
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioactivity of the cannabigerol cannabinoid and its analogues – the role of 3-dimensional conformation†
Mohammed
Salha
 abc,
Henry
Adenusi
de,
John H.
Dupuis
f,
Enrico
Bodo
c,
Bruno
Botta
g,
Iain
McKenzie
h,
Rickey Y.
Yada
f,
David H.
Farrar
a,
Jakob
Magolan
ab,
Kun V.
Tian
*afg and
Gregory A.
Chass
*afi
abc,
Henry
Adenusi
de,
John H.
Dupuis
f,
Enrico
Bodo
c,
Bruno
Botta
g,
Iain
McKenzie
h,
Rickey Y.
Yada
f,
David H.
Farrar
a,
Jakob
Magolan
ab,
Kun V.
Tian
*afg and
Gregory A.
Chass
*afi
aDepartment of Chemistry and Chemical Biology, McMaster University, Hamilton, Ontario L8S 4M1, Canada. E-mail: kun.tian@uniroma1.it; g.chass@qmul.ac.uk
bMichael G. DeGroote Institute of Infectious Disease Research, McMaster University, Hamilton, Ontario L8N 3Z5, Canada
cDepartment of Chemistry, Sapienza University of Rome, Piazzale Aldo Moro 5, 00185, Rome, Italy
dHong Kong Quantum AI Lab, 17 Science Park West Avenue, Hong Kong, China
eDepartment of Chemistry, Hong Kong University, Hong Kong, China
fFaculty of Land and Food Systems, The University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada
gDepartment of Chemical Science and Pharmaceutical Technologies, Sapienza University of Rome, Piazzale Aldo Moro 5, 00185, Rome, Italy
hCentre for Molecular and Materials Science, TRIUMF, 4004 Wesbrook Mall, Vancouver, BC V6T 2A3, Canada
iSchool of Physical and Chemical Sciences, Queen Mary University of London, London E1 4NS, UK
First published on 24th May 2023
Abstract
Cannabinoids are naturally occurring bioactive compounds with the potential to help treat chronic illnesses including epilepsy, Parkinson's disease, dementia and multiple sclerosis. Their general structures and efficient syntheses are well documented in the literature, yet their quantitative structure–activity relationships (QSARs), particularly 3-dimensional (3-D) conformation-specific bioactivities, are not fully resolved. Cannabigerol (CBG), an antibacterial precursor molecule for the most abundant phytocannabinoids, was characterised herein using density functional theory (DFT), together with selected analogues, to ascertain the influence of the 3D structure on their activity and stability. Results showed that the CBG family's geranyl chains tend to coil around the central phenol ring while its alkyl side-chains form H-bonds with the para-substituted hydroxyl groups as well as CH⋯π interactions with the aromatic density of the ring itself, among other interactions. Although weakly polar, these interactions are structurally and dynamically influential, effectively ‘stapling’ the ends of the chains to the central ring structure. Molecular docking of the differing 3-D poses of CBG to cytochrome P450 3A4 resulted in lowered inhibitory action by the coiled conformers, relative to their fully-extended counterparts, helping explain the trends in the inhibition of the metabolic activity of the CYP450 3A4. The approach detailed herein represents an effective method for the characterisation of other bioactive molecules, towards improved understanding of their QSARs and in guiding the rational design and synthesis of related compounds.
Introduction
Cannabinoid bioactivity
Work toward the characterisation of naturally occurring cannabinoids and their metabolites has accelerated in the past few decades due in part to their therapeutic potential in treating epilepsy, Parkinson's disease, multiple sclerosis and a wide range of other neurological disorders.1–5 Coupled with the recent advances in their efficient syntheses and evolving understanding of their antibacterial properties, cannabinoids represent a useful and cost-effective addition to the medicinal chemist's toolkit.6Cannabigerol (CBG), a geranylated phenol (Fig. 1) and naturally occurring cannabinoid, has been proven to be effective in inhibiting undesirable biofilm formation and in the eradication of preformed biofilms in antibiotic resistant bacteria, including methicillin resistant Staphylococcus aureus (MRSA).7–14 Jentsch et al. have successfully synthesised CBG in one step from inexpensive 5-alkyl-resorcinols, olivetol and orcinol while preserving regioselectivity through the innovative use of alumina-promoted regioselective aromatic allylation reactions.15 However, the mechanism of this process has yet to be quantitatively resolved, driving further work on resolving the reactive selectivity in cannabinoids.15
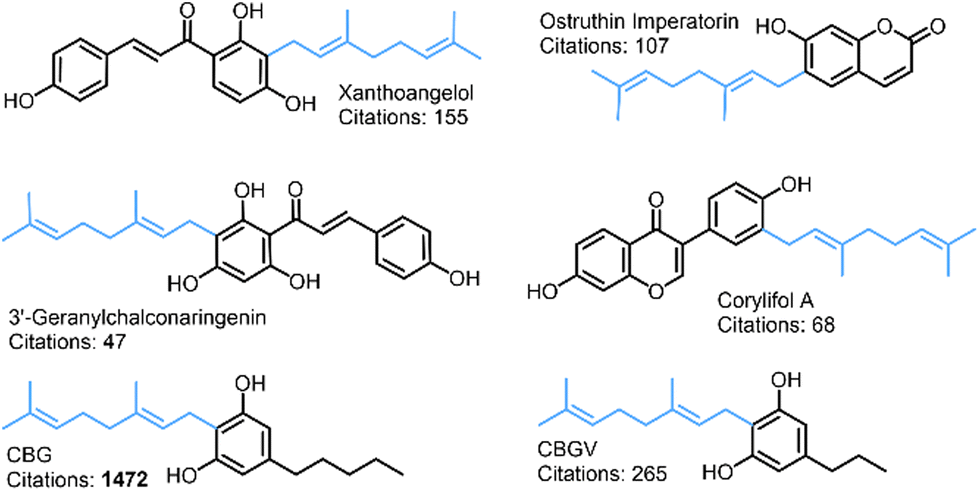 | ||
| Fig. 1 Examples of naturally occurring geranylated phenols. The 10-carbon geranyl alkyl chain (C10H17) is highlighted in blue. | ||
This ambiguity extends to the structure and bioactivity of cannabinoids. Although cannabinoids are described as inhibitors of the major cytochrome p450 (CYP450) enzymes, a standardised quantitative explanation of the inhibition mechanism is lacking.16–18 Preliminary studies have revealed that selected cannabinoid derivatives did not inhibit the in vivo metabolism of triozolam, nifedipine or testosterone (Fig. 2) in CYP3A4;19 another work found that CBD is a potent inhibitor of human CYP3A-mediated oxidation of diltiazem.20 Therein, the two phenolic hydroxyl groups were shown to be the key contributors to this bioactivity, as olivetol showed partial inhibition while D-limonene showed none.
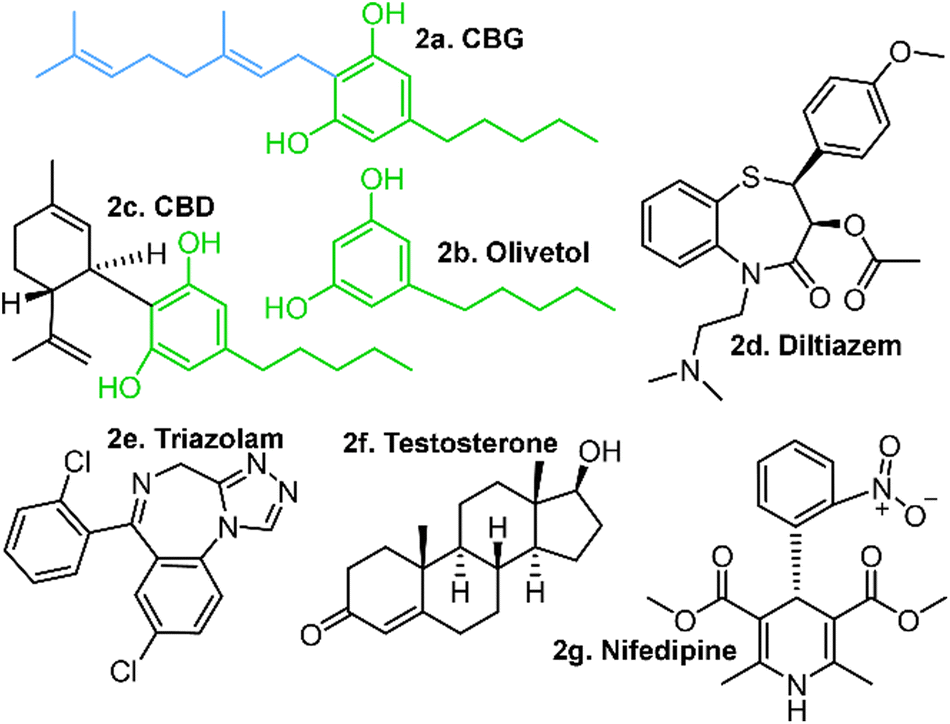 | ||
| Fig. 2 Structures of potential cytochrome P450 inhibitors, including: CBG, olivetol and CBD (a, b & c, respectively) with common structural features highlighted in green. Cannabinoids are known to inhibit the in vitro metabolism of diltiazem (d), yet show no significant inhibition of triazolam (e), testosterone (f) or nifedipine (g). Note the geranyl chain in CBG, which is missing in olivetol and partially cyclised in CBD. | ||
Molecular docking can be used to quantitatively characterise the influence of a molecule on an enzyme's ability to bind a substrate molecule (i.e. drug) to form an enzyme–substrate complex (E–S). Hindrance or retardation of E–S formation by the molecule renders it an inhibitor. As inhibition is substrate dependent, the interplay between the molecular inhibitor and the enzyme must be explicitly considered in the presence of the different substrates towards resolving each quantitative structure–activity relationship (QSAR) emerging from the specific combinations.21–23
The results of an IC50 study present a promising lead for cannabinoid-dependent inhibition given that a range of concentrations were tested against the diltiazem substrate.20 A competing cannabinoid molecule might inhibit diltiazem; yet others such as testosterone, nifedipine and triozolam (Fig. 2) may not, making a case for drug–drug interactions (DDIs) between selected substrates and cannabinoids.24 Cannabinoids could be expected to be metabolised by CYP enzymes, in large part due to their resorcinol moieties.25 Most phenols are prone to oxidative metabolism by CYP enzymes, which target the hydroxyl groups, transforming them to quinones or ethers. These are then further transformed to more water soluble forms prior to filtering via the kidneys and subsequent excretion.26,27 Hence, phenols are not associated with “drug-likeness” and less so with inactive inhibition of another drug.28 This raises the possibility for some means (likely intramolecular) by which these phenolic hydroxyl groups are insulated from metabolic action, in turn raising questions about the 3-dimensional structures (conformations) and their relevance to stability, activity and selectivity in these cannabinoids, conformation-specific activity and selectivity in particular.
