyrazolines. Isolated yields. a3.75 mmol scale; b3.06 mmol scale, 5 F; c2.62 mmol scale; d2.94 mmol scale; e1.5 mmol scale; f4 F; g4.5 F; h1 M NaBr (aq.); i2 F; j1 M NaX (aq., X− = Cl−/I− (1:1)); k5 F; l2.5 F; m6.5 F. | ||
The applicability of the more substituted 1,3,5-triarylpyrazolines was then tested under these conditions. Although 1H-4,5-dihydro-1,3,5-triphenylpyrazole was converted smoothly using optimized reaction conditions, yielding the corresponding pyrazole 5s in 88%, the chosen substrate concentration of 0.75 M (relative to the organic layer) proved too high for the three fold substituted 1H-1,3,5-triaryl-4,5-dihydro-pyrazolines used. Therefore, the concentration of substituted 1H-1,3,5-triaryl-4,5-dihydro-pyrazolines was lowered to 0.3 M. Variation of the 3-position on the aryl ring showed some general trends. Whilst 1H-1,3,5-triphenylpyrazole (5s) was obtained in a good yield of 88%, the yields dropped with increasing electron-donating character of the para substituents: p-methyl (5t, 28%), p-pivaloylamino (5r, 24%). As an exception, the p-tert-butyl derivative 5u was obtained in 41% yield, most likely due to steric aspects. In contrast, substrates bearing electron-donating groups in meta-position undergo the reaction well, as shown by pyrazole 5p (72%). Furthermore, electron-withdrawing substituents in para-position gave satisfying yields of 61% and 71% for p-NO2 (5q) and p-CF3 (5o), respectively. For chloro-substituted pyrazoles 5i–5l a general trend for the increase in yield for meta (5j, 45%) < ortho (5i, 50%) < para (5l, 88%) was observed. To our delight, para-fluoro (5k, 60%), para-bromo (5m, 87%), and even para-iodo (5n, 46%) derivatives were obtained; the iodo derivative result was especially pleasing as it is prone to undergo side reactions resulting from oxidation of the iodoarene.49 Limitations of the method were found for 3- and 5-thien-2-yl-substituted pyrazoles 5l′ (8%) and 5k′ (12%), whereby degradation of the substrates was predominantly observed. Interestingly, conversion of a 5-furan-2-yl substituted pyrazoline was successful, giving 31% of the corresponding pyrazole 5w.
Further investigation into the influence of different substituents on the aryl ring at the 1-position was conducted. Analogous to the results for para-substituted arenes in the 3-position, electron-withdrawing groups proved beneficial for the oxidation of pyrazolines. Therefore, para-cyano functionalization gave pyrazole 5b′ in a very good yield of 85%. Again, para-halogenated derivatives gave moderate to very good yields (pyrazoles 5x–5z, F 56% < Br 84% < Cl 90%). Indeed, the yields and findings are in accordance with those obtained for the para-halo substituted arenes in the 3-position. However, for successful conversion of these halogenated pyrazolines a change of the supporting electrolyte was necessary. Whilst fluorinated pyrazole 5x as well as 2,5-dichloro substituted pyrazole 5a′ (51%) gave the best results when 1 M NaBr (aq.) was used, a 1:1 mixture of NaCl (aq.) and NaI (aq.) with respect to a total halide concentration of 1 M had to be employed for chloro (5y) and bromo (5z) derivatives. The influence of different supporting electrolytes was investigated for the conversion of 1-(p-methylphenyl) substituted pyrazoline 4c′. Although the application of solely sodium chloride led to side product formation and degradation of the starting material, the corresponding pyrazole 5c′ was obtained in a very good yield of 89% when the mixed aqueous supporting electrolyte NaCl/NaI (1:1, total halide concentration 1 M) was employed. To gain insight into the influence of halogen species in this specific oxidation, experiments with elemental iodine and iodine monochloride were carried out. Pyrazole 5c′ was only obtained in 57% (NMR yield) when iodine was directly used. However, application of iodine monochloride gave pyrazole 5c′ in 92% (NMR yield), which is indeed comparable to the 89% isolated yield achieved using our electrolysis technique. Hence, we propose that the use of the mixed NaCl/NaI electrolyte results in the in situ formation of iodine monochloride, which then serves as the oxidizing agent. Additionally, the application of 1 M NaBr (aq.) in the electrolysis was investigated. Interestingly, after 3.25 F both the desired pyrazole as well as the monobrominated compound 5d′ were obtained. Furthermore, 5d′ was obtained selectively in 99% yield after 5 F of charge was applied. A control experiment employing elemental bromine yielded both the pyrazole 5c′ (55% NMR yield) and the monobrominated compound 5d′ (35%). The electrochemical bromination of pyrazoles using aqueous sodium bromide electrolytes has also been reported in literature.34,35 Hence, our results are in accordance with literature findings and the presented method could also be extended to electrochemical bromination reactions of pyrazoles.
Sequential anodic synthesis from hydrazones and scale-up
To emphasise the application potential of the presented reaction, a telescoped synthesis of pyrazole 5a starting from ethyl glyoxylate phenyl hydrazone (6) was carried out (Scheme 3). In the first step, the corresponding pyrazoline was synthesized according to a recently published protocol.44 After the first electrolysis, styrene and excess iodine were removed and the crude product was subjected to a second electrolysis step: oxidative aromatization of the obtained pyrazoline. To our delight, when using a telescoped two-step electrosynthesis we obtained pyrazole 5a in 60% yield. Furthermore, when each step was conducted with product isolation both steps gave 77% yield, with an identical overall yield of 60%. Hence, pyrazoles can be obtained via this two-step sequential electrolysis, wherein isolation of intermediate pyrazolines is not required. | ||
| Scheme 3 Telescoped double electrochemical conversion of hydrazones to pyrazoles. | ||
Furthermore, the multi-gram scale synthesis of pyrazole 5a was performed to an excellent yield of 89% (6.82 g, 23.3 mmol; Table 2). This clearly demonstrates the robustness of the established electrochemical conversion and indicates its application potential for synthesis on technically relevant scales.


Application in the synthesis of pyraclostrobin
The oxidation of pyrazolinone 4m′ as a key step in the synthesis of pyraclostrobin (2) (BASF SE, sales volume 735 million. US$ (2009)51) was chosen to illustrate a potential application of the presented reaction. On 3.75 mmol scale, the reaction was shown to proceed smoothly, yielding 73% of the desired pyrazole 5m′ (Scheme 4). Together with the observed robustness in scale-up, this emphasises the relevance of this set-up for further investigation and extension towards technical or industrial scale. Due to the simple work-up and usage of widely employed techniques such as distillation and crystallization, it is easy to see this process as transferrable to and used on larger scales.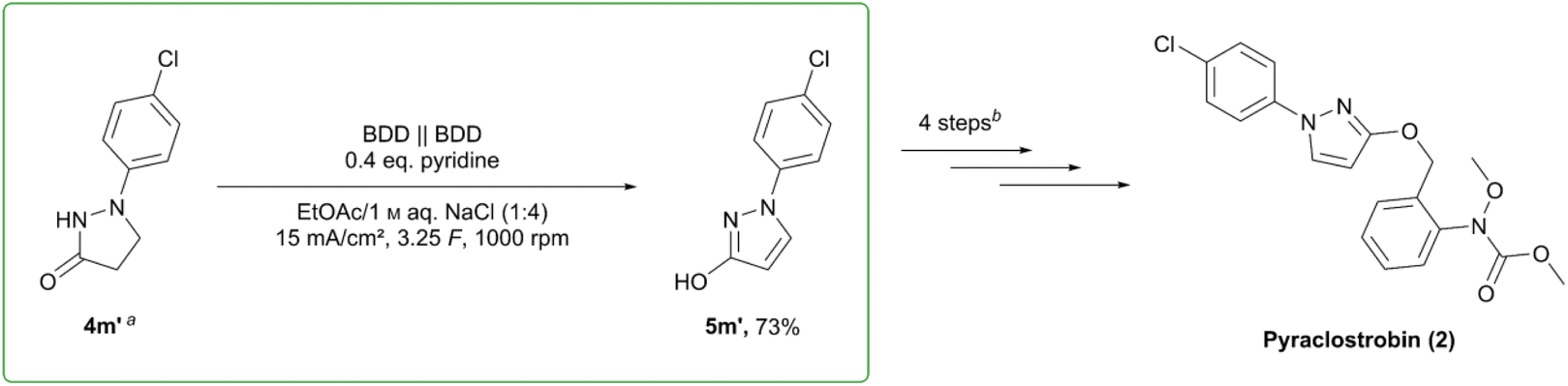 | ||
| Scheme 4 Electrochemical key step in the synthesis of pyraclostrobin (2). aSynthesis of 4m′ from 4-chlorophenylhydrazine hydrochloride and methyl acrylate, MeOH, NaOMe, Δ. b(1) base, 2-nitrobenzyl chloride; (2) H2/Pt; (3) methyl chloroformate; (4) base, dimethyl sulfate.50,51 | ||
Mechanistic considerations
As a result of the broad functional group tolerance and previous investigations into biphasic electrochemical reactions involving halogens as mediators,44 we propose an ionic reaction mechanism (Scheme 5). In the aqueous layer, anodic oxidation of chloride ions initially forms adsorbed or dissolved chlorine. In aqueous media, further reaction with water occurs immediately, forming hypochlorite. However, further oxidation to chlorine species in higher oxidation states, e.g. chlorate, is possible.52 Fast stirring and sufficient mixing of organic and aqueous layer proved key for successful conversion of the pyrazolines. Thus, chlorination of pyrazoline 4a at the nitrogen in position 2 occurs most likely at or close to the phase boundary, forming positively charged intermediate I. This is also supported by the observation that addition of tetraalkylammonium salts with longer carbon chains had no effect on the pyrazole formation. Subsequent base-promoted proton elimination results in chlorinated intermediate II. Formal elimination of HCl in the final step results in aromatization and, consequently, formation of pyrazole 5a. As hydrogen evolution serves as the cathodic counter reaction, stoichiometric amounts of hydroxide ions are formed during the reaction. However, addition of sub-stoichiometric amounts of pyridine (0.4 eq.) to the reaction mixture promoted product formation and improved the yields. Therefore, we assume that the addition of pyridine as a base supports the proton elimination and, as a result, the product formation in the beginning of the reaction. Additionally, pyridine may also interact with intermediates I and II within the organic layer and not just at the phase boundary, thereby promoting a fast conversion to the corresponding pyrazole. Chlorinated pyrazolines, e.g., in 5-position, were never observed by any analytical method, even when the organic layer was subjected to GC-MS analysis before the theoretical amount of charge (2 F) was applied. As a consequence, we propose the oxidative aromatization occurs via initial chlorination at the nitrogen in 2-position in accordance with the literature.24,34,53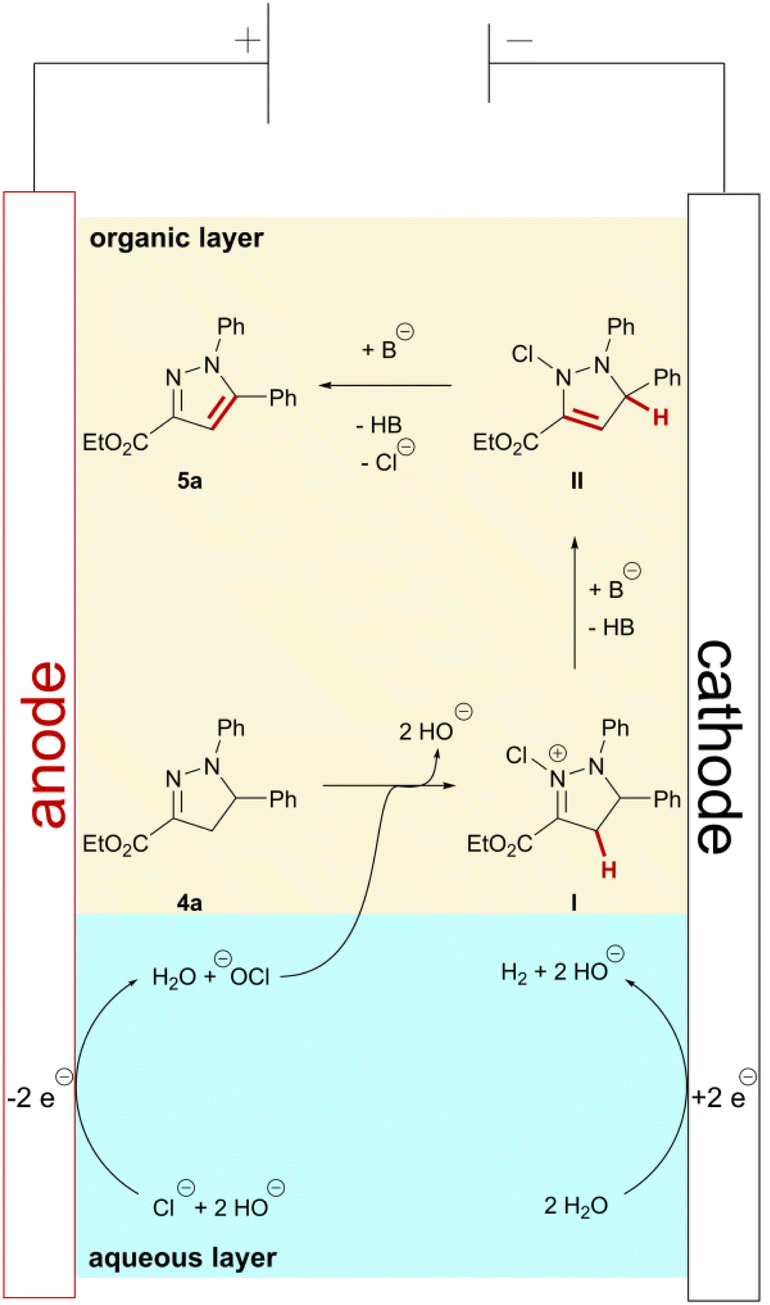 | ||
| Scheme 5 Postulated reaction mechanism. Hydroxide or pyridine both may serve as the base (B). | ||
Conclusions
A new method for the synthesis of pharmaceutically and agrochemically relevant pyrazoles was established. Starting from readily available pyrazolines, e.g., by condensation of chalcones with hydrazines, more than 35 examples were synthesized in moderate to excellent yields up to 93%.The reaction features a broad functional group tolerance and was demonstrated to be perfectly scalable to multi-gram scale. Ubiquitous and inexpensive sodium chloride was used in a double role as redox mediator and supporting electrolyte. This allows for facile work-up strategies and simplifies solvent recovery as well as waste management, since sodium chloride is commonly tolerated in sewage. Moreover, a sequential electrochemical synthesis from hydrazones without intermittent work-up and no loss in yield was performed. This allows for telescoped electrosynthetic approaches by switching the aqueous layers. Relevance of the demonstrated reaction was further underlined by a key step oxidation in the synthesis of the industrially relevant fungicide pyraclostrobin (2). In addition, the established system can be extended to oxidative aromatization and in situ bromination, as shown by the synthesis of a 4-brominated pyrazole in quantitative yield. Therefore, the reaction can potentially be used to access building blocks for coupling reactions. Finally, carefully chosen control experiments allowed the proposal of a convincing reaction mechanism which is in agreement with literature findings.
Due to the simple experimental set-up featuring sustainable carbon-based electrode materials, inexpensive and environmentally benign solvents and electrolytes, the demonstrated reaction is a viable green alternative to previous conventional approaches for pyrazoline oxidation.
Author contributions
S. H. and M. L. contributed equally to this work. S. H. and M. L. established the reaction, analysed experimental data, and performed scope and scale-up reactions. S. H. and J. N. conducted the experiments for initial reaction optimization and DoE. M. L., S. H., and F. N. W synthesized starting materials and carried out batch-type electrolyses. S. H., M. L., and S. R. W. wrote the manuscript. All authors discussed the results and agreed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr R. Hamill for her assistance in editing the manuscript. Financial support by the Deutsche Forschungsgemeinschaft (Wa 1276/17-2) is highly appreciated. Endorsement by SusInnoScience in frame of the Forschungsinitiative Rheinland-Pfalz was very helpful.References
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC.
- R. F. Costa, L. C. Turones, K. V. N. Cavalcante, I. A. Rosa Júnior, C. H. Xavier, L. P. Rosseto, H. B. Napolitano, P. F. S. Da Castro, M. L. F. Neto, G. M. Galvão, R. Menegatti, G. R. Pedrino, E. A. Costa, J. L. R. Martins and J. O. Fajemiroye, Front. Pharmacol., 2021, 12, 666725 CrossRef CAS PubMed.
- A. Samat, B. Tomlinson, S. Taheri and G. N. Thomas, Recent Pat. Cardiovasc. Drug Discovery, 2008, 3, 187 CrossRef CAS PubMed.
- K. R. A. Abdellatif, E. K. A. Abdelall, H. A. H. Elshemy, J. N. Philoppes, E. H. M. Hassanein and N. M. Kahk, Bioorg. Chem., 2021, 114, 105122 CrossRef CAS PubMed.
- J.-H. Jung, I. H. Jang, Y. O. Kim, S. Kim, M. H. Yoon and Y.-C. Kim, Drug Dev. Res., 2022, 83, 1600 CrossRef CAS PubMed.
- R. Verma, S. K. Verma, K. P. Rakesh, Y. R. Girish, M. Ashrafizadeh, K. S. Sharath Kumar and K. S. Rangappa, Eur. J. Med. Chem., 2021, 212, 113134 CrossRef CAS PubMed.
- G. Li, Y. Cheng, C. Han, C. Song, N. Huang and Y. Du, RSC Med. Chem., 2022, 13, 1300 RSC.
- S. Basu, D. A. Barawkar, V. Ramdas, M. Patel, Y. Waman, A. Panmand, S. Kumar, S. Thorat, M. Naykodi, A. Goswami, B. S. Reddy, V. Prasad, S. Chaturvedi, A. Quraishi, S. Menon, S. Paliwal, A. Kulkarni, V. Karande, I. Ghosh, S. Mustafa, S. De, V. Jain, E. R. Banerjee, S. R. Rouduri, V. P. Palle, A. Chugh and K. A. Mookhtiar, Eur. J. Med. Chem., 2017, 134, 218 CrossRef CAS PubMed.
- I. F. Florentino, D. P. B. Silva, C. S. Cardoso, R. Menegatti, F. S. de Carvalho, L. M. Lião, P. M. Pinto, S. Peigneur, E. A. Costa and J. Tytgat, Biomed. Pharmacother., 2019, 115, 108915 CrossRef CAS PubMed.
- (a) H. Aziz, A. F. Zahoor and S. Ahmad, J. Chil. Chem. Soc., 2020, 65, 4746 CrossRef CAS; (b) F. E. Bennani, L. Doudach, Y. Cherrah, Y. Ramli, K. Karrouchi, M. Ansar and M. E. A. Faouzi, Bioorg. Chem., 2020, 97, 103470 CrossRef CAS PubMed.
- J. Joshi, S. Sharma and K. N. Guruprasad, Pestic. Biochem. Physiol., 2014, 114, 61 CrossRef CAS PubMed.
- W. C. Quayle, D. P. Oliver, S. Zrna and A. Fattore, J. Agric. Food Chem., 2007, 55, 5199 CrossRef CAS PubMed.
- (a) S. T. Heller and S. R. Natarajan, Org. Lett., 2006, 8, 2675 CrossRef CAS PubMed; (b) S. Fustero, A. Simón-Fuentes and J. F. Sanz-Cervera, Org. Prep. Proced. Int., 2009, 41, 253 CrossRef CAS.
- D. B. Grotjahn, S. Van, D. Combs, D. A. Lev, C. Schneider, M. Rideout, C. Meyer, G. Hernandez and L. Mejorado, J. Org. Chem., 2002, 67, 9200 CrossRef CAS PubMed.
- S. B. Ötvös, Á. Georgiádes, D. Ozsvár and F. Fülöp, RSC Adv., 2019, 9, 8197 RSC.
- (a) M. Breugst, R. Huisgen and H.-U. Reissig, Eur. J. Org. Chem., 2018, 2477 CrossRef CAS; (b) M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293 CrossRef CAS PubMed.
- (a) M. Tang, Y. Wang, H. Wang and Y. Kong, Synthesis, 2016, 3065 CrossRef CAS; (b) J. Shao, K. Shu, S. Liu, H. Zhu, J. Zhang, C. Zhang, L.-H. Zeng and W. Chen, Synlett, 2021, 316 CrossRef CAS; (c) D. Y. Li, X. F. Mao, H. J. Chen, G. R. Chen and P. N. Liu, Org. Lett., 2014, 16, 3476 CrossRef CAS PubMed; (d) J. Hu, S. Chen, Y. Sun, J. Yang and Y. Rao, Org. Lett., 2012, 14, 5030 CrossRef CAS PubMed.
- Z. Fan, J. Feng, Y. Hou, M. Rao and J. Cheng, Org. Lett., 2020, 22, 7981 CrossRef CAS PubMed.
- G. C. Senadi, W.-P. Hu, T.-Y. Lu, A. M. Garkhedkar, J. K. Vandavasi and J.-J. Wang, Org. Lett., 2015, 17, 1521 CrossRef CAS PubMed.
- (a) X. Deng and N. S. Mani, Org. Lett., 2006, 8, 3505 CrossRef CAS PubMed; (b) X. Deng and N. S. Mani, J. Org. Chem., 2008, 73, 2412 CrossRef CAS PubMed.
- (a) L. Han, B. Zhang, M. Zhu and J. Yan, Tetrahedron Lett., 2014, 55, 2308 CrossRef CAS; (b) X. Zhang, J. Kang, P. Niu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2014, 79, 10170 CrossRef CAS PubMed; (c) Y. Kawashita and M. Hayashi, Molecules, 2009, 14, 3073 CrossRef CAS PubMed.
- S. T. Kolla, R. Somanaboina and C. R. Bhimapaka, Synth. Commun., 2021, 51, 1425 CrossRef CAS.
- (a) J. P. Waldo, S. Mehta and R. C. Larock, J. Org. Chem., 2008, 73, 6666 CrossRef CAS PubMed; (b) N. Nakamichi, Y. Kawashita and M. Hayashi, Synthesis, 2004, 1015 CAS.
- K. Khosravi, Res. Chem. Intermed., 2015, 41, 5253 CrossRef CAS.
- N. Nakamichi, Y. Kawashita and M. Hayashi, Org. Lett., 2002, 4, 3955 CrossRef CAS PubMed.
- (a) G. Suez, M. Grabarnick and A. Frenklah, WO2016113741A1, 2016; (b) U. J. Vogelbacher, M. Keil, R. Klintz, J. Wahl, H. Wingert, H. König, M. Rack, R. Götz and J. H. Teles, US6040458A, 1998; (c) P. Zhang, A. Guan, X. Xia, X. Sun, S. Wei, J. Yang, J. Wang, Z. Li, J. Lan and C. Liu, J. Agric. Food Chem., 2019, 67, 11893 CrossRef CAS PubMed; (d) Y. Liu, G. He, K. Chen, Y. Jin, Y. Li and H. Zhu, Eur. J. Org. Chem., 2011, 5323 CrossRef CAS; (e) J. V. Mercader, C. Agulló, A. Abad-Somovilla and A. Abad-Fuentes, Org. Biomol. Chem., 2011, 9, 1443 RSC.
- (a) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386 RSC; (b) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC; (c) A. Shatskiy, H. Lundberg and M. D. Kärkäs, ChemElectroChem, 2019, 6, 4067 CrossRef CAS; (d) K. Lam, S. D. Minteer and D. L. Poole, Org. Biomol. Chem., 2023, 21, 221 RSC; (e) K. Lam, Synlett, 2022, 1953 CrossRef CAS.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (b) E. Rodrigo, H. Baunis, E. Suna and S. R. Waldvogel, Chem. Commun., 2019, 55, 12255 RSC; (c) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef PubMed; (d) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed.
- (a) Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed; (b) S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750 CrossRef CAS PubMed.
- (a) J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7, E42 CrossRef; (b) K. Lam and K. M. P. Wheelhouse, Org. Process Res. Dev., 2021, 25, 2579 CrossRef CAS.
- (a) S. Lips and S. R. Waldvogel, ChemElectroChem, 2019, 6, 1649 CrossRef CAS; (b) S. R. Waldvogel, S. Mentizi and A. Kirste, Top. Curr. Chem., 2012, 320, 1 CAS.
- B. V. Lyalin, V. L. Sigacheva, A. S. Kudinova, S. V. Neverov, V. A. Kokorekin and V. A. Petrosyan, Molecules, 2021, 26, 4749 CrossRef CAS PubMed.
- B. V. Lyalin, V. A. Petrosyan and B. I. Ugrak, Russ. J. Electrochem., 2008, 44, 1320 CrossRef CAS.
- B. V. Lyalin and V. A. Petrosyan, Chem. Heterocycl. Compd., 2014, 49, 1599 CrossRef CAS.
- S. Zandi and F. Nikpour, Z. Naturforsch., B: J. Chem. Sci., 2022, 77, 35 CrossRef CAS.
- B. V. Lyalin and V. A. Petrosyan, Russ. Chem. Bull., 2014, 63, 360 CrossRef CAS.
- R.-N. Yi, Z.-L. Wu, W.-T. Ouyang, W.-F. Wang and W.-M. He, Tetrahedron Lett., 2021, 77, 153257 CrossRef CAS.
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS PubMed.
- J. Feng, Y. Wang, L. Gao, Y. Yu, J. B. Baell and F. Huang, J. Org. Chem., 2022, 87, 13138 CrossRef CAS PubMed.
- S. A. Paveliev, O. O. Segida, O. V. Bityukov, H.-T. Tang, Y.-M. Pan, G. I. Nikishin and A. O. Terent'ev, Adv. Synth. Catal., 2022, 364, 3910 CrossRef CAS.
- (a) T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317 CrossRef CAS PubMed; (b) T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 9437 CrossRef CAS PubMed.
- F. Lecoutre and G. Barbey, Electrochim. Acta, 1983, 28, 1017 CrossRef CAS.
- I. Tabaković, M. Laćan and S. Damoni, Electrochim. Acta, 1976, 21, 621 CrossRef.
- M. Linden, S. Hofmann, A. Herman, N. Ehler, R. M. Bär and S. R. Waldvogel, Angew. Chem., Int. Ed., 2023, e202214820 CAS.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- M. Klein and S. R. Waldvogel, Angew. Chem., Int. Ed., 2022, 61, e202204140 CrossRef CAS PubMed.
- (a) M. Santi, J. Seitz, R. Cicala, T. Hardwick, N. Ahmed and T. Wirth, Chem. – Eur. J., 2019, 25, 16230 CrossRef CAS PubMed; (b) M. Dörr, M. M. Hielscher, J. Proppe and S. R. Waldvogel, ChemElectroChem, 2021, 8, 2621 CrossRef; (c) M. Hielscher, E. K. Oehl, B. Gleede, J. Buchholz and S. R. Waldvogel, ChemElectroChem, 2021, 8, 3904 CrossRef CAS.
- (a) R. Francke, Curr. Opin. Electrochem., 2019, 15, 83 CrossRef CAS; (b) T. Wirth, Curr. Opin. Electrochem., 2021, 28, 100701 CrossRef CAS.
- M. Dochnahl and B. Müller, WO2011113884A1, 2011.
- H. Sauter, in Modern Crop Protection Compounds, ed. P. Jeschke, W. Krämer and U. Schirmer, Wiley VCH Imprint, Weinheim, 2nd edn, 2012, pp. 584–627 Search PubMed.
- A. M. Polcaro, A. Vacca, M. Mascia, S. Palmas and J. Rodriguez Ruiz, J. Appl. Electrochem., 2009, 39, 2083 CrossRef CAS.
- S. V. Gamapwar, N. P. Tale and N. N. Karade, Synth. Commun., 2012, 42, 2617 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00671a |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |