
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the accessibility, synthetic utility, and biological applications of aziridines
Christian
Dank
a and
Laura
Ielo
 *b
*b
aInstitute of Organic Chemistry, University of Vienna, Währinger Strasse 38, 1090 Vienna, Austria
bDepartment of Chemistry, University of Turin, Via P. Giuria 7, 10125 Torino, Italy. E-mail: laura.ielo@unito.it
First published on 15th May 2023
Abstract
Compounds featuring aziridine moieties are widely known and extensively reported in the literature. Due to their great potential from both synthetic and pharmacological points of view, many researchers have focused their efforts on the development of new methodologies for the preparation and transformation of these interesting compounds. Over the years, more and more ways to obtain molecules bearing these three-membered functional groups, which are challenging due to their inherent reactivity, have been described. Among them, several are more sustainable. In this review, we report the recent advances in the biological and chemical evolution of aziridine derivatives, in particular, the variety of methodologies described for the synthesis of aziridines and their chemical transformations leading to the formation of interesting derivatives, such as 4–7 membered heterocycles of pharmaceutical interest due to their promising biological activities.
 Christian Dank | Christian Dank pursued studies in chemistry at the University of Vienna, Austria. After obtaining the Master of Science (M.Sc.) degree in the group of Prof. Johann Mulzer in the field of total synthesis, during his Ph.D., he worked on the syntheses of novel antimalarials, supervised by Dr Hubert Gstach and Prof. Walther Schmid. After his Ph.D., he joined the medicinal chemistry department of Boehringer Ingelheim in Vienna, working on oncology projects for two and a half years. During his postdoctoral fellowship in the Lautens group in Toronto, Ontario, Canada, he contributed to his fields of expertise, total synthesis and medicinal chemistry, and explored the field of metal catalysis. Currently, he is teaching at his alma mater, the University of Vienna, as a senior lecturer. His current research interests are the design and synthesis of probes to explore pathways of biomolecules as well as methodological studies. |
 Laura Ielo | Laura Ielo graduated cum laude in Pharmaceutical Chemistry and Technology in 2015 from the University of Messina (Italy) where she also completed her doctoral studies with Prof. De Luca. She received her doctoral degree in 2018 from the University of Messina. She was a visiting PhD student at the University of Vienna (Austria) under the supervision of Prof. Vittorio Pace, where she also conducted her postdoctoral studies. In 2021, she took up an Assistant Professor position at the University of Turin (Italy), and in 2022 obtained the Habilitation for Associate Professor of Organic Chemistry. Her main research activities are focused on the development of new synthetic strategies in homologation reactions with lithium carbenoids and the design and synthesis of biologically active compounds, especially tyrosinase and proteasome inhibitors. |
1. Introduction
Aziridines are the simplest class of three-membered saturated nitrogen-containing heterocycles. They are less basic than alkylamines but more basic than arylamines. The strain caused by the geometric constraints of the trigonal ring confers high reactivity.1 Indeed, aziridines enable various intriguing synthetic opportunities that can be harnessed. Among these are reactions such as ring-opening, ring enlargement, tandem2 and multicomponent reactions.3 In the past, aziridines were considered to be difficult compounds to synthesize due to their instability, which is caused by their reactivity. Nevertheless, a variety of methodologies have been reported for preparing these interesting compounds including catalysis, annulation, and cycloaddition reactions, and organometallic chemistry. Lately, more sustainable conditions have been developed in good agreement with the principles of green chemistry including flow chemistry applications. Furthermore, aziridines are useful building blocks for the preparation of more complex heterocycles or amino acids which are also interesting from biological and pharmacological points of view due to their various biological activities.4 For these reasons, the interest of the scientific community in these small and fascinating compounds is ever increasing. Therefore, we herein report an overview of the recently developed methodologies regarding the syntheses, synthetic uses, and biological applications of aziridines. We have focused our attention on the past three years, trying to describe the most important breakthroughs that have been made after the publication of other reviews covering this field.52. Preparations of aziridines
2.1 Aziridination of olefins
Several methodologies have been described for the preparation of aziridine derivatives. Kappe and co-workers reported a continuous flow bromodimethylsulfonium bromide (2, BDMS) generation/alkene sulfobromination/aziridination sequence for preparing functionalized aziridines (4). BDMS (2) is a versatile reagent employed in organic synthesis as a stoichiometric reagent or as a catalyst. However, upon storage, it has to be treated as a hazard since it is corrosive and sensitive to heat and moisture, releasing molecular bromine (Br2) upon contact with water. For this reason, the authors decided to develop a continuous flow procedure for the safe generation and utilization of BDMS (2). In addition, by using a flow chemistry approach, the potential hazard from the accumulation of this reagent in batch can be overcome, as the reagent is immediately consumed upon generation. Thus, BDMS (2) was prepared for the first time in flow, starting from HBr and DMSO, and was then further reacted with primary amines (3) to yield 2-phenyl aziridines (4) in a 3-step sequential process (Scheme 1).6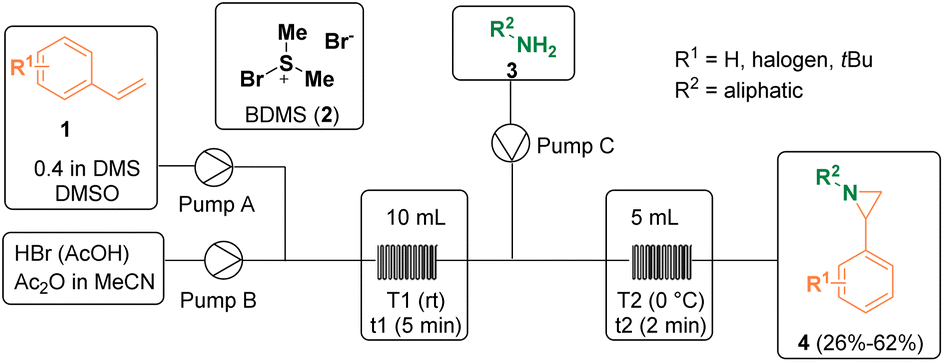 | ||
| Scheme 1 Flow BDMS generation/alkene sulfobromination/aziridination sequence. | ||
Gonnade and co-workers documented the syntheses of aziridine 7 which was then used for the preparation of the well-known Tamiflu (8), also called oseltamivir phosphate, which is used as a medicine to cure both influenza A and B and to prevent the spread of influenza. In one of the described procedures, cis-aziridine (7) was employed as a chiral synthon (Scheme 2).7
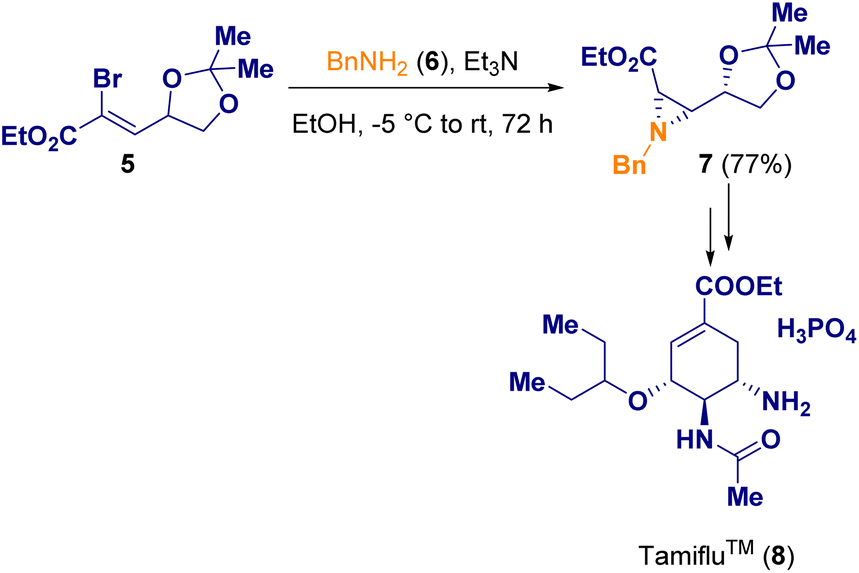 | ||
| Scheme 2 Aziridination of vinyl bromide 5 for preparing Tamiflu (8). | ||
Watson and co-workers reported in 2022 the asymmetric synthesis of aziridines (13) via enantioselective protonation of catalytically generated enamines by using chiral Brønsted acids such as 11 ((S)-TCYP). The aziridine ring (13) is achieved after treating the so-formed α-chloroamine 12 with a base in a one-pot process (Scheme 3).8
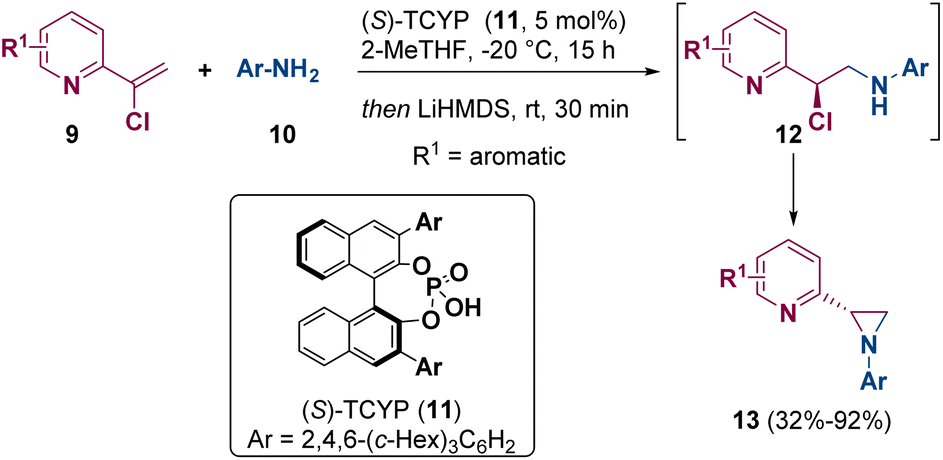 | ||
| Scheme 3 Asymmetric synthesis of aziridines. | ||
Catalysis is one of the most used methodologies for preparing aziridine derivatives. Indeed, a lot of pathways have been described by using different catalysts.9 Among them, nickel was employed and in 2022 Wu and co-workers reported a nickel-catalyzed aminofluoroalkylative cyclization of unactive alkenes (15) with iododifluoromethyl ketones (14) to afford versatile difluoroalkylated nitrogen-containing hetorocycles including aziridines (18), as shown in Scheme 4.10 According to the authors, the transformation proceeds through a radical mechanism. Upon treatment with the Ni-catalyst and base, iododifluoromethyl ketones (14) are transformed into intermediates such as 16, which react with N-allyl anilines (15) to give radical species 17, from which the aziridine products (18) are obtained.
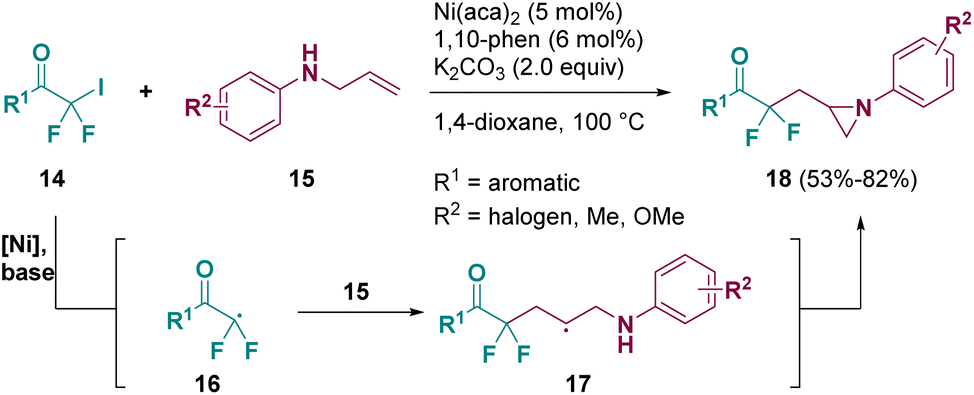 | ||
| Scheme 4 Nickel catalyzed aziridination of unactive alkenes. | ||
Berhal et al. documented the iron-catalyzed reaction between alkenes (20) and hydroxylamine derivatives (19) to give aziridines 22. In particular, they used simple iron(II) sources and readily available ligands rendering the reaction conditions more sustainable (Scheme 5).11 Considering the mechanism of the reaction, the in situ generated iron catalyst [Fe] provides an iron–nitrene intermediate (24 or 25) after reacting with the hydroxylamine derivative 19, releasing an equivalent amount of carboxylic acid (23). Since metal–nitrene complexes exist in two different spin states, the authors consider two possible reaction pathways. If the metal–nitrene complex predominantly exists in its singlet state (24), a concerted (2 + 1) cycloaddition could take place, leading to a stereospecific process (Scheme 5 – path a). On the other hand, if the iron–nitrene complex is in its triplet state (25), a radical addition followed by a radical-based ring closure could occur (Scheme 5 – path b). In this case, due to the multistep procedure, no stereoselectivity should be observed.
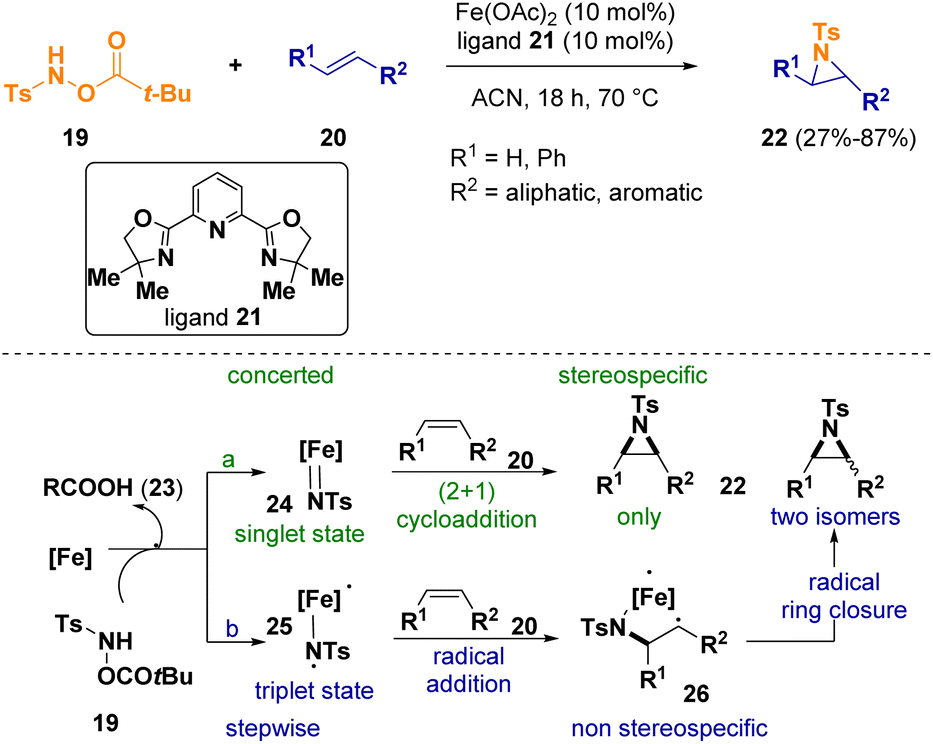 | ||
| Scheme 5 Reaction of alkenes with hydroxylamines for preparing aziridines. | ||
Driver et al. described an intermolecular Rh2(II)-catalyzed aziridination of olefins (28) using anilines (27) as non-activated nitrogen atom precursors and an iodine(III) reagent (30) as the stoichiometric oxidant. During the process, the N-aryl nitrene fragment is transferred from the intermediate iminoiodinane (31) to the Rh(II) carboxylate catalyst 29. The reaction proved to be stereospecific and chemo- and diastereoselective to produce N-aryl aziridine 33 as the only amination product (Scheme 6).12