Molecular structure and geometric conformation
A fully extended molecular structure has marked structural differences relative to a helical or coiled one, including its solvent exposed surface area. This places disparate functional chemical moieties in contact with the solvent; thus there is potential for them to be involved (or not) in intra- and inter-molecular interactions. It is therefore essential to have some knowledge of the conformational flexibility and energetically preferred poses of a molecule, particularly for structures with multiple single bonds around which rotations can occur to populate differing rotamers with negligible energetic barriers to the interconversion.29 Most often, conventional spectroscopic techniques are technically limited in the explicit differentiation of differing conformations (e.g. insufficient resolution, non-active dynamic modes, etc.) and low time-resolution with respect to the fast resonance between differing conformations. Nuclear magnetic resonance (NMR) is prone to temporal limitations, where the spectra for a selected molecule (i.e. cannabinoid) represents an average of several 3-D structures resulting from multiple conformations being populated over the time of the measurement.30 Refining a structure's conformation with NMR can require low temperatures and the use of multiple solvents, with the difficulty increasing with increasing molecular size, flexibility and floppiness, as well as its propensity to form stabilising intramolecular interactions.31Modern computational techniques can effectively and economically provide complementary information on these bioactive molecular systems and their 3-D structures, with ever growing accuracy, as has been accomplished for amino acids, oligo-/poly-peptides and proteins in particular since the 1970s.32 These have scaled-up since then, facilitating the full characterisation of systems with ∼300 atoms or less, in a relatively short time on personal computers (PCs) or modest workstations. The requisite raw computational power is no longer the principal limitation; instead, the number of (meaningful) structures that can be generated, the ‘dimensionality’ of the problem and the number of results that can be analysed and properly interpreted are.33 A systematic approach of indexing and accounting for the contributions of each molecular component and their individual degrees of freedom allows one to more effectively generate a non-degenerate set of topologically possible conformations (poses) towards resolving the structurally probable set. Such an approach has been successfully implemented for peptides,34 lipids,35,36 anti-oxidants,37,38 bioactive molecules,39–42 antibiotics43 and relevant polyphenols.44 Each manifests their own unique set of conformational poses, stabilised by unique intramolecular interactions45 and systemic energy management.46 We now apply this methodology to characterise the structure and stability of differing conformations of cannabinoids, specifically CBG, CBGV and cannabigerocin, with the confidence that approaches advanced and lessons learned will be applicable to the study of a wide range of naturally occurring geranylated phenols.
Methods
Molecular computations
With the Gaussian 09 (G09) program package,47 the conformers were optimised using the B3LYP DFT-method with D3 dispersion correction and the 6-31G(d,p) Pople basis set48 employing the six cartesian d-orbitals (in lieu of the spherical five).49 The Polarizable Continuum Model (PCM)50 for ethanol was used to simulate solvent effects.51 Analytical frequencies computed on each geometry-optimised structure confirmed that each structure resided at a true minimum on their respective potential energy hypersurfaces (PEHSs). Zero-point energy (ZPE), thermal corrections and entropic contributions were used to quantify the free energy (ΔG) of each structure. Towards confirming any intra-atomic interactions that might explain the relative stability of the given conformers, Bader's Atoms in Molecules (AIM) analyses52 were carried out on the wavefunctions of each conformer. Electronic density topological analyses of the electronic wavefunctions generated molecular graphs replete with bond critical points (BCP) confirming the existence of an inter-atomic sharing of electrons (ē). Quantification of electronic densities (ρb) at each BCP in ē per cubic-volume (ē bohr−3) provided accurate evaluations of the relative bond strength.52Biomolecular docking
The optimised conformations of CBG and its analogues with the lowest free-energies (CBG, CBD and olivetol) were the fully extended straight-chain geometries. These were each docked onto human cytochrome P450-3A4 (PDB: 1W0E)53 in the GOLD 2022 1.0 molecular docking suite.54 Furthermore, differing conformations of the CBG system were docked and scored relative to the fully-extended geometry to resolve the influence of conformation on bioactivity. The dockings of CBD and olivetol were used as controls to determine the ligand's preference for binding to the active site at the phenolic OHs, in accordance with the literature.20,55 The main binding site was defined and a cavity was created for all atoms within 10 Å of the Fe atom on HEM1501 (HEM). The same ligands were also docked at proximal potential binding sites centred at the following residues to probe inactive inhibition: ARG375, ARG372, ARG105, LEU373, LEU364 and ILE369. A ligand preferentially binding to an alternative site with respect to the active site (HEM) could indicate a form of inactive inhibition, wherein the ligand is not necessarily metabolised, yet still occupies the region around the active site. Molecular docking programmes produce poses ranked by binding affinity between a substrate and a site on the enzyme. The pose is a visual representation of conformation, literally showing the orientation of a substrate with respect to the enzyme. Docking poses were generated using the GOLD's default genetic algorithm (GA) search54 and ranked using Chemscore, an indicator of binding potential of a substrate to a protein.Molecular conformations
CBG contains a geranyl chain (C10H17) and a para-substituted sidechain on the central benzene ring, with several single C–C bonds with rotational freedom effectively giving rise to a relatively flat PEHS (Fig. 3) making CBG structural flexible and able to populate many stable 3D structural conformations. To reduce workload and demand on resources, one could consider a reduced set of possible conformations, yet could miss ones that are either lower in energy, higher in activity or with pronounced specificity; for example trialing a limited number of idealised poses, or ones with traditional, ‘expected’ or easily visualised structures (i.e. extended, or with traditional H-bonds only).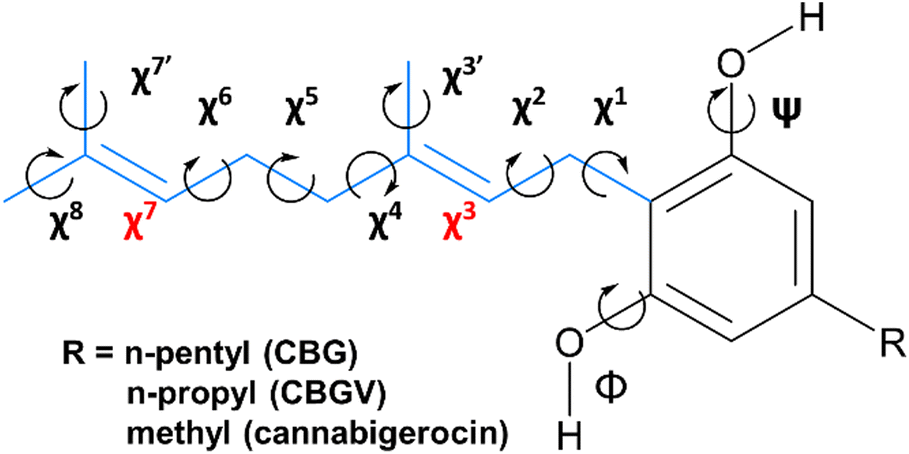 | ||
Fig. 3 CBG and its n-propyl CBGV and methyl cannabigerocin analogues. Torsional degrees of rotational freedom are indexed (χ1–8) along the geranyl chain (blue) and at the two hydroxyl positions (φ and ψ). The latter have energetic preference to orientate co-planar to the central ring and point syn or anti (towards or away from geranyl, respectively); both anti as-shown (labelled ‘aa’). The torsions about the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (χ3 and χ7, in red) are not easily rotatable and thus are fixed in one of the two isomers (cis or trans). C bonds (χ3 and χ7, in red) are not easily rotatable and thus are fixed in one of the two isomers (cis or trans). | ||
To avoid this pitfall and to tackle such complex multi-dimensional conformational analysis,56 we employed analytical definitions to generate, analyse and interpret the conformations, as previously employed for other bioactive molecules.35 Therein, a systematic definition was employed to describe the constituent atoms and their related spatial positions and orientations arising from rotations about flexible dihedral angles (torsional motions of extremities of 4 bound atoms). This was achieved for geranial and its para-substituted alkyl chain (Fig. 3), labelling each with progressive increments of χn, where n defined the dihedral angle n-dihedrals away from the central phenol ring, with three possible rotamers for each dihedral angle, as per relative orientations defined by fundamental Newman projections (gauche+, anti, gauche−).35 The single mid-chain and two terminal methyl rotors were labelled χ3, χ′7 and χ8, respectively.
With respect to rotational flexibility, even the simpler cannabigerocin system has numerous structural conformations it can adopt arising from the flexibility of the 5 single C–C rotors in the alkyl-chain and 2 –OH rotors, with 2187 theoretically possible conformers (35 × 32). To tackle this large structural phase-space, we explored the χ2 rotor in anti-conformation, while arranging the methyl rotors to be bisecting in the starting geometries (χ′3, χ7 and χ′7 = 180° = anti).
E and Z isomerism at the χ3 rotor was tackled by modelling the cis and trans isomers for each of the conformations. Despite trans isomers predominantly being more stable in most (bio)chemical and biological systems, there are some exceptions where the cis isomers are equally or even more stable.57 In summary, the two isomers together, single rotors explored (χ1, χ4 and χ5) in 3 differing geometries (gauche+, anti, and gauche−) and the 2 –OH rotors explored in-plane with their parent ring (anti = 180° and syn = 0°), produced 108 possible conformers (2 × 33 × 22 = 2 × 27 × 2). The conformational space was then further expanded with selected poses of the geranyl chain (see Fig. S1 and S2†).
Results and discussion
Molecular docking
A total of 42 key amino acid groups were identified in CYP450 3A4 as potential binding sites, using Genetic Optimisation for Ligand Docking software from the Cambridge Crystallographic Data Centre (GOLD, CCDC, Cambridge, UK). Among these, the active site containing a porphyrin ring is indexed as HEM. An inhibitory molecule can act without binding directly to the active site.57 Thus, each ligand (CBG, CBD, and olivetol) was also docked to the 6 amino acid groups identified near the porphyrin ring (Fig. 4A). Ligands were docked into various poses and each pose was given a Chemscore in accordance with how well they bind to the enzyme. The aims here were twofold: to determine whether CBG binds more favourably to the porphyrin ring or another site in its vicinity and then establish the orientation of the molecule once it is docked (Fig. 4A–E).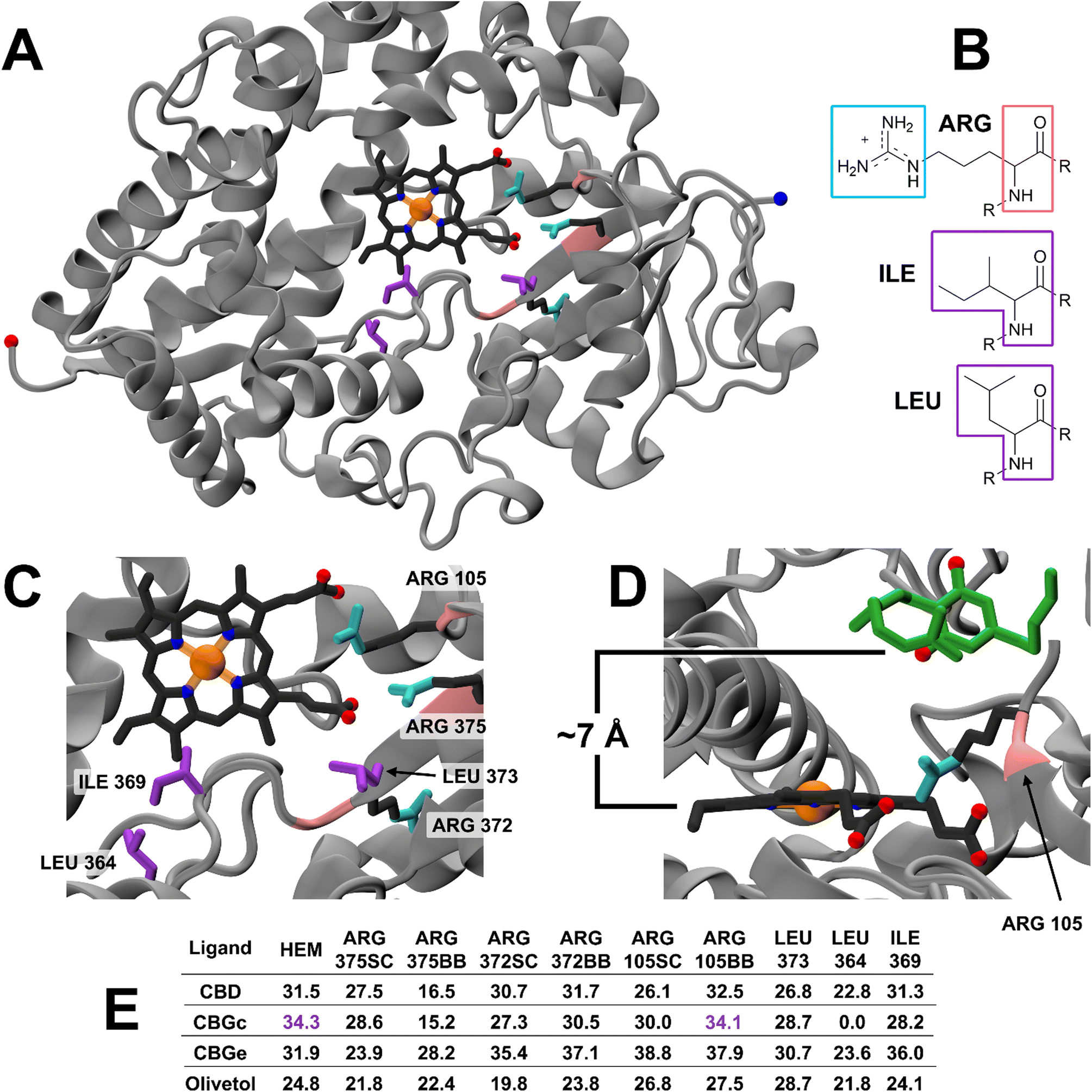 | ||
| Fig. 4 Molecular docking of cannabinoid substrates to CYP450 3A4 enzyme, using GOLD. (A) Global view of the CYP450 3A4 enzyme with active site HEM (dark grey, with the complexed Fe, N and O atoms indicated by orange, blue and red balls, respectively). Main binding and additional sites are coloured based on groups used for docking: purple – Ile and Leu; cyan – Arg sidechain guanidinium group (ARGSC); and pink – Arg backbone N–C–CO atoms (ARGBB). N- and C-termini of the enzyme are included as large blue and red balls. (B) Chemical structures of Arg, Ile, and Leu residues. (C) Close-up view of the HEM and binding site with peptide residues (numbered accordingly). (D) Close-up view of the highest scoring pose between CYP450 3A4 HEM and CBGc (green) in a coiled conformation (Chemscore 34.3), the pose produced is very similar to ARG105BB CBGc (Chemscore 34.1) leaving a 7 Å gap between CBGc and HEM. CBGc O-atoms represented by red balls. (E) List of Chemscore predictions for each docking pose between ligands and binding sites made by GOLD. Purple scores are those for the conformational pose highlighted in D. CGBc = coiled; CBGe = extended. | ||
Fig. 4E displays the main findings of the docking calculations using GOLD. Conversely, docking olivetol to the active site leads to structural poses that make use of its phenolic hydroxyls, as suggested by IC50 studies.20 When CBD is docked into the HEM and ARG105, very similar Chemscore and binding modes are produced (Fig. 5), as identified in other studies.58,59 Given that both cases are not indicative of any positive interaction between the ligand and the active site (HEM), it is likely that CBD inhibits metabolic action by binding closer to the ARG105 site, while obstructing the active site indirectly. Coiled-CBG → HEM (Chemscore 34.3) is nearly equivalent to coiled-CBG → ARG105 (34.1). Both cases left a 7 Å gap between the active site and CBG, indicating that the ligand interacts with the arginine and other amino acids nearby (Fig. 4D). This gap is not present when docking the extended, straight-chain conformation of CBG (Fig. 5, right), cementing the importance of molecular conformation in this case. Binding of diltiazem, nifedipine, testosterone and triazolam (Fig. 2) to the active site via sequential docking occurs in the gap left behind by the main binding pose of coiled-CBG (Fig. 6). Diltiazem struggles to dock well, given the steric hindrance involved in its pose, along with the odd arrangement of hydrogens to CBG-oxygens. Diltiazem's negative Chemscore, while not directly comparable, can be contrasted with the positive scores produced by docking testosterone and triazolam. Thus, the position and conformation of CBG in Fig. 6 accurately replicates trends seen in the literature, in that it inhibits diltiazem binding but not testosterone or triazolam.19 These poses generated by the docking (Fig. 6) would not be very probable in the presence of a fully extended CBG conformer. Future work involving more robust modelling of the DDI between nifedipine and CBG would be an asset to complement the trends uncovered in this work and authors would look either to tackle this themselves or to the wider community to perhaps help in this endeavour soon. The likelihood of CBG existing in the coiled form as opposed to the fully extended form can be assessed via conformational studies.
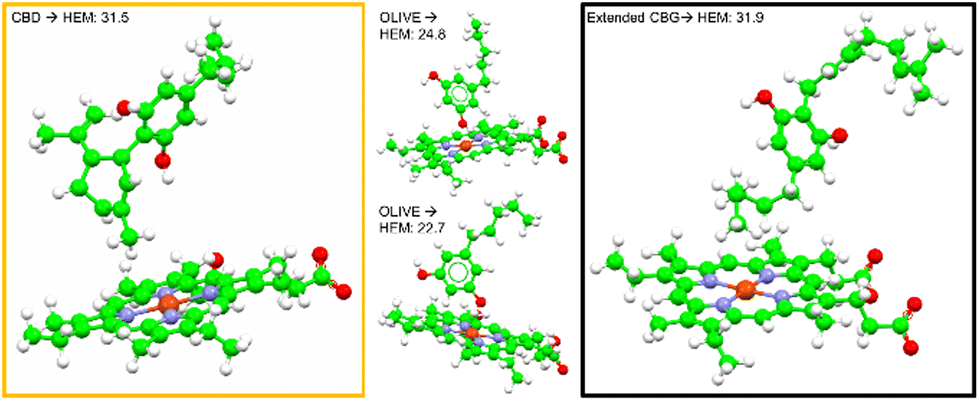 | ||
| Fig. 5 Molecular docking poses for CBD, olivetol and extended CBG to the HEM active site. | ||
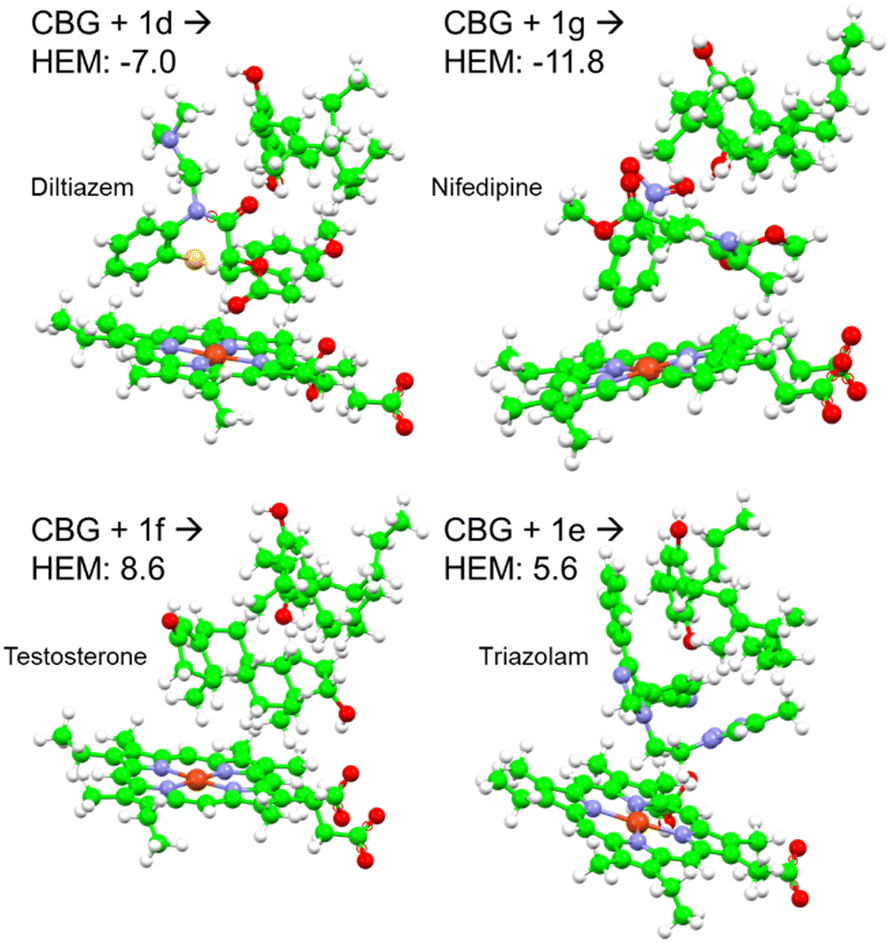 | ||
| Fig. 6 Sequential docking of coiled CBG + drug (diltiazem, nifedipine, testosterone and triazolam) to the CYP450 3A4 active site, using their labels from Fig. 2. Negative Chemscores are highlighted in red. | ||
Key conformers/energetic spread
Trends in free energy of cannabigerocin conformations were identified and correlated with the number of positive interactions. Ramachandran plots of all the possible conformers (ESI Fig. S1†) revealed adjacent structures with near-identical free energies, indicating ‘moats’ in the potential energy surface (PES). When high energy geometries resided near a local minima, they tended to rotate strained dihedrals in order to lower their free energy, falling into a PES ‘moat’. This may be evidenced by the seemingly distinct conformers ss1+3c4a5+ and ss1+3c4–5+ for example, whose final actual dihedral angles χ4 were identical (Table 1).| Expected (°) | Actual (°) | Actual χ4 | |
|---|---|---|---|
| a Each number indicates the relevant torsional angle χ, followed by the angle's position: gauche+ (+), anti (a) or gauche− (−), with cis (c) and trans (t) labels only relevant to E/Z isomerism at χ3 (i.e., 3c = 3 cis). Ss corresponds to both OH rotors syn and aa to both OH rotors anti. | |||
| χ 4ss1+3c4a5+a | 120–240 | 246.4 | g- |
| χ 4ss1+3c4–5+ | 240–36 | 246.4 | g- |
Fig. 7 presents some of the key unique poses for cannabigerocin. Of these key poses, cis and trans both occupy similar points in the range of +0–3.6 kcal relative to the lowest energy conformer (green). Interestingly, the fully extended conformers that are more in line with typical 2D representations of CBG are among the less stable (solid blue, red, purple). The main contributor to stability is indeed the number of positive interactions formed. In the cis conformers (right), these mainly manifest as hydrogen bonds between the Φ OH and the alkyl hydrogens. Whereas the trans conformers (and the most stable conformer) show OH-pi bonding between the Φ OH and the geranyl CC double bonds. Plotting the complete spread of free energies of all 108 cannabigerocin isomers (Fig. 8) highlights the large number of conformers that fall into degenerate structures, as shown by the clustering of points around 2–3 kcal in the Boltzmann distribution fashion. Despite this spread, the highest and lowest energy conformers can be filtered out to separate unfavourable modes (ss1-3c green line) from favourable ones (ss1a3t orange line). The trend in energy along shifting χ4 and χ5 is generally conserved; yet, the large gap in energy between ss1-3c and ss1a3t cannot be attributed to χ1 or χ3 alone. Instead, the stability of conformers is related to specific geometries that achieve a higher number of positive interactions coinciding with identical dihedral angles. Laws of adjacent rotors, whether eclipsing or gauche to one another, come secondary to the larger interactive forces at play.
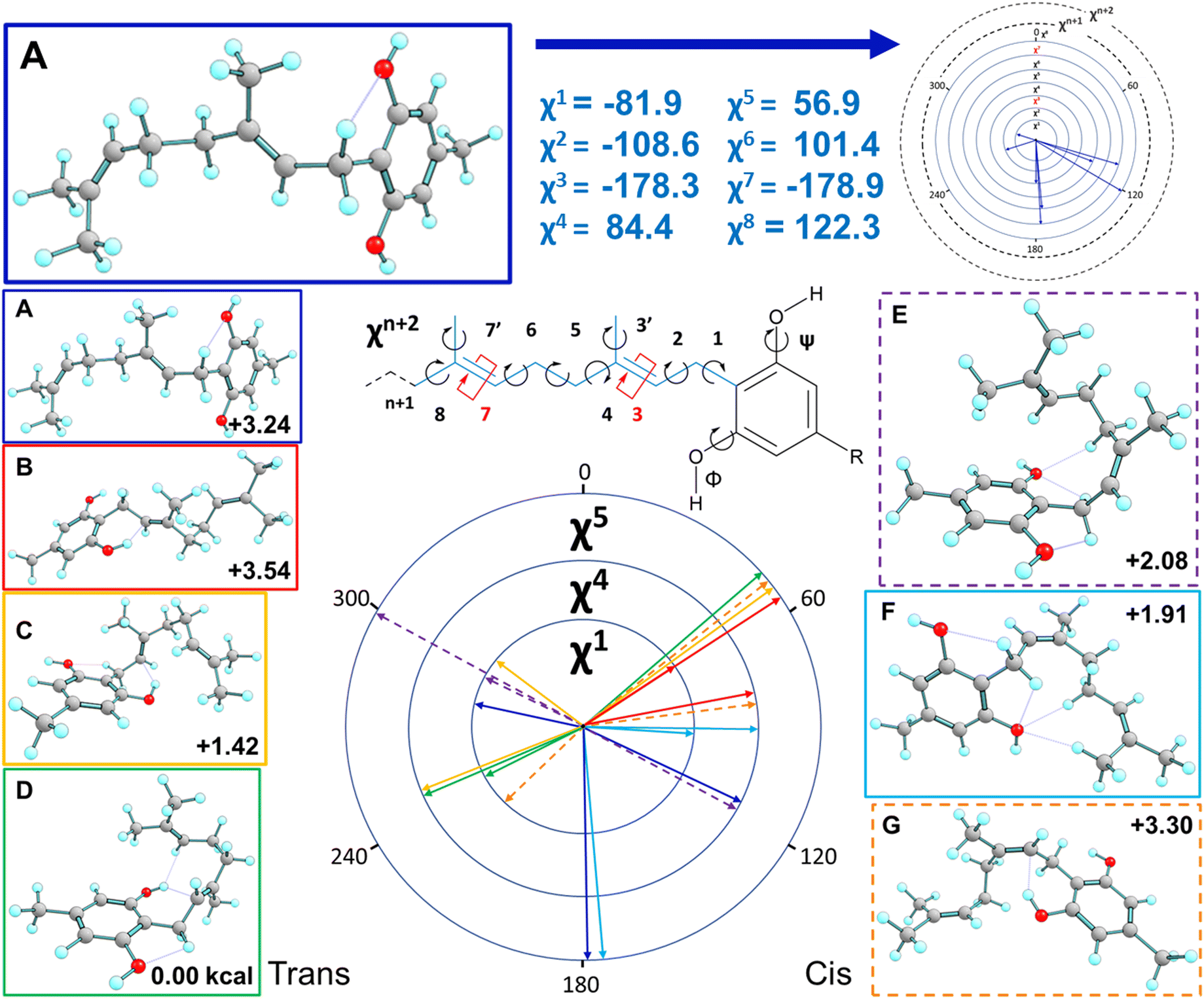 | ||
| Fig. 7 Highlights of the main cannabigerocin conformers. Dihedral ‘wheel’ maps the three key torsional angles χ1, χ4 and χ5, colour coded to show the resulting 3D conformations. Each conformation is labelled with its free energy in kcal relative to the lowest energy conformer (D). Conformers are divided into trans (left) and cis (right) columns depending on which geometric isomer they adopt as a result of switching χ3. Structure A is repeated at the top of the figure as a detailed exemplar to aid interpretation. | ||
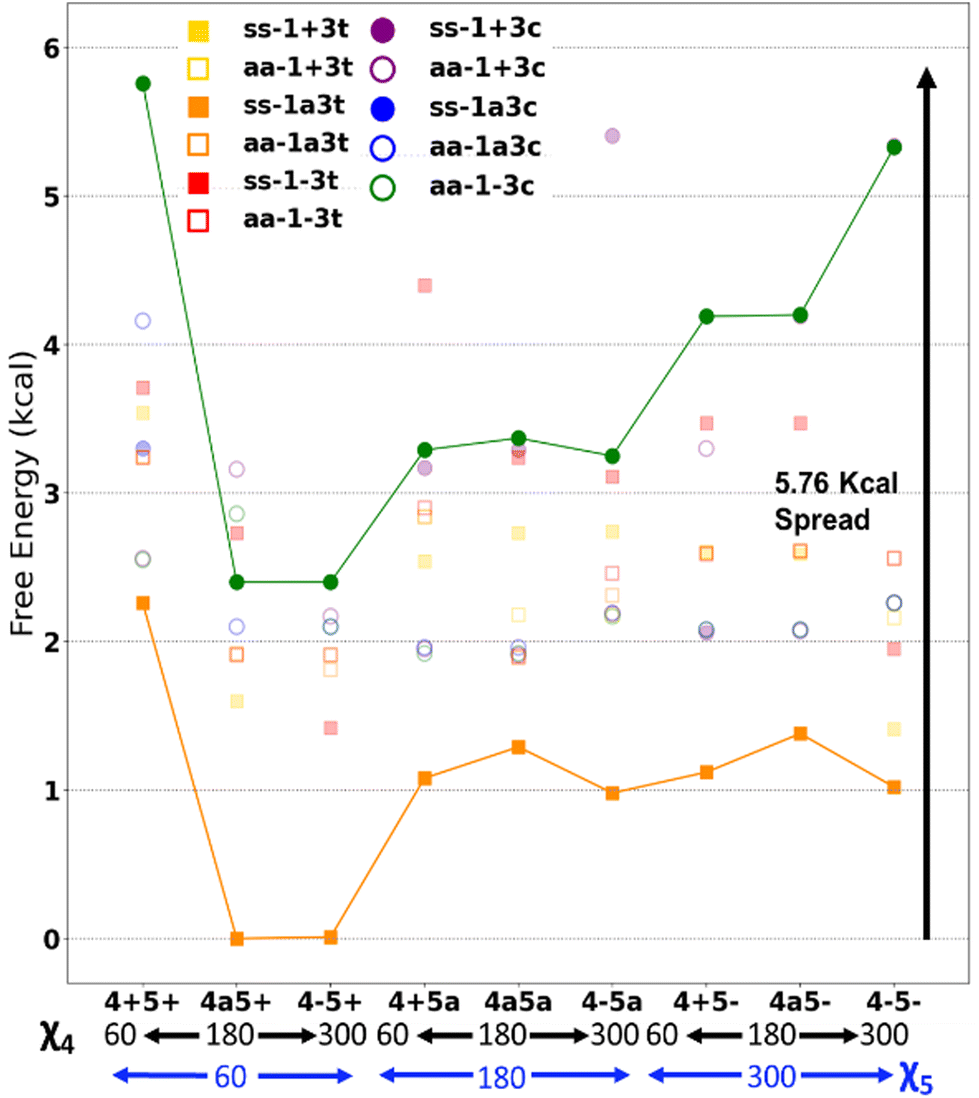 | ||
| Fig. 8 Free energies of all cannabigerocin isomers, with cis (circles) and trans (squares) distinguished; these are relative to the lowest energy isomer (ss1a3t4a5+). | ||
E/Z isomers
Fig. 7 displays key χ3cis conformers that are as/more stable than their trans isomeric counterparts, with their relative energies better reflected in Fig. 8. cis conformers (circles in Fig. 8) are not highlighted with a fixed energy cost but are instead interlaced with ‘usual suspects’. While trans conformers are generally considered to be more stable, there are system-specific exceptions. Some interactions have a greater stabilising effect than the steric hindrance that trans conformers normally provide. Most of the stable cis conformers are anti with OH rotors, facilitating up to 3 H-bonds from the geranyl C–H⋯OH. When considering E/Z isomerism, E (trans) preference is understood as reducing steric hindrance between higher priority (typically larger) substituents, but what is often overlooked is that certain substituted alkenes have their own mechanism of reducing steric clash between lower priority groups. Specific orientation of the alpha hydrogens in space can further lower the energy of cis conformers. By offsetting the dihedral angle formed across an H atom on one alpha carbon to the next, we show how H′′′ almost bisects the H–C–H angle on the opposite end of the double bond, falling into the space between H′ and H′′ (Fig. 9). This lack of co-planarity between opposite alpha hydrogens tends to result in more stable structures than their neighbouring conformers by an average of 0.3 kcal (Fig. 8).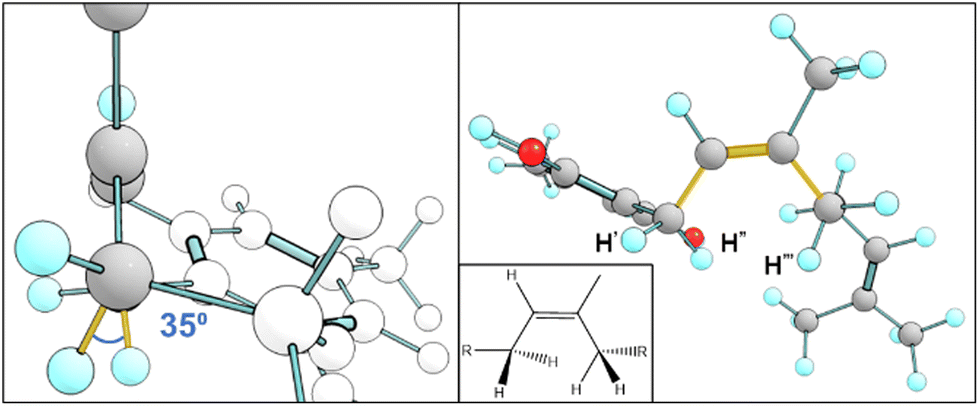 | ||
| Fig. 9 Model aa1-3c4a5a an example of a cis isomer with bisecting hydrogens. View along the double bond (left) shows the dihedral angle between the two alpha hydrogens that would normally overlap H′′–Cα–Cα–H′′′, highlighted in yellow. The same structure is shown in 2D and 3D (right). | ||
An intramolecular ‘staple’
Fig. 10 reports the key conformers of CBG from this search revealing that the side chain plays a moderate role in stability. The key finding of the conformational study for CBG, CBGV and cannabigerocin was that the coiled pose for the geranyl chain was consistently identified in the most stable conformers. The introduction of the propyl/pentyl side chain presented more spatial combinations in conjunction with the geranyl chain. The set of side chain conformers were built using the most stable orientation of the geranyl chain (Fig. 7D) which had one syn OH rotor and χ3 set to trans. From here we could observe if different poses of the side chain would reinforce or remove the geranyl's “coiling” (Fig. 10, right). In the interest of removing this one geranyl pose as the limiting variable, the same side chain poses were also calculated in conjunction with the most stable χ3cis conformers (Fig. 10, left).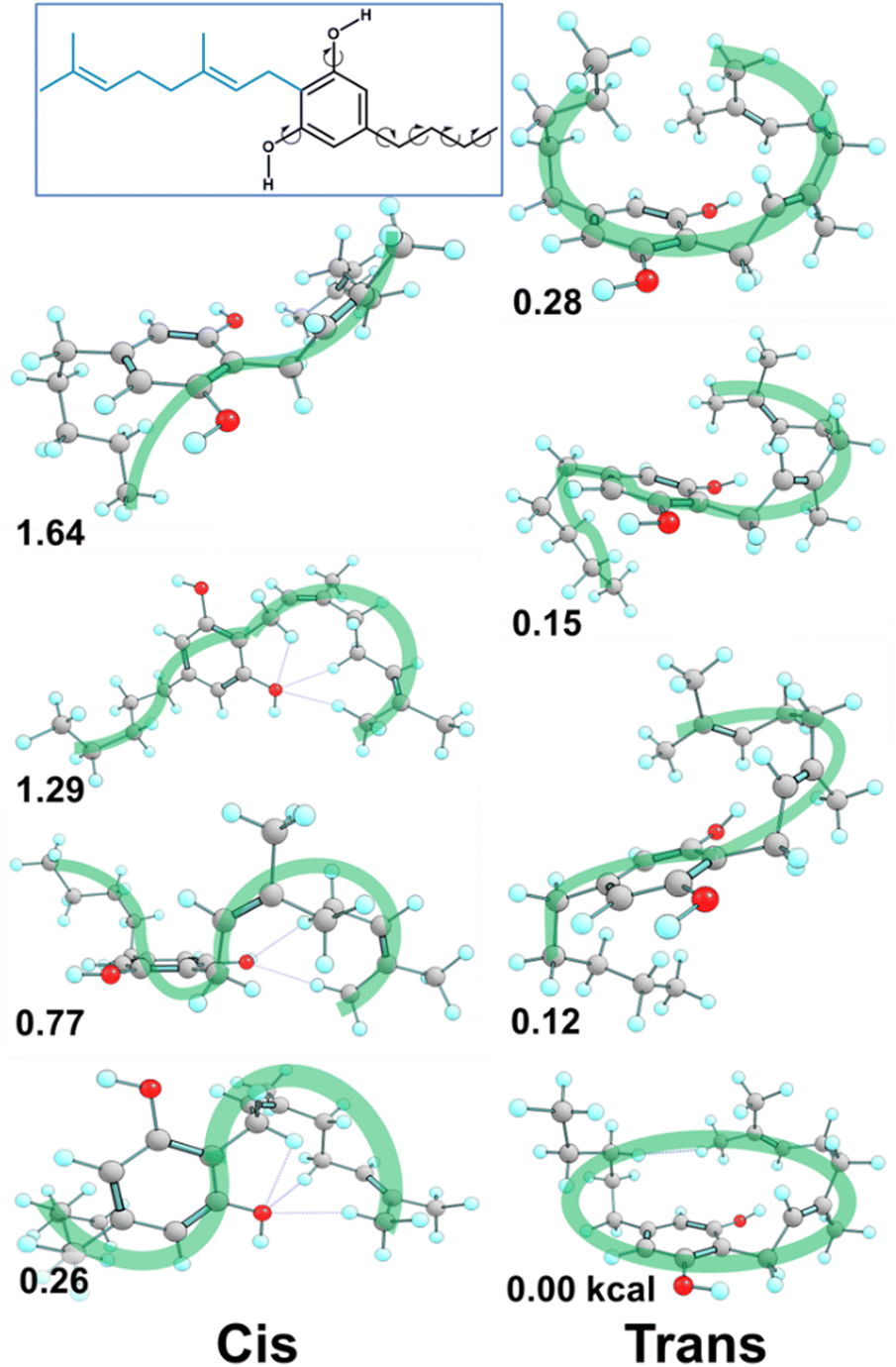 | ||
| Fig. 10 Methodology extended to CBG, spatial variations of the pentyl side chain on previously identified lowest energy conformers produced the most stable (bottom) structures for cis (left) and trans (right) χ3. CBGV conformers are reported in Fig. S5.† | ||
This intramolecular ‘staple’ results in a CBG molecule that wraps its chains over and around the ring. The 114.6° angle between the second and fourth carbon on the pentyl side chain pushes the third carbon inwards, leaving its hydrogens in proximity to the terminal hydrogen atom on the geranyl chain (Fig. 11). In CBGV, we expect the same H–H interaction to occur between the terminal methyl on the propyl side chain and the end of the geranyl chain. However, the propyl side chain has the plane of its C–C bonds running parallel to the benzene ring, keeping this terminal methyl just out of reach of the geranyl's tail (Fig. 11). Thus, the most stable conformer for CBGV defaults to having its side chain extended out and away from the ring (ESI Fig. S5†). There are further noteworthy variations in conformation among the lowest energy structures. For instance, Fig. 12 shows the bottom –OH group forming an H–H interaction with –CH2. Switching this OH rotor to the anti-position turns this interaction into a hydrogen bond from –CH2⋯O(H) but ultimately keeps the topology the same (Fig. 8 top left vs. bottom left).
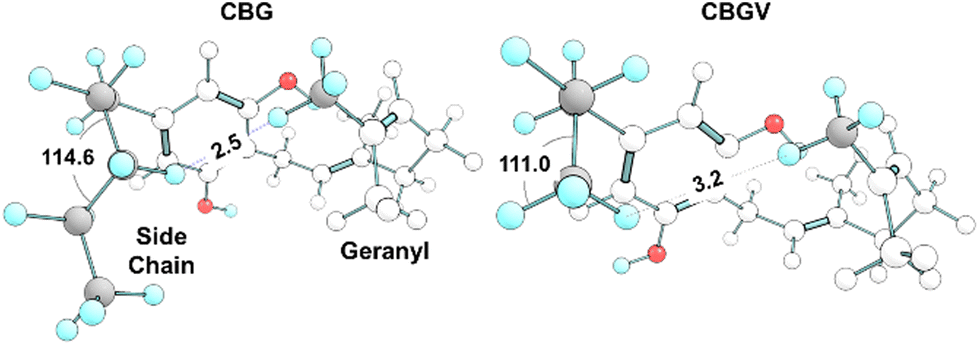 | ||
| Fig. 11 Comparison of CBG and CBGV side chains. CBG's pentyl side chain kinks at C3, allowing its hydrogen to reach within 2.5 Å of the geranyl's tail, forming the H–H interaction shown in Fig. 9E. CBGV lacks this ‘intramolecular staple’ and thus avoids full coiling. | ||
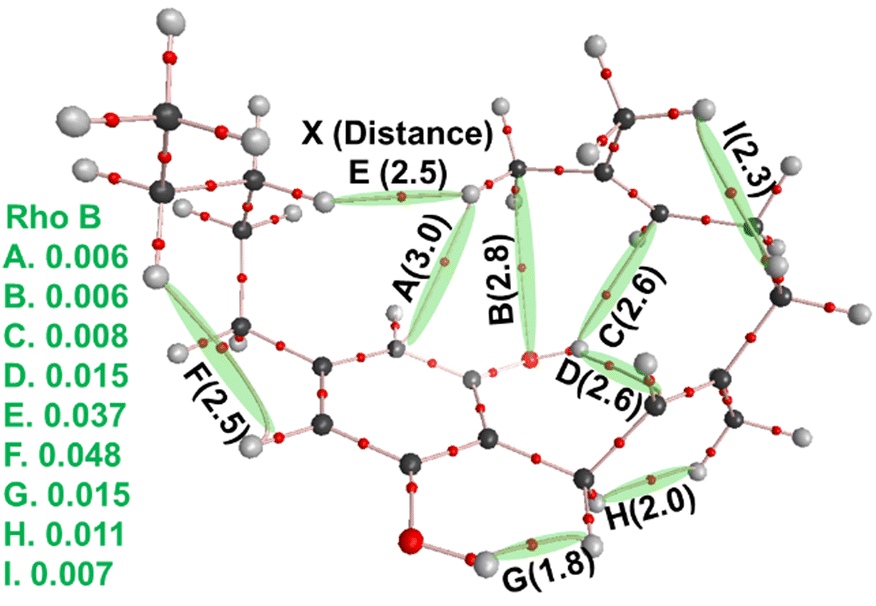 | ||
| Fig. 12 Molecular graph of the most stable CBG isomer (ss-CBG-e2) built up from the cannabigerocin conformers. Bader's atoms-in-molecules (AIM) analyses of the wavefunction of the stable geometry and the bond critical points (BCPs, smaller red) and bond paths (pink) identified. The BCPs evidence sharing of electronic density and thus inter-atomic interaction or bonding (selected distances provided in parentheses in black), helping to stabilise this specific 3D geometry (conformation). Electron density values per volume-space (ρb/ē bohr−3) are listed at left (green). | ||
The principal source of stabilisation in this case arises from the intramolecular interactions including hydrogen bonds formed between phenolic hydroxyl groups and alkyl hydrogens. Thus, one can expect anti conformations (OH facing away from the alkyl chain) to be present in the majority of the most stable conformers. However, the most stable conformers for cannabigerocin and CBG have one –OH group orientated syn and the other anti to the chain. Wavefunction analyses of the electronic structure revealed that as the geranyl chain coils inwards towards the central aromatic ring, OH⋯π interactions are formed between the polarised H-atom and the adjacent CC double bonds. Subsequently, an H-bond is formed between the exposed oxygen and an H-atom from one of the terminal methyl groups. Albeit not particularly strong, these interactions are directing in nature locking the cannabinoid molecules into these 3D poses, lowering the configurational entropy and thus the overall free-energy.60 When the –OH rotors are anti to the geranyl chain, this affords space for H-bonds to form with optimal geometry (i.e. OH⋯X bond angles of 180°, with X as the acceptor), albeit at the expense of the –OH⋯π or weaker –CH⋯π ones.61
The most stable conformers for CBG (Fig. 10, right) are stabilised by –OH⋯π interactions (Fig. 12C and D), maintaining the geranyl chain wrapped around the central phenol ring. The side chain further contributes to stabilising this 3D ‘wrapping’ via a weakly polar H–H interaction (Fig. 12E), which are not unknown.62,63 We label these interactions intramolecular ‘staples’, effectively binding one end of the molecule to the other.
The greatest implication is the topology change of CBG: the ring is now enveloped on one side by these weak/loose interactions, resulting in the whole structure taking up much less volume than the fully extended conformer. The biological implications of this conformation are outlined above, with coiling greatly reducing the inhibitory potential of CBG docked to CYP450 3A4.
Conclusions
We have identified the most stable conformations for the CBG cannabinoid and selected analogues. The intramolecular staple formed via the geranyl chain coiling around the phenolic hydroxyl group is further supported by Bader's AIM wavefunction analyses of the electronic structure. This is perhaps common to a wide range of natural phenolic compounds containing flexible alkyl chains. Molecular docking with CYP450 3A4 revealed that the coiled conformer presents less inhibitory potential and likely reduces the number of drug–drug interactions (DDIs) with respect to an extended conformer. Noteworthy is the ∼20% of cis isomers that structurally reduce repulsion between alpha H-atoms, stabilising these geometries by 0.2–0.3 kcal in free-energy, with respect to similar geometries for the trans CC isomers. Although the trans isomers do dominate the overall distribution of geometries, the cis isomers cannot be ignored and must be considered. However, relevant isomerisation mechanisms during synthesis or in vivo are as yet not established and their exploration could form the bases of future empirical and computational works.
Future endeavours to rationally optimise the synthesis of bioactive compounds could build upon warehoused data and information on the structural preferences of these cannabinoid and related bioactive systems. This would allow formulation of structure–property relationships, particularly when combined with spectroscopic data, including state-of-art neutron scattering (i.e. QENS, INS, NCS) to map dynamic-structuring as well as coherent-THz measurements to characterise conformation-specific vibrational density of states (VDOS), low-energy cooperative vibrational dynamics in particular which are linked to functionality.64 Such trends could assist in the formulation of a series of rules for the design of systems with conformation-specific bioactivity and specificity, as observed for cannabinoids and other flexible bioactive molecules and the receptors they bind with.65,66
Author contributions
Conceptualisation: G. A. C.; computation: M. S. and G. A. C.; data dissemination & graphics: M. S., H. A., J. H. D., K. V. T., and G. A. C.; writing – original draft: M. S. and G. A. C.; writing – review & editing: H. A., J. H. D., B. B., I. M., R. Y. Y., D. H. F., J. M., K. V. T., and G. A. C.; and funding acquisition: E. B., R. Y. Y., D. H. F., J. M., and G. A. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MS acknowledges the Michael DeGroote Centre for Learning and Discovery, in particular the McArthur Lab, for hosting and providing computational resources related to this work. HA acknowledges the Hong Kong Quantum AI Lab Limited, AIR@InnoHK for supporting his fellowship. Furthermore, funding to support RYY through the Natural Sciences and Engineering Research Council, Canada (RGPIN 04598), is gratefully acknowledged. DHF, KVT and GAC acknowledge and thank TRIUMF, Canada, for funding to support work on cannabinoids and related bioactive systems (M2081, M2082, and M2168). KVT thanks the Department of Chemistry (McMaster University) and the Faculty of Land and Food Systems (University of British Columbia, UBC) for support. GAC acknowledges EU-Horizon 2020 and BEIS, UK for supporting the FUNMIN project (ACT, No. 299668) and thanks McMaster University and University of British Columbia, Canada for supporting his adjunct and affiliate professorships, respectively. MS, KVT and GAC thank the GIOCOMMS institute (Toronto, CA; Rome, IT) for supporting the work.References
- N. M. Kogan and R. Mechoulam, Dialogues Clin. Neurosci., 2007, 9, 413–430 CrossRef PubMed.
- P. F. Whiting, R. F. Wolff, S. Deshpande, M. Di Nisio, S. Duffy, A. V. Hernandez, J. C. Keurentjes, S. Lang, K. Misso, S. Ryder, S. Schmidlkofer, M. Westwood and J. Kleijnen, J. Am. Med. Assoc., 2015, 313, 2456–2473 CrossRef CAS PubMed.
- A. Ameri, Prog. Neurobiol., 1999, 58, 315–348 CrossRef CAS PubMed.
- R. Mechoulam and L. r. Hanuš, Chem. Phys. Lipids, 2000, 108, 1–13 CrossRef CAS PubMed.
- J. Gonçalves, T. Rosado, S. Soares, A. Y. Simão, D. Caramelo, Â. Luís, N. Fernández, M. Barroso, E. Gallardo and A. P. Duarte, Medicines, 2019, 6, 31 CrossRef PubMed.
- E. Perez, J. R. Fernandez, C. Fitzgerald, K. Rouzard, M. Tamura and C. Savile, Molecules, 2022, 27, 491 CrossRef CAS PubMed.
- M. A. Farha, O. M. El-Halfawy, R. T. Gale, C. R. MacNair, L. A. Carfrae, X. Zhang, N. G. Jentsch, J. Magolan and E. D. Brown, ACS Infect. Dis., 2020, 6, 338–346 CrossRef CAS PubMed.
- M. Aqawi, R. Gallily, R. V. Sionov, B. Zaks, M. Friedman and D. Steinberg, Front. Microbiol., 2020, 11, 858 CrossRef PubMed.
- R. Nachnani, W. M. Raup-Konsavage and K. E. Vrana, J. Pharmacol. Exp. Ther., 2021, 376, 204–212 CrossRef CAS.
- C. P. Anokwuru, F. L. Makolo, M. Sandasi, S. Y. Tankeu, I. L. Elisha, C. Agoni, S. Combrinck and A. Viljoen, Phytochem. Rev., 2022, 21, 1523–1547 CrossRef CAS.
- A. Jastrząb, I. Jarocka-Karpowicz and E. Skrzydlewska, Int. J. Mol. Sci., 2022, 23, 7929 CrossRef PubMed.
- G. Messina, F. Rovelli and P. Lissoni, Clin. Rev. Cases., 2022, 4, 1–4 Search PubMed.
- T. T. Lah, M. Novak, M. A. Pena Almidon, O. Marinelli, B. Žvar Baškovič, B. Majc, M. Mlinar, R. Bošnjak, B. Breznik, R. Zomer and M. Nabissi, Cells, 2021, 10, 340 CrossRef CAS PubMed.
- S. Mammana, E. Cavalli, A. Gugliandolo, S. Silvestro, F. Pollastro, P. Bramanti and E. Mazzon, Medicina, 2019, 55, 747 CrossRef PubMed.
- N. G. Jentsch, X. Zhang and J. Magolan, J. Nat. Prod., 2020, 83, 2587–2591 CrossRef CAS PubMed.
- O. Zendulka, G. Dovrtelova, K. Nosková, M. Turjap, A. Sulcova, L. Hanus and J. Jurica, Curr. Drug Metab., 2016, 17, 206–226 CrossRef CAS PubMed.
- S. M. M. Alsanosi, C. Skiffington and S. Padmanabhan, in Handbook of Pharmacogenomics and Stratified Medicine, ed. S. Padmanabhan, Academic Press, 2014, pp. 341–364. DOI:10.1016/b978-0-12-386882-4.00017-7.
- S. Nasrin, C. J. W. Watson, Y. X. Perez-Paramo and P. Lazarus, Drug Metab. Dispos., 2021, 49, 1070–1080 CrossRef CAS PubMed.
- P. T. Doohan, L. D. Oldfield, J. C. Arnold and L. L. Anderson, AAPS J., 2021, 23, 91 CrossRef CAS PubMed.
- S. Yamaori, J. Ebisawa, Y. Okushima, I. Yamamoto and K. Watanabe, Life Sci., 2011, 88, 730–736 CrossRef CAS PubMed.
- J. H. Lin, Adv. Drug Delivery Rev., 2003, 55, 53–81 CrossRef CAS PubMed.
- L. Zhao, J. L.-S. Au and M. G. Wientjes, Front. Biosci., Elite Ed., 2010, 2, 241–249 Search PubMed.
- I. F. Sevrioukova and T. L. Poulos, Dalton Trans., 2013, 42, 3116–3126 RSC.
- S. Moradi, H. Gholami, C. Karami, N. Farhadian, F. Balaei, M. Ansari and M. Shahlaei, Int. J. Biol. Macromol., 2020, 165, 2855–2868 CrossRef CAS PubMed.
- J. L. Beers, D. Fu and K. D. Jackson, Drug Metab. Dispos., 2021, 49, 882–891 CrossRef CAS PubMed.
- T. B. Hughes and S. J. Swamidass, Chem. Res. Toxicol., 2017, 30, 642–656 Search PubMed.
- L. Chai, H. Zhang, F. Guo, R. Song, H. Yu and L. Ji, Chem. Res. Toxicol., 2022, 35, 440–449 Search PubMed.
- W. P. Walters, A. A. Murcko and M. A. Murcko, Curr. Opin. Chem. Biol., 1999, 3, 384–387 CrossRef CAS.
- W. H. Okamura, M. M. Midland, M. W. Hammond, N. Abd Rahman, M. C. Dormanen, I. Nemere and A. W. Norman, J. Steroid Biochem. Mol. Biol., 1995, 53, 603–613 CrossRef CAS PubMed.
- E. L. Eliel and S. H. Wilen, Stereochemistry of organic compounds, John Wiley & Sons, 1994 Search PubMed.
- C. F. Tormena, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 96, 73–88 CrossRef CAS PubMed.
- G. A. Chasse, A. M. Rodriguez, M. L. Mak, E. Deretey, A. Perczel, C. P. Sosa, R. D. Enriz and I. G. Csizmadia, J. Mol. Struct.: THEOCHEM, 2001, 537, 319–361 CrossRef CAS.
- T. P. Straatsma, E. J. Bylaska, H. J. J. van Dam, N. Govind, W. A. de Jong, K. Kowalski and M. Valiev, in Annual Reports in Computational Chemistry, ed. R. A. Wheeler, Elsevier, 2011, vol. 7, pp. 151–177 Search PubMed.
- G. A. Chass, M. A. Sahai, J. M. S. Law, S. Lovas, Ö. Farkas, A. Perczel, J.-L. Rivail and I. G. Csizmadia, Int. J. Quantum Chem., 2002, 90, 933–968 CrossRef CAS.
- J. M. S. Law, G. A. Chass, L. L. Torday, A. Varro and J. G. Papp, J. Mol. Struct.: THEOCHEM, 2002, 619, 1–20 CrossRef CAS.
- J. M. S. Law, D. H. Setiadi, G. A. Chass, I. G. Csizmadia and B. Viskolcz, J. Phys. Chem. A, 2005, 109, 520–533 CrossRef CAS PubMed.
- D. H. Setiadi, G. A. Chass, L. L. Torday, A. Varro, J. G. Papp and I. G. Csizmadia, Eur. Phys. J. D, 2002, 20, 609–618 CrossRef CAS.
- D. H. Setiadi, G. A. Chass, L. L. Torday, A. Varro and J. G. Papp, J. Mol. Struct.: THEOCHEM, 2002, 594, 161–172 CrossRef CAS.
- H. T. Vu, F. V. Song, K. V. Tian, H. Su and G. A. Chass, Org. Biomol. Chem., 2019, 17, 9942–9950 RSC.
- T. J. Sull, G. A. Chass, A. Varro and J. G. Papp, J. Mol. Struct.: THEOCHEM, 2003, 623, 51–62 CrossRef CAS.
- L. F. Pisterzi, D. R. P. Almeida, G. A. Chass, L. L. Torday, J. G. Papp, A. Varro and I. G. Csizmadia, Chem. Phys. Lett., 2002, 365, 542–551 CrossRef CAS.
- M. A. Sahai, D. H. Setiadi, G. A. Chass, E. F. Pai, B. Penke and I. G. Csizmadia, J. Mol. Struct.: THEOCHEM, 2003, 666–667, 311–319 CrossRef CAS.
- Z. Mucsi, G. A. Chass, P. Ábrányi-Balogh, B. Jójárt, D.-C. Fang, A. J. Ramirez-Cuesta, B. Viskolcz and I. G. Csizmadia, Phys. Chem. Chem. Phys., 2013, 15, 20447–20455 RSC.
- Z. Mucsi, G. A. Chass and I. G. Csizmadia, J. Phys. Chem. B, 2009, 113, 10308–10314 CrossRef CAS.
- K. S. Lau, A. Mantas, G. A. Chass, F. H. Ferretti, M. Estrada, G. Zamarbide and I. G. Csizmadia, Can. J. Chem., 2002, 80, 845–855 CrossRef CAS.
- G. A. Chasse, K. P. Chasse, A. Kucsman, L. L. Torday and J. G. Papp, J. Mol. Struct.: THEOCHEM, 2001, 571, 7–26 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and a. D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- W.-H. Mu, G. A. Chasse and D.-C. Fang, Int. J. Quantum Chem., 2008, 108, 1422–1434 CrossRef CAS.
- Y.-M. Chen, G. A. Chass and D.-C. Fang, Phys. Chem. Chem. Phys., 2014, 16, 1078–1083 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- F. Li, P. F. Egea, A. J. Vecchio, I. Asial, M. Gupta, J. Paulino, R. Bajaj, M. S. Dickinson, S. Ferguson-Miller, B. C. Monk and R. M. Stroud, J. Biol. Chem., 2021, 296, 100557 CrossRef CAS PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- M. D. Eldridge, C. W. Murray, T. R. Auton, G. V. Paolini and R. P. Mee, J. Comput.-Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS.
- A. Perczel, J. G. Angyan, M. Kajtar, W. Viviani, J. L. Rivail, J. F. Marcoccia and I. G. Csizmadia, J. Am. Chem. Soc., 1991, 113, 6256–6265 CrossRef CAS.
- G. A. Chasse, M. L. Mak, E. Deretey, I. Farkas, L. L. Torday, J. G. Papp, D. S. R. Sarma, A. Agarwal, S. Chakravarthi, S. Agarwal and A. V. Rao, J. Mol. Struct.: THEOCHEM, 2001, 571, 27–37 CrossRef CAS.
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins, 2003, 52, 609–623 CrossRef CAS PubMed.
- F. Sapundzhi, K. Prodanova and M. Lazarova, AIP Conf. Proc., 2019, 2172, 100008 CrossRef CAS.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- G. A. Chass, S. Lovas, R. F. Murphy and I. G. Csizamdia, Eur. Phys. J. D, 2002, 20, 481–497 CrossRef CAS.
- D. J. Wolstenholme and T. S. Cameron, J. Phys. Chem. A, 2006, 110, 8970–8978 CrossRef CAS PubMed.
- C. F. Matta, J. Hernández-Trujillo, T.-H. Tang and R. F. W. Bader, Chem. – Eur. J., 2003, 9, 1940–1951 CrossRef CAS PubMed.
- F. Song, B. Yang, D. Di Tommaso, R. S. Donnan, G. A. Chass, R. Y. Yada, D. H. Farrar and K. V. Tian, Mater. Adv., 2022, 3, 4982–4990 RSC.
- A. R. Keimowitz, B. R. Martin, R. K. Razdan, P. J. Crocker, S. W. Mascarella and B. F. Thomas, J. Med. Chem. A, 2000, 43, 59–70 CrossRef CAS PubMed.
- P. T. F. Williamson, A. Verhoeven, K. W. Miller and A. Watts, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18031–18036 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00383c |
| This journal is © The Royal Society of Chemistry 2023 |