
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of both enantiomers of the biosurfactant aureosurfactin via bidirectional synthesis with a chiral Horner–Wittig building block†
Fabia
Mittendorf
,
Moritz
Quambusch
 and
Stefan F.
Kirsch
*
and
Stefan F.
Kirsch
*
University of Wuppertal, Department of Organic Chemistry, Germany. E-mail: sfkirsch@uni-wuppertal.de
First published on 24th May 2023
Abstract
Aureosurfactin is a novel biosurfactant that exhibits similar surface tension activity to known biosurfactants. In this work, we now report a facile synthesis for aureosurfactin using a bidirectional synthetic strategy. Both enantiomers of the target compound were accessed from the (S)-building block, derived from the same chiral pool starting material.
Surfactants are distinguished by their amphiphilic nature:1 their structure consists of both hydrophobic and hydrophilic moieties, enabling them to reduce surface and interfacial tension by accumulation of the respective hydro- or lipophilic parts on the surface of one phase of two immiscible fluids.2–4 Surfactants are widely used in cosmetic, textile, pharmaceutical and food industries, making them indispensable for daily use.5 However, the wide range of applications of mainly petroleum-based surfactants also contributes to environmental pollution, as they are typically not fully biodegradable.6,7 As a result, biosurfactants gained in importance in recent years, and the investigation of non-toxic, biodegradable and thus environmentally friendly biosurfactants is currently of utmost interest for the respective chemical industries.1b,8–10 In 2016, Yun et al. isolated the novel biosurfactant aureosurfactin (1) from Aureobasidium pullulans, with a surface tension activity similar to those of known biosurfactants with commercial potential such as rhamnolipid.8,11 Aureosurfactin (1) was characterized as a methyl ester of an acyclic dimer of either (3R,5R)- or (3S,5S)-3,5-dihydroxydecanoic acid (Scheme 1). The relative stereochemistry of the natural product was determined by comparison of 13C NMR spectra with those of (3R,5R)-3,5-dihydroxydecanoic acid, its trimer exophilin A12 and the related halymecins.13 The absolute configuration of 1 was not elucidated and no values for optical rotation were provided. However, based on the isolation of (3R,5R)-3,5-dihydroxy decanoic acid from Aureobasidium pullulans14 and its abundance as structural unit of natural products such as (+)-(3R,5R)-3-hydroxy-5-decanolide,15 exophilin A,12 halymecins13 and the cyclic dimer verbalactone16 we consider the (3R,5R,3′R,5′R)-configuration of aureosurfactin (1b) as being the more likely one.
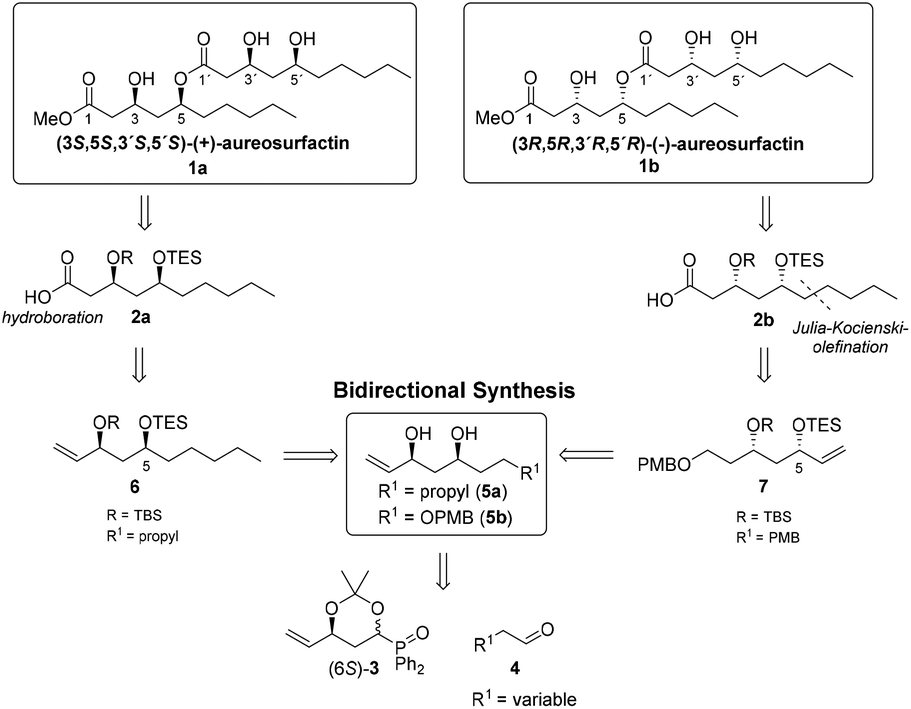 | ||
| Scheme 1 Access to both enantiomers of aureosurfactin (1) via bidirectional synthesis starting from a Horner–Wittig building block. | ||
Due to the uncertainty with regard to the absolute configuration of the natural surfactant, we decided to develop a de novo synthesis for both enantiomers of aureosurfactin (1). We point out that several syntheses of (3R,5R)-3,5-dihydroxydecanoic acid have been described in connection with the synthetic entries toward verbalactone,17 The synthesis of methyl (3S,5S)-3,5-dihydroxydecanoate has been reported by Mineeva.18 For the construction of (3S,5S,3′S,5′S)- and (3R,5R,3′R,5′R)-aureosurfactin (1), respectively, we envisioned the protected 3,5-dihydroxydecanoic acids 2a und 2b as key intermediates where the 5-OH bears a triethylsilyl-(TES) group that can be selectively removed over the tert-butyldimethylsilyl-(TBS) group attached at the 3-OH, thus making the coupling of both fragments via ester formation possible. On the other hand, global removal of all remaining silylether protecting groups in one step, following the connection of the fragments, was also possible.
For the synthesis of both enantiomers 2a and 2b we selected our recently developed modular strategy for the diastereoselective synthesis of syn- and anti-1,3-polyol units based on the chiral diphenylphosphane oxide (6S)-3 (Scheme 1),19,20 which can be easily prepared from readily available and inexpensive 2-deoxy-D-ribose in seven steps.21 A Horner–Wittig reaction with aldehydes 4a (R = nPr) and 4b (R = OPMB), respectively, and a subsequent syn-reduction should provide the 1,3-diols 5a and 5b. The preparation of (3S,5S)-3,5-dihydroxydecaonic acid derivative 2a should be obtained from 5avia sequential silyl protection. The resulting alkene 6 should afford the carboxylic acid 2a by hydroboration of the double bond and subsequent oxidation of the resulting primary alcohol. The retrosynthetic plan for the construction of the (3R,5R)-enantiomer 2b included first a sequential silyl protection of 5b to give the alkene 7. The prolongation of the vinyl- to a 1-pentenyl side chain proceeded via Lemieux–Johnson oxidation and a subsequent Julia–Kocienski olefination. Following simultaneous hydrogenation of the olefinic double bond and PMB-deprotection, acid 2b should be obtained by oxidation of the primary alcohol.
This synthetic strategy for the first total synthesis of (+)- and (−)-aureosurfactin (1), starting with a mutual chiral pool-derived building block (6S)-3, demonstrates another facet of how to easily access 1,3-polyol-containing structures with the use of Horner–Wittig methodologies: chiral diphenylphosphane oxide 3 is rapidly elaborated in both directions, thus providing a fully controlled entry to both enantiomeric series of the desired target compounds.
The synthesis of (3S,5S,3′S,5′S)-aureosurfactin (1a) started with the Horner–Wittig reaction of valeraldehyde 4a and chiral building block (6S)-3 to obtain β-hydroxyketone 8 in 86% yield (Scheme 2). Subsequent Narasaka–Prasad reduction22 furnished syn-diol 5a in 81% yield with an excellent diastereoselectivity (d.r. >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), after purification. Selective TBS-protection of the allylic alcohol gave rise to silyl ether 9 in 67% yield.23 Protection of the remaining secondary alcohol using TESCl yielded silyl ether 6, and hydroboration of the double bond followed by oxidation then provided the primary alcohol 10 in 75% over two steps. We then used an oxidation method developed by Paterson et al.24 to obtain the carboxylic acid 2a in 70% yield in one step from alcohol 10a, employing ruthenium dioxide and sodium periodate. Subsequent methylation with iodomethane and potassium carbonate following the protocol of Nakata et al.25 gave methyl ester 11a in high yield. Next, we explored different conditions for the selective TES-deprotection: the use of standard conditions like PPTS, CSA or TFA led only to undesired cyclization products, while using 5 equivalents of the less acidic HF·pyridine complex26 at 0 °C gave the desired product 12a in excellent 94% yield (Scheme 2).
:1), after purification. Selective TBS-protection of the allylic alcohol gave rise to silyl ether 9 in 67% yield.23 Protection of the remaining secondary alcohol using TESCl yielded silyl ether 6, and hydroboration of the double bond followed by oxidation then provided the primary alcohol 10 in 75% over two steps. We then used an oxidation method developed by Paterson et al.24 to obtain the carboxylic acid 2a in 70% yield in one step from alcohol 10a, employing ruthenium dioxide and sodium periodate. Subsequent methylation with iodomethane and potassium carbonate following the protocol of Nakata et al.25 gave methyl ester 11a in high yield. Next, we explored different conditions for the selective TES-deprotection: the use of standard conditions like PPTS, CSA or TFA led only to undesired cyclization products, while using 5 equivalents of the less acidic HF·pyridine complex26 at 0 °C gave the desired product 12a in excellent 94% yield (Scheme 2).
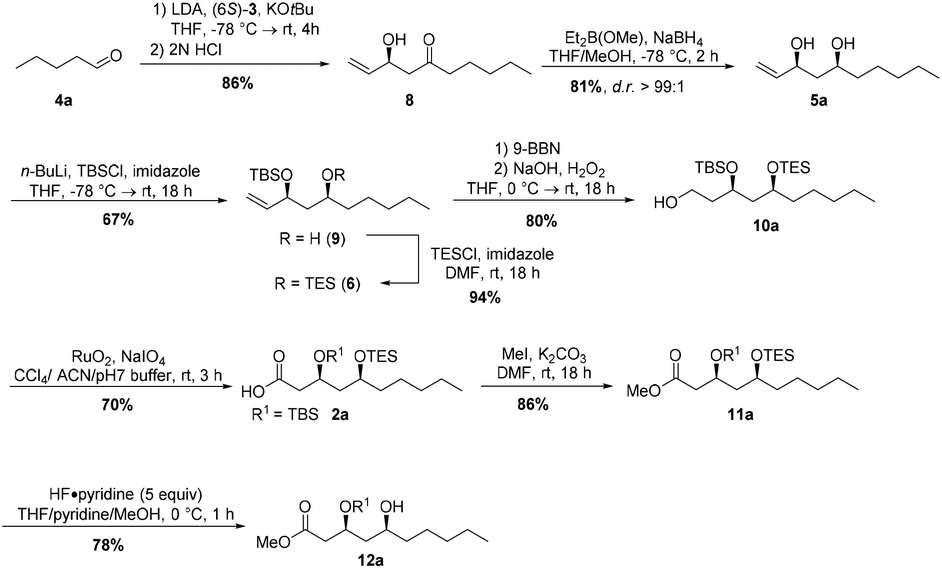 | ||
| Scheme 2 Synthesis of the intermediates 2a and 12a of (+)-aureosurfactin (1a). | ||
With acid 2a and alcohol 12a in hand, we planned to connect the fragments with classical Yamaguchi esterification conditions.27 Gratifyingly, the reaction proceeded smoothly to provide the envisaged ester product 13a in 92% yield (Scheme 3). The following global deprotection of the silyl ethers with 20 equivalents of HF·pyridine complex26 gave the desired (3S,5S,3′S,5′S)-(+)-aureosurfactin (1a) in almost quantitative yield. Hence, we were able to synthesize 39 mg of 1a, with an overall yield of 15% through the longest linear sequence of 10 steps (Scheme 3).
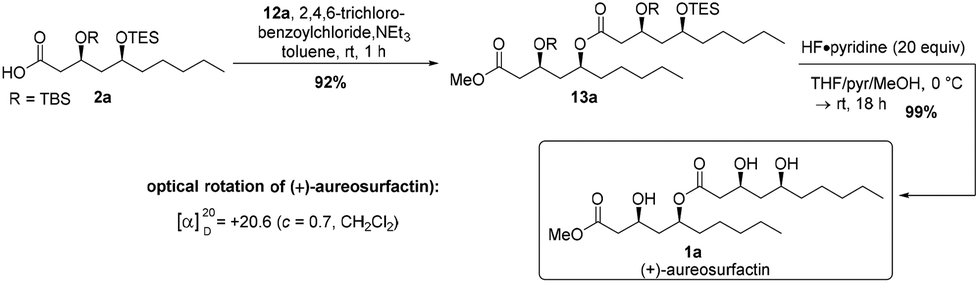 | ||
| Scheme 3 Final steps of the total synthesis of (+)-aureosurfactin (1a). | ||
Next, we envisaged the total synthesis of the other enantiomer, (3R,5R,3′R,5′R)-aureosurfactin (1b). A corresponding synthesis using the respective (6R)-enantiomer of building block 3 was inapplicable, due to the high costs for 2-deoxy-L-ribose that would be required as starting material for building block synthesis. Instead, our retrosynthetic approach for (3R,5R,3′R,5′R)-1 envisioned the above-described bidirectional synthesis involving a Horner–Wittig reaction of (6S)-3 with 2-((4-methoxybenzyl)-oxy)-acetaldehyde (4b),28 which gave rise to the β-hydroxyketone 14 in 89% yield (Scheme 4). Subsequent reduction of 14 by employing Narasaka-Prasad conditions yielded syn-diol 5b in 96% yield and with good diastereoselectivity (d.r. >99:1). The following selective TES-protection of the allylic alcohol was accomplished in moderate 58% yield with TESCl in the presence of 2,6-lutidine and DMAP.29 After TBS-protection, Lemieux–Johnson oxidation of the resulting silyl ether 7 gave the aldehyde 16 in 90% yield over two steps. Julia–Kocienski olefination with sulfone 17, which was generated over two steps starting from n-butanol (see ESI†), and subsequent hydrogenation gave primary alcohol 10b in 67% over two steps. Oxidation of the hydroxy group using ruthenium dioxide and sodium periodate afforded carboxylic acid 2b in high yield. In analogy to the other enantiomer the methyl ester fragment 12b of (3R,5R,3′R,5′R)-1b was obtained via methylation of the acid with iodomethane and potassium carbonate and a selective TES-deprotection with five equivalents of HF·pyridine complex26 at 0 °C.
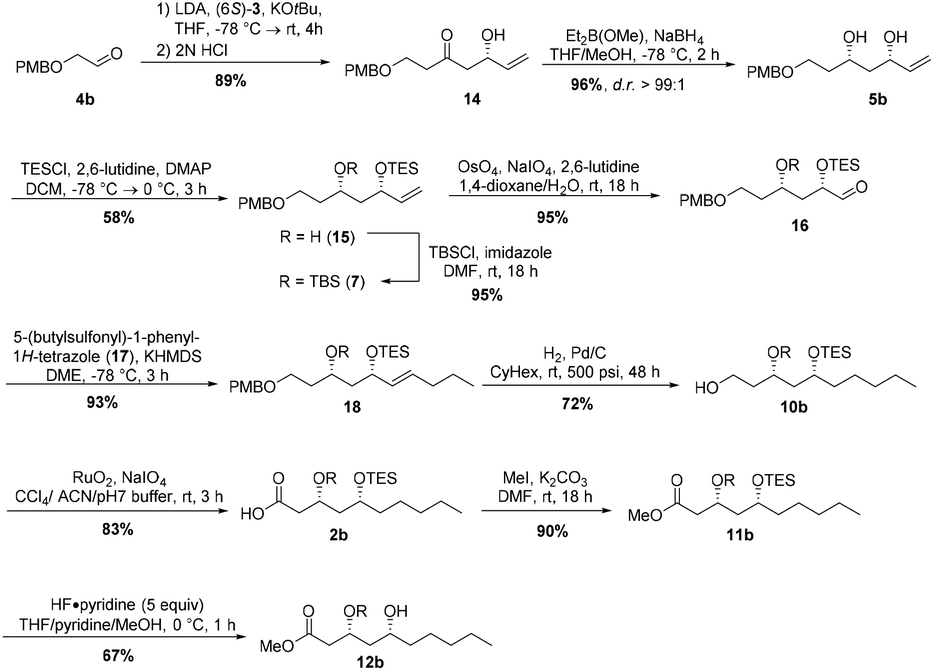 | ||
| Scheme 4 Synthesis of the intermediates 2b and 12b of (−)-aureosurfactin (1b). | ||
After Yamaguchi esterification27 of carboxylic acid 2b with alcohol 12b and global deprotection with HF·pyridine complex,26 we obtained 50 mg of the desired product (3R,5R,3′R,5′R)-(−)-aureosurfactin (1b) in 73% over two steps (Scheme 5). In total, (−)-aureosurfactin (1b) was successfully obtained in an overall yield of 11% through the longest linear sequence of 12 steps.
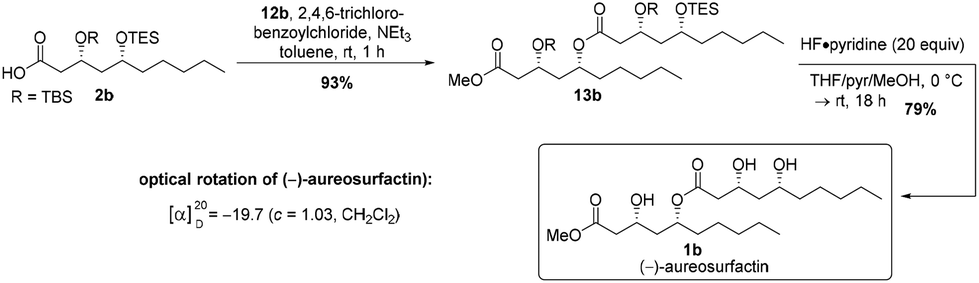 | ||
| Scheme 5 Final steps of the total synthesis of (−)-aureosurfactin (1b). | ||
In a simple experimental setting, we verified the described surface tension activity of aureosurfactin (1): to this end, the synthetic compound was dissolved in distilled water at a concentration of 1 mg per 100 mL, and 50 μL of the resulting solution were carefully placed on Parafilm and the degree of spreading was measured across the diameter (Fig. 1; 50 μL of distilled water were used as control). The reproducible results showed that the solutions containing either enantiomer have indeed a lower surface tension than the water.11 Surprisingly for us, the racemic mixture of the two aureosurfactin enantiomers disrupts the surface tension of water to an even greater extent, an effect that may be further studied by others.
 | ||
| Fig. 1 First studies for the comparison of surface tension activity of 1a and 1b and the racemic mixture of 1. | ||
Conclusions
In conclusion, we have described a bidirectional synthesis of both enantiomers of aureosurfactin (1), starting from the chiral Horner–Wittig building block (6S)-3. While (3S,5S,3′S,5′S)-(+)-1a could be synthesized in a ten-step sequence from valeraldehyde, with an overall yield of 18%, the synthesis of (3R,5R,3′R,5′R)-(−)-1b required 12 synthetic steps with an overall yield of 13.5%. The structure and the relative stereochemistry proposed by Yun et al.11 were confirmed, making both enantiomers accessible through de novo-synthesis.Conflicts of interest
There are no conflicts to declare.References
- (a) J. D. Van Hamme, A. Singh and O. P. Ward, Biotechnol. Adv., 2006, 24, 604–620 CrossRef CAS PubMed; (b) A. Singh, J. D. Van Hamme and O. P. Ward, Biotechnol. Adv., 2007, 25, 99–121 CrossRef CAS PubMed.
- E. Z. Ron and E. Rosenberg, Environ. Microbiol., 2001, 3, 229–236 CrossRef CAS PubMed.
- Y. Zhang and H. Zhao, Langmuir, 2016, 32, 3567–3575 CrossRef CAS PubMed.
- S. Hosseinpour, V. Götz and W. Peukert, Angew. Chem., Int. Ed., 2021, 60, 25143–25150 CrossRef CAS PubMed.
- (a) D. B. Tripathy, A. Mishra, J. Clark and T. Farmer, C. R. Chim., 2018, 21, 112–130 CrossRef CAS; (b) M. I. Van Dyke, H. Lee and J. T. Trevors, Biotechnol. Adv., 1991, 9, 241–252 CrossRef CAS PubMed.
- S. Shah, A. Bhattarai and S. Chatterjee, BIBECHANA, 2011, 7, 61–64 CrossRef.
- S. De, S. Malik, A. Ghosh, R. Saha and B. Saha, RSC Adv., 2015, 5, 65757–65767 RSC.
- J. D. Desai and I. M. Banat, Microbiol. Mol. Biol. Rev., 1997, 61, 47–64 CAS.
- D. Kitamoto, H. Isoda and T. Nakahara, J. Biosci. Bioeng., 2002, 94, 187–201 CrossRef CAS PubMed.
- C. N. Mulligan, Environ. Pollut., 2005, 133, 183–198 CrossRef CAS PubMed.
- J.-S. Kim, I.-K. Lee, D.-W. Kim and B.-S. Yun, J. Antibiot., 2016, 69, 759–761 CrossRef CAS PubMed.
- J. Doshida, H. Hasegawa, H. Onuki and N. Shimidzu, J. Antibiot., 1996, 49, 1105–1109 CrossRef CAS PubMed.
- C. Chen, N. Imamura, M. Nishijima, K. Adachi, M. Sakai and H. Sano, J. Antibiot., 1996, 49, 998–1005 CrossRef CAS PubMed.
- H. G. Choi, J. W. Kim, H. Choi, K. S. Kang and S. H. Shim, Molecules, 2019, 24, 4051–4062 CrossRef CAS PubMed.
- Y. Romeyke, M. Keller, H. Kluge, S. Grabley and P. Hammann, Tetrahedron, 1991, 47, 3335–3346 CrossRef CAS.
- P. Magiatis, D. Spanakis, S. Mitaku, E. Tsitsa, A. Mentis and C. Harvala, J. Nat. Prod., 2001, 64, 1093–1094 CrossRef CAS PubMed.
- (a) S. Gogoi, N. C. Barua and B. Kalita, Tetrahedron Lett., 2004, 45, 5577–5579 CrossRef CAS; (b) G. V. M. Sharma and C. A. Govardhan Reddy, Tetrahedron Lett., 2004, 45, 7483–7485 CrossRef CAS; (c) F. Allais, M.-C. Louvel and J. Cossy, Synlett, 2007, 451–452 CAS; (d) J.-Z. Wu, J. Gao, G.-B. Ren, Z.-B. Zhen, Y. Zhang and Y. Wu, Tetrahedron, 2009, 65, 289–299 CrossRef CAS; (e) G. B. Salunke, I. Shivakumar and M. K. Gurjar, Tetrahedron Lett., 2009, 50, 2048–2049 CrossRef CAS; (f) B. Das, K. Laxminarayana, M. Krishnaiah and D. N. A. Kumar, Helv. Chim. Acta, 2009, 92, 1840–1844 CrossRef CAS; (g) A. Garg and V. A. Singh, Tetrahedron, 2009, 65, 8677–8682 CrossRef CAS; (h) L. Carosi and D. G. Hall, Can. J. Chem., 2009, 87, 650–661 CrossRef CAS; (i) A. Harbindu and P. Kumar, Synthesis, 2011, 1954–1959 CAS; (j) I. V. Mineeva, Russ. J. Org. Chem., 2012, 48, 977–981 CrossRef CAS; (k) A. Venkatesham, R. S. Rao and K. Nagaiah, Tetrahedron: Asymmetry, 2012, 23, 381–387 CrossRef CAS; (l) M. Madala, B. Raman, K. V. Sastry and S. A. Musulla, Monatsh. Chem., 2016, 147, 1985–1990 CrossRef CAS; (m) S. Vanjivaka, K. Ramanakumar, M. Rajeswari, J. Vantikommu, G. Sridhar and S. Palle, ARKIVOC, 2018, 7, 50–57 Search PubMed.
- I. V. Mineeva, Russ. J. Org. Chem., 2013, 49, 1647–1654 CrossRef CAS.
- (a) A. Bredenkamp, Z.-B. Zhu and S. F. Kirsch, Eur. J. Org. Chem., 2016, 252–254 CrossRef CAS; (b) A. Bredenkamp, M. Wegener, S. Hummel, A. P. Häring and S. F. Kirsch, Chem. Commun., 2016, 52, 1875–1878 RSC.
- F. Ballaschk, Y. Özkaya and S. F. Kirsch, Eur. J. Org. Chem., 2020, 6078–6080 CrossRef CAS.
- F. Mittendorf, I. E. Celik and S. F. Kirsch, J. Org. Chem., 2022, 87, 14899–14908 CrossRef CAS PubMed.
- (a) K. Narasaka and F.-C. Pai, Chem. Lett., 1980, 1415–1418 CrossRef CAS; (b) K. Narasaka and F.-C. Pai, Tetrahedron, 1984, 40, 2233–2238 CrossRef CAS; (c) K. M. Chen, G. E. Hardtmann, K. Prasad, O. Repic and M. J. Shapiro, Tetrahedron Lett., 1987, 28, 155–158 CrossRef CAS; (d) K. M. Chen, K. G. Gunderson, G. E. Hardtmann, K. Prasad, O. Repic and M. J. Shapiro, Chem. Lett., 1987, 10, 1923–1926 CrossRef.
- W. R. Roush, H. R. Gillis and A. P. Essenfeld, J. Org. Chem., 1984, 49, 4674–4682 CrossRef CAS.
- S. M. Dalby, J. Goodwin-Tindall and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 6517–6521 CrossRef CAS PubMed.
- Y. Matsuda, T. Koyama, M. Kato, T. Kawaguchi, Y. Saikawa and M. Nakata, Tetrahedron, 2015, 71, 2134–2148 CrossRef CAS.
- B. C. Wilcock, M. M. Endo, B. E. Uno and M. D. Burke, J. Am. Chem. Soc., 2013, 135, 8488–8491 CrossRef CAS PubMed.
- J. Inanga, K. Hirata, H. Saeki, T. Katsuki and M. A. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef.
- S. Ghilagaber, W. N. Huntera and R. Marquez, Org. Biomol. Chem., 2007, 5, 97–102 RSC.
- J. R. Dunetz and W. R. Roush, Org. Lett., 2008, 10, 2059–2062 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00650f |
| This journal is © The Royal Society of Chemistry 2023 |