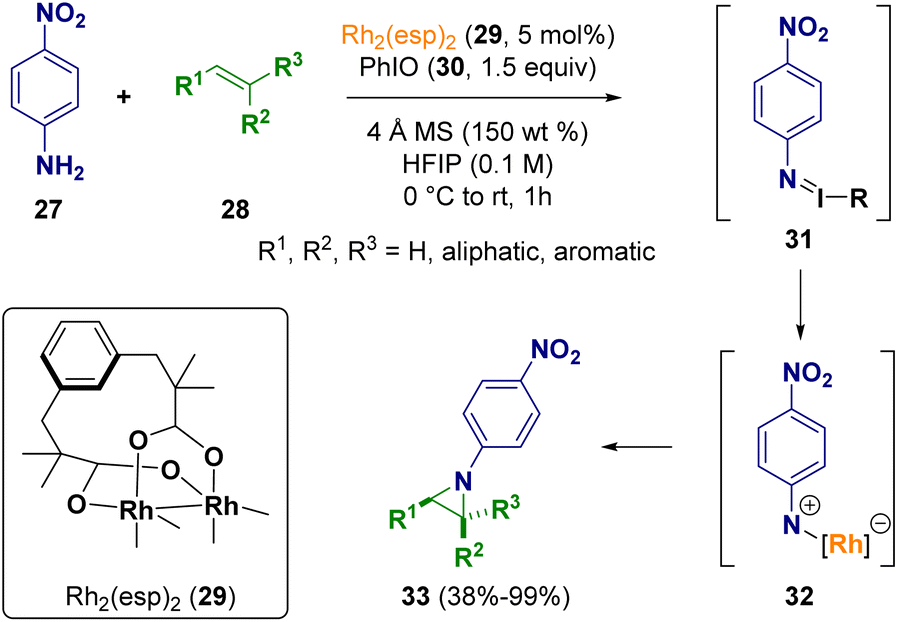 | ||
| Scheme 6 Intermolecular Rh2(II)-catalyzed aziridination of olefins. | ||
After describing in 2022 the use of (NHC)M (M = Cu, Ag, Au) cores as catalysts for the olefin aziridination reaction,13 recently, Pérez and co-workers reported the copper-catalyzed aziridination of olefins.14 The authors highlighted the important role of the halide characterizing the copper catalyst (Scheme 7 – path a). Indeed, they demonstrated via mechanistic studies that the employed copper(I) complexes (TTM)CuCl (35) and [(TTM)Cu-(NCMe)]PF6 (36) (TTM = tris(triazolyl)methane ligand) possess different behaviors, from catalytic and mechanistic points of view, depending on the presence or absence of the chloride ligand bonded to the metal center. If coordination is present, the limiting step of the reaction concerns the formation of the carbon–nitrene bond (39). In case the chlorine atom is not present, the highest barrier corresponds to the formation of the copper–nitrene intermediate (40). Chen and co-workers selected bis(pyrazolyl)borate Cu(I) complexes (43) as catalysts for the aziridination of olefins (Scheme 7 – path b). The reaction was carried out starting from a suitable styrene (41) and [N-(sulfonyl)imino]phenyliodinane (42). During the catalytic process, a nitrene is generated and added to the double bond.15
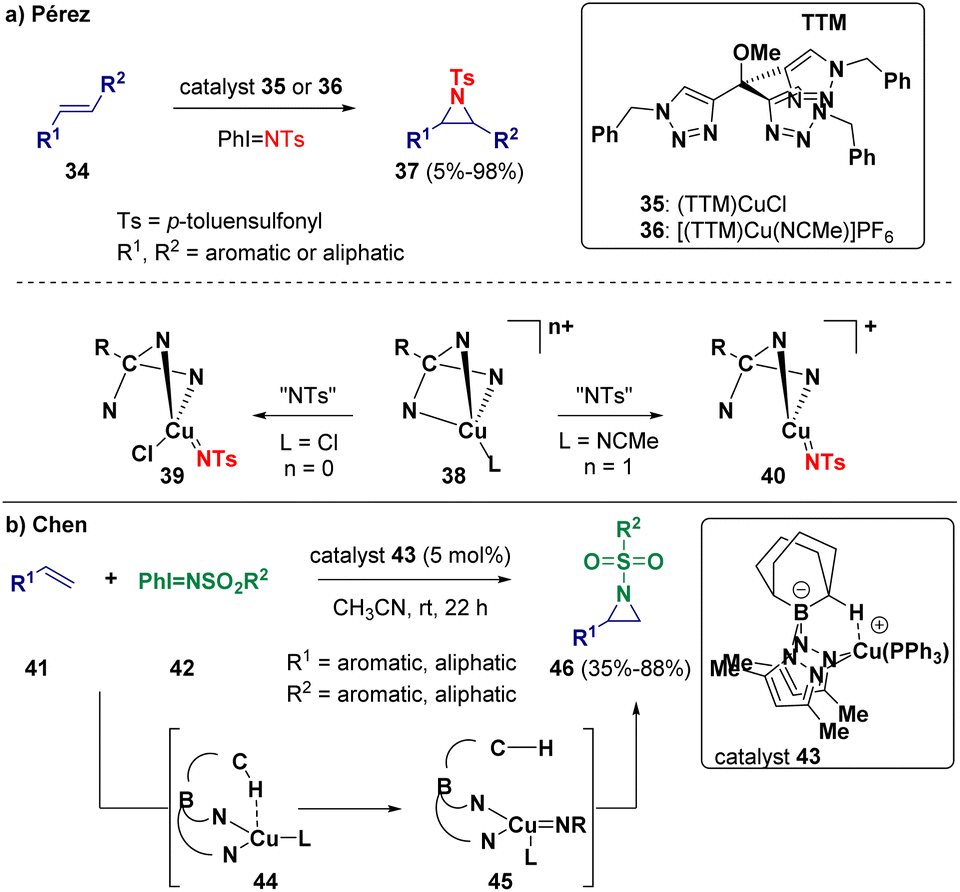 | ||
| Scheme 7 Copper-catalyzed olefin aziridination. | ||
Dauban et al. employed C4-symmetrical dirhodium(II) tetracarboxylates (48) as catalysts for the asymmetric intermolecular aziridination of substituted alkenes (47) with p-tBu-phenylsulfamates 49 (TBPhsNH2) (Scheme 8). The authors proposed a two-spin-state mechanism, involving a triplet Rh–nitrene species as the key intermediate (50) to direct the approach with stereocontrol and for the activation of the substrate. DFT studies support the proposed mechanism. An enantiomeric excess of up to 99% was observed.16
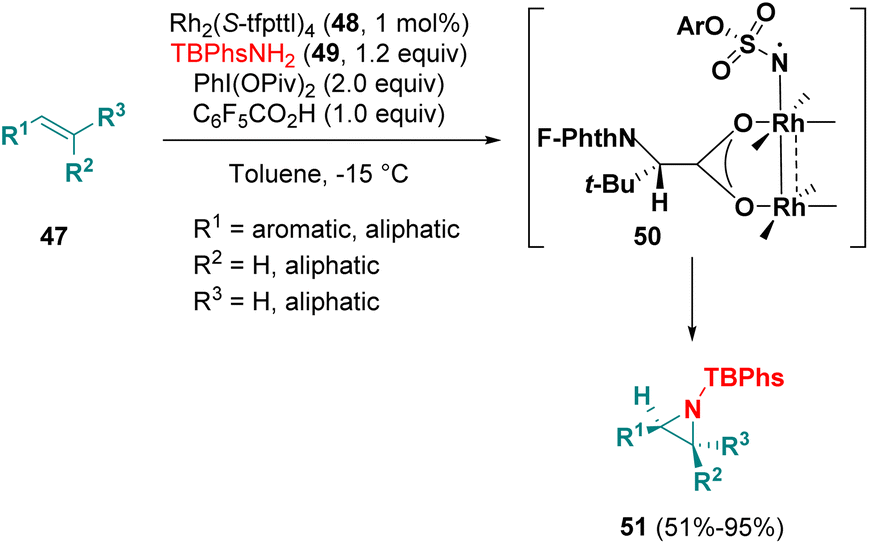 | ||
| Scheme 8 Asymmetric intermolecular aziridination of substituted alkenes. | ||
Zirconium has also been employed as a catalyst for the synthesis of aziridine derivatives. Moura-Letts and co-workers described the aziridination of alkenes (41) by using chloramine T17 (52) as the quantitative source of nitrogen (Scheme 9). Supported by kinetics and model reaction studies, the authors propose that the reaction mechanism involves the formation of a zirconooxaziridine complex (53) as the active catalyst.18
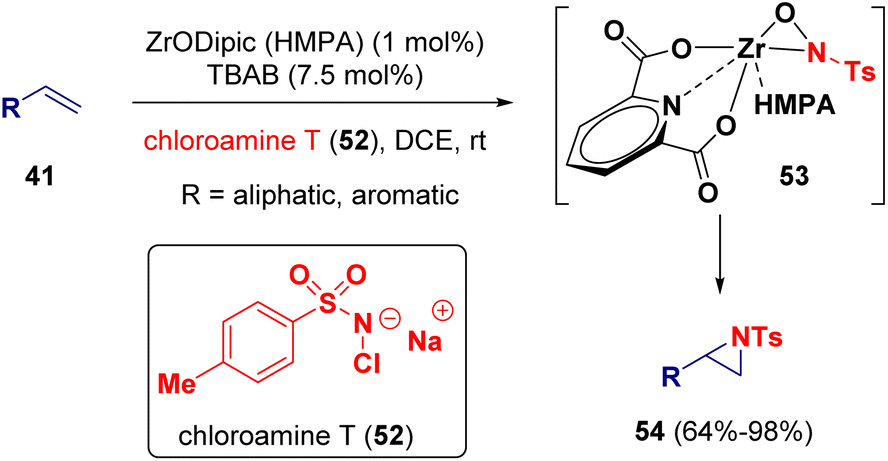 | ||
| Scheme 9 Zirconium catalyzed synthesis of aziridines. | ||
Jat and co-workers reported the iron(II) catalyzed direct N–H/N–Me aziridination of olefins (55) employing O-arylsulfonyl hydroxylamines (56). The one-pot methodology proved to be stereo- and regioselective, yielding a variety of unactivated aziridines (57) in good to excellent yields (Scheme 10).19
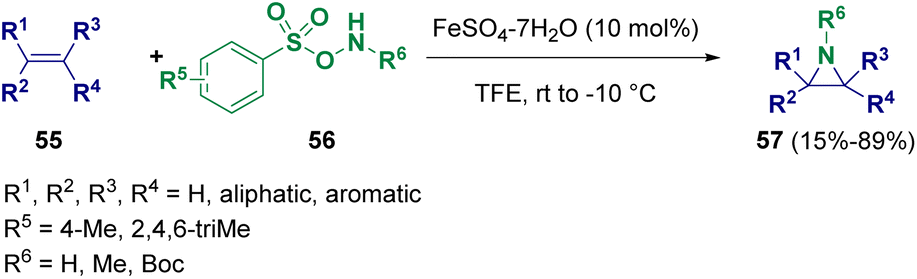 | ||
| Scheme 10 Iron(II) catalyzed direct N–H/N–Me aziridination of olefins. | ||
Mixed approaches of photo- and metal-catalysis have also been reported for the synthesis of aziridines.20 Koenigs et al. described the preparation of trifluoromethylated aziridines (62) starting from fluorinated olefins (58) and iodinanes (59), which undergo oxidative quenching in the presence of a Ru(bpy)3Cl2 catalyst (60), releasing a nitrene radical anion (61) (Scheme 11). Computational studies confirmed that the nitrene radical (61) serves as a reactive intermediate in direct aziridination reactions.21
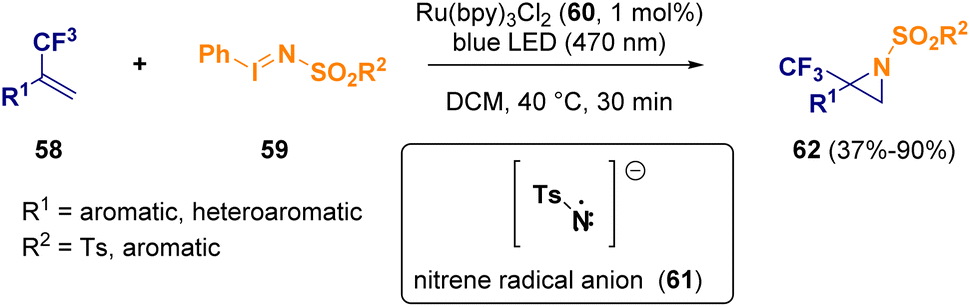 | ||
| Scheme 11 Photocatalytic amination reaction for preparing trifluoromethylated aziridines. | ||
Zhang and co-workers employed the carbonyl azide TrocN3 (65, 2,2,2-trichloroethoxycarbonyl azide), which is a potent nitrogen radical precursor for the aziridination of olefins (64) via Co(II)-based metalloradical catalysis (65) (Scheme 12). Chiral N-carbonyl aziridines (66) were prepared at room temperature in high yields with excellent enantioselectivities. The obtained N-Troc-aziridines (66) can be opened by different nucleophiles, achieving a variety of chiral amines with excellent stereospecificity (89–100% es).22
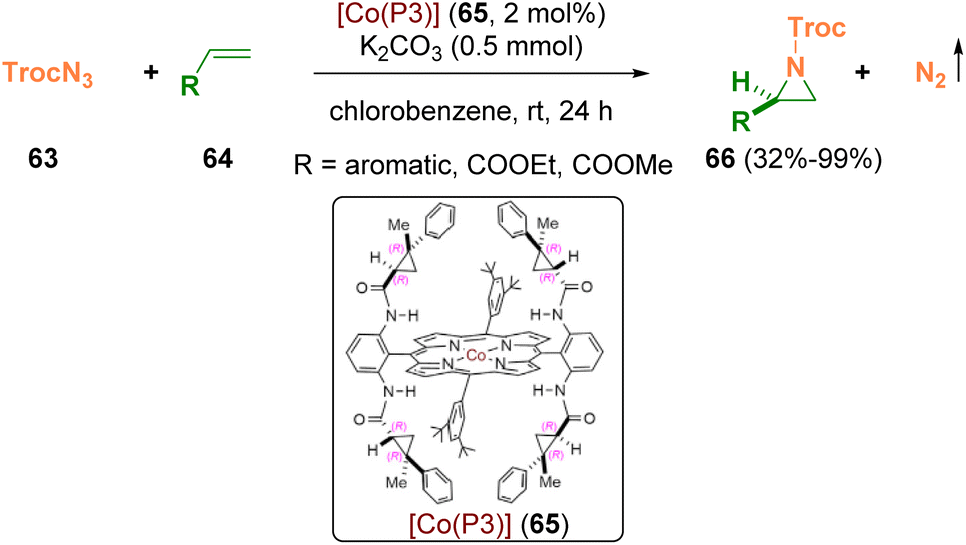 | ||
| Scheme 12 Olefin aziridination via Co(II)-based metalloradical catalysis. | ||
The major part of the reported methodologies for synthesizing aziridines rely on the transition-metal catalyzed reactions of alkenes with nitrene precursors or imines with carbene precursors.23 However, these pathways are limited by the use of hazardous and explosive carbene and nitrene precursors as well as additional steps for diazo synthesis and transition-metal residue removal. For these reasons, the development of more sustainable methodologies has been pursued by scientists.24 Different pathways employing electrochemical activation to facilitate oxidative cyclization were developed.25 Nevertheless, significant limitations and challenges are still to be overcome. Recently, the employment of thianthrenium salts was reported to be an alternative methodology.26 In 2021, Wickens et al. documented the electrochemical transformation of non-activated alkenes (67) into metastable, dicationic intermediates 69 and 70 that undergo aziridination with primary amines (71) under basic conditions (Scheme 13). This new approach allows the preparation of diverse aziridine building blocks (72) bearing sensitive functional groups, such as allyl and cyano groups, that are challenging to access through more conventional approaches.26
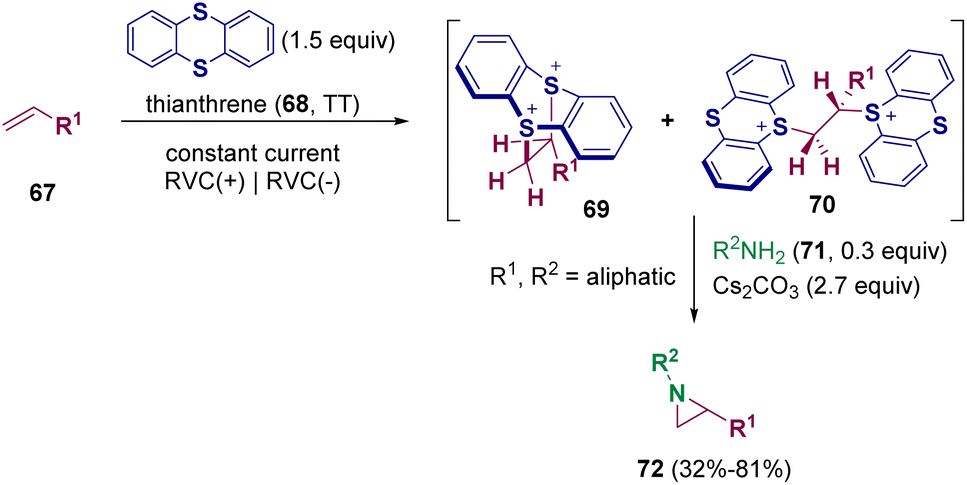 | ||
| Scheme 13 Electrochemical transformation of unactivated alkenes into aziridines. | ||
In 2022, Shu and co-workers developed a straightforward aziridination pathway using primary amines (73) with alkenes substituted with thianthrenes (74). The methodology works well for terminal, internal, aromatic, and aliphatic alkenes (Scheme 14).27 In comparison with the electrochemical thianthrene-mediated aziridine formation, which only allows functionalization of terminal olefins with primary amines26 (Scheme 13), this conventional approach allows aziridination of both terminal and internal alkenes. Furthermore it is not limited to the use of primary amines, but also tolerates primary amides, carbamates, and sulfonamides. The use of active methylenes instead leads to cyclopropanation, making the methodology even more versatile.27
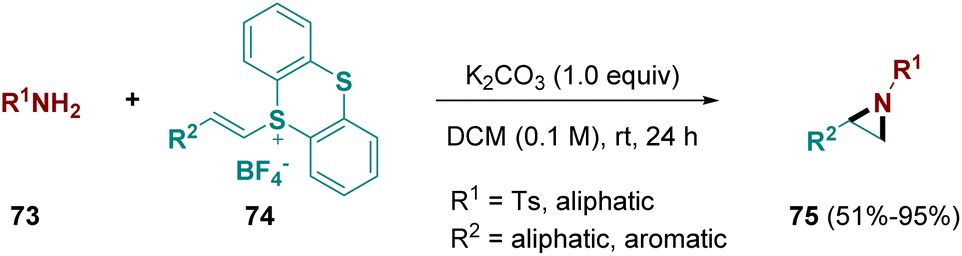 | ||
| Scheme 14 Aziridination pathway using free primary amines and alkenes substituted with thianthrenes. | ||
The chemoselective aziridination of styrenes (76) performed in the presence of hydroxylamine derivatives (77) via cobalt single-atom catalysis was reported by Tang and co-workers in 2023 (Scheme 15). The developed methodology is carried out under mild conditions and has a wide scope and high atom economy. Catalyst 78 is recyclable and not air-sensitive. Several natural products and drug-derived olefins have also been subjected to aziridination.28
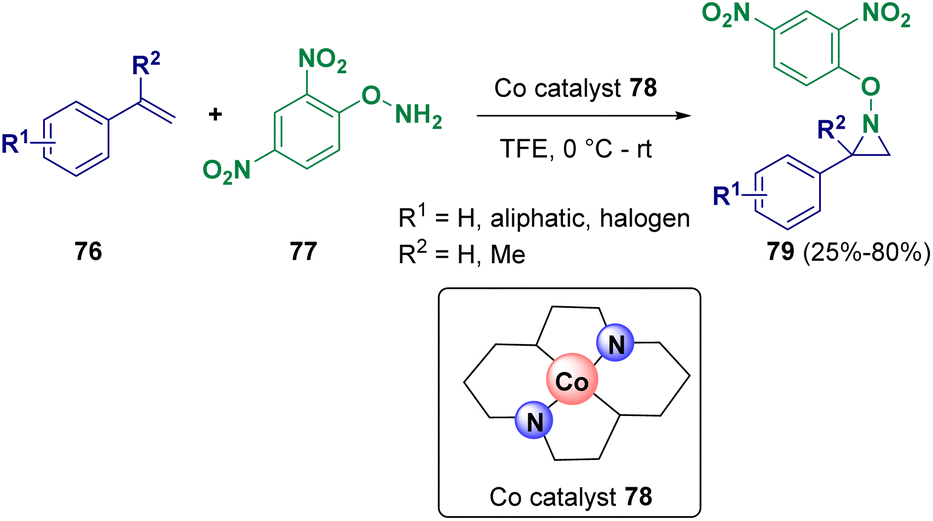 | ||
| Scheme 15 Aziridination pathway via cobalt single atom catalysis. | ||
Díez-González and co-workers reported the preparation of aziridines (82) from readily available azides (80) and alkenes (81). The reaction was carried out without any further additive by using technical solvents without the need for an inert atmosphere as the reaction can proceed in reaction vessels open to air (Scheme 16). The so-prepared aziridines (82) were then subjected to ring opening and ring enlargement reactions.29
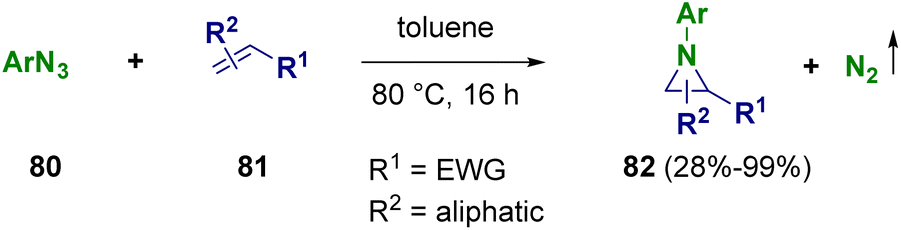 | ||
| Scheme 16 Preparation of aziridines from azides and alkenes. | ||
2.2 Aziridination of imines
Another common way for synthesizing aziridines is the addition of organometallics or other reagents to imine surrogates or amines.30 Different methodologies have been reported and some of them allowed the preparation of interesting heterocycles under even more sustainable conditions.31The presence of fluorine atoms within an organic compound can modulate its physico-chemical properties.32 Therefore, the employment of fluorinated derivatives in medicinal chemistry is very common and the development of new methodologies for the preparation of such interesting molecules is a central topic within the scientific community. In 2021, Njardarson and co-workers described the preparation of trisubstituted trifluoromethylthiolated (SCF3) aziridines (85) via the Darzens pathway. In particular, trisubstituted acetophenone nucleophiles (83) bearing SCF3 and bromine substituents at the α position undergo reactions with tosyl-protected imines (84) under mild conditions to achieve the desired aziridine derivatives (85) (Scheme 17).33
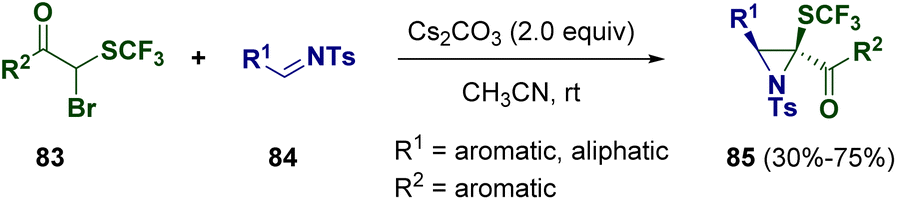 | ||
| Scheme 17 Preparation of trisubstituted trifluoromethylthiolated (SCF3) aziridines via the Darzens pathway. | ||
In 2022, Njandarson and co-workers documented a reaction for preparing aziridines (88) starting from benzothiophene 1,1-dioxide (86) and imines (87). This vinylogous aza-Darzens reaction is base dependent (Cs2CO3 is employed) and γ-selective, favouring the formation of trans-aziridines (Scheme 18).34
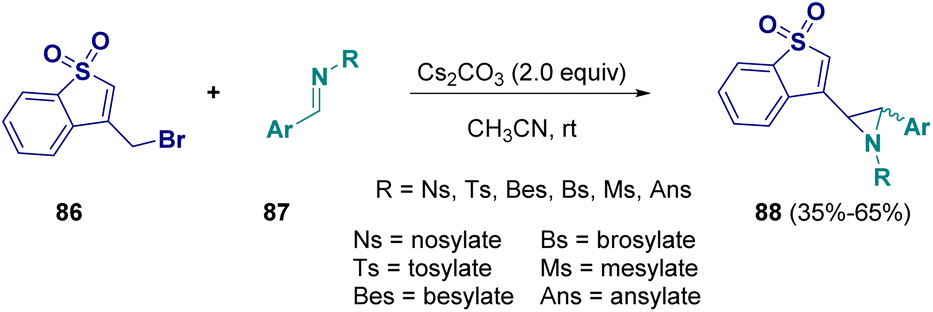 | ||
| Scheme 18 The aza-Darzens reaction for the preparation of aziridines. | ||
A telescoped reaction for the preparation of aziridines was described by Kürti and co-workers. Various electron-deficient O-sulfonyl oximes (90) were reacted with α,α-disubstituted acetophenone-derived enolates (89) to afford highly substituted aziridines (91) via an aza-quasi-Favorskii rearrangement (Scheme 19). This methodology was supported by computational studies which suggested a rearrangement pathway. The reaction of enolate 92 with imine 90 generates the N-activated β-aminoketone 93 which cyclizes to the N-activated azetidine 94. The so-formed azetidine 94 undergoes an aza-quasi-Favorskii rearrangement to yield the highly strained and substituted aziridines 91.35
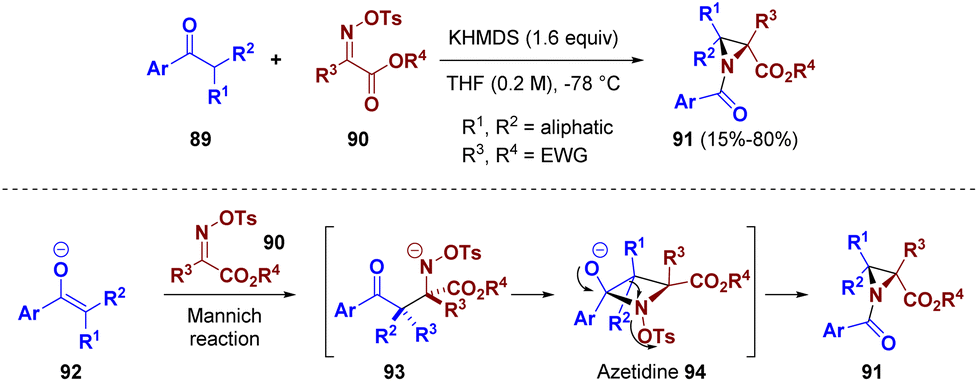 | ||
| Scheme 19 The aza-quasi-Favorskii rearrangement for the synthesis of aziridines. | ||
Pace and co-workers reported an unprecedent homologation reaction via lithium carbenoids for synthesizing mono- (96) and bis-homologated (97) trifluoromethyl-aziridine derivatives. The potency of this methodology relies on the selectivity of the reaction. Indeed, just by adjusting the stoichiometry of the employed carbenoid by using either 1.2 or 2.8 equivalents, the authors were able to selectively obtain chloro(trifluoromethyl)- (96) or chloromethyl(trifluoromethyl)aziridine derivatives (97), respectively.36 A wide scope and full chemocontrol was observed by using LiCH2Cl or LiCH2F as the homologating agent, prepared via lithium-halogen exchange starting from ICH2Cl or ICH2F and MeLi-LiBr (Scheme 20 – path a).37 Later on, the same group, in collaboration with Luisi and co-workers, described the preparation of rare α-fluoroaziridines (100) by using the unknown LiCHFI as the homologating agent. In this case, it was prepared via lithium-proton exchange (deprotonation) in the presence of the lithium amide base LiN(i-Pr)Cy and ICH2F. After the homologation of the imine derivatives (98) with LiCHFI, a series of highly functionalized β-fluoroiodoamines (99) were isolated, which were subjected to deprotonation with NaH and after ring closure the desired α-fluoroaziridines (100) were achieved (Scheme 20 – path b). Only in the case of 102, the direct formation of the aziridine ring was observed (Scheme 20 – path c).38
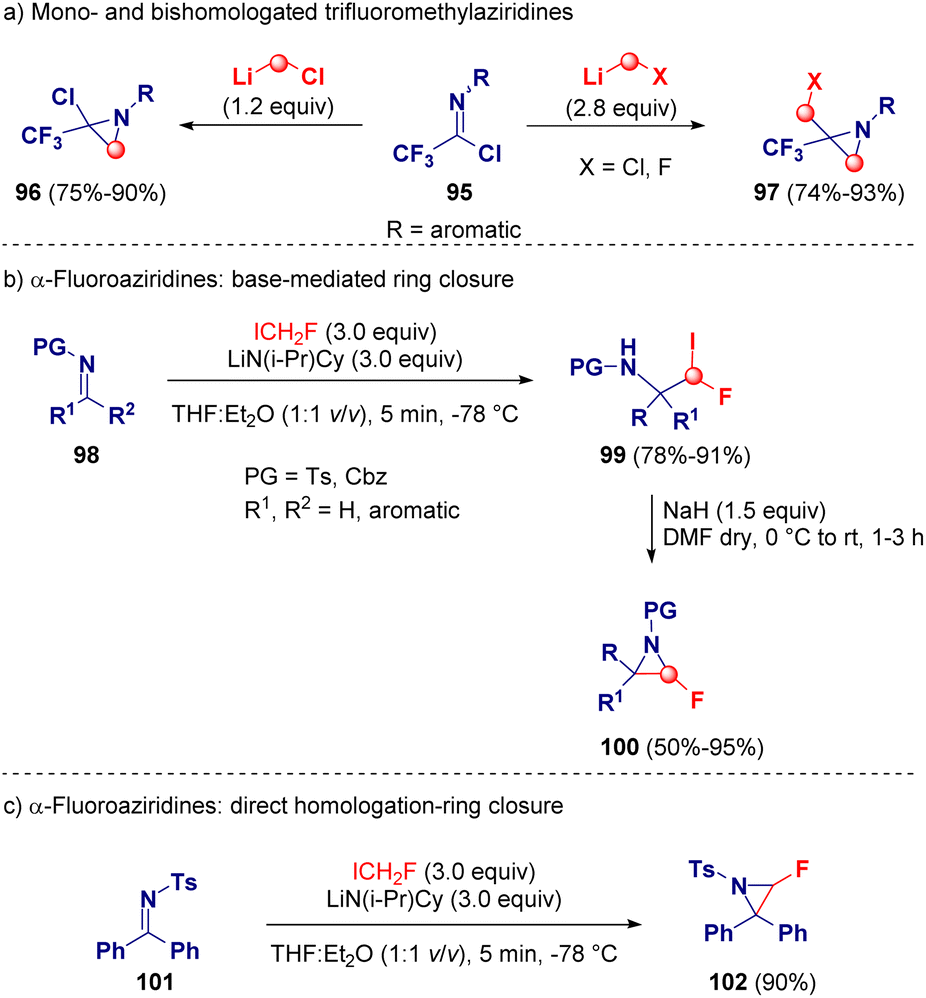 | ||
| Scheme 20 Aziridine preparation via lithium carbenoids homologations. | ||
In 2023, Aggarwal and co-workers described a two-step one-pot preparation of spirocyclic aziridines containing a cyclobutane motif. In the initial step, a bicyclo[1.1.0]butyl sulfoxide (104) is lithiated and added to a suitable imine (103). Afterward, the resulting intermediate (106) is cross-coupled with an aryl triflate (107) through a C–C σ-bond alkoxy- or aminopalladation, resulting in the related aziridine (108) formation (Scheme 21).39
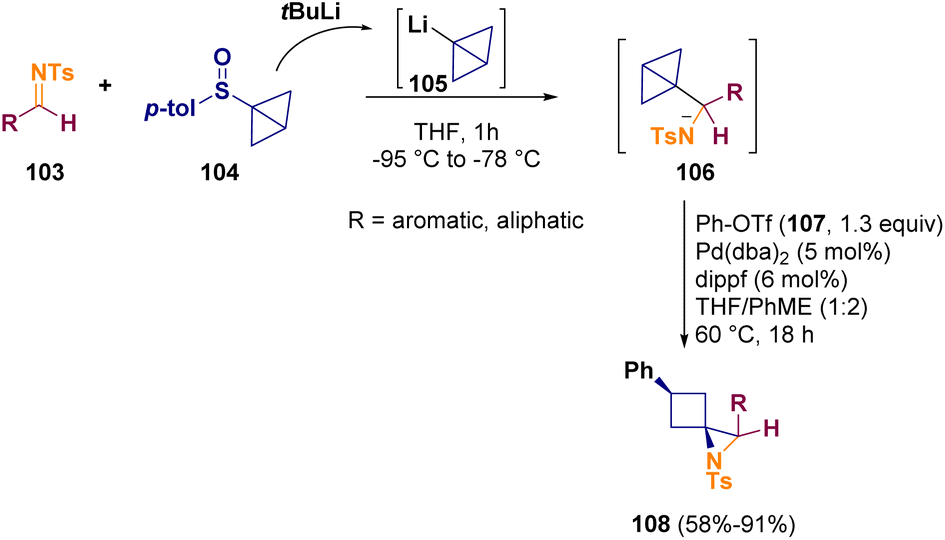 | ||
| Scheme 21 Preparation of spirocyclic aziridines containing a cyclobutane motif. | ||
2.3 Miscellaneous intramolecular aziridinations
In 2021, Luisi et al. documented a sustainable mixed flow-batch approach for preparing functionalized NH-aziridines (112) from vinyl azides (109). The first part of the procedure was conducted under continuous flow conditions using CPME (cyclopentyl methyl ether) as the green solvent for obtaining a variety of 2H-azirines (110). Afterwards, the 2H-azirine (110) solution from the microfluidic system was collected in a round bottom flask, cooled to −78 °C and reacted with various lithiated species (e.g., phenyllithium, hexyllithium, n-butyllithium, and i-butyllithium, 111), affording the desired NH-aziridines (112) (Scheme 22).40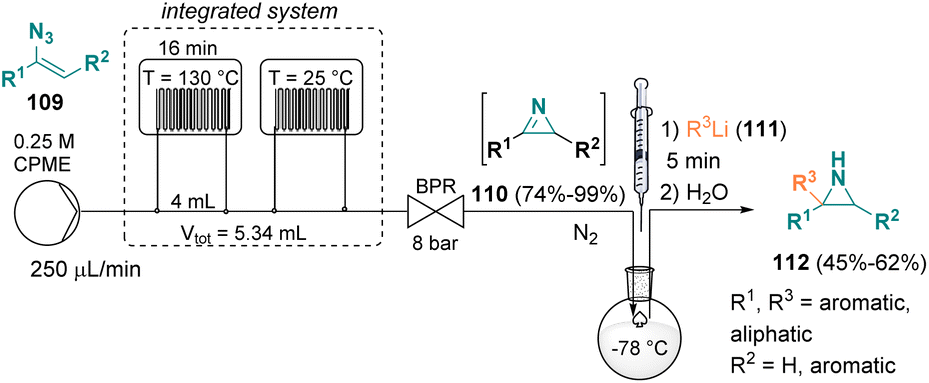 | ||
| Scheme 22 Mixed flow-batch approach for preparing functionalized NH-aziridines. | ||
Gnanaprakasam et al. described a mixed continuous flow/batch approach for the preparation of spiro-aziridines (116). In the first step of the reaction, a variety of spirooxindole 2H-azirines (114) were synthesized via intramolecular oxidative cyclization of 3-(amino(phenyl)methylene)-indolin-2-one derivatives (113) in the presence of I2 and Cs2CO3. The so-prepared spirooxindole 2H-azirines (114) were transformed into spiroaziridine derivatives (116) via the addition of Grignard reagents (115) (Scheme 23).41
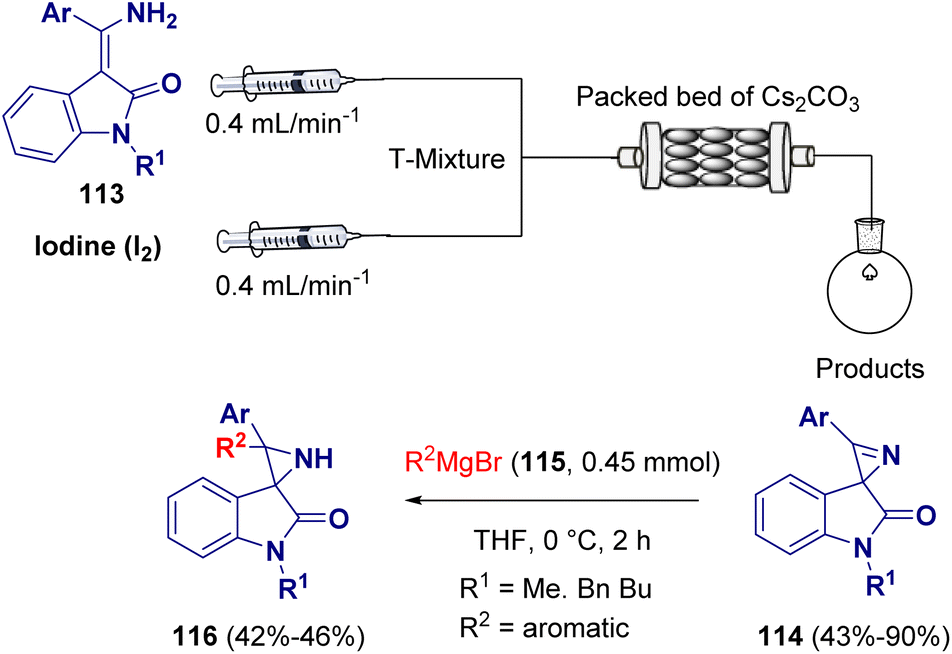 | ||
| Scheme 23 Mixed continuous flow/batch approach for preparing aziridines. | ||
In 2022, Uchiyama et al. reported the aziridination of L-Val (117) by using the non-heme iron enzyme Fe(II)/α-ketoglutarate-dependent oxygenase (118, TqaL) as catalyst, via cyclization proceeding through β-hydrogen abstraction (Scheme 24 – path a). This pathway proceeds through an unusual, diverse stereochemical route implying both retention (121) and inversion (122) of the C3(Cβ) stereocenter.42 Nishiyama et al. described the biosynthesis of the aziridine derivative vazabitide A (125) catalyzed by Vzb10/11 via sulfate elimination to give aziridine 124 (Scheme 24 – path b). Vazabitide A (125) has a similar structure to azinomycin B, which shows antitumoral activity by alkylating the DNA via aziridine ring opening. Through structural analysis, the authors were able to elucidate the biosynthetic reaction mechanism.43
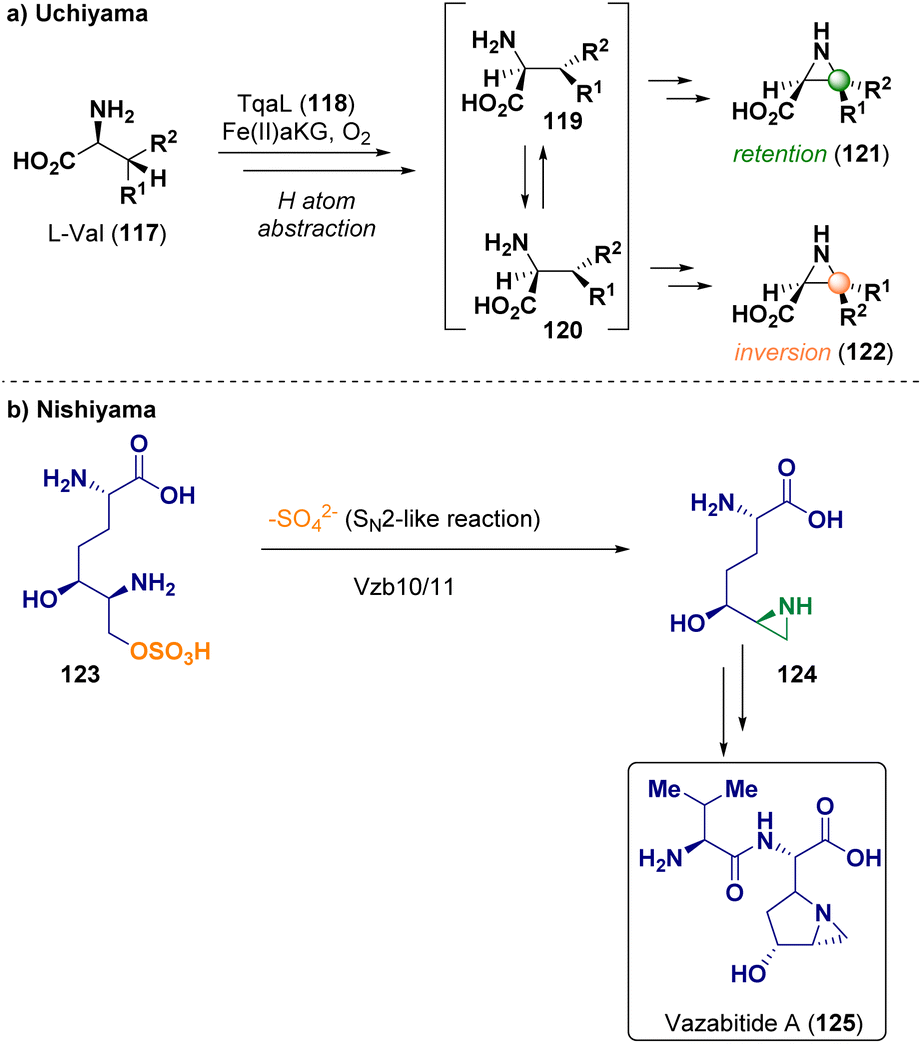 | ||
| Scheme 24 Aziridination via biocatalysis. | ||
With regard to copper-catalyzed aziridine formation, Oestreich et al. reported in 2023 the preparation of C-silylated unprotected aziridines (130) via an enantioselective copper-catalyzed (127) addition of a silicon nucleophile (129) to 3-substituted 2H-azirines (126). They employed an Si–B reagent44 and in particular, a silyl boronic ester (129) as a silicon pronucleophile (Scheme 25).45
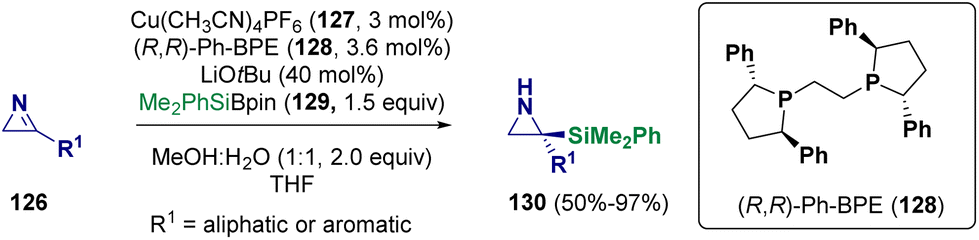 | ||
| Scheme 25 Preparation of C-silylated unprotected aziridines via an enantioselective copper-catalyzed addition. | ||
3. Reactions of aziridines
3.1 Cycloadditions
Aziridine derivatives are very important substrates and due to their high reactivity, they are largely employed in cycloaddition reactions46 for the preparation of heterocycles and molecules that are important from a biological point of view.47 Different methodologies have been applied and in recent years, more sustainable conditions have been reported for the preparation of such interesting derivatives.48Several chiral trans-substituted imidazolidines (134) were synthesized by Feng and co-workers via the enantioselective reaction of donor–acceptor aziridines (131) with N-aryl protected imines (132) using Ni(ClO4)2·6H2O/N,N′-dioxide (133) as the catalytic system (Scheme 26).49 The transformation appears to proceed through transition states such as 135, where the ring formation starts with the attack from the imine from the si-face as the re-face is blocked by the N-arylsulfonyl moiety. Ring closure occurs subsequently by the attack on the bond that is broken, forming a bond with the carbon bearing the aryl group.
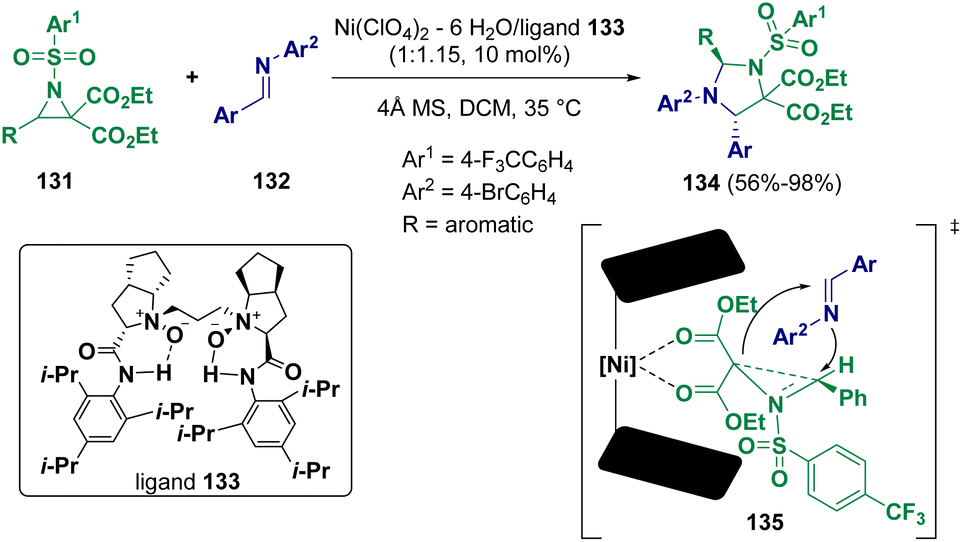 | ||
| Scheme 26 Enantioselective [3 + 2] cycloaddition of aziridines with N-aryl protected imines. | ||
Usually, the activation of aziridines requires the employment of either a strong Lewis acid or transition metals. Recently, Wang and co-workers described a cycloaddition reaction of a weakly bonded aziridine–selenide complex with non-activated alkenes (137) by using phosphonium selenide-based chalcogen bonding catalysts (138) (Scheme 27 – path a). This study was supported by computational calculation and NMR, demonstrating that an activation mode (145) involving the cooperative Se–O and Se–N interactions is involved. The scope of the methodology was extended to alkynes (141) (Scheme 27 – path b) and ketones (143) (Scheme 27 – path c).50
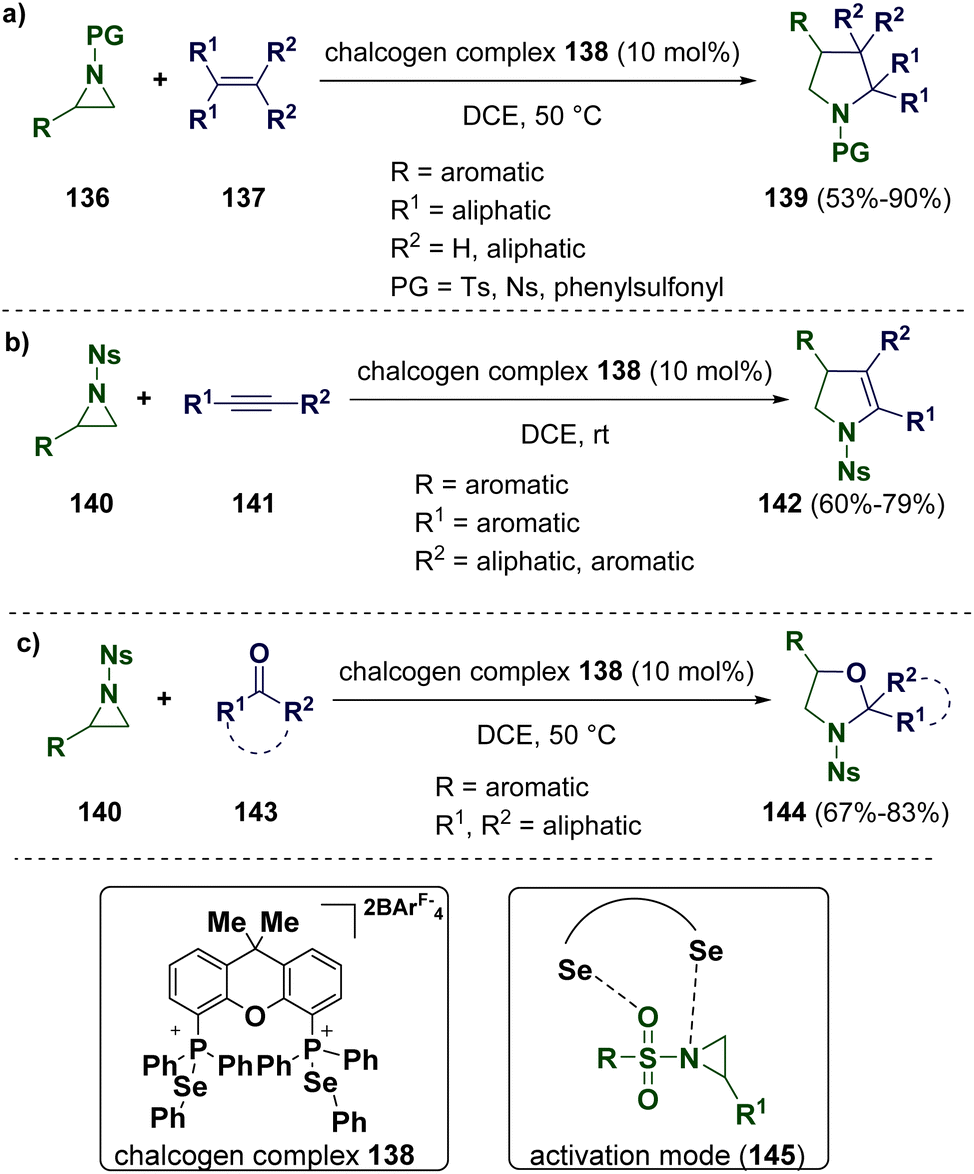 | ||
| Scheme 27 Cycloaddition of weakly bonded aziridine-selenide complexes with non-activated alkenes (a), alkynes (b), and ketones (c). | ||
3.2 Ring enlargements
Ring-size manipulation has emerged as a powerful strategy to convert readily available cyclic structures into ring-expanded or ring-contracted compounds, which are more difficult to synthesize. Many methodologies have been described for the preparation of interesting heterocycles starting from aziridines.51Ye et al. reported in 2020 the synthesis of 4-benzoxazepinones (150) via a N-heterocyclic carbene/copper co-catalyzed reaction of salicylic aldehydes (146) with aziridines (147). The applied strategy allowed the authors to obtain compounds of this class, which are also interesting from a pharmaceutical viewpoint, in good yield and with full regioselectivity (Scheme 28).52
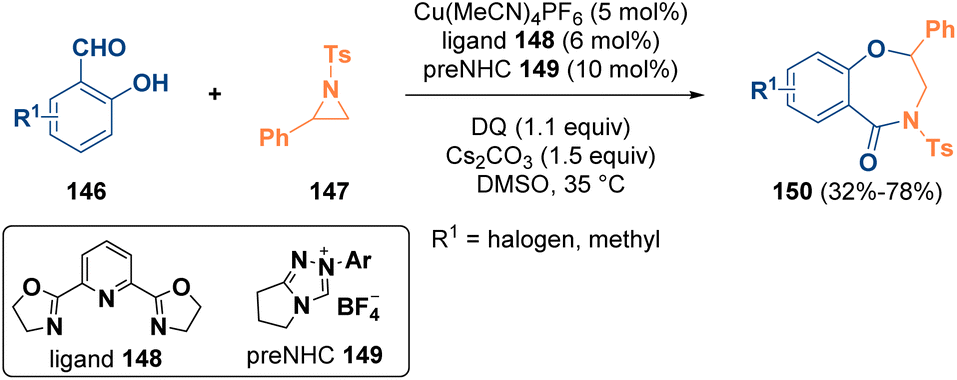 | ||
| Scheme 28 Synthesis of 4-benzoxazepinones. | ||
The spirooxindole-featuring azacycles have gained interest in the fields of synthetic as well as medicinal chemistry due to their peculiar three-dimensional architecture and interesting biological profiles. In 2022, Hajra and co-workers documented the Brønsted acid- and/or Lewis acid-catalyzed selective C3-allylation of spiro-aziridine oxindoles (151) with allyl silanes (152 and 153) or allyl Grignard reagents (154) to access 3-allyl-3-aminomethyl oxindoles (155) and 5-silyl methyl spiro[pyrrolidine-3,3′-oxindoles] (156), respectively (Scheme 29).53 When chiral spiroaziridines were used, the methodology did not show any stereoselectivity. In contrast, the catalyst-free reaction of nonracemic spiroaziridines with allyl-Grignard reagents provided 3-allyl-3-aminomethyl oxindoles (155) with good stereoselectivity (ee up to 80%). The pathway was applied for preparing coerulescine (157) and various 5′-substituted spiro[pyrrolidine-3,3′-oxindoles] (156).
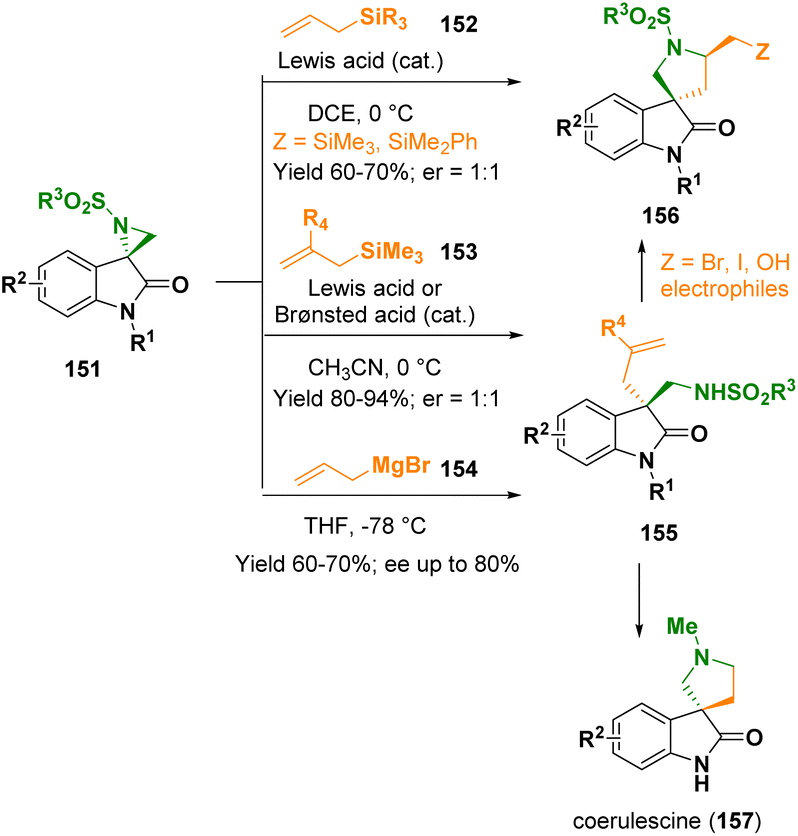 | ||
| Scheme 29 Selective C3-allylation of spiro-aziridine oxindoles with allyl silanes and allyl Grignard reagents. | ||
A ring enlargement pathway was described as a useful method for the preparation of β-lactams (160) by Li and co-workers. They reported a palladium-catalyzed ring expansion reaction of vinyl aziridines (158) with commercially available sodium 2-chloro-2,2-difluoroacetate (ClCF2COONa, 159), which serves as the carbonyl source of a difluorocarbene precursor. The authors reported a difluorocarbene-involved (161) reaction of π-allyl Pd(II) complexes (Scheme 30 – path a).54
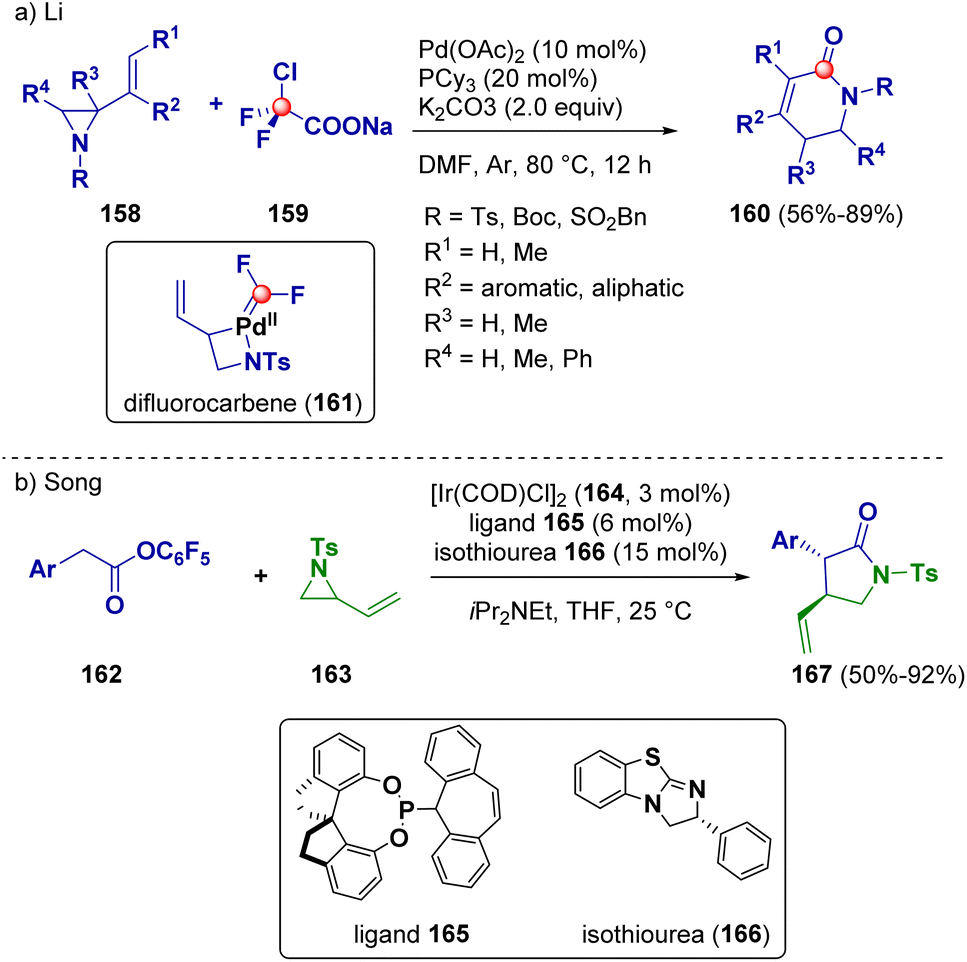 | ||
| Scheme 30 Ring enlargement pathways for preparing β- and γ-lactams. | ||
In 2023, Song et al. synthesized chiral γ-lactams (167) through an isothiourea (166, ITU)/iridium (164) co-catalyzed reaction of vinyl aziridines (163) with pentafluorophenyl esters (162). This methodology allowed the achievement of several optically active compounds in good yields and with high asymmetric induction (up to 98% ee) (Scheme 30 – path b).55
Pyridinium 1,4-zwitterionic thiolates (168) were employed by Chen and co-workers for the regioselective and stereospecific ring enlargement of aziridines (169). 3,4-Dihydro-2H-1,4-thiazines (170 and 171) were prepared via a domino SN2 ring-opening/N-Michael addition cyclization/retro-Michael addition/pyridine extrusion procedure under mild conditions without metal mediation or the need for a strong base (Scheme 31).108
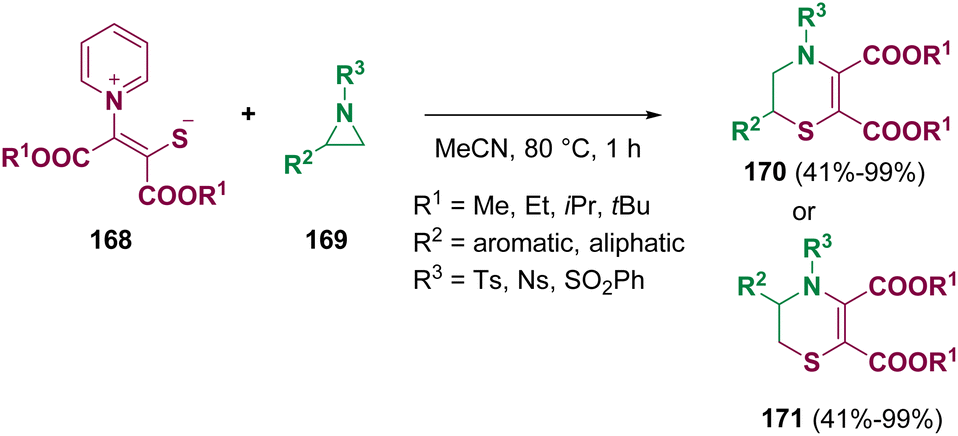 | ||
| Scheme 31 Ring enlargement of aziridines with pyridinium 1,4-zwitterionic thiolates. | ||
Unprotected guanidine derivatives (176 and 177) were prepared via ring expansion of 2-substituted aziridines (172) and N-tosyl cyanamides (173) in a domino regioselective ring-opening/5-exo-dig cyclization. This metal-free methodology works well even with weaker bases such as cesium fluoride (CsF). Furthermore, the so-obtained compounds could be subjected to hydrolysis in order to obtain highly biologically interesting urea analogs (178 and 179) (Scheme 32).56
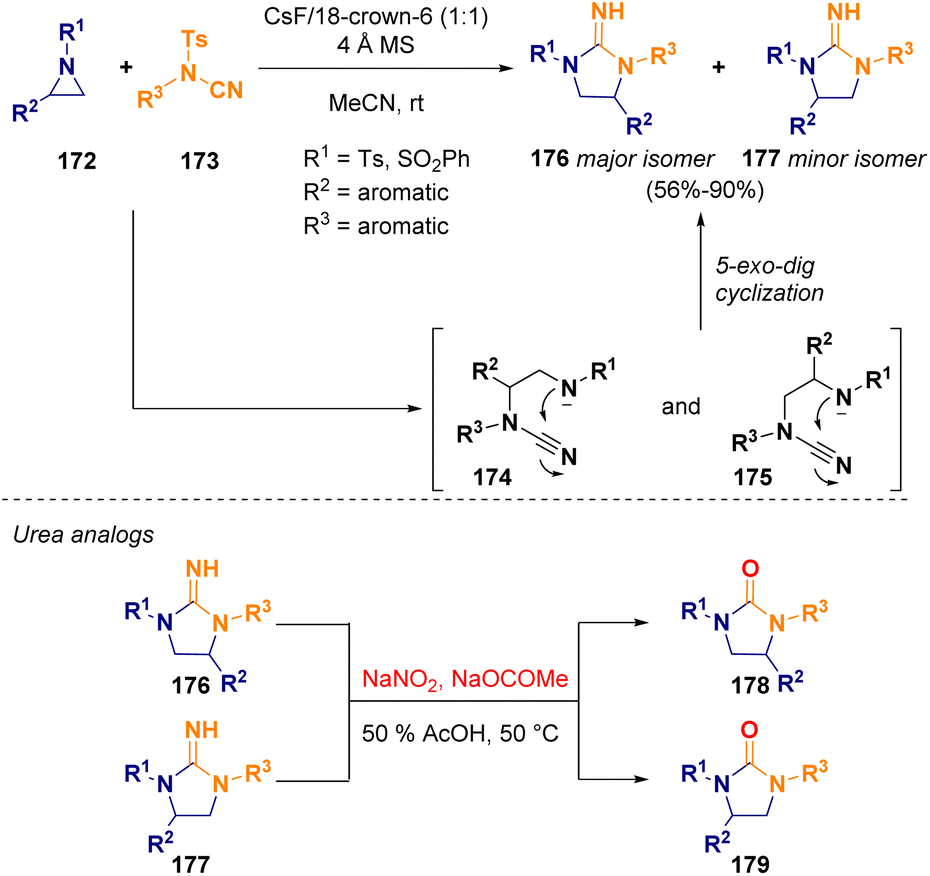 | ||
| Scheme 32 Aziridine ring enlargements for preparing unprotected guanidine derivatives. | ||
Schomaker et al. reported a [3 + 3] ring expansion of bicyclic aziridines (180) and rhodium-bound vinyl carbenes to synthesize a variety of dehydropiperidines (184). Supported by mechanistic studies, the authors supposed that the pathway proceeds via the formation of a vinyl aziridinium ylide (183) which undergoes a pseudo-[1,4]-sigmatropic rearrangement to yield heterocyclic products (184) with net retention of the configuration at the new CC bond (Scheme 33).57
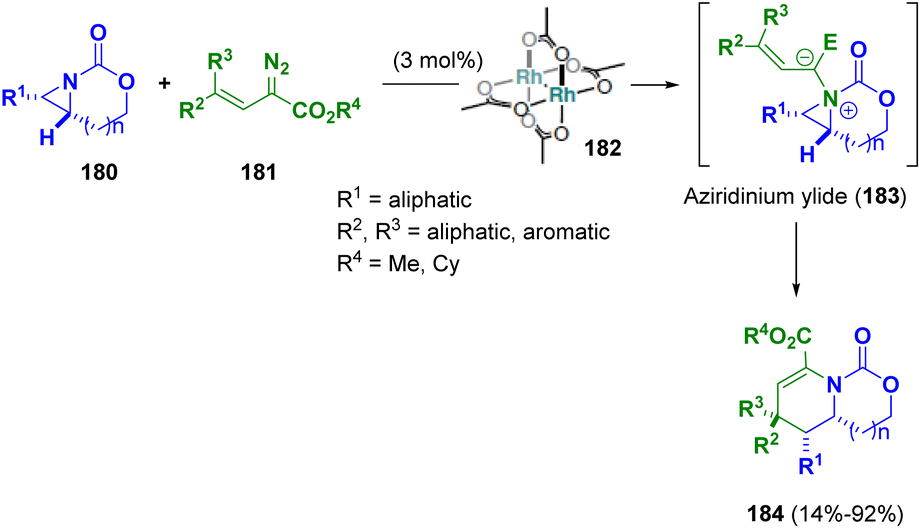 | ||
| Scheme 33 Preparation of dehydropiperidines. | ||
In 2022, Feng et al. documented the ring enlargement of racemic donor–acceptor (D–A) aziridines (185) with isocyanides (186) catalyzed by a chiral N,N-dioxide (187)/Mg(II) complex, proceeding through ring opening via intermediates, such as 188, to yield enantioenriched exo-imido azetidines (189) (Scheme 34).109
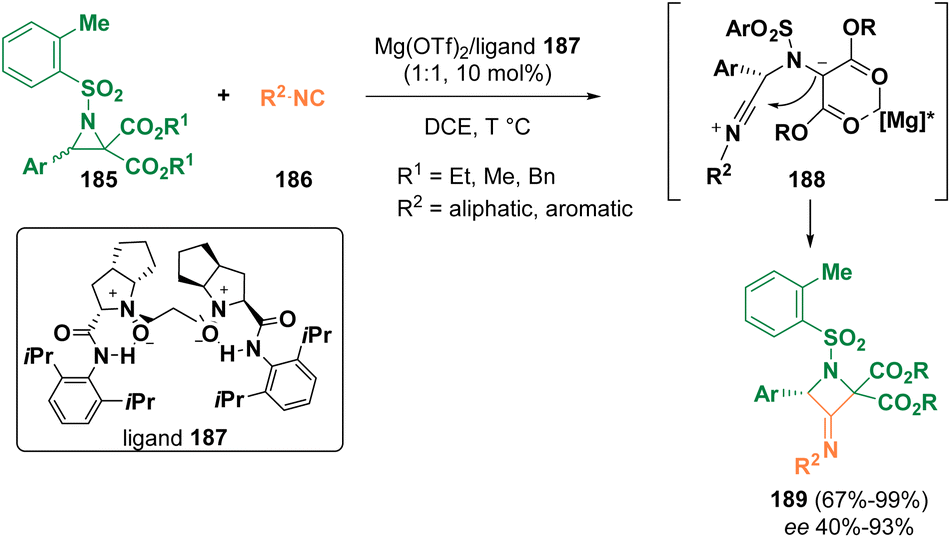 | ||
| Scheme 34 Exo-imido azetidine preparation. | ||
The preparation of 2-(2-oxoalkylidene)-1,3-oxazolidine derivatives (193) was described by Xu and co-workers in 2021 via the catalyst-free electrophilic ring expansion of N-unprotected aziridines (191) and the ketene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond of α-oxoketenes (192).58 Products 192 were prepared in situ through microwave-assisted Wolff rearrangement of 2-diazo-1,3-diketones (190) (Scheme 35). Products 193 were obtained with an E configuration via an SN1 mechanism.
O double bond of α-oxoketenes (192).58 Products 192 were prepared in situ through microwave-assisted Wolff rearrangement of 2-diazo-1,3-diketones (190) (Scheme 35). Products 193 were obtained with an E configuration via an SN1 mechanism.
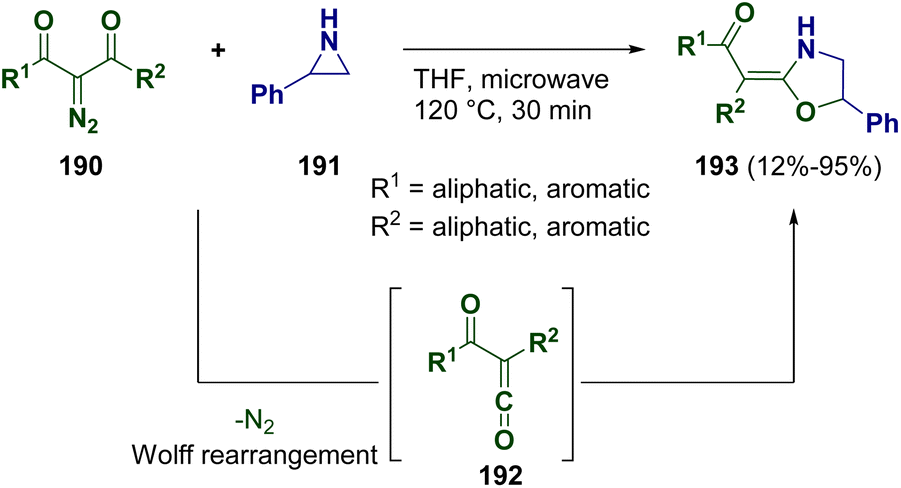 | ||
| Scheme 35 Catalyst-free ring expansion of N-unprotected aziridines for preparing 2-(2-oxoalkylidene)-1,3-oxazolidine derivatives. | ||
Alkoxycarbonylketenes (196), generated from alkyl 2-diazo-3-oxoalkanoates (194), were employed as substrates for the electrophilic ring expansion of aziridines (195) in order to obtain alkyl 2-(oxazolin-2-yl)alkanoates (197) under microwave irradiation.59 The corresponding final compounds (197) were obtained in good yields; in all cases, the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of diastereomeric products was observed (Scheme 36).
:1 mixtures of diastereomeric products was observed (Scheme 36).
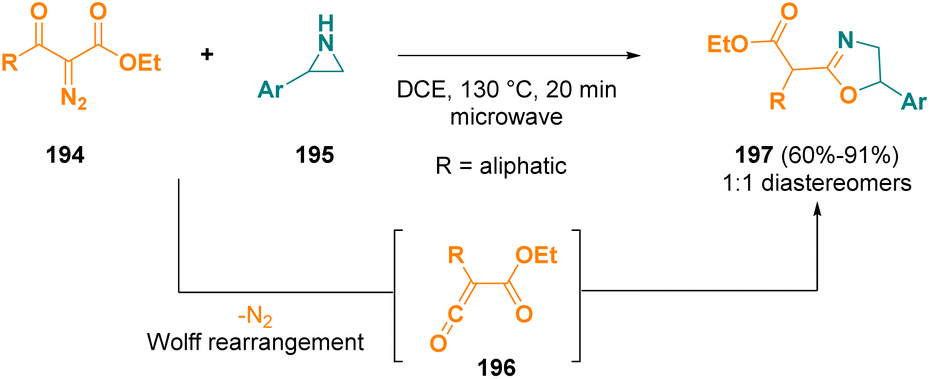 | ||
| Scheme 36 Microwave-assisted ring expansion of aziridines to obtain alkyl 2-(oxazolin-2-yl)alkanoates. | ||
In 2022, Arnold and co-workers documented the biocatalytic enantioselective one-carbon ring expansion of aziridines (198) to yield azetidines (202) via [1,2]-Stevens rearrangement by using “carbene transferase” as enzymes (Scheme 37). The employment of biocatalysts was crucial for controlling the reactivity of the formed aziridinium ylide intermediates (201), which could be not controlled by using other catalyst classes.60
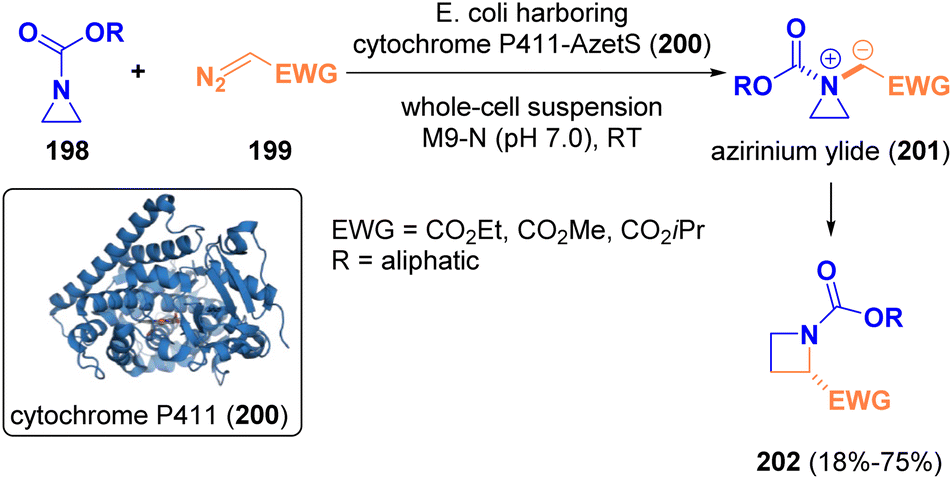 | ||
| Scheme 37 Azetidines from aziridines via ring enlargement. | ||
A typical ring enlargement reaction concerning aziridines is the one with CO2 (204).61 Indeed, different methodologies were described by using diverse catalysts.62 Among them, Caselli and co-workers reported the preparation of 5-substituted 1,3-oxazolidin-2-ones (206) catalysed by ammonium ferrates (205) (Scheme 38 – path a). Theoretical calculations clarified that the reaction mechanism involves just one ferrate molecule and the rate determining step is the 1,3-oxazolidin-2-one ring closure.63 Zhao and co-workers documented the reaction of CO2 (204) and aziridines (207) catalyzed by MOFs (metal–organic frameworks). In particular, they prepared three-dimensional cluster-based MOF {(NH2Me2)[Co3(μ3-OH)(BTB)2(H2O)]·9H2O·5DMF}n (208) with large pores assembled by BTB ligands (209, BTB = 1,3,5-tri(4-carboxyphenyl)benzene) and [Co3] clusters (Scheme 38 – path b). With this methodology they achieved oxazolidinones (210 and 211) in up to 99% yield.64 Recently, Venkatasubbaiah et al. described the solvent-free reaction of aziridines (212) and carbon dioxide (204), catalyzed by a zinc-salen having a B–N coordinated phenanthroimidazole motif (213) as a photocatalyst, for the synthesis of oxazolidinones (214) (Scheme 38 – path c).65
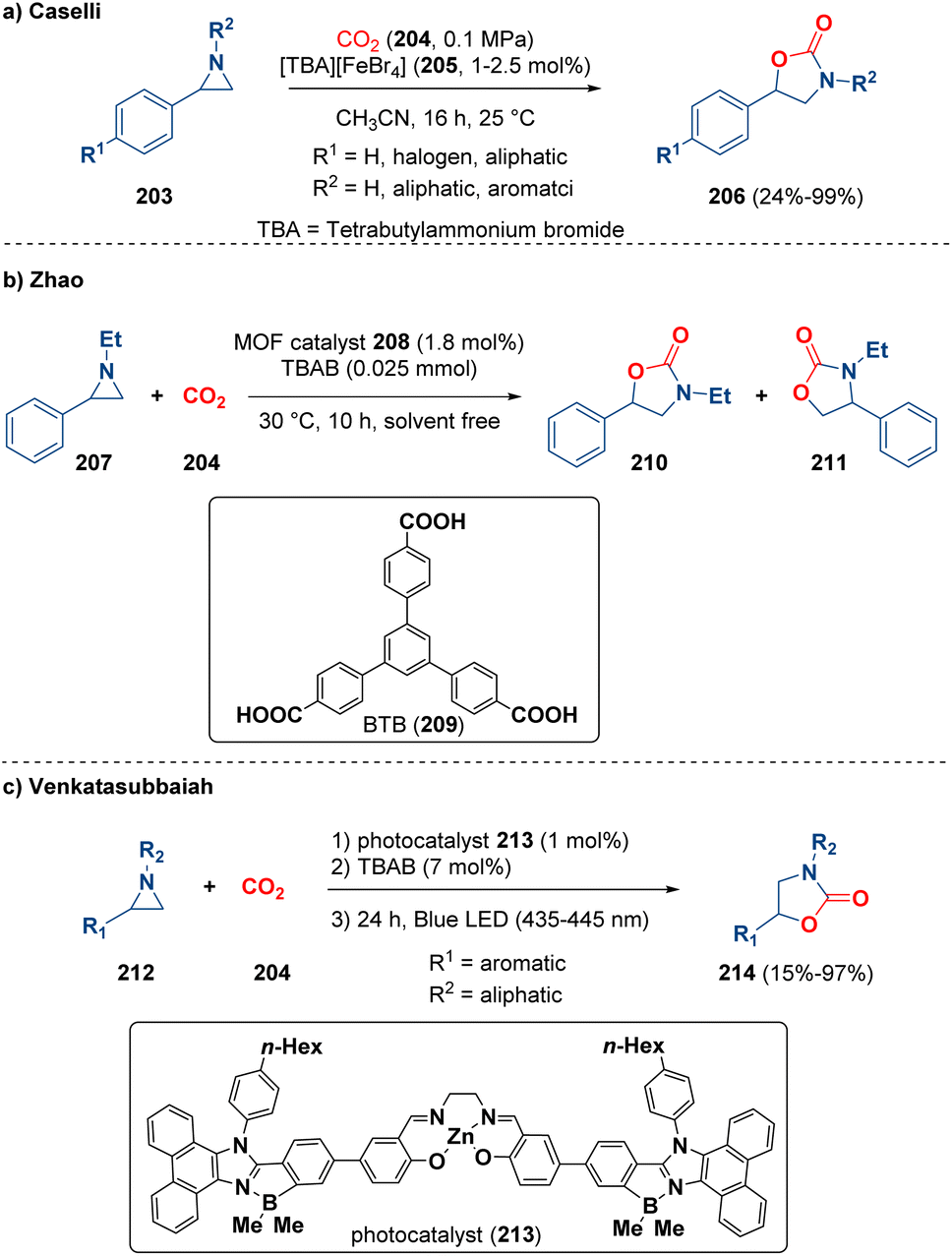 | ||
| Scheme 38 Ring enlargement by the reaction of CO2 and aziridines for preparing oxazolidinones. | ||
Schomaker et al. described a Rh catalyzed ring expansion of aziridines (215) and N-sulfonyl-1,2,3-triazoles (216). Instead of the expected dehydropiperazines (218), the authors observed the formation of [3,9]-bicyclic aziridines (217), see Scheme 39. The structure was confirmed by computational calculation and X-ray studies.66
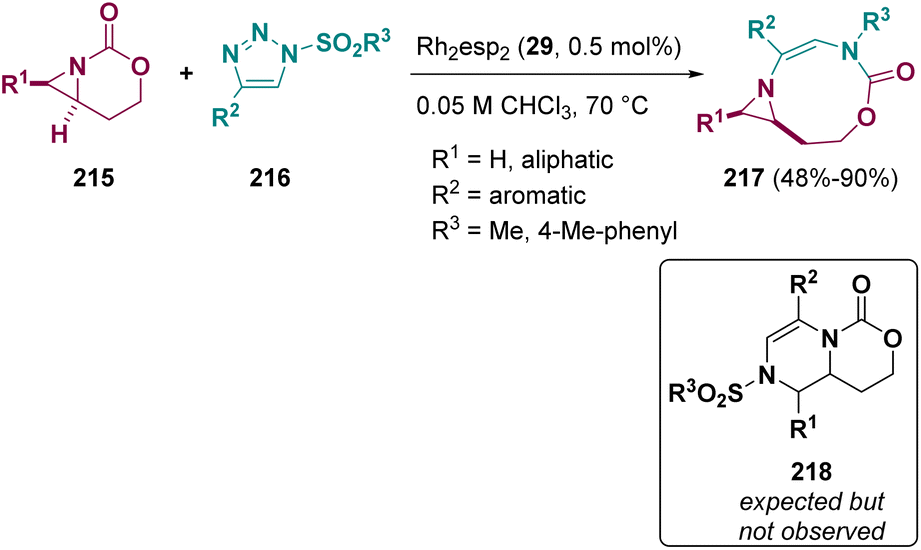 | ||
| Scheme 39 Rh catalyzed ring expansion of aziridines and N-sulfonyl-1,2,3-triazoles. | ||
A one-pot reaction for preparing benzooxepino-fused pyrrole derivatives (221) starting from substituted alkynyl aziridines (219) was reported by Sridhar and co-workers. In this metal-free procedure, two new CC bonds were established via the initial cleavage of the CC bond of the aziridine ring by the in situ generated azomethine ylides (220) (Scheme 40).67
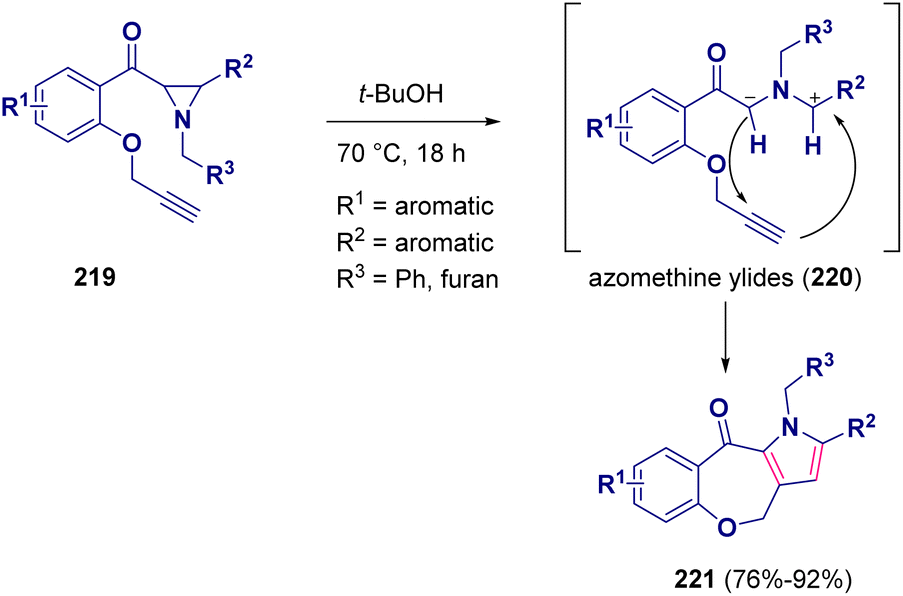 | ||
| Scheme 40 Preparation of benzooxepino-fused pyrrole derivatives. | ||
3.3 Ring openings
Ring opening reactions are amongst the most important synthetic pathways of aziridines68 as they can be transformed into many interesting compounds such as amino acids and natural products69 that are very often useful for pharmacological applications.Various N-(2,2-diphenylvinyl)-β-oxoamides (225) were prepared by Xu and co-workers, via a microwave-assisted catalyst-free methodology, starting from 2-diazo-1,3-dicarbonyl compounds (222) through an electrophilic ring opening of N-alkyl-2,2-diphenylaziridines (223).70 α-Oxoketenes (224), which then reacted with aziridines 223, were generated from 2-diazo-1,3-dicarbonyl derivatives (222) via a Wolff rearrangement (Scheme 41). The so-obtained N-(2,2-diphenylvinyl)-β-oxoamides (225) are useful molecules as synthons for the preparation of β-lactams but also common structural motifs in biologically active compounds.
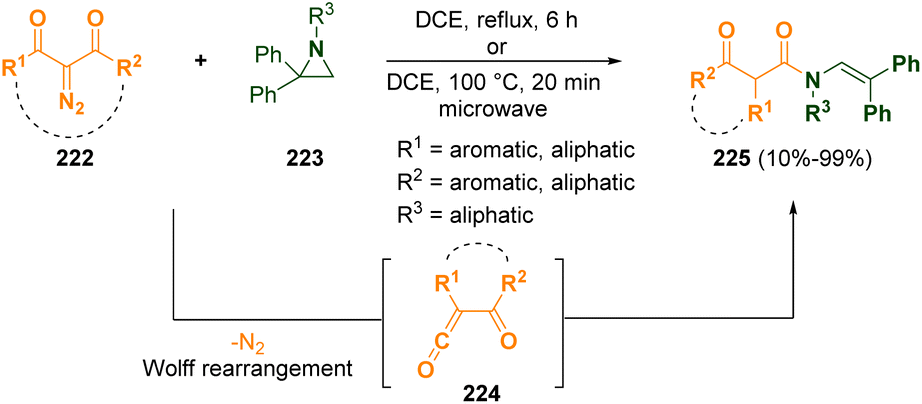 | ||
| Scheme 41 N-(2,2-Diphenylvinyl)-β-oxoamide generation from 2-diazo-1,3-dicarbonyl compounds and N-alkyl-2,2-diphenylaziridines. | ||
Zhu and co-workers reported the synthesis of 4-spiroannulated tetrahydroisoquinolines (228) via a multistep procedure involving a sequential ring opening of aziridines 226 and the subsequent Pictet–Spengler reaction.71 The reaction proceeds under mild conditions with a broad scope. Considering the reaction mechanism, the TBS group is eliminated from derivative 229 by treatment with TBAF and the so-obtained free OH group (230) promoted an internal aziridine ring opening. The newly formed intermediate 231 reacted with formaldehyde (227) under acidic conditions to generate the iminium ion 232, which undergoes electrophilic cyclization to afford the final product 233 (Scheme 42).
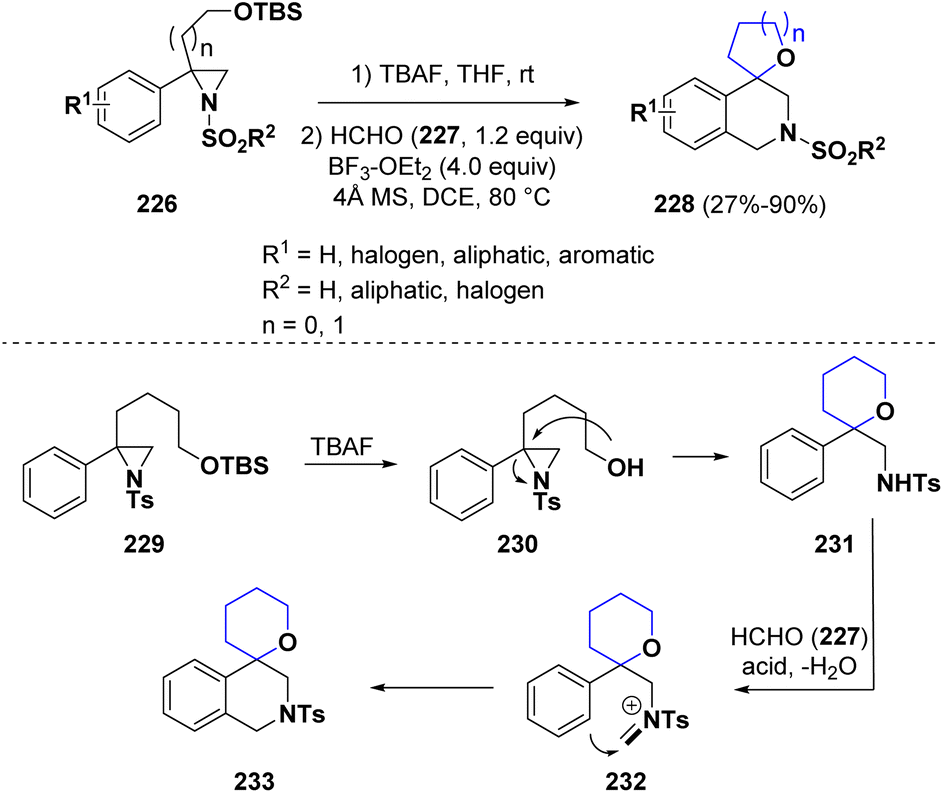 | ||
| Scheme 42 Synthesis of 4-spiroannulated tetrahydroisoquinolines via a sequential ring opening of aziridines and the Pictet–Spengler reaction. | ||
C-Glycosyl-aminoethyl sulfide derivatives (246–249) were synthesized by Zawisza et al. via a reaction between tributyltin derivatives of glycals (234–235) and aziridinecarbaldehyde (236) and the regioselective ring opening of chiral aziridines (241–244) with thiophenol (245) (Scheme 43).72 Glycoconjugates are very interesting derivatives due to their participation in many important biochemical processes.
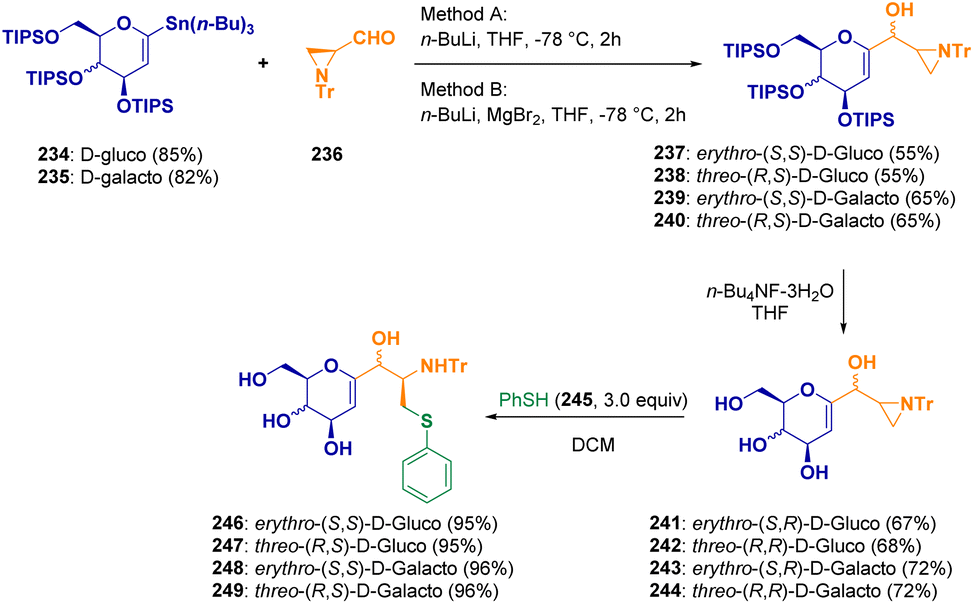 | ||
| Scheme 43 Preparation of C-glycosyl-aminoethyl sulfide derivatives. | ||
In 2022, Tang et al. described a methodology for the preparation of β-trifluoromethoxylated amines (252). This fascinating class of compounds was achieved via a silver-catalyzed ring opening of tosyl-aziridines (250) in the presence of trifluoromethyl arylsulfonate (251) (Scheme 44 – path a). A good chemo- and regioselectivity was observed under mild conditions.110 The same group accessed a series of β-trifluoromethylthiolated isothiocyanates (253) and amines (254) by using AgSCF3 and different iodine sources (TBAI, KI) (Scheme 44 – path b).73
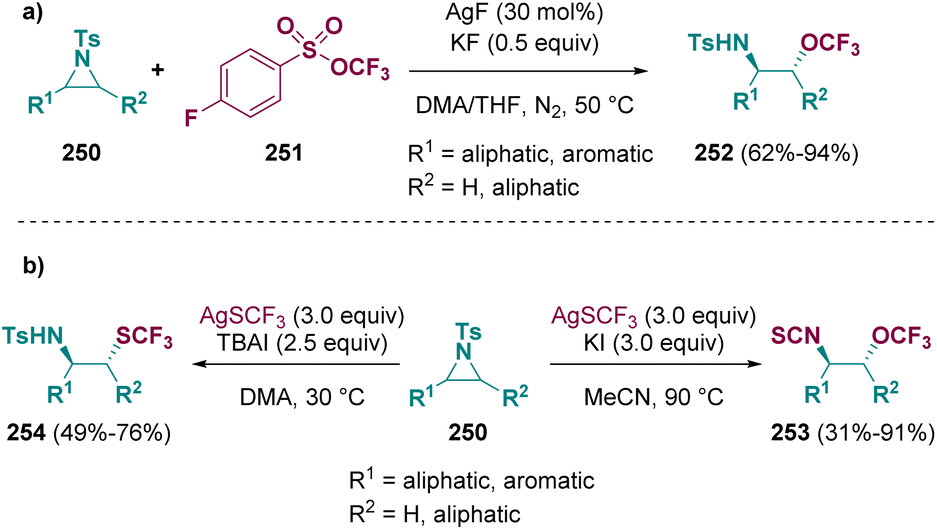 | ||
| Scheme 44 Silver-catalyzed aziridine ring opening. | ||
In 2022, Doyle and co-workers reported an aziridine ring opening procedure with methyl/1°/2° aliphatic alcohols activated as benzaldehyde dialkyl acetals (256) via a Ni/photoredox cross-coupling.74 The activation of the benzaldehyde dialkyl acetal (256) is carried out through hydrogen atom abstraction and β-scission via a bromine radical. The authors demonstrated that aziridine activation proceeds through oxidative addition to Ni(I) rather than a Ni(II)azametallacycle, as reported previously (Scheme 45).75
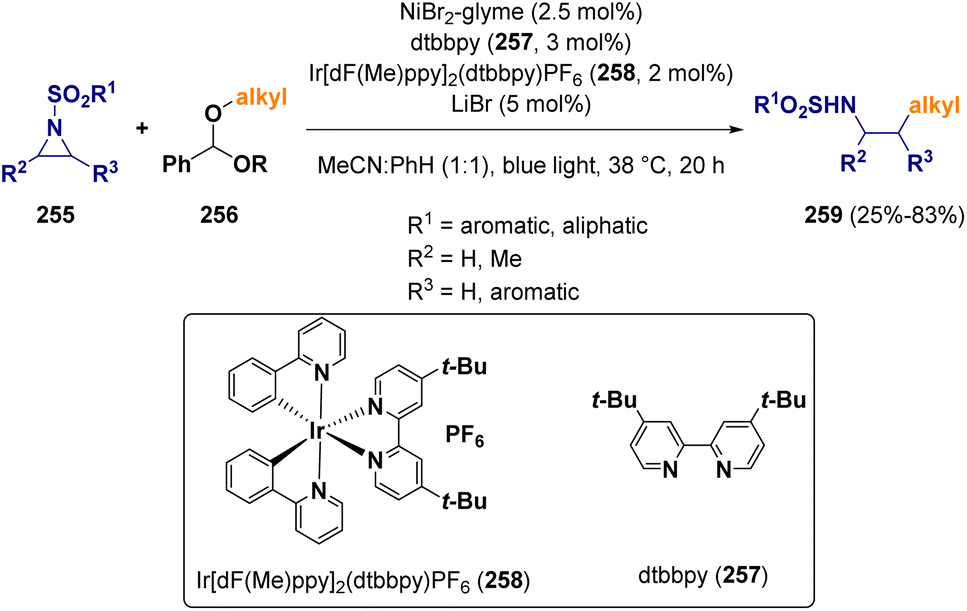 | ||
| Scheme 45 Aziridines ring opening via a Ni/photoredox cross-coupling. | ||
Palladium is the catalyst employed by Zhou and co-workers in a three-component Catellani reaction76 starting from aryl iodides (260), typically for Catellani reactions with substituents at the ortho-position, aziridines (261), and (triisopropylsilyl)acetylene (262) as the building blocks. This first step of the reaction was useful for the preparation of the 2′-alkynylaryl-2-ethylamines (265), which were further reacted via a multistep procedure for preparing 1,3-trans-disubstituted tetrahydroisoquinolines 266 (THIQ), which is important from a biological point of view (Scheme 46).77
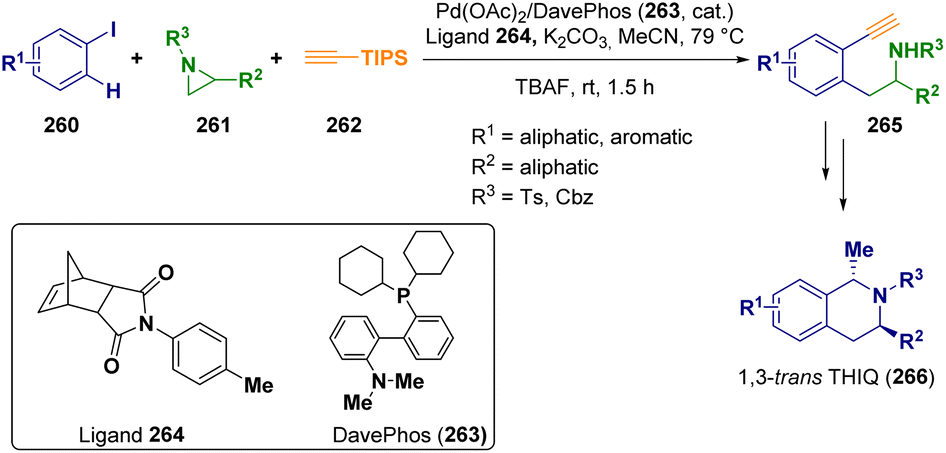 | ||
| Scheme 46 Synthesis of 1,3-trans-disubstituted tetrahydroisoquinolines. | ||
Han et al. instead documented the copper-catalyzed regioselective and stereospecific ring opening of aziridines with pyrydinyl Grignard nucleophiles. With this methodology, the preparation of β-pyridylethylamines (269) was achieved, which are potential scaffolds for the synthesis of biologically active molecules. Challenging chiral dihydroazaindoles (270) were prepared via a mild one-pot aziridine opening followed by nucleophilic cyclization (Scheme 47).78
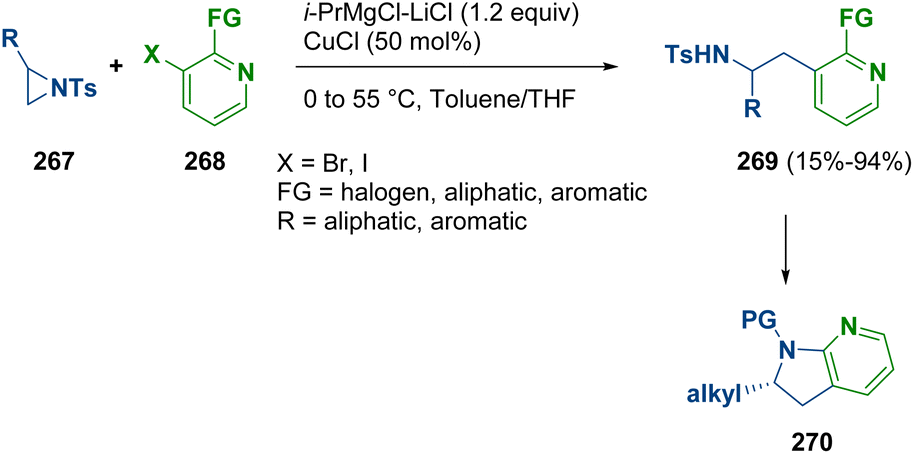 | ||
| Scheme 47 Aziridine ring opening with pyridyl Grignard nucleophiles. | ||
Hayla and co-workers described a catalyst-free regioselective ring-opening of aziridines (274), including spiroaziridine oxindoles (271), with commercially available 50% aqueous hydrogen peroxide (272). The reaction, which can be carried out without any additional organic solvents and reagents, gives rise to secondary benzylic β-hydroperoxy amines (275) and tertiary 3-hydroperoxy oxindoles (273) (Scheme 48).79 The importance of this methodology relies on the utility of organic hydroperoxides, which are prevalent motifs in various biosynthetic intermediates, natural products, and bioactive compounds.80
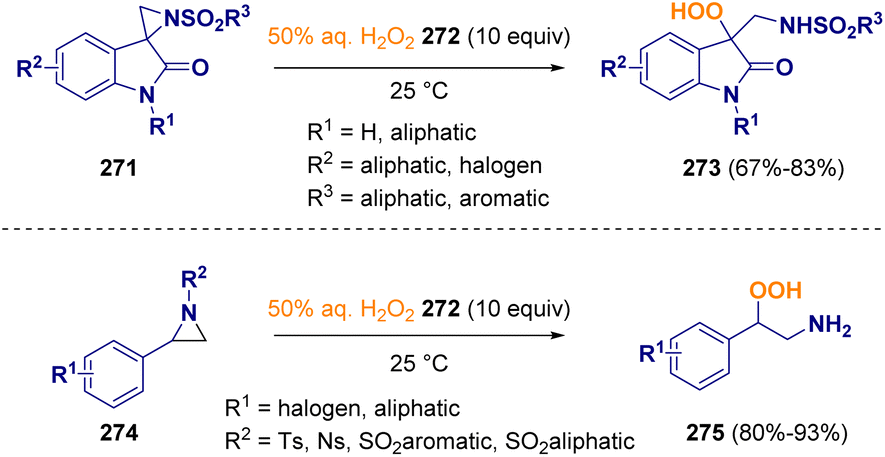 | ||
| Scheme 48 Aziridine ring opening by using 50% aqueous H2O2. | ||
Hou et al. reported the ring opening of diastereomerically pure 2-oxazolidinone-fused aziridines (276) via fluoride anions. This methodology allows the preparation of optically active, primary, secondary, and tertiary organofluorides (277), which are precursors of interesting compounds such as fluorinated amino acids (Scheme 49).81
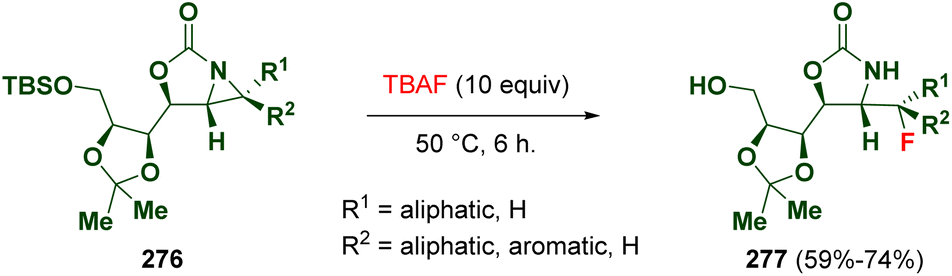 | ||
| Scheme 49 Ring opening of 2-oxazolidinone-fused aziridines via a fluoride anion. | ||
3.4 Catalysis
Most drugs are small molecules featuring heterocycles; therefore, they are ubiquitous in medicinal chemistry. Among them, indole derivatives are common scaffolds used for the design of plausible “drug-like” compounds. For these reasons, scientists around the world have put in efforts for the development of novel methodologies that can be useful for preparing and modifying these heterocyclic compounds. Koley and co-workers reported in 2022 a palladium catalyzed cross-coupling of aliphatic aziridines (279 and 282) with indoles (278), indolines, tetrahydroquinolines, and anilines (281) for preparing β-arylethylamine derivatives (280 and 283) with a wide scope (Scheme 50).82 These transformations proceed via CH activation.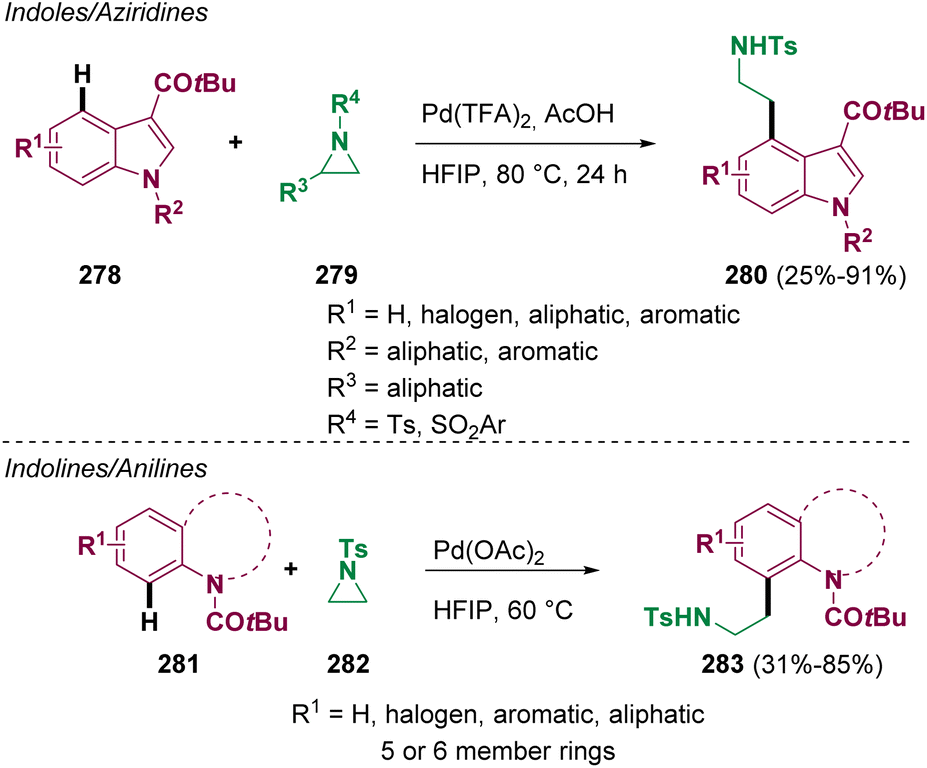 | ||
| Scheme 50 Preparation of β-arylethylamine derivatives. | ||
Another important aspect is the preparation of amino acid derivatives from aziridine substrates. In recent years, the synthesis of functional quaternary amino acid derivatives has attracted considerable attention since they are key components in many active pharmaceutical ingredients. Among them, in 2022, Gao and co-workers reported the preparation of functionalized precursors of quaternary allylic amino acids(287) via a palladium-catalyzed allylic alkylation reaction of azalactones (284) with vinyl aziridine (285) (Scheme 51).83
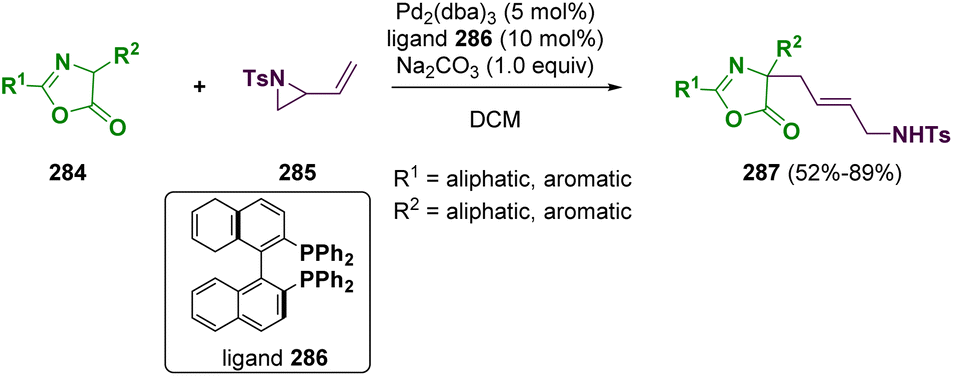 | ||
| Scheme 51 Preparation of functionalized quaternary allylic amino acid precursors. | ||
The preparation of chiral pyridine-oxazolines (290) starting from (meso)-N-(2-picolinoyl)-aziridines (288) catalyzed by a chiral ytterbium(III)-N,N′-dioxide (289) complex was reported by Liu and Feng in 2022. The reaction proceeds via an asymmetric Heine reaction, giving excellent yields with very good enantioselectivities (Scheme 52).84
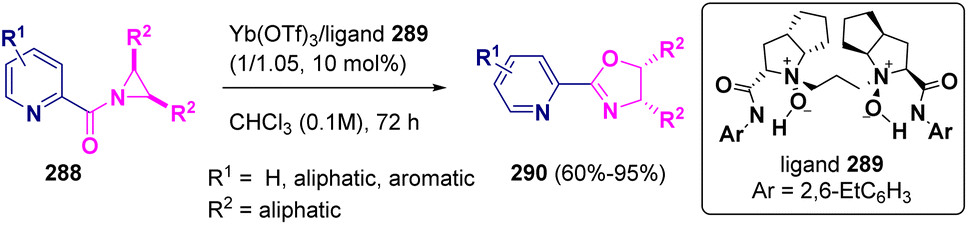 | ||
| Scheme 52 Ytterbium catalyzed synthesis of chiral pyridine-oxazolines. | ||
Martin and co-workers reported a nickel-catalyzed reductive carboxylation of N-substituted aziridines (291) with CO2 (204) at atmospheric pressure for accessing β-amino acids (293) (Scheme 53 – path a). The procedure works under mild conditions and exhibits high chemo- and regioselectivity.85 Wang et al. applied a combined nickel/photoco-catalyzed hydrogen-atom-transfer in order to achieve the ring opening of N-tosyl styrenyl aziridines (295) with aldehydes (294). This method is a novel and atom-economical synthetic path towards a variety of β-amino ketones (297) with complete regiocontrol (Scheme 53 – path b). With this strategy, the difficult coupling between aldehydes and aziridines, which are both electrophilic species, can be facilitated.86
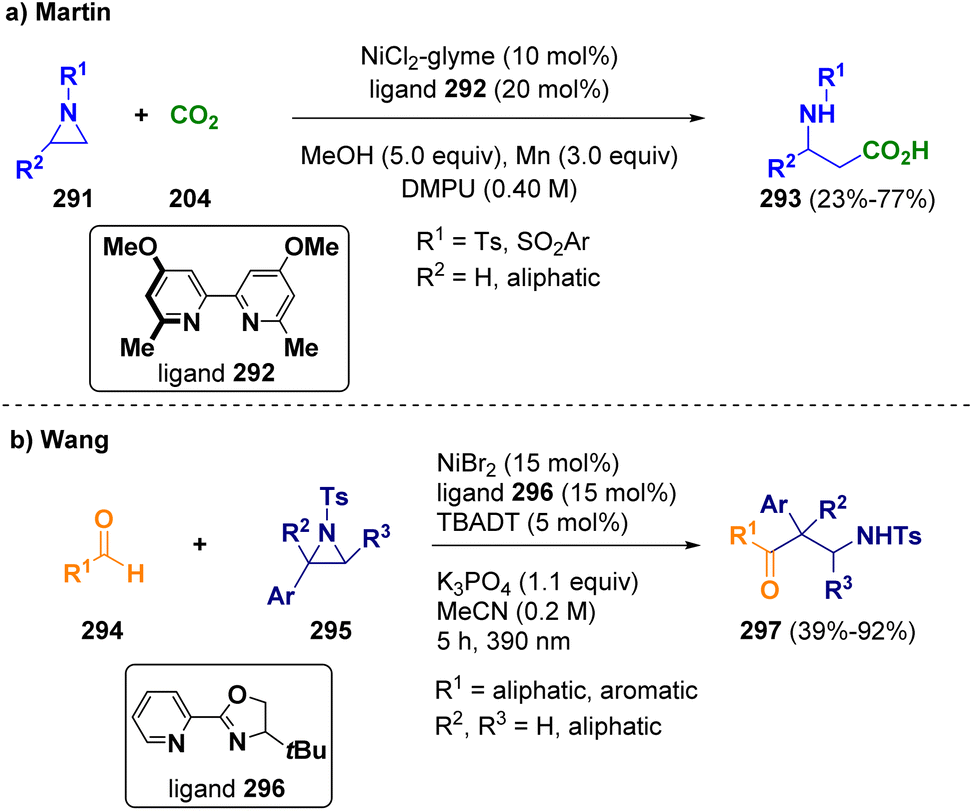 | ||
| Scheme 53 Nickel-catalyzed preparation of β-amino acids. | ||
Anderson and co-workers described the preparation of 1,3-disubstituted bicyclo[1.1.1]pentylamines (302, BCPAs) based on a radical functionalization strategy. In particular, sulfonamidyl radicals, obtained via α-iodoaziridine (298) fragmentation, undergo initial addition with [1.1.1]propellane (299) to afford iodo-BCPAs (300). Afterwards, the so-formed CI bond is functionalized via a silyl-mediated Giese reaction (Scheme 54 – path a).87 Xu et al. reported a radical addition/elimination strategy for preparing fluorinated allenes (305) starting from fluoroalkyl halides (304 and 306) and alkynyl aziridines (303) under visible-light irradiation (Scheme 54 – path b).88
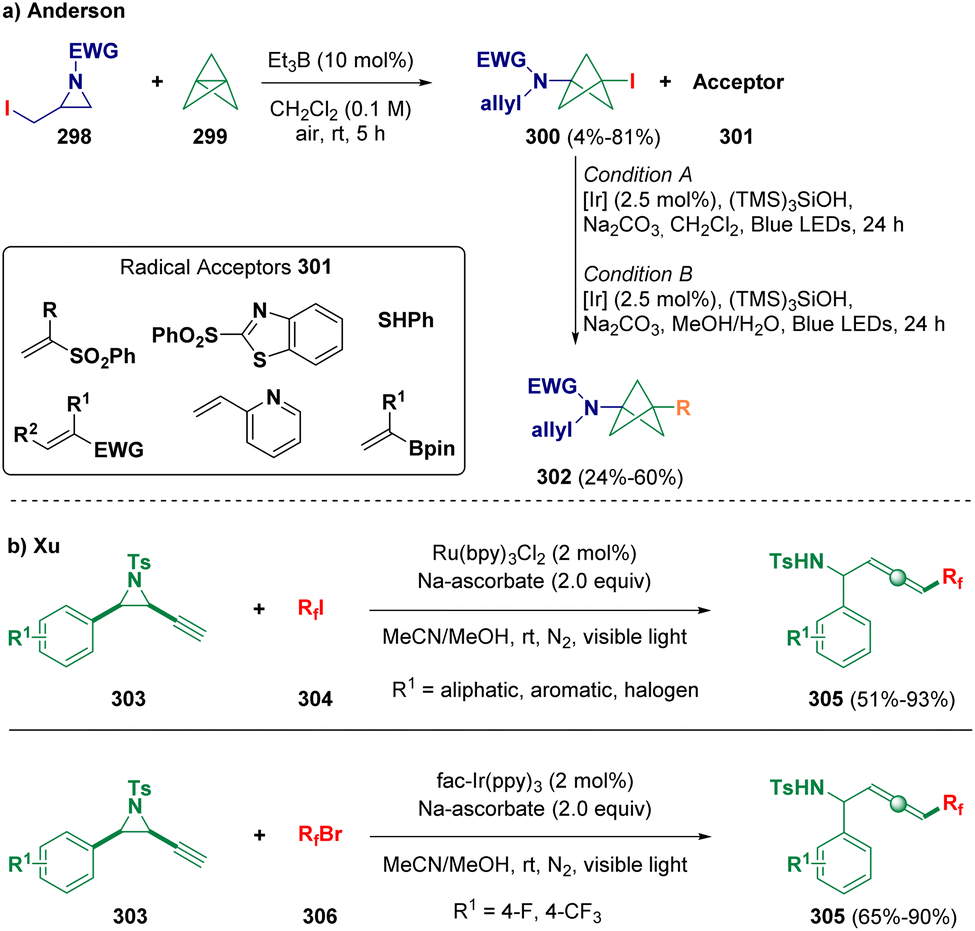 | ||
| Scheme 54 Preparation of 1,3-disubstituted bicyclo[1.1.1]pentylamines. | ||
Recently, the ring formation by the reaction of tosyl-aziridines (308) and indole derivatives (307) via [Cp*RhCl2]2 catalysis (309) was described by Zhu and co-workers. A series of cis-1,4-disubstitued tetrahydro-γ-carbolines (312) was formed in high yields and excellent cis-diastereoselectivity under mild conditions. The authors postulated a stepwise mechanism, proceeding through the rhodacyclic intermediate 310, which undergoes ring opening upon coordination of silver phosphate with nitrogen. The resulting intermediate 311 undergoes Michael addition to form a six-membered ring (Scheme 55).89 According to the authors, only the formations of the cis-1,4-diastereomers was observed.
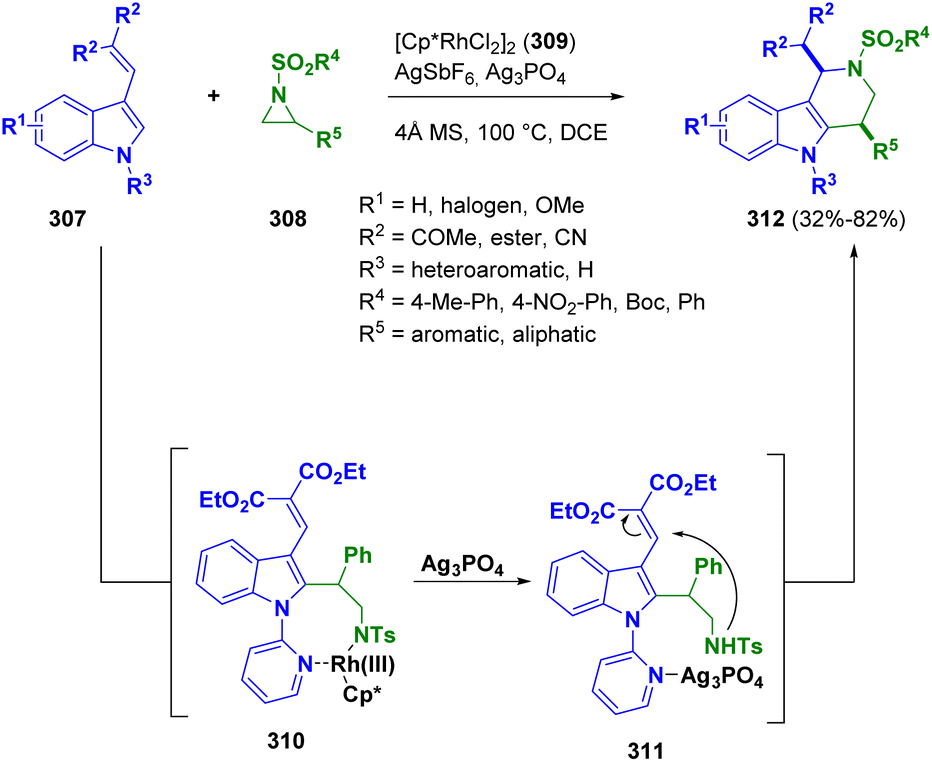 | ||
| Scheme 55 Preparation of cis-1,4-disubstitued tetrahydro-γ-carbolines from aziridines and 3-vinyl indoles substituted with EWGs. | ||
4. Aziridines as reaction intermediates and catalysts
Aziridines are very interesting reactions intermediates. Due to the ring strain, they are highly reactive and largely employed in multi-step reactions. Aziridines are useful for the preparation of fascinating organic compounds as well as for the synthesis of biologically active molecules.90In 2021, Pelletier et al. documented the synthesis of imidazo-[1,2-a]pyridines (317) by using 2-chloropyridines (313) and 2H-azirines (314) in the presence of triflic anhydride (315, Tf2O). An electrophilic 1-trifloyl-aziridin-2-yl triflate species is formed as the reaction intermediate which reacts in situ with the 2-halopyridines, forming transient pyridinium salts (316). These salts were treated in the same pot with triethylamine (Et3N), leading to the selective formation of the desired compounds (317) (Scheme 56).91
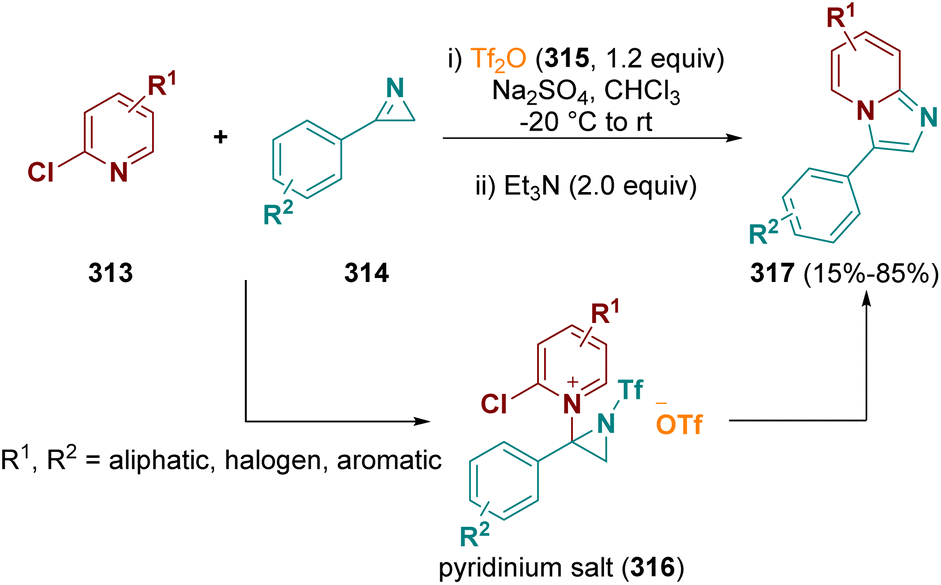 | ||
| Scheme 56 Aziridines used as intermediates. | ||
Another interesting example that highlights the use of aziridines as intermediates was reported in 2022 by Powers and co-workers. The authors described an olefin aziridination procedure by employing N-aminopyridinium reagents (319) as the activating group to afford N-pyridinium aziridines (320). The so-formed aziridines (320) then were subjected to a nickel-catalyzed C–N cross-coupling reaction with aryl boronic acids (321). The N-pyridinium aziridine intermediates (320) also participate in ring-opening chemistry with different nucleophiles to achieve 1,2-aminofunctionalization products. Mechanistic studies denote that the aziridine cross-coupling proceeds via a noncanonical mechanism involving initial aziridine opening (322), promoted by the bromide counterion of the nickel catalyst, and finally the formation of the aziridine moiety (323) by a ring-closing step (Scheme 57).92
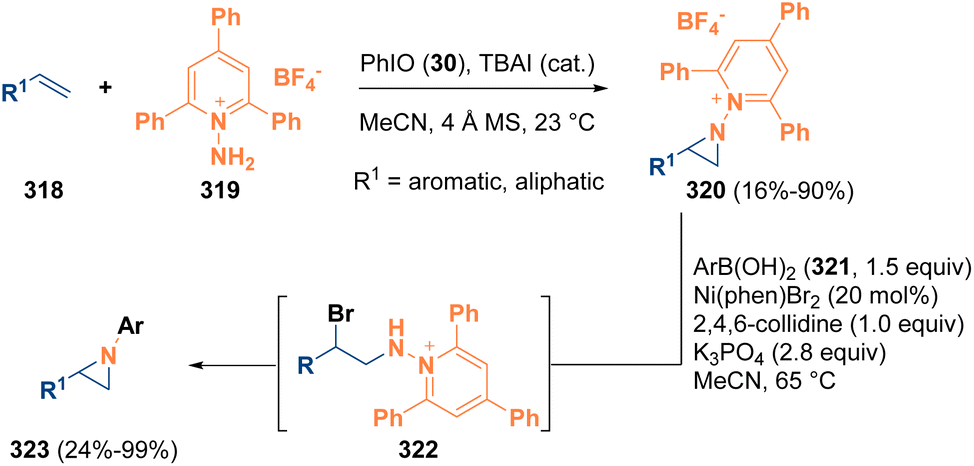 | ||
| Scheme 57 Aziridinations of alkenes and nickel-catalyzed cross-coupling. | ||
In 2023, Bower and co-workers reported the preparation of stereochemically complex polyheterocyclic ring systems (327) via an aziridine intermediate (326). In particular, an intramolecular stereospecific aza-Prilezhaev aziridination occurs which is followed by a CN bond cleavage operated by a pendant nucleophile (Scheme 58). With this methodology, a variety of alkene anti-1,2-difunctionalizations (i.e., diaminations, amino-oxygenations and amino-arylations) can be facilitated. The majority of the obtained structures are relevant in medicinal chemistry.93
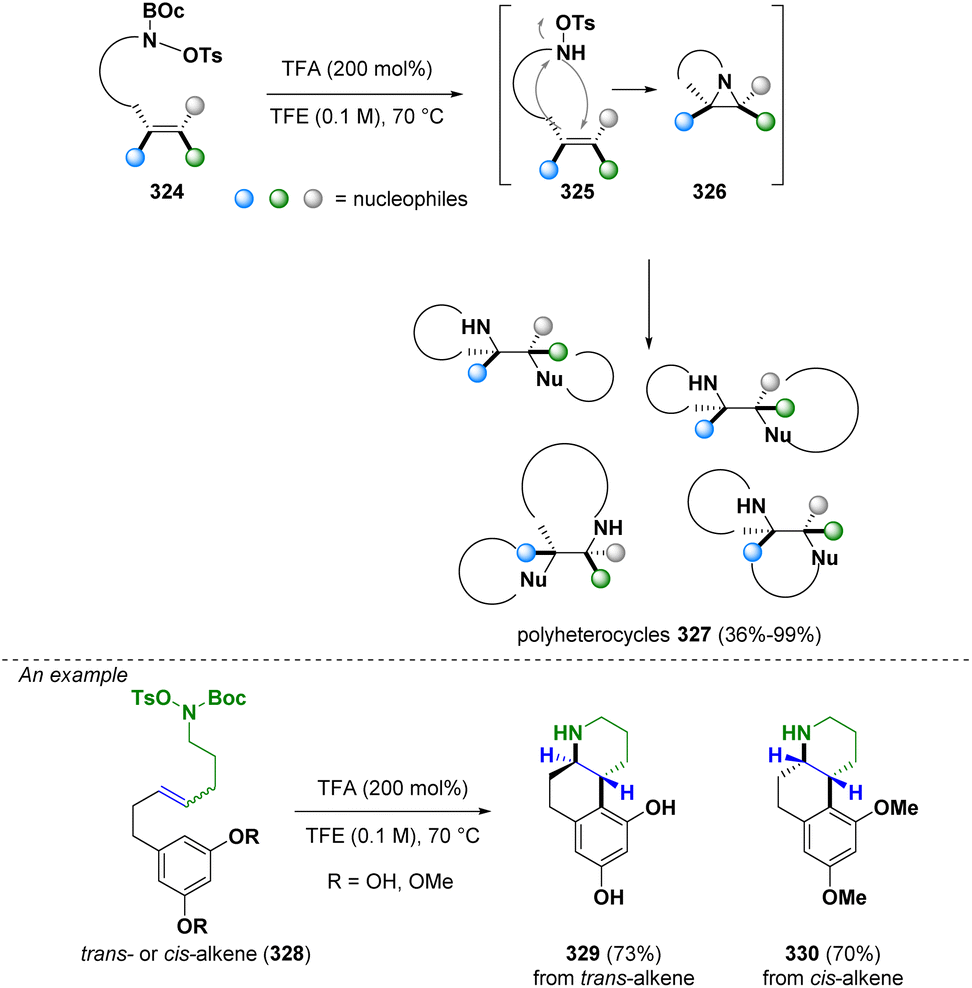 | ||
| Scheme 58 Preparation of stereochemically complex polyheterocycles. | ||
Luisi et al. employed aziridines as chiral nucleophiles in the enantioselective synthesis of oxaspirohexane sulfonamide derivatives (338). In particular, (S)-N-t-butylsulfonyl-2-phenylaziridine (333) was prepared starting from (S)-phenyl-glycinol (332). The lithiated intermediate 334 was trapped with 3-phenylcyclobutanone (331) to give a 90:10 cis/trans diastereomeric mixture of aziridino cyclobutanol (335 and 336). The cis-stereoisomer underwent Payne rearrangement under basic conditions, leading to the desired 1-azaspiro[2,3]hexane (338) (Scheme 59).94
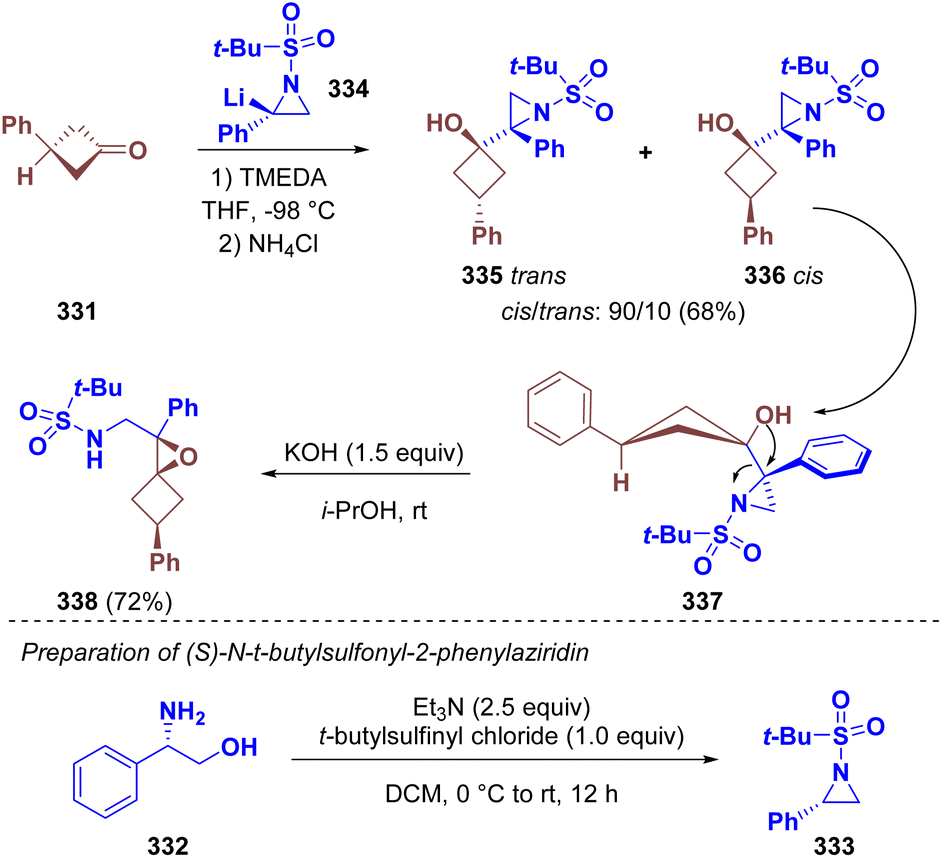 | ||
| Scheme 59 Synthesis of oxaspirohexane sulfonamide derivatives. | ||
Aziridines were also reported to be useful catalysts.95 Rachwalski and co-workers described an asymmetric Morita–Baylis–Hillman reaction of methyl vinyl ketone and methyl acrylate (339) with various aromatic aldehydes (340) using chiral aziridine-phosphines (341) as chiral catalysts (Scheme 60).96
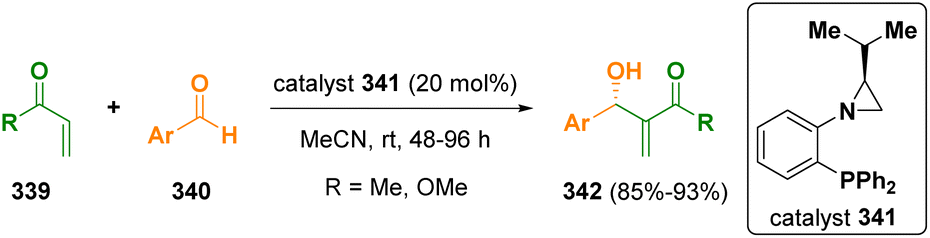 | ||
| Scheme 60 Chiral aziridines as chiral catalysts. | ||
5. Biological activities of aziridines
Aziridine derivatives possess different biological activities. Among them a large variety of compounds are active against various types of oncological diseases.97 However, several molecules displayed inhibitory activity towards other biological targets such proving to be good glucocerebrosidase (GBA),98 cysteine proteases,99 tuberculosis,100 microbial,101 and Leishmania102 inhibitors (Fig. 1).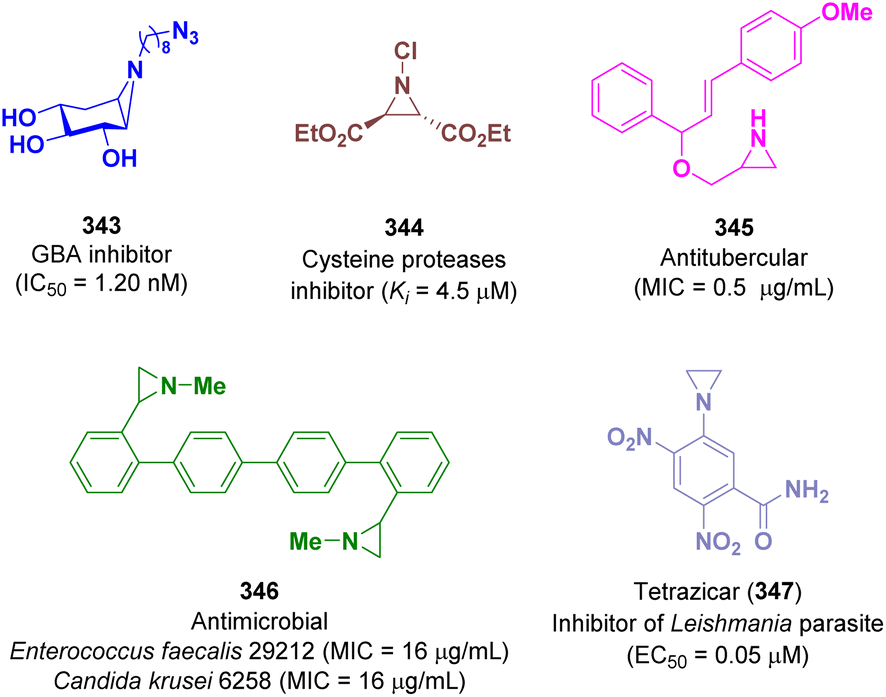 | ||
| Fig. 1 Biological activity of aziridine derivatives. | ||
5.1 Aziridines showing activity against GBA
Kuo and co-workers discovered a series of cyclophellitol aziridine derivatives as promising selective GBA inhibitors. Genetic defects in GBA determine Gaucher disease (GD) and also represent a risk factor for developing Parkinson's disease. One of the most active compound of the series is derivative 343 with an IC50 value of 1.20 nM on human GBA (Fig. 1).985.2 Aziridines showing activity against cysteine proteases
Schirmeister et al. reported a series of aziridine-2,3-dicarboxylate derivatives as potent irreversible inhibitors of cysteine proteases, which are involved in the life cycle of parasites that cause tropical disease such as malaria. The best activity was shown by compound 344 with a Ki value of 4.5 μM (Fig. 1).995.3 Aziridines showing activity against tuberculosis
The in vitro antitubercular activity of aziridine derivatives against Mycobacterium tuberculosis was evaluated by Gandhimathi et al. in 2021. Promising activity was reported for derivative 345 with a minimum inhibitory concentration (MIC) value of 0.5 μg mL−1 (Fig. 1).1005.4 Aziridines showing antimicrobial activity
The antimicrobial activity of functionalized 2-arylaziridines was identified by Luisi and co-workers. The most interesting compound is derivative 346 which showed a selective antibacterial activity against Enterococcus faecalis 29212 (MIC = 16 μg mL−1) and an interesting antifungal action against Candida krusei 6258 (MIC = 16 μg mL−1) (Fig. 1).101a5.5 Aziridines showing activity against Leishmania
Sharlow et al. described tretazicar (CB1954, 5-(aziridin-1-yl)-2,4-dinitrobenzamide, 347) as a potent inhibitor of the Leishmania parasite. In vivo studies showed promising activity for curing cutaneous leishmaniasis, which is the most common form affecting humans (EC50 = 0.05 μM in L. major cell-based amastigote) (Fig. 1).1025.6 Aziridines showing activity against oncological targets
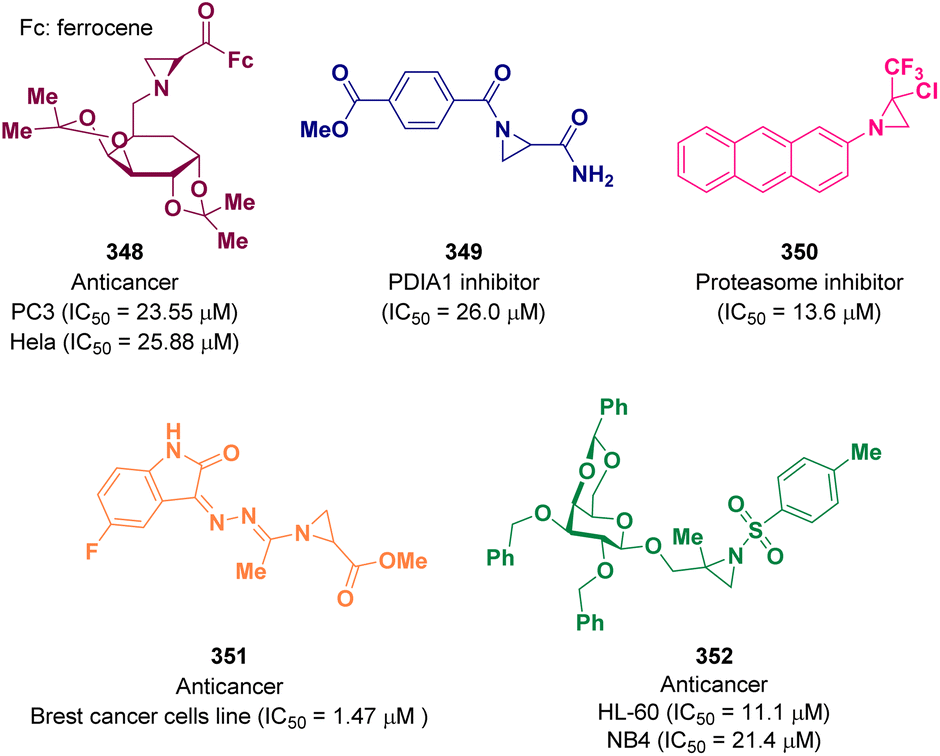
Aziridine derivatives are well-known for possessing anti-cancer activity. Great efforts over the years have made it possible to discover potent active molecules.In 2021, Bulut and co-workers reported for the first time the synthesis of N-sugar substituted chiral aziridines via the Gabriel–Cromwell reaction. Among the newly prepared compounds were promising prodrug candidates for prostate (PC3) and cervical (HeLa) cancers. In particular, derivative 348 showed good activity with an IC50 value of 23.55 μM for PC3 and 25.88 μM for HeLa (Fig. 2).103
 | ||
| Fig. 2 Examples of aziridine derivatives with promising anticancer activity. | ||
Kalvins et al. documented a class of acyl derivatives of aziridine-2-carboxylic acid as weak to moderately active PDIA1 (protein disulfide isomerase) inhibitors. Derivative 349 showed good inhibitory activity with an IC50 value of 26.0 μM. The in vitro cytotoxicity value toward a panel of cells was also evaluated and promising results were achieved (Fig. 2).104
In 2022, a series of trifluromethyl-aziridine derivatives were reported as proteasome inhibitors, selective for the β5 subunit. The in vitro biological activity, both enzymatic inhibition and anti-proliferative profile against two leukemia cells lines, was evaluated and promising results were achieved. The best result was obtained for derivative 350 with an IC50 value of 13.6 μM against the β5 subunit and 25.45 μM against drug-sensitive acute lymphocytic leukemia cells (CCRF-CEM) and 24.08 μM against a multidrug-resistant leukemia sub-cell line (CEM/ADR5000) (Fig. 2).105
Other anticancer aziridine bearing molecules were reported by Cheke et al. as inhibitors of the stem cell growth factor receptor often known as the c-KIT kinase domain. This is one of the 20 subfamilies of human receptor tyrosine kinases (RTKs) which is one of the main studied targets to fight cancer. The synthesized compounds were tested against the NCI-60 human cancer cell lines for a single-dose concentration. Derivative 351, which was one of the most promising ones, was evaluated for a five-dose anticancer study showing an IC50 value of 1.47 μM against different breast cancer cell lines (Fig. 2).106
Aziridines β-D-galactopyranoside derivatives were studied as anticancer agents by Calderón-Montaño and co-workers. The best result was obtained for compound 352, which proved to induce DNA damage. The authors suggested that 352 has an anticancer therapeutic potential future since it showed selective cytotoxicity against different malignant cells in comparison with normal cells. In particular, the highest selectivity was observed for two acute promyelocytic leukemia cell lines, human acute promyelocytic leukemia cells (HL-60) and human acute promyelocytic leukemia cells (NB4) with an IC50 value of 11.1 and 21.4 μM, respectively (Fig. 2).107
6. Conclusions
In conclusion, this overview of the recent advances in the chemistry of aziridine derivatives highlights the syntheses and chemical transformations of compounds bearing aziridine moieties. The variety of methodologies developed to prepare aziridines in the last three years speak for the importance of these compounds. Aziridines have been used as building blocks, for example as precursors for the preparation of more complex heterocycles or amino acids which are interesting from biological and pharmacological points of view due to various biological applications related to this class of compounds. Furthermore, several aziridine-bearing compounds themselves show promising biological activities. For these reasons, the interest in aziridines will endure and occupy scientists in the future too. Thus, we can expect more scientific breakthroughs reported in a rapid manner and in large numbers.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the University of Turin and the University of Vienna for financial support, Professor Vittorio Pace for his support, and Randy Sanichar for proofreading. The authors acknowledge support from Project CH4.0 under the MUR (Italian Ministry for the University) program “Dipartimenti di Eccellenza 2023–2027” (CUP: D13C22003520001).References
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC.
- (a) Y.-X. Cao, P. Silalai, C.-F. Liu, K.-Y. Yu, X. Bao, X.-H. Zhao, R. Saeeng and C.-A. Fan, Chem. – Eur. J., 2021, 27, 8473–8478 CrossRef CAS PubMed; (b) S. Park, J. Koo, W. Kim and H. G. Lee, Chem. Commun., 2022, 58, 3767–3770 RSC; (c) Y. I. Lichtenstein, I. S. Golovanov, S. L. Ioffe and A. A. Tabolin, Tetrahedron, 2022, 110, 132693 CrossRef CAS.
- (a) Y. Zhou, A. K. Gupta, M. Mukherjee, L. Zheng and W. D. Wulff, J. Org. Chem., 2017, 82, 13121–13140 CrossRef CAS PubMed; (b) M. Mukherjee, Y. Zhou, A. K. Gupta, Y. Guan and W. D. Wulff, Eur. J. Org. Chem., 2014, 1386–1390 CrossRef CAS; (c) Y. Zhou, M. Mukherjee, A. K. Gupta and W. D. Wulff, Org. Lett., 2017, 19, 2230–2233 CrossRef CAS PubMed; (d) X. Feng, Y. Song and W. Lin, J. Am. Chem. Soc., 2021, 143, 8184–8192 CrossRef CAS PubMed.
- G. S. Singh, M. D'Hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080–2135 CrossRef CAS PubMed.
- T. Qadir, A. Amin, D. Sarkar and P. K. Sharma, Curr. Org. Chem., 2021, 25, 1868–1893 CrossRef CAS.
- M. Prieschl, D. Cantillo and C. O. Kappe, J. Flow Chem., 2021, 11, 117–125 CrossRef CAS.
- (a) S. P. Chavan, S. A. Kawale and R. G. Gonnade, Eur. J. Org. Chem., 2022, e202200384 CAS; (b) S. P. Chavan, A. L. Kadam, S. S. Shinde and R. G. Gonnade, Chem. – Asian J., 2020, 15, 415–424 CrossRef CAS PubMed.
- L. A. McLean, M. W. Ashford, J. W. B. Fyfe, A. M. Z. Slawin, A. G. Leach and A. J. B. Watson, Chem. – Eur. J., 2022, 28, e202200060 CrossRef CAS PubMed.
- (a) I. Suzuki, Y. Takenaka, Y. Morishita and I. Shibata, Chem. Lett., 2022, 51, 9–12 CrossRef CAS; (b) J. L. Jat, A. K. Yadav, C. B. Pandey, D. Chandra and B. Tiwari, J. Org. Chem., 2022, 87, 3751–3757 CrossRef CAS PubMed.
- X. Fu, T. Zhang, J. Wu, Y. Sun and F. Wu, Eur. J. Org. Chem., 2022, e202101205 CAS.
- G. Kirby, L. Grimaud, M. R. Vitale, G. Prestat and F. Berhal, Green Chem., 2021, 23, 9428–9432 RSC.
- T. Deng, W. Mazumdar, Y. Yoshinaga, P. B. Patel, D. Malo, T. Malo, D. J. Wink and T. G. Driver, J. Am. Chem. Soc., 2021, 143, 19149–19159 CrossRef CAS PubMed.
- J. Pérez-Ruíz, P. J. Pérez and M. M. Díaz-Requejo, Organometallics, 2022, 41, 3349–3355 CrossRef.
- M. R. Rodríguez, A. M. Rodríguez, S. López-Resano, M. A. Pericàs, M. M. Díaz-Requejo, F. Maseras and P. J. Pérez, ACS Catal., 2023, 13, 706–713 CrossRef.
- Q. Zhao, Q.-Y. Yao, Y.-J. Zhang, T. Xu, J. Zhang and X. Chen, Eur. J. Org. Chem., 2022, e202200790 CAS.
- V. Boquet, A. Nasrallah, A. L. Dana, E. Brunard, P. H. Di Chenna, F. J. Duran, P. Retailleau, B. Darses, M. Sircoglou and P. Dauban, J. Am. Chem. Soc., 2022, 144, 17156–17164 CrossRef CAS PubMed.
- D. K. Wolgemuth, S. D. Elmore, J. D. Cope, P. E. Sheridan, S. L. Stokes and J. P. Emerson, Catal. Commun., 2021, 150, 106275 CrossRef CAS.
- A. A. Pinarci, N. Daniecki, T. M. TenHoeve, B. Dellosso, R. Madiu, L. Mejia, S. E. Bektas and G. Moura-Letts, Chem. Commun., 2022, 58, 4909–4912 RSC.
- D. Chandra, A. K. Yadav, V. Singh, B. Tiwari and J. L. Jat, ChemistrySelect, 2021, 6, 10524–10526 CrossRef CAS.
- X. Cheng, B.-G. Cai, H. Mao, J. Lu, L. Li, K. Wang and J. Xuan, Org. Lett., 2021, 23, 4109–4114 CrossRef CAS PubMed.
- Y. Guo, C. Pei, S. Jana and R. M. Koenigs, ACS Catal., 2021, 11, 337–342 CrossRef CAS.
- X. Riart-Ferrer, P. Sang, J. Tao, H. Xu, L.-M. Jin, H. Lu, X. Cui, L. Wojtas and X. P. Zhang, Chem, 2021, 7, 1120–1134 CAS.
- (a) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed; (b) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS PubMed.
- J. L. Jat, D. Chandra, P. Kumar, V. Singh and B. Tiwari, Synthesis, 2022, 4513–4520 CrossRef CAS.
- (a) J. Chen, W.-Q. Yan, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, Org. Lett., 2015, 17, 986–989 CrossRef CAS PubMed; (b) M. Ošeka, G. Laudadio, N. P. van Leest, M. Dyga, A. D. A. Bartolomeu, L. J. Gooßen, B. de Bruin, K. T. de Oliveira and T. Noël, Chem, 2021, 7, 255–266 CrossRef.
- D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74–79 CrossRef CAS PubMed.
- M.-S. Liu, H.-W. Du, J.-F. Cui and W. Shu, Angew. Chem., Int. Ed., 2022, 61, e202209929 CAS.
- W. Xue, Z. Zhu, S. Chen, B. You and C. Tang, J. Am. Chem. Soc., 2023, 145, 4142–4149 CrossRef CAS PubMed.
- F. Sebest, L. Radtanajiravong, S. Kaukver, A. J. P. White and S. Díez-González, Chem. Commun., 2022, 58, 3681–3684 RSC.
- (a) M. A. Marsini, J. T. Reeves, J.-N. Desrosiers, M. A. Herbage, J. Savoie, Z. Li, K. R. Fandrick, C. A. Sader, B. McKibben, D. A. Gao, J. Cui, N. C. Gonnella, H. Lee, X. Wei, F. Roschangar, B. Z. Lu and C. H. Senanayake, Org. Lett., 2015, 17, 5614–5617 CrossRef CAS PubMed; (b) T. Boultwood, D. P. Affron and J. A. Bull, Tetrahedron, 2015, 71, 4949–4957 CrossRef CAS; (c) M. J. Kerner, C. A. Kuttruff, M. Chevliakov, F. G. Buono, D. A. Gao, M. Krawiec, C. A. Busacca, C. H. Senanayake, P. Wipf and J. T. Reeves, Org. Lett., 2021, 23, 4396–4399 CrossRef CAS PubMed.
- A. Giovine, B. Musio, L. Degennaro, A. Falcicchio, A. Nagaki, J.-I. Yoshida and R. Luisi, Chem. – Eur. J., 2013, 19, 1872–1876 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- M. D. Delost and J. T. Njardarson, Org. Lett., 2021, 23, 6121–6125 CrossRef CAS PubMed.
- M. D. Delost and J. T. Njardarson, Org. Lett., 2020, 22, 6917–6921 CrossRef CAS PubMed.
- P. V. Kattamuri, J. Zhao, T. K. Das, J. H. Siitonen, N. Morgan, D. H. Ess and L. Kürti, J. Am. Chem. Soc., 2022, 144, 10943–10949 CrossRef CAS PubMed.
- L. Ielo, S. Touqeer, A. Roller, T. Langer, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2019, 58, 2479–2484 CrossRef CAS PubMed.
- G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648–13651 CrossRef CAS PubMed.
- S. Monticelli, M. Colella, V. Pillari, A. Tota, T. Langer, W. Holzer, L. Degennaro, R. Luisi and V. Pace, Org. Lett., 2019, 21, 584–588 CrossRef CAS PubMed.
- B. Wölfl, N. Winter, J. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2023, 62, e202217064 CrossRef PubMed.
- M. Andresini, L. Degannaro and R. Luisi, Beilstein J. Org. Chem., 2021, 17, 203–209 CrossRef CAS PubMed.
- G. S. Bisht, T. D. Dunchu and B. Gnanaprakasam, Chem. – Asian J., 2021, 16, 656–665 CrossRef CAS PubMed.
- H. Tao, R. Ushimaru, T. Awakawa, T. Mori, M. Uchiyama and I. Abe, J. Am. Chem. Soc., 2022, 144, 21512–21520 CrossRef CAS PubMed.
- S. Kurosawa, F. Hasebe, H. Okamura, A. Yoshida, K. Matsuda, Y. Sone, T. Tomita, T. Shinada, H. Takikawa, T. Kuzuyama, S. Kosono and M. Nishiyama, J. Am. Chem. Soc., 2022, 144, 16164–16170 CrossRef CAS PubMed.
- (a) A. Hensel, K. Nagura, L. B. Delvos and M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 4964–4967 CrossRef CAS PubMed; (b) T. Mita, M. Sugawara, K. Saito and Y. Sato, Org. Lett., 2014, 16, 3028–3031 CrossRef CAS PubMed; (c) C. Zhao, C. Jiang, J. Wang, C. Wu, Q.-W. Zhang and W. He, Asian J. Org. Chem., 2014, 3, 851–855 CrossRef CAS.
- Z.-Y. Zhao, M. Cui, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2023, 62, e202215032 CAS.
- (a) A. P. Molchanov, M. M. Efremova, M. A. Kryukova and M. A. Kuznetsov, Beilstein J. Org. Chem., 2020, 16, 2679–2686 CrossRef PubMed; (b) S. Hajra and A. Biswas, Org. Lett., 2020, 22, 4990–4994 CrossRef CAS PubMed; (c) A. Viceriat, I. Marchand, S. Carret and J.-F. Poisson, Org. Lett., 2021, 23, 2449–2454 CrossRef CAS PubMed; (d) Y. Chen, J. Ling, A. B. Keto, Y. He, K.-H. Low, E. H. Krenske and P. Chiu, Angew. Chem., Int. Ed., 2022, 61, e202116099 CAS; (e) T. Zhang, S. Wang, D. Zuo, J. Zhao, W. Luo, C. Wang and P. Li, J. Org. Chem., 2022, 87, 10408–10415 CrossRef CAS PubMed.
- (a) A. Biswas and S. Hajra, Adv. Synth. Catal., 2022, 364, 3035–3042 CrossRef CAS; (b) S. Xing, Y. Wang, C. Jin, S. Shi, Y. Zhang, Z. Liao, K. Wang and B. Zhu, J. Org. Chem., 2022, 87, 6426–6431 CrossRef CAS PubMed.
- Y. Hu, L. Yang and X. Liu, Molecules, 2023, 28, 242 CrossRef CAS PubMed.
- J. Qiao, S. Wang, X. Liu and X. Feng, Chem. – Eur. J., 2023, 29, e202203757 CrossRef CAS PubMed.
- H. Zhu, P.-P. Zhou and Y. Wang, Nat. Commun., 2022, 13, 3563 CrossRef CAS PubMed.
- (a) R. Li, B. Li, H. Zhang, C.-W. Ju, Y. Qin, X.-S. Xue and D. Zhao, Nat. Chem., 2021, 13, 1006–1016 CrossRef CAS PubMed; (b) Q. Wu and J. Xu, Chem. Commun., 2022, 58, 2714–2717 RSC.
- Y.-F. Han, Z.-H. Gao, C.-L. Zhang and S. Ye, Org. Lett., 2020, 22, 8396–8400 CrossRef CAS PubMed.
- S. K. A. Saleh, A. Hazra, M. S. Singh and S. Hajra, J. Org. Chem., 2022, 87, 8656–8671 CrossRef CAS PubMed.
- D. Zuo, T. Zhang, J. Zhao, W. Luo, C. Wang and P. Li, Org. Lett., 2022, 24, 4630–4634 CrossRef CAS PubMed.
- Q. Wang, T. Fan and J. Song, Org. Lett., 2023, 25, 1246–1251 CrossRef CAS PubMed.
- C.-C. Wang, X.-L. Wang, Q.-L. Zhang, J. Liu, Z.-W. Ma, Z.-J. Liu and Y.-J. Chen, Org. Chem. Front., 2022, 9, 1574–1579 RSC.
- J. Eshon, K. A. Nicastri, S. C. Schmid, W. T. Raskopf, I. A. Guzei, I. Fernández and J. M. Schomaker, Nat. Commun., 2020, 11, 1273 CrossRef CAS PubMed.
- X. Chen, Z. Huang and J. Xu, Adv. Synth. Catal., 2021, 363, 3098–3108 CrossRef CAS.
- Y. Lei and J. Xu, Beilstein J. Org. Chem., 2022, 18, 70–76 CrossRef CAS PubMed.
- D. C. Miller, R. G. Lal, L. A. Marchetti and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 4739–4745 CrossRef CAS PubMed.
- P. Sonzini, N. Berthet, C. Damiano, V. Dufaud and E. Gallo, J. Catal., 2022, 414, 143–154 CrossRef CAS.
- M. Cavalleri, C. Damiano, G. Manca and E. Gallo, Chem. – Eur. J., 2023, 29, e202202729 CrossRef CAS PubMed.
- N. Panza, M. Alberti, S. Galiè, C. Damiano, F. Cargnoni, M. Italo Trioni and A. Caselli, Eur. J. Org. Chem., 2022, e202200908 CAS.
- X.-R. Tian, X.-L. Jiang, S.-L. Hou, Z.-H. Jiao, J. Han and B. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202200123 CAS.
- P. Nayak, A. Chandrasekar Murali, V. Rao Velpuri, V. Chandrasekhar and K. Venkatasubbaiah, Adv. Synth. Catal., 2023, 365, 230–237 CrossRef CAS.
- H. J. Dequina, J. Eshon, S. C. Schmid, W. T. Raskopf, K. M. Sanders, I. Fernández and J. M. Schomaker, J. Org. Chem., 2022, 87, 10902–10907 CrossRef CAS PubMed.
- G. V. Karunakar, K. Sunil, P. Bharath kumar, M. Chandrappa, R. Guduru, S. Kantevari and B. Sridhar, Chem. – Asian J., 2023, 18, e202201071 CrossRef CAS PubMed.
- (a) S. Hajra, A. Hazra, S. K. A. Saleh and A. S. Mondal, Org. Lett., 2019, 21, 10154–10158 CrossRef CAS PubMed; (b) C.-Y. Liu, V. Angamuthu, W.-C. Chen and D.-R. Hou, Org. Lett., 2020, 22, 2246–2250 CrossRef CAS PubMed; (c) D. A. Vasilenko, S. E. Dronov, Y. K. Grishin and E. B. Averina, Asian J. Org. Chem., 2022, 11, e202200355 CrossRef CAS; (d) H. M. Holst, J. T. Floreancig, C. B. Ritts and N. J. Race, Org. Lett., 2022, 24, 501–505 CrossRef CAS PubMed.
- (a) S. Nagamalla, D. Paul, J. T. Mague and S. Sathyamoorthi, Org. Lett., 2022, 24, 6202–6207 CrossRef CAS PubMed; (b) I. A. Wani, S. Sk, A. Mal, A. Sengupta and M. K. Ghorai, Org. Lett., 2022, 24, 7867–7872 CrossRef CAS PubMed.
- X. Chen, Y. Lei, D. Fu and J. Xu, Org. Biomol. Chem., 2021, 19, 7678–7689 RSC.
- S. Xing, C. Wang, T. Gao, Y. Wang, H. Wang, H. Wang, K. Wang and B. Zhu, New J. Chem., 2022, 46, 2553–2558 RSC.
- A. Tracz, M. Malinowska, S. Leśniak and A. Zawisza, Molecules, 2022, 27, 1764 CrossRef CAS PubMed.
- J. Xin, T. Chen and P. Tang, Org. Lett., 2022, 24, 2035–2039 CrossRef CAS PubMed.
- S. Dongbang and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 20067–20077 CrossRef CAS PubMed.
- K. L. Jensen, E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2014, 136, 11145–11152 CrossRef CAS PubMed.
- S. Pache and M. Lautens, Org. Lett., 2003, 5, 4827–4830 CrossRef CAS PubMed.
- M. Bai, S. Jia, J. Zhang, H.-G. Cheng, H. Cong, S. Liu, Z. Huang, Y. Huang, X. Chen and Q. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202205245 CAS.
- J. Lee, X. Ju, M. Lee, Q. Jiang, H. Jang, W. S. Kim, L. Wu, S. Williams, X.-J. Wang, X. Zeng, J. Payne and Z. S. Han, Org. Lett., 2022, 24, 2655–2659 CrossRef CAS PubMed.
- S. A. Saleh, A. Hazra and S. Hajra, Adv. Synth. Catal., 2022, 364, 391–404 CrossRef CAS.
- V. M. Dembitsky, Eur. J. Med. Chem., 2008, 43, 223–251 CrossRef CAS PubMed.
- H.-C. Chen, C.-Y. Liu, V. Angamuthu, W.-C. Chen, C.-S. Wen and D.-R. Hou, Org. Lett., 2023, 25, 190–194 CrossRef CAS PubMed.
- A. Ahmad, H. S. Dutta, M. Kumar, S. Raziullah, M. K. Gangwar and D. Koley, Org. Lett., 2022, 24, 2783–2787 CrossRef CAS PubMed.
- K.-X. Huang, Z.-Y. Chen, X.-G. Liu, H.-Y. Ye and W.-C. Gao, Synthesis, 2022, 4495–4502 CrossRef CAS.
- Y. Li, W.-Y. Li, X. Tang, X. Liu and X. Feng, Org. Chem. Front., 2022, 9, 1531–1535 RSC.
- J. Davies, D. Janssen-Müller, D. P. Zimin, C. S. Day, T. Yanagi, J. Elfert and R. Martin, J. Am. Chem. Soc., 2021, 143, 4949–4954 CrossRef CAS PubMed.
- P. Fan, Y. Jin, J. Liu, R. Wang and C. Wang, Org. Lett., 2021, 23, 7364–7369 CrossRef CAS PubMed.
- H. D. Pickford, J. Nugent, B. Owen, J. J. Mousseau, R. C. Smith and E. A. Anderson, J. Am. Chem. Soc., 2021, 143, 9729–9736 CrossRef CAS PubMed.
- T. Song, L. Zhu, H. Li, C.-H. Tung, Y. Lan and Z. Xu, Org. Lett., 2020, 22, 2419–2424 CrossRef CAS PubMed.
- S. Xing, T. Gao, C. Jin, H. Dong, X. Shao, K. Wang and B. Zhu, Adv. Synth. Catal., 2023, 365, 104–109 CrossRef CAS.
- (a) G. Coin, O. de Ferrier de Montal, P. Dubourdeaux and J.-M. Latour, Eur. J. Org. Chem., 2021, 443–448 CrossRef CAS; (b) A. S. Pankova, J. Org. Chem., 2022, 87, 11121–11130 CrossRef CAS PubMed; (c) L. Song, X. Tian, K. Farshadfar, F. Shiri, F. Rominger, A. Ariafard and A. S. K. Hashmi, Nat. Commun., 2023, 14, 831 CrossRef CAS PubMed.
- F. Vuillermet, J. Bourret and G. Pelletier, J. Org. Chem., 2021, 86, 388–402 CrossRef CAS PubMed.
- H. Tan, S. Samanta, A. Maity, P. Roychowdhury and D. C. Powers, Nat. Commun., 2022, 13, 3341 CrossRef CAS PubMed.
- Y. Zhu, M. J. S. Smith, W. Tu and J. Bower, Angew. Chem., Int. Ed., 2023, 62, e202301262 CrossRef CAS PubMed.
- A. Cocco, M. G. Rubanu, M. L. Sechi, A. Frongia, P. Mastrorilli, L. Degennaro, M. Colella, R. Luisi and F. Secci, Org. Biomol. Chem., 2021, 19, 1945–1949 RSC.
- A. Buchcic-Szychowska, J. Adamczyk, L. Marciniak, A. M. Pieczonka, A. Zawisza, S. Leśniak and M. Rachwalski, Catalysts, 2021, 11, 968 CrossRef CAS.
- A. Buchcic-Szychowska, A. Zawisza, S. Leśniak and M. Rachwalski, Catalysts, 2022, 12, 394 CrossRef CAS.
- (a) K. C. Nicolaou, Y. G. Shelke, B. D. Dherange, A. Kempema, B. Lin, C. Gu, J. Sandoval, M. Hammond, M. Aujay and J. Gavrilyuk, J. Org. Chem., 2020, 85, 2865–2917 CrossRef CAS PubMed; (b) S. Lin, Y. Liang, J. Cheng, F. Pan and Y. Wang, Eur. J. Med. Chem., 2021, 214, 113256 CrossRef CAS PubMed; (c) V. Carramiñana, A. M. Ochoa de Retana, F. Palacios and J. M. de los Santos, Molecules, 2021, 26, 4265 CrossRef PubMed.
- R. J. Rowland, Y. Chen, I. Breen, L. Wu, W. A. Offen, T. J. Beenakker, Q. Su, A. M. C. H. van den Nieuwendijk, J. M. F. G. Aerts, M. Artola, H. S. Overkleeft and G. J. Davies, Chem. – Eur. J., 2021, 27, 16377–16388 CrossRef CAS PubMed.
- V. Buback, M. Mladenovic, B. Engels and T. Schirmeister, J. Phys. Chem. B, 2009, 113, 5282–5289 CrossRef CAS PubMed.
- P. Sarojini, M. Jeyachandran, D. Sriram, P. Ranganathan and S. Gandhimathi, J. Mol. Struct., 2021, 1233, 130038 CrossRef CAS.
- (a) A. Giovine, M. Muraglia, M. A. Florio, A. Rosato, F. Corbo, C. Franchini, B. Musio, L. Degennaro and R. Luisi, Molecules, 2014, 19, 11505–11519 CrossRef PubMed; (b) H. K. Santra, S. Maity and D. Banerjee, Molecules, 2022, 27, 1459 CrossRef CAS PubMed; (c) M. Pitscheider, N. Mäusbacher and S. A. Sieber, Chem. Sci., 2012, 3, 2035–2041 RSC; (d) S. Ghannay, A. Kadri and K. Aouadi, Monatsh. Chem., 2020, 151, 267–280 CrossRef CAS.
- D. Caridha, R. J. Sciotti, J. Sousa, B. Vesely, T. Teshome, G. Bonkoungou, C. Vuong, S. Leed, M. Khraiwesh, E. Penn, M. Kreishman-Deitrick, P. Lee, B. Pybus, J. S. Lazo and E. R. Sharlow, ACS Infect. Dis., 2021, 7, 506–517 CrossRef CAS PubMed.
- M. Sert, Ö. Işılar, A. S. Yaglioglu and A. Bulut, Carbohydr. Res., 2021, 509, 108430 CrossRef CAS PubMed.
- I. Leite, V. Andrianov, D. Zelencova-Gopejenko, E. Loza, I. Kazhoka-Lapsa, I. Domracheva, M. Stoyak, S. Chlopicki and I. Kalvins, Chem. Heterocycl. Compd., 2021, 57, 1086–1106 CrossRef CAS.
- L. Ielo, V. Patamia, A. Citarella, T. Efferth, N. Shahhamzehei, T. Schirmeister, C. Stagno, T. Langer, A. Rescifina, N. Micale and V. Pace, Int. J. Mol. Sci., 2022, 23, 12363 CrossRef CAS PubMed.
- P. J. Chaudhari, S. B. Bari, S. J. Surana, A. A. Shirkhedkar, C. G. Bonde, S. C. Khadse, V. G. Ugale, A. A. Nagar and R. S. Cheke, ACS Omega, 2022, 7, 17270–17294 CrossRef CAS PubMed.
- E. Burgos-Morón, N. Pastor, M. L. Orta, J. J. Jiménez-Alonso, C. Palo-Nieto, M. Vega-Holm, J. M. Vega-Pérez, F. Iglesias-Guerra, S. Mateos, M. López-Lázaro and J. M. Calderón-Montaño, Biomedicines, 2022, 10, 41 CrossRef PubMed.
- C. C. Wang, Y. T. Yang, Q. L. Wang, X. H. Liu and Y. J. Chen, Org. Chem. Front., 2022, 9, 4271–4276 RSC.
- F. Zhang, X. Sang, Y. Zhou, W. Cao and X. Feng, Org. Lett., 2022, 24, 1513–1517 CrossRef CAS PubMed.
- J. Xin, X. Deng and P. Tang, Org. Lett., 2022, 24, 881–885 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |