
Visible light-assisted chemistry of vinyl azides and its applications in organic synthesis
Barakha
Saxena
,
Roshan I.
Patel
,
Jaya
Tripathi
and
Anuj
Sharma
 *
*
Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee-247667, India. E-mail: anujsharma.mcl@gmail.com; anuj.sharma@cy.iitr.ac.in
First published on 18th May 2023
Abstract
Vinyl azides have emerged as highly versatile precursors in organic synthesis due to their rich reactivity driven by the excellent leaving-group ability of molecular nitrogen. Over the years, significant advancement has been achieved in the manipulation of vinyl azides for the construction of C–C and C–X bonds. Typical methods involve the application of transition metals and strong oxidants for the conversion of vinyl azides into useful compounds employing harsh reaction conditions coupled with intense product purification. In this regard, visible light chemistry has become one of the most exciting fields in organic synthesis for being mild, sustainable, and often orthogonal to conventional approaches. Visible light-induced reactions involving vinyl azides generate either 2H-azirines or iminyl radicals as key intermediates, which may undergo further useful transformations to form the desired cyclic or acyclic products. Herein, we provide the most significant transformations of vinyl azides as versatile synthetic precursors or transient intermediates for compounds of synthetic and biological significance under visible light photocatalysis. We have classified this review into two parts: (i) formation of an iminyl radical intermediate and (ii) formation of 2H-azirine intermediate-based reactions.
 Barakha Saxena | Barakha Saxena obtained her BSc (2015) and MSc (2017) degrees in Organic Chemistry from Mahatma Jyotiba Phule Rohilkhand University, Bareilly, Uttar Pradesh, India. She received the Junior Research Fellowship Award (CSIR-JRF) just after completing her master's degree. She joined as a PhD scholar in the research group of Prof. Anuj Sharma in the Department of Chemistry, Indian Institute of Technology (IIT), Roorkee, India. She has received the Senior Research Fellowship Award (CSIR-SRF). Her current research is focused on novel strategies for C–C and C–X bond formation. |
 Roshan I. Patel | Roshan I. Patel obtained his BSc (2015) and MSc (2017) degrees in Organic Chemistry from Sardar Patel University, India. He was a gold medalist in his BSc. After receiving the prestigious Junior Research Fellowship award, he joined as a PhD scholar in the research group of Prof. Anuj Sharma in the Department of Chemistry, Indian Institute of Technology, Roorkee, India. He has received the Senior Research Fellowship Award (UGC-SRF). His current research is focused on efficient methods for C–H/X functionalization using contemporary methods. |
 Jaya Tripathi | Jaya Tripathi pursued her Bachelor of Science and Master of Science degrees in Chemistry from Chhatrapati Shahu Ji Maharaj University, Kanpur, Uttar Pradesh, India. Currently, she is a PhD scholar in the research group of Prof. Anuj Sharma in the Department of Chemistry, Indian Institute of Technology, Roorkee, India. Her research interests include photocatalytic manipulations in the design of novel synthetic molecules. |
 Anuj Sharma | Prof. Anuj Sharma earned his PhD from the Institute of Himalayan Bioresource Technology (IHBT) in 2006. Afterward, he undertook two short postdoctoral assignments in UFSM, Santa Maria, Brazil, in 2006, and KU Leuven, Belgium, in 2007 before moving to the University of Arizona as a NIH postdoctoral fellow in Prof. Laurence Hurley's group. He is currently working as a professor in the Department of Chemistry, Indian Institute of Technology (IIT), Roorkee, India. His group focuses on developing green organic synthetic methodologies, including the use of visible light for C–H functionalization. |
1. Introduction
Organic azides are potentially active substances that are present in a wide range of biologically active compounds and have attracted significant interest in recent decades.1 Among them, vinyl azides are characterized by the presence of an azide (N3) group on a double bond, which acts as a highly versatile three-atom synthon by a facile release of nitrogen molecules. These vinyl azides are powerful building blocks in organic synthesis and have been widely used to construct numerous acyclic and cyclic N-heterocyclic compounds.2 Vinyl azides can function as nucleophiles, electrophiles, and radical acceptors, as well as participate in cycloaddition reactions.3 Thus, several valuable and unprecedented chemical transformations have been demonstrated starting from vinyl azides for the synthesis of N-containing heterocyclic compounds and their derivatives (Fig. 1).2,3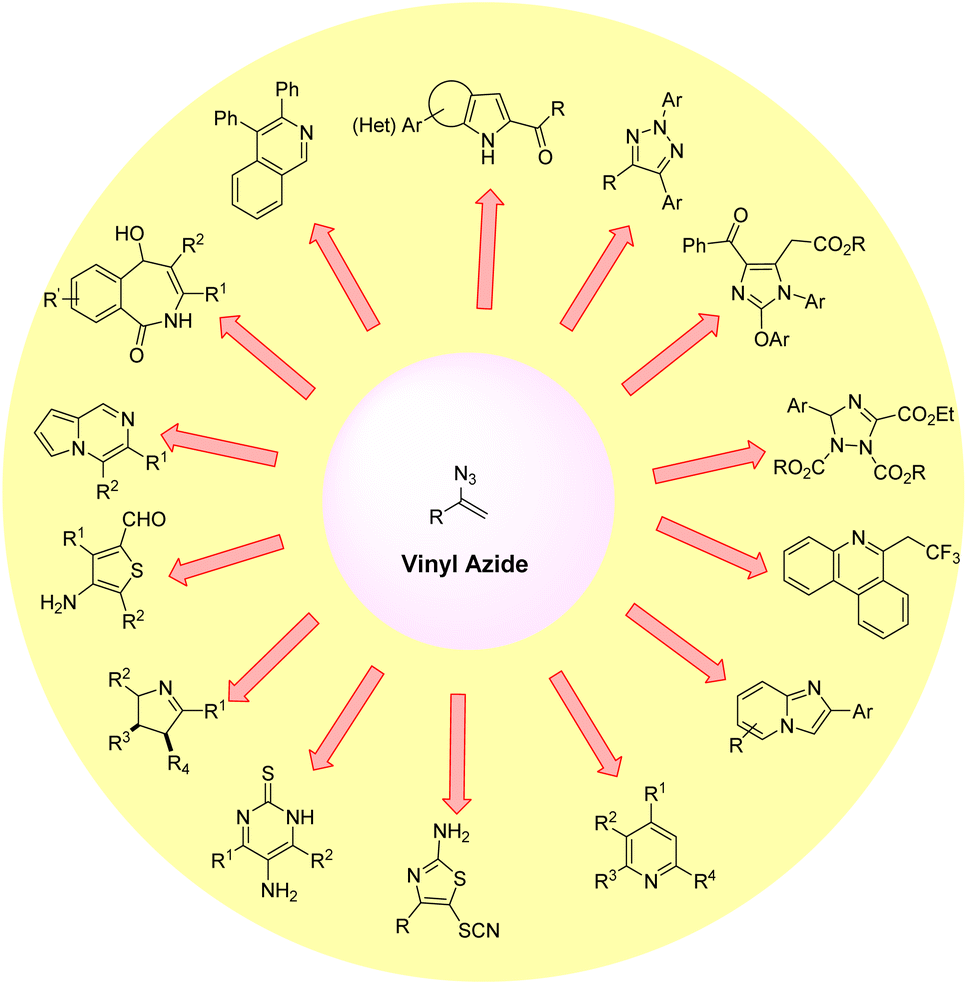 | ||
| Fig. 1 Classical representative transformations of vinyl azides. | ||
One of the most popular methods for vinyl azide synthesis is the hydroazidation of alkynes. This involves the reaction of TMSN3 as an azide source with terminal alkynes, catalyzed by AgN3 (Scheme 1).4 This strategy allows access to vinyl azides in good yields. Next, with a slight change in the reaction conditions, vinyl azides can also be easily synthesized by the application of Ag2CO3 as a catalyst.5 Besides, terminal alkenes can also be transformed into useful vinyl azide derivatives by the reaction with IN3 as a reagent in the presence of a strong base.6 This method requires the application of very low reaction temperatures for product formation.
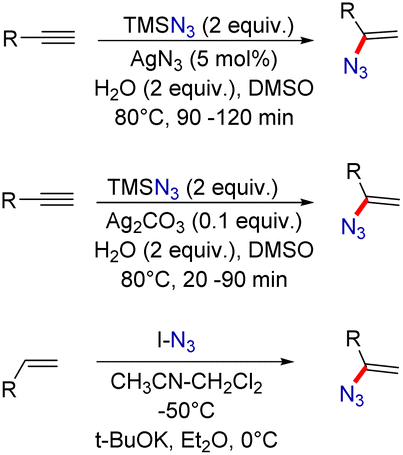 | ||
| Scheme 1 Preparative methods of vinyl azides mentioned in this review. | ||
There have been impressive efforts devoted to the manipulations of vinyl azides for the construction of cyclic and acyclic compounds.2,7 This can be seen in a series of outstanding reviews dedicated to this area. Some of the notable ones are by Hu and DiMagno (2015), who discussed the synthesis of complex and diverse N-heterocyclic compounds from vinyl azides.7a The application of heat, transition metals, Lewis-acid coordination, etc. on vinyl azides and their novel transformation was nicely discussed in their review. Later, Chiba and co-workers (2017) penned an excellent article on the applications of vinyl azides as radical acceptors and enamine-type nucleophiles for the construction of nitrogen-containing compounds.8 Thereafter, Anderson, Bi, and co-workers in their review article disclosed the alkene functionalization of α-substituted vinyl azides.9 The generation of nitrilium ions, imino-diazonium ions, metal emaminyl radicals, and iminyl radicals from vinyl azides and their useful transformations were discussed in their review. Recently, Mahdavi and co-workers wrote an article on the overview of vinyl azides in organic synthesis. The useful conversion of iminyl ions, nitrilium ions, iminyl metal complexes, 2H-azirines and iminyl radical intermediates from vinyl azides was discussed in their paper.7b
The use of light in organic reactions has been a constant area of research over the past decades among the synthetic community. However, classical photochemistry has revolved chiefly around high-energy UV radiation, which resulted in photodecomposition and diminished product yields. It therefore, later, shifted to visible light alternatives due to safety concerns and avoided the possibilities of side product formation associated with high-energy UV radiation.10 In the last decade, state-of-the-art visible light-assisted photo-redox methodologies have become a flourishing area of research in the field of organic synthesis. Substantial progress has been made in this area by exploiting transition metal photoredox catalysts,11 organic dye-based photocatalysts,12 semiconductor based photocatalysts,13 and visible light-assisted EDA-complex based reactions,14 to name a few, for the construction of C–C and C–X bonds. The structure and redox potential of all photocatalysts mentioned in this article are listed in Fig. 2 and Table 1, respectively.
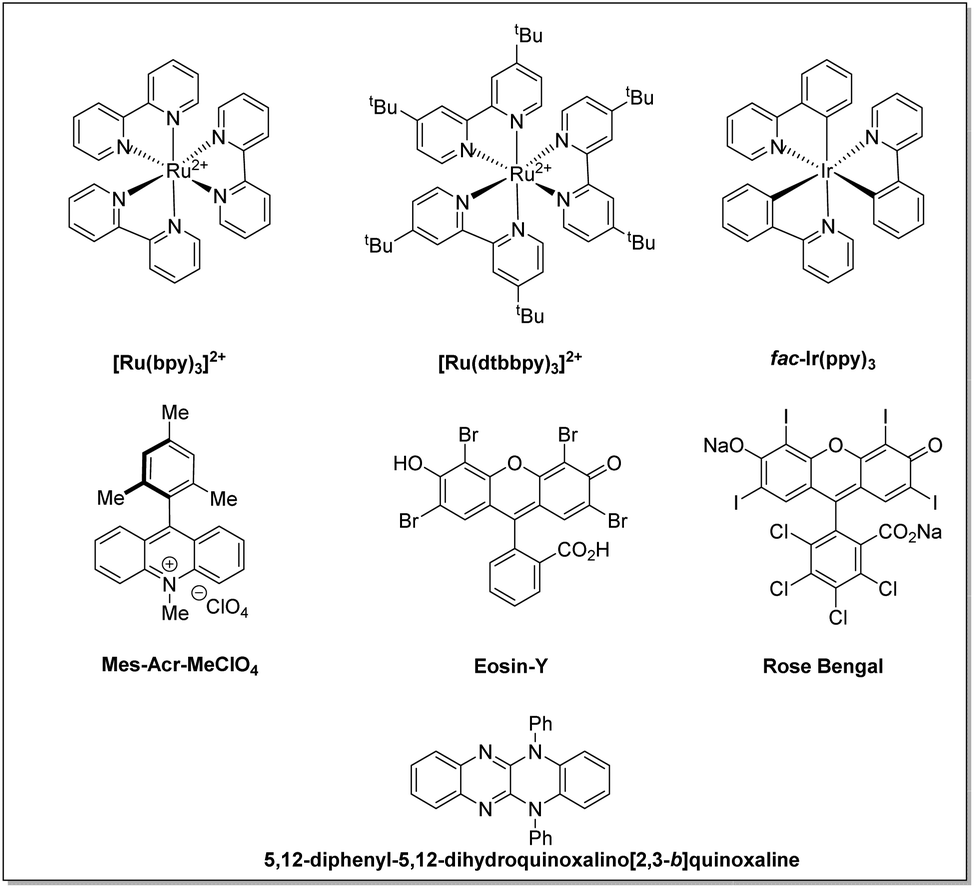 | ||
| Fig. 2 Structures of photocatalysts discussed in this review. | ||
| S. no. | Photoredox-catalysts | PC*/PC˙− | PC/PC˙− | PC˙+/PC | PC˙+/PC* | Ref. |
|---|---|---|---|---|---|---|
| 1 | [Ru(bpy)3]2+ | +0.77 | −1.33 | +1.29 | −0.81 | 11a |
| 2 | [Ru(dtbbpy)3]2+ | — | — | — | — | 11a |
| 3 | fac-Ir(ppy)3 | +0.31 | −2.19 | +0.77 | −1.73 | 11a |
| 4 | Eosin-Y | +1.18 | −1.14 | +0.72 | −1.60 | 12a |
| 5 | Rose Bengal | +0.9 | −0.78 | +1.09 | −0.68 | 12a |
| 6 | Mes-Acr-MeCIO4 | +2.08 | −0.57 | — | — | 12a |
| 7 | 5,12-Diphenyl-5,12-dihydroquinoxalino[2,3-b]quinoxaline | — | — | +0.68 | −1.61 | 11d |
As a corollary, there has been significant growth in the application of visible light photochemistry in the manipulation of vinyl azides for the formation of acyclic and cyclic compounds. This eliminates the use of harsh and toxic reagents for the activation of vinyl azides, which is usually associated with extensive product purification.
Generally, vinyl azides do not absorb visible light; however, they can undergo photochemical transformations under visible light irradiation in the presence of photocatalysts. Many transition metal complexes (Ir and Ru-complexes) and organic dyes (rose bengal, Eosin-Y, Mes-acr+, etc.) have been used as photocatalysts in these transformations. These approaches take advantage of the reducing/oxidizing properties of the photocatalysts in their excited state, which can effectively transform vinyl azides into useful compounds.
The photoexcited catalyst can then choose one of the two modes for substrate activation viz. single electron transfer (SET) or energy transfer (EnT) (Scheme 2). The former makes use of the high redox potential of the excited photocatalyst and the substrate, initiating a single electron transfer (SET) process. In this case, under visible light irradiation, the photocatalyst (PC) gets excited to its higher excited state PC* and generates a radical species (R˙) from the corresponding substrate via a reductive or oxidative quenching process with the substrate. This radical species (R˙) undergoes an addition reaction with the terminal double bond of the vinyl azide, resulting in the generation of the key iminyl radical intermediate via an α-azido radical along with the simultaneous release of dinitrogen. Depending on the reaction conditions, the resulting iminyl radical intermediate is then converted into the desired product.
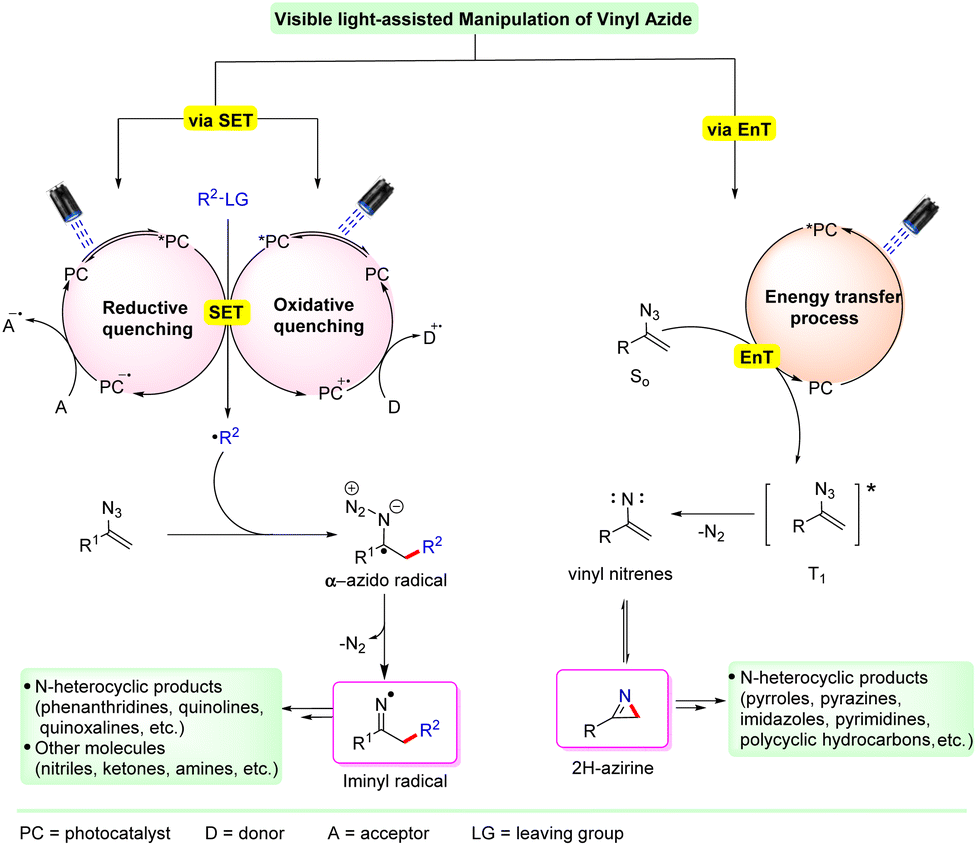 | ||
| Scheme 2 Transformations of vinyl azides under visible light irradiation. | ||
On the other hand, an energy transfer (EnT) mode of activation is also possible in some cases. This involves the activation of the photocatalyst from its ground state to its excited state PC*. This excited catalyst PC* further interacts with the ground state vinyl azide (S0) substrate and forms a triplet state vinyl azide (T0) via energy transfer (EnT). This triplet-vinyl azide undergoes photodecomposition with the subsequent release of N2 to produce a highly strained three-membered cyclic 2H-azirine intermediate, via a nitrene intermediate. This 2H-azirine can serve as a versatile intermediate and can further undergo reaction to form a cyclic or acyclic compound depending on the reaction conditions.
Moreover, there are some cases in which the vinyl azide can transform into a cyclic or acyclic compound without the need for an external photocatalyst. In such cases, vinyl azides bearing an extended conjunction can absorb visible light irradiation and convert into their triplet excited state. This triplet excited state undergoes photodecomposition to form the 2H-azirine intermediate. This intermediate can further react and transform into the desired product.
Photoactivation of vinyl azides by the methods explained above offers several benefits over traditional protocols, which suffer from the need for rigorous reaction conditions (such as BR3 as a radical initiator),15 expensive toxic metals and ligands,16 over-stoichiometric amounts of oxidants (e.g., PhI(OAc)2 and TBPB),17 high reaction temperature, and high energy UV-irradiation.18 The exceptionally mild reaction conditions (room temperature and household lamp or direct sunlight irradiation) by which these reactive intermediates can be obtained using visible light photocatalysis provide high functional group compatibility.
Despite the significant progress in this area in the last few years, to the best of our knowledge, there has not been any review article dealing exclusively with visible light-assisted transformations of vinyl azides and this is the inspiration for the current article. In this article, we have endeavored to highlight the visible light-mediated synthesis of organic compounds from vinyl azides. The purpose of this review is to bring the readers up to date on the research on visible light photoredox catalyzed or sensitized vinyl azide-based organic transformations. Based on the above discussion, the current review is broadly categorized into two parts: (i) iminyl radical-based transformation and (ii) 2H-azirine intermediate-based transformation.
2. Iminyl radical intermediate based transformation
Vinyl azides can be activated by a radical species to form an iminyl radical by the release of molecular nitrogen under photoredox conditions. In this section, we highlight the visible light-mediated activation of vinyl azides by a radical species. This section is further subdivided into 2-subsections.2.1. Formation of cyclic compounds
Phenanthridines are the core structures of several naturally occurring and biologically relevant compounds.19 Traditional cross-coupling methodologies require transition metals such as Pd, In, and Cu-catalysts under elevated temperatures for their synthesis.20 These reactions are usually associated with the application of toxic and expensive catalysts and intense follow-up purification processes. The use of visible light photoredox catalysis helps overcome some of the disadvantages associated with organometallic methodologies. In this context, significant effort has been made to develop various approaches based on photocatalysis for the synthesis of phenanthridine derivatives.In 2016, Yu and co-workers demonstrated an Ir-photoredox catalyzed synthesis of 6-alkylated-phenanthridine derivatives 3 under visible light illumination (Scheme 3).21 The authors utilized alkyl bromides 2 as a radical precursor with fac-Ir(ppy)3 as a photocatalyst and Na2HPO4 as a base in DMF under white LEDs. With the optimized reaction conditions in hand, various substituted vinyl azides 1 having electron-withdrawing and electron-donating groups were tested with electron-deficient bromides to obtain the required 6-alkylated phenanthridine derivatives 3 in 49–90% yields. Besides, a series of bromoesters 2 having linear alkyl-, branched- or saturated alkyl chains also reacted well under the optimized conditions.
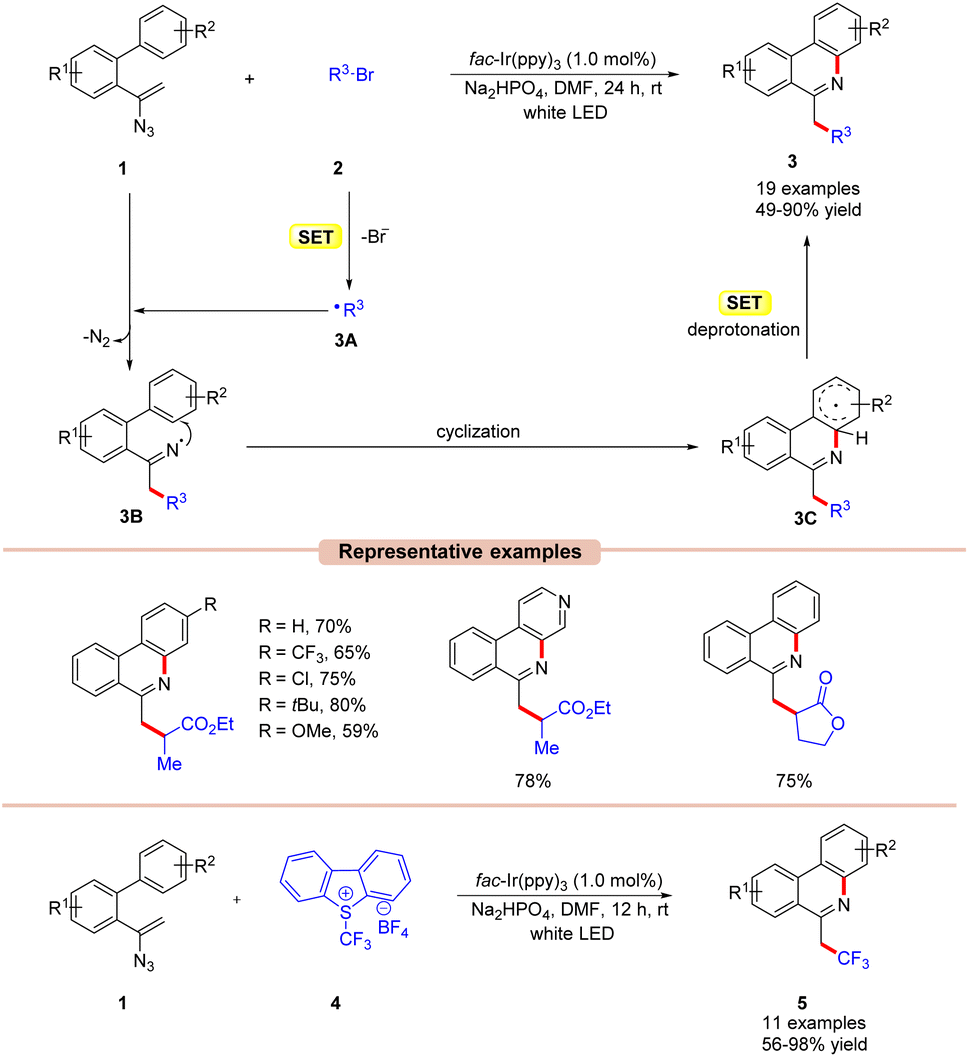 | ||
| Scheme 3 Ir-photoredox catalyzed synthesis of 6-alkylated-, 6-(fluoro)alkylated phenanthridines from vinyl azides. | ||
Unfortunately, bromoamides and electron-deficient chlorides proved to be inactive substrates for this transformation. The presence of a radical scavenger, such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), significantly suppressed the progress of the reaction. Furthermore, the TEMPO adducts were detected by 19F-NMR analysis, thereby confirming the involvement of a radical pathway. Moreover, luminescence quenching experiments showed that the excited photoredox catalyst was effectively quenched by ethyl 2-bromopropanoate 2via a single electron transfer (SET) event. Based on the findings, the authors proposed that the photoexcited catalyst *Ir(III) reduces the alkyl bromides 2via the SET process, generating the electrophilic radical intermediate 3A. Furthermore, 3A adds to the double bond of vinyl azide 1 resulting in the iminyl radical intermediate 3B with the release of molecular nitrogen. Subsequently, radical 3B undergoes cyclization, followed by SET/deprotonation to yield the desired product 3.
In addition, the authors further illustrated the compatibility of the reaction for the synthesis of 6-(fluoro)alkylated phenanthridine derivatives 5, using Umemoto's reagent 4 as a trifluoromethyl radical source and a range of substituted vinyl azide derivatives 1.
Sulfonyl-containing compounds have been demonstrated to be essential structures in pharmaceuticals and agrochemicals.22
The sulfonylation/cyclization of vinyl azides 1 for the synthesis of 6-(sulfonylmethyl)-phenanthridine derivatives 7 was achieved by Mao, Zhou, and co-workers in 2017, under visible light irradiation (Scheme 4a).23a
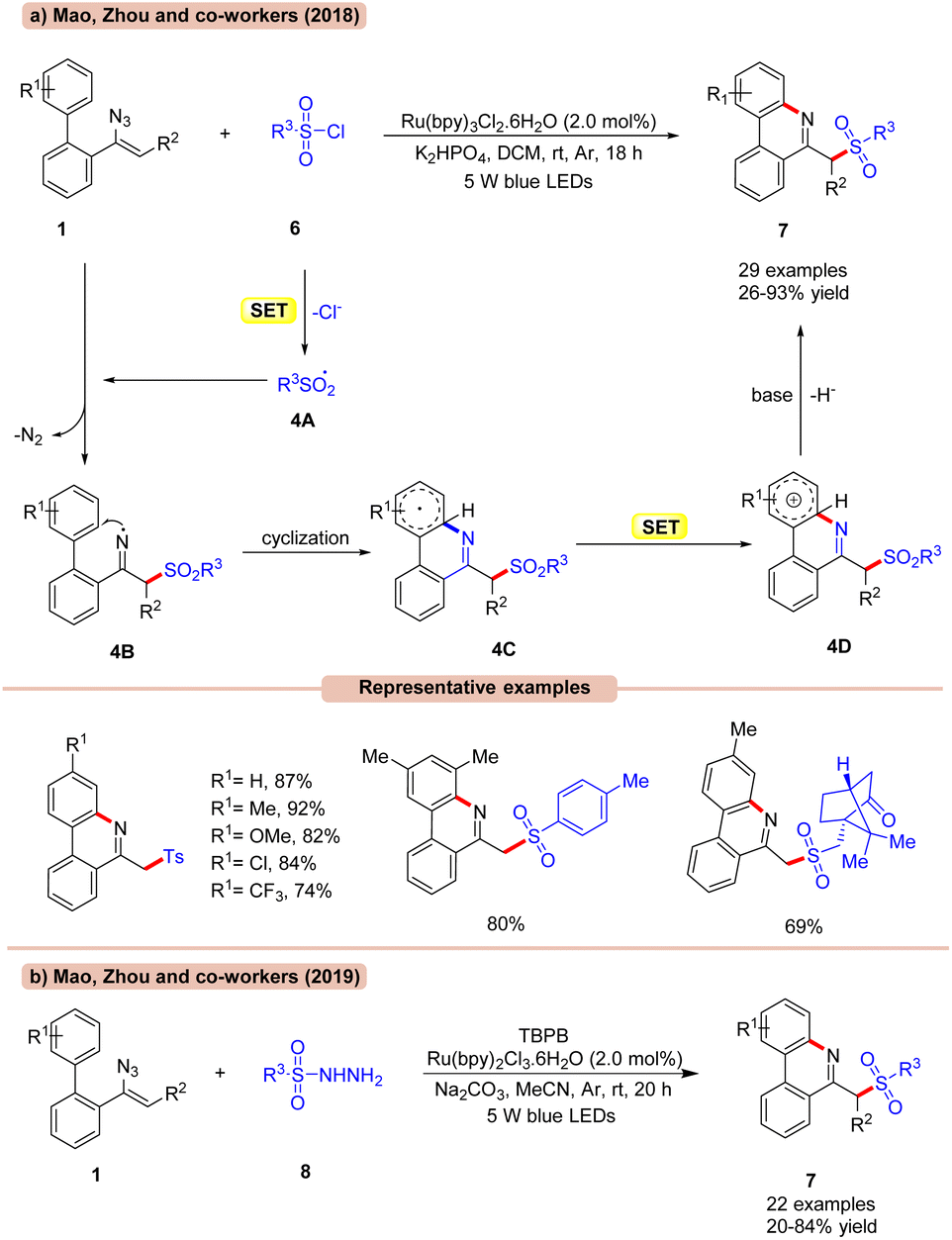 | ||
| Scheme 4 Ru-photoredox catalyzed synthesis of 6-(sulfonylmethyl)-phenanthridine. | ||
Out of the various photocatalysts screened, Ru(bpy)3Cl2 displayed the highest efficiency in catalyzing the reaction with K2HPO4 as a base in DCM under visible light illumination. A library of biphenyl vinyl azide compounds 1 bearing electron-donating and electron-withdrawing functionalities smoothly reacted with p-toluene sulfonyl chlorides 6 to afford the desired products 7 in 72–92% yields. Besides, aryl- and alkyl sulfonyl chlorides 6 bearing various functional groups such as methyl-, benzyl- and D-(+)-10-camphor-, and cyclopropane groups were well tolerated under the optimized conditions, affording the required products 7 in 26–91% yields. Radical trapping experiments with TEMPO and 1,1-diphenylethylene suggested the involvement of a radical mechanistic pathway. Mechanistically, RSO2Cl 6 is reduced by the photoexcited catalyst *Ru(II) to give the radical intermediate 4A, which upon addition to vinyl azide 1 generates the iminyl radical intermediate 4B. Thereafter, the iminyl radical undergoes subsequent cyclization, followed by SET/deprotonation to yield the desired product 7.
Furthermore, extending their previous methodology, the same group in 2019, disclosed the synthesis of 6-(sulfonylmethyl)-phenanthridine derivatives 7 by the reaction between vinyl azides 1 and sulfonyl hydrazides 8 under visible light illumination (Scheme 4b).23b With a slight change in the reaction conditions, readily available sulfonyl hydrazides 8 were proven to be efficient sulfonylating reagents for the formation of 6-(sulfonylmethyl)-phenanthridine derivatives 7. Moreover, the reaction could also be calibrated on a gram scale.
Alternatively, the work (Scheme 4a) appears better in terms of a higher reaction yield of the desired product and wider substrate scope compared to their previous work (Scheme 4b), which required an external oxidant for its successful execution.
In 2018, Yang and co-workers disclosed a visible light-mediated phosphorylation/cyclization of vinyl azides 1 for the synthesis of phosphorus phenanthridine derivatives 10 (Scheme 5).24 The reaction parameters comprised Ru(bpy)3Cl2·6H2O as a photocatalyst, tert-butyl peroxybenzoate (TBPB) as an oxidant, diphenylphosphine oxides 9 as a phosphinoyl radical source, and Na2CO3 as a base. The biphenyl vinyl azide compounds 1 with electron-donating and electron-withdrawing groups reacted with diphenylphosphine oxides 9 to afford the corresponding phosphorylated phenanthridine derivatives 10 in 24–94% yields. Mechanistically, the TBPB is reduced to the tert-butoxy radical 5Avia the SET process. This resulting radical 5A then abstracts an H-atom from diphenylphosphine oxide 9, generating the phosphinoyl radical 5B. This radical intermediate 5B upon addition to the vinyl azide 1 results in the iminyl radical intermediate 5C. Moreover, the nitrogen-centred radical 5C undergoes an intramolecular cyclization followed by a concomitant SET process in the presence of a base, which results in the phosphorylated phenanthridine product 10. The compatibility of this protocol was further demonstrated under gram-scale reaction conditions, affording the desired product 10 in 71% yield.
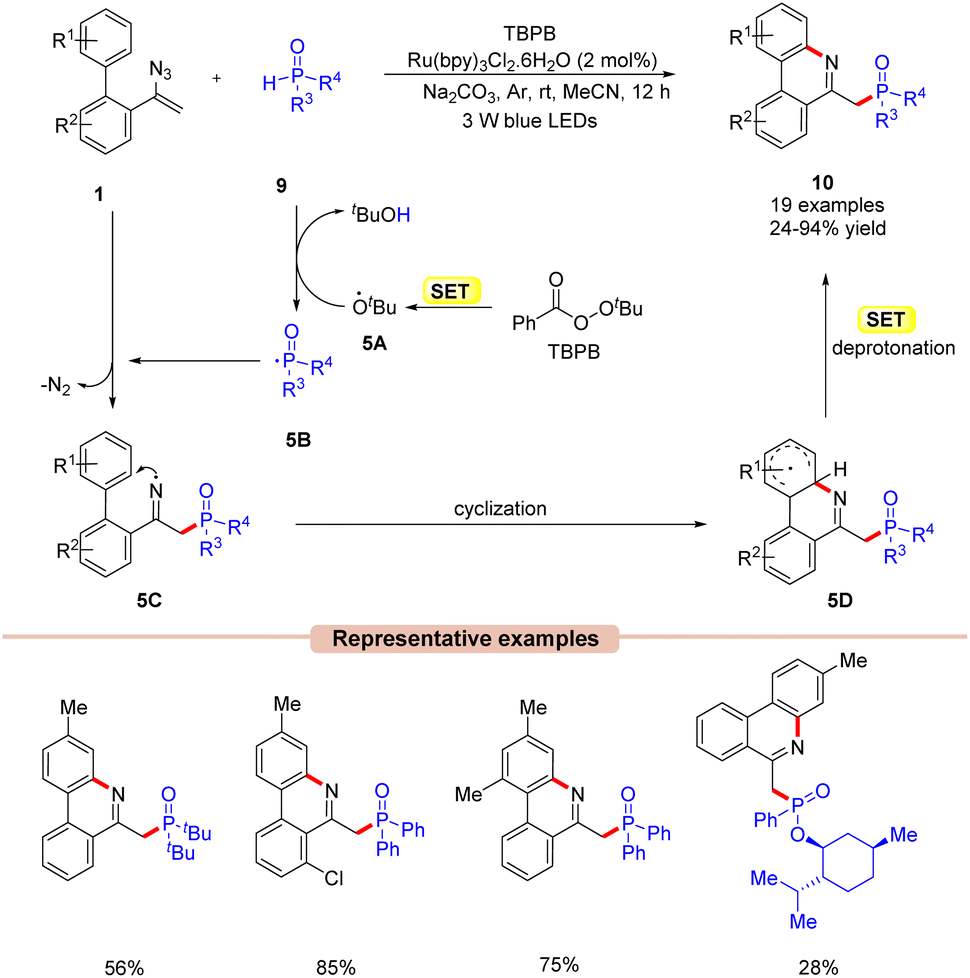 | ||
| Scheme 5 Ru-photoredox catalyzed phosphorylation of vinyl azides. | ||
Guo and co-workers in 2018, developed a visible light-induced decarboxylative cyclization of N-acyloxylphthalimides 11 with vinyl azides 1 to access substituted alkyl-phenanthridines 3 (Scheme 6).25 The authors employed Eosin-Y as a photocatalyst with TMEDA as a base in DMF at room temperature under visible light illumination. A variety of biphenyl vinyl azides 1 with different functionalities at the C3 position such as methyl, fluoro, chloro, bromo, and cyano-groups reacted well under the given reaction conditions and gave the corresponding phenanthridine products 3 in moderate to good yields (47–78%). Notably, sterically hindered ortho-substituted vinyl azides 1 reacted with NHP ester 11 to give the corresponding product in 72% yield. The reaction was suppressed upon using the radical scavengers TEMPO and BHT (butylated hydroxytoluene). Moreover, radical-clock experiments with the cyclopropyl acetic acid-derived NHP ester indicated the involvement of a radical intermediate in the present reaction. Based upon these findings, it was proposed that the photoexcited catalyst *EY was oxidatively quenched by the NHP ester 11via SET, generating the radical anion 6A, followed by N–O bond cleavage and subsequent decarboxylation to give the alkyl radical 6B. Next, the radical addition of 6B to vinyl azide 1 generates the iminyl radical intermediate 6C. Lastly, the cyclization of 6C followed by SET/deprotonation forms the desired phenanthridine product 3. Intermediate 6D can also undergo SET with 11, generating intermediate 6A, thus propagating the chain (as evident from the quantum yield experiment ϕ = 14.9).
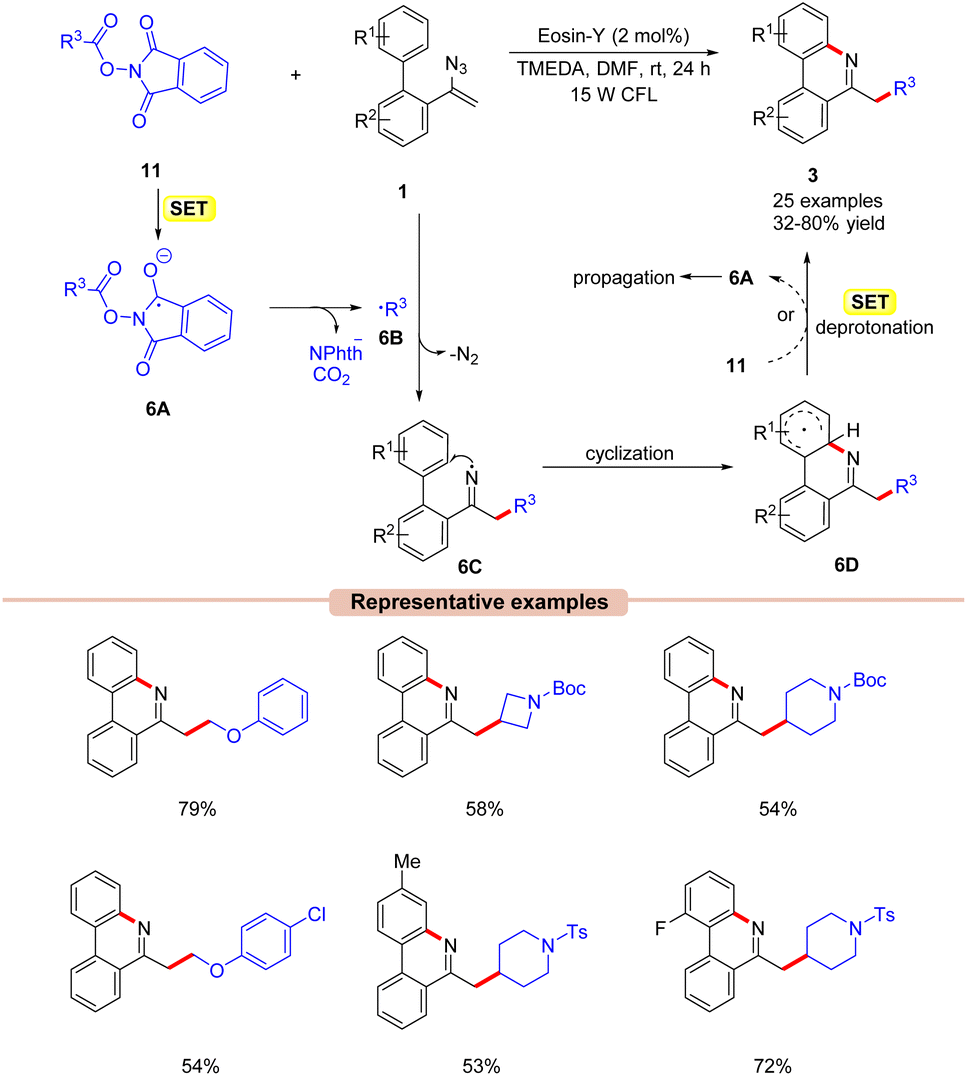 | ||
| Scheme 6 Visible light Eosin-Y mediated decarboxylative cyclization of vinyl azides with N-acyloxyphthalimides. | ||
The key feature of this method is the use of a metal-free inexpensive organo-photocatalyst under mild reaction conditions.
Thereafter, Xu and co-workers in 2021, reported the synthesis of the 1-tetralones 14via visible light-assisted radical cyclization of vinyl azides 12 with alkyl bromides 13 (Scheme 7).26 Here, the authors employed a highly reducing organic photocatalyst under blue LEDs. The substrate scope was expanded to a wide range of vinyl azides 12 bearing electron-rich and electron-poor functionalities. Notably, vinyl azides 12 bearing electron-rich groups at the para-position of the phenyl ring displayed higher yields than those having electron-poor groups under the given reaction parameters. Furthermore, this protocol was compatible at the gram scale. Radical trapping experiments with BHT and TEMPO revealed the involvement of a radical pathway. Mechanistically, the photoexcited catalyst PC* reduces the alkyl bromide 13via SET oxidative quenching, resulting in the radical species 7A. This radical is then trapped by vinyl azides 12 to give the imine radical intermediate 7B. Next, intermediate 7B undergoes a 1,5-hydrogen-atom-transfer followed by intramolecular cyclization and subsequent SET/deprotonation to afford intermediate 7E. Lastly, the desired product 14 is obtained upon hydrolysis. Moreover, the authors demonstrated the transformation of the synthesized product 14 into relevantly useful biological derivatives.
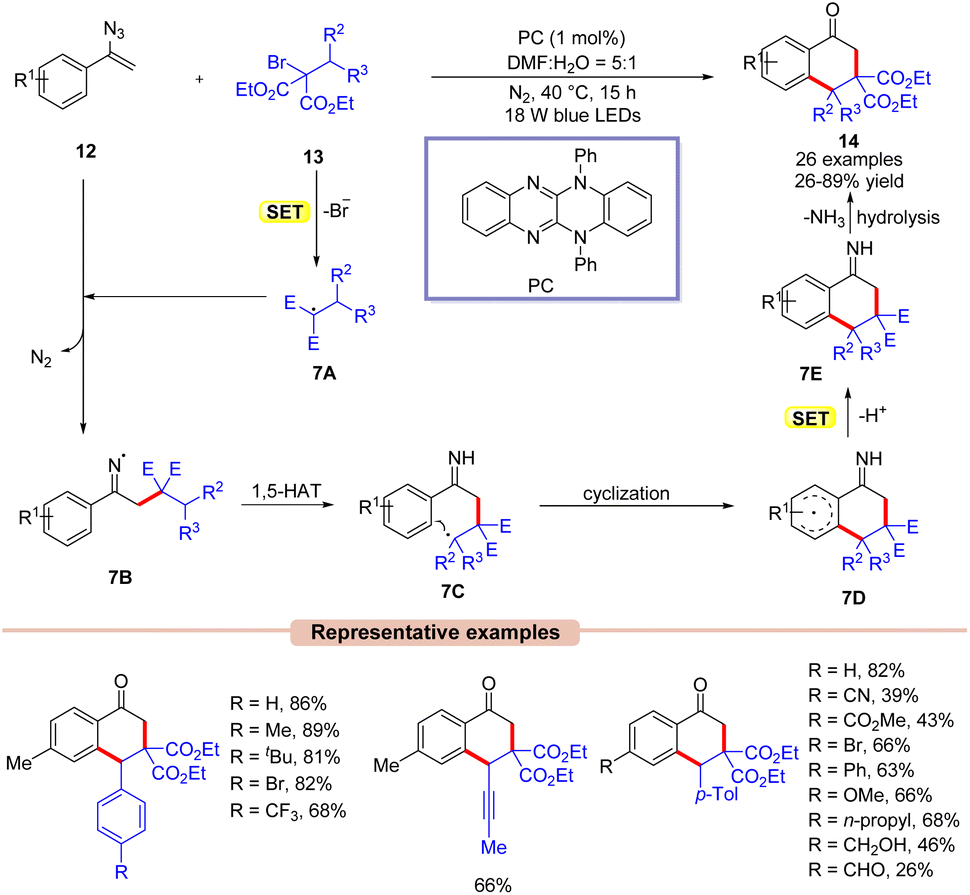 | ||
| Scheme 7 Visible light-mediated synthesis of tetralones with vinyl azides and alkyl bromides. | ||
Quinoline is one of the most widely explored scaffolds by synthetic chemists because of its broad range of biological activities.27 Zhou and co-workers in 2015, reported an interesting approach towards the synthesis of poly-substituted quinolines 16via a visible light Ir-photoredox catalyzed reaction of vinyl azides 12 and α-carbonyl benzyl bromides 15 (Scheme 8).28 The reaction parameters comprised Ir(ppy)3 as a photocatalyst, K2HPO4 as a base, and 18-crown-6 as an additive in MeCN/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). An array of substituted vinyl azides 12 reacted well with a wide range of α-carbonyl benzyl bromide derivatives 15, affording the desired products 16 in 40–78% yields. In contrast, alkyl-substituted vinyl azides failed to deliver the desired products 16 due to instability under visible light illumination. Stern–Volmer quenching and control experimental studies revealed that the excited Ir-photocatalyst is capable of reducing the α-carbonyl benzyl bromides 15. As shown in Scheme 8, the authors proposed that the α-carbonyl benzyl bromide derivative 15 was reduced by the excited catalyst *Ir(III) via SET, generating the carbon-centered radical intermediate 8A. Next, the radical addition of the carbon radical 8A to vinyl azide 12, led to the key iminyl radical intermediate 8B with the extrusion of N2. This intermediate 8B undergoes cyclization, accompanied by subsequent SET/deprotonation to give 2H-quinoline 8C. Furthermore, the base-mediated deprotonation of 8C led to another intermediate 8D, which may tentatively form an electron donor–acceptor (EDA)-complex with α-carbonyl benzyl bromide 15 to afford intermediates 8A and 8E. Thereafter, another subsequent SET-oxidation of 8E followed by deprotonation gives the desired product 16.
:1). An array of substituted vinyl azides 12 reacted well with a wide range of α-carbonyl benzyl bromide derivatives 15, affording the desired products 16 in 40–78% yields. In contrast, alkyl-substituted vinyl azides failed to deliver the desired products 16 due to instability under visible light illumination. Stern–Volmer quenching and control experimental studies revealed that the excited Ir-photocatalyst is capable of reducing the α-carbonyl benzyl bromides 15. As shown in Scheme 8, the authors proposed that the α-carbonyl benzyl bromide derivative 15 was reduced by the excited catalyst *Ir(III) via SET, generating the carbon-centered radical intermediate 8A. Next, the radical addition of the carbon radical 8A to vinyl azide 12, led to the key iminyl radical intermediate 8B with the extrusion of N2. This intermediate 8B undergoes cyclization, accompanied by subsequent SET/deprotonation to give 2H-quinoline 8C. Furthermore, the base-mediated deprotonation of 8C led to another intermediate 8D, which may tentatively form an electron donor–acceptor (EDA)-complex with α-carbonyl benzyl bromide 15 to afford intermediates 8A and 8E. Thereafter, another subsequent SET-oxidation of 8E followed by deprotonation gives the desired product 16.
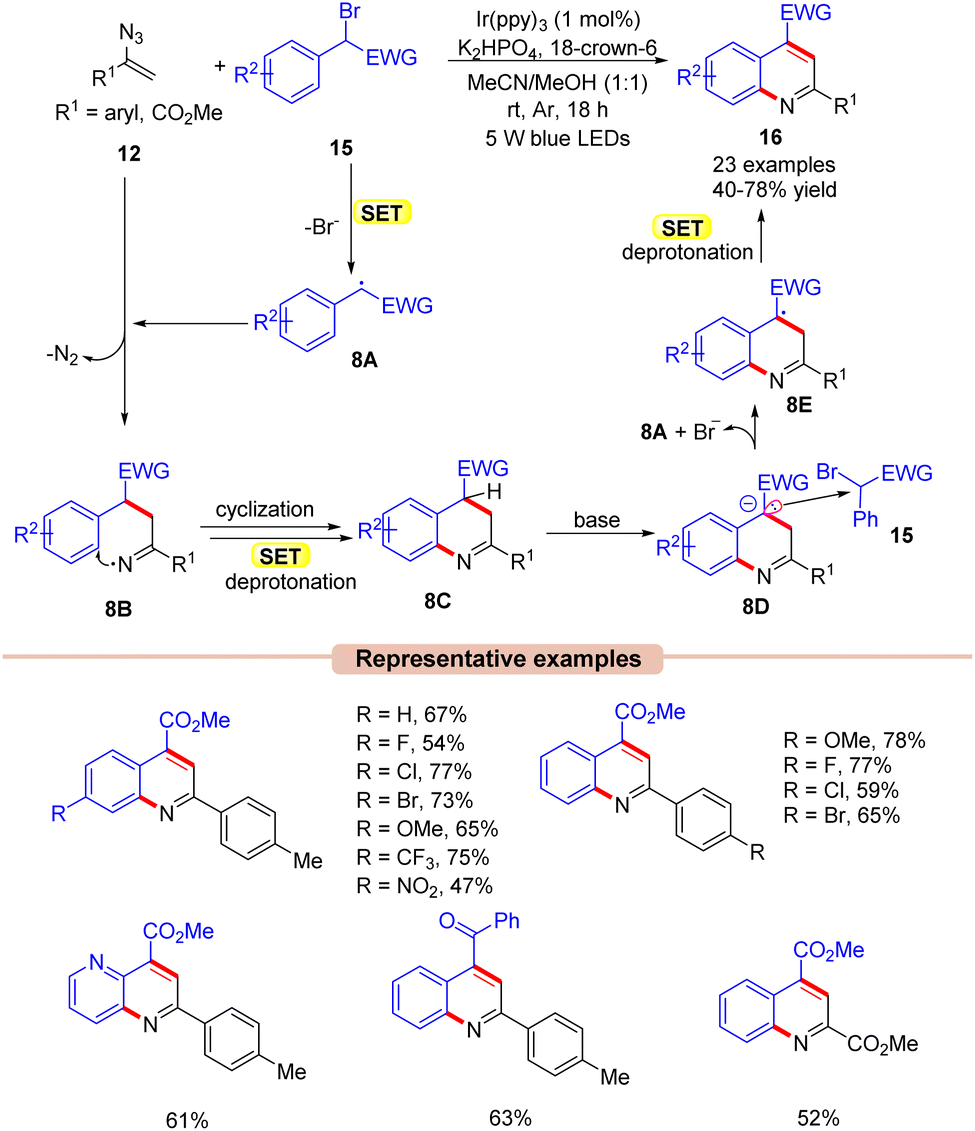 | ||
| Scheme 8 Ir-photoredox catalyzed synthesis of quinoline derivatives by the reaction of vinyl azides and α-carbonyl benzyl bromides. | ||
Thereafter, Meggers and co-workers in 2017,developed a visible light-mediated-asymmetric [2 + 3] cycloaddition of alkenes 17 with vinyl azides 12 producing a series of pyrrolines 18 in excellent yields (Scheme 9).29 Different substituents on the chiral auxiliary were screened under the given conditions. Amongst them, 3-(4-methoxyphenyl)pyrazole turned out to be the optimum one. The synthetic applicability of this approach was further demonstrated by the [2 + 3] photocycloaddition with a vinyl azide derived from ethisterone. Furthermore, this reaction could be scaled-up to a gram scale. In this protocol, the quantum efficiency ϕ = 0.19 suggested that the reaction did not involve a chain process. The general mechanistic approach for this asymmetric [2 + 3] cycloaddition reaction is shown in Scheme 9. Initially, the chiral chelate intermediate 9A is formed by the coordination of substrate 17 with the chiral Ru-catalyst. After photoexcitation, this intermediate forms the excited triplet state 9B. At this stage, it is trapped by vinyl azide 12 to deliver the catalyst-bound diradical intermediate 9C, which undergoes stereoselective cyclization to produce the desired pyrroline species 18.
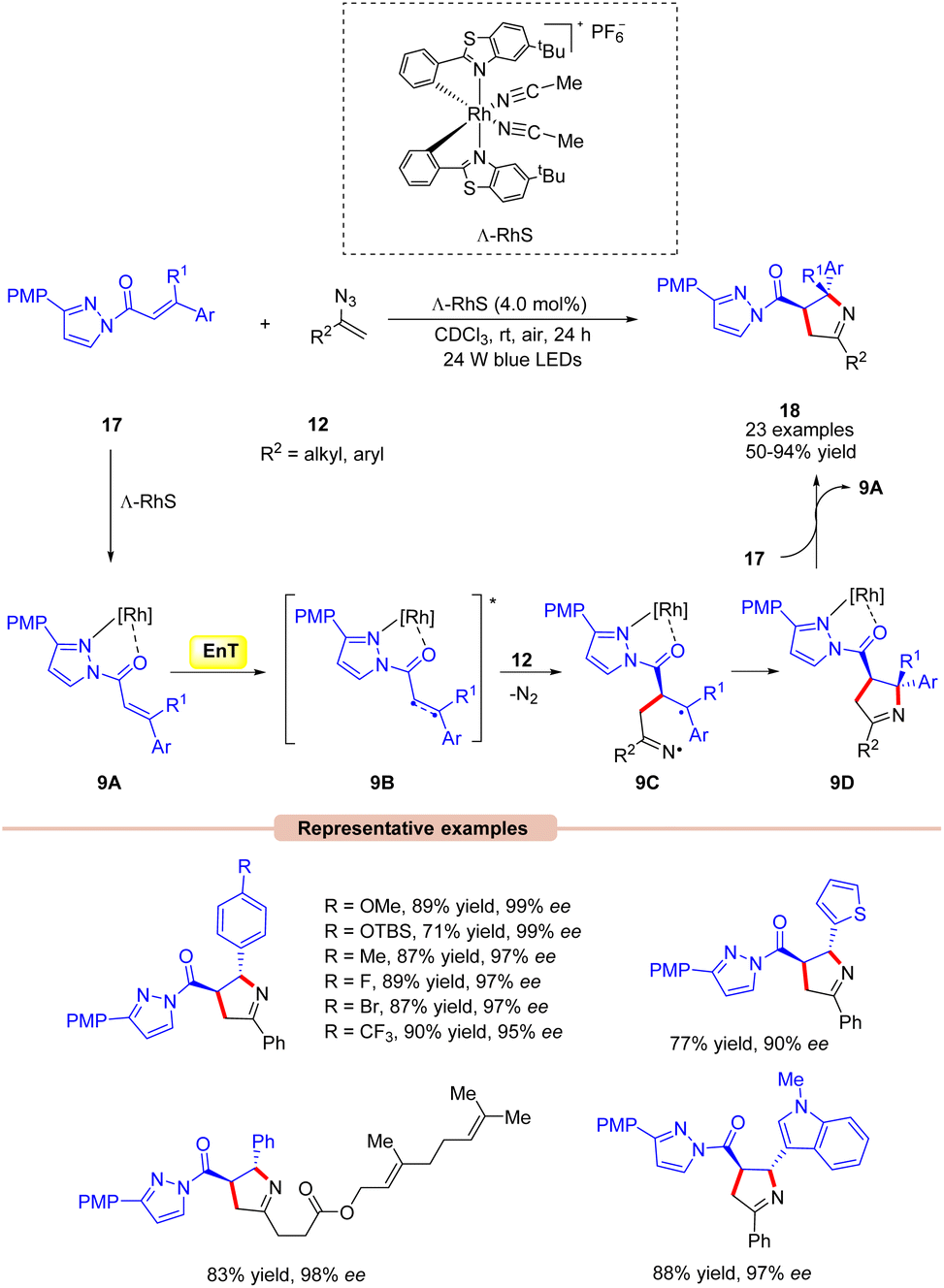 | ||
| Scheme 9 Chiral Rh-catalyzed [2 + 3] photocycloaddition reaction for the synthesis of pyrrolines. | ||
In 2022, Majee and co-workers developed a visible light-Rose Bengal catalyzed synthesis of 1-oxa-3-aza-spirooxazolines 21 by the reaction between quinones 19 with vinyl azides 20 (Scheme 10).30 In this protocol, a variety of α-phenyl vinyl azides 20 were readily coupled with benzoquinone and naphthoquinone 19 to afford product 21 in moderate to excellent yields. Moreover, the reaction was also carried out on a gram scale with good yields. Radical scavenging experiments confirmed that the reaction proceeds via a radical pathway. Based on these results, it was proposed that the photo-excited Rose Bengal transfers its energy to benzoquinone 19 to form the intermediate 10A. This intermediate 10A reacts with the vinyl azide to give the iminyl radical intermediate 10B, which undergoes an intramolecular radical coupling to give the final product 21.
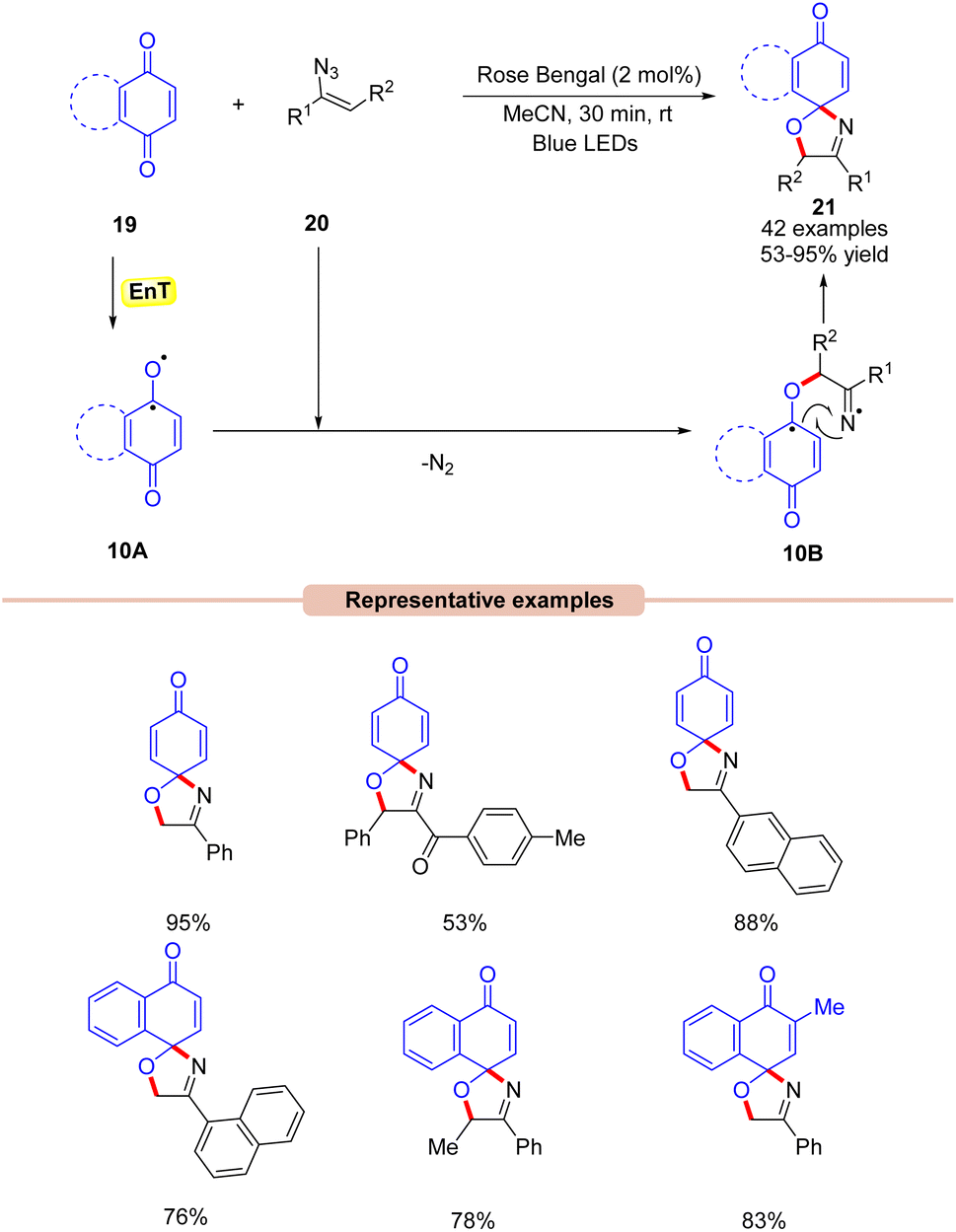 | ||
| Scheme 10 Rose bengal-catalyzed synthesis of 1-oxa-3-aza-spirooxazolines from vinyl azides. | ||
Under mild reaction conditions, this method provides access to 1-oxa-3-aza-spirooxazolines through subsequent C–O and C–N bond formation.
2.2. Formation of acyclic compounds
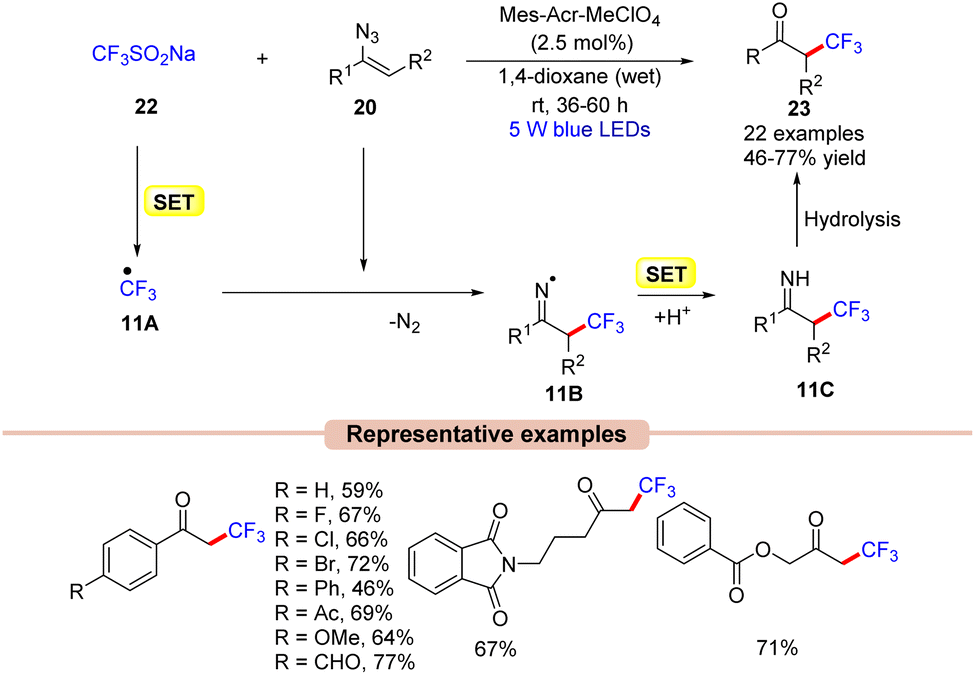
Recently, much attention has been given to the development of methods to incorporate fluorine into organic molecules because of its significant impact on biological properties, such as lipophilicity, binding affinity, and metabolic stability.31In this context, Liu and co-workers in 2017 disclosed an efficient strategy for the radical trifluoromethylation of vinyl azides 20 by the reaction of the Langlois reagent (CF3SO2Na) 22 using N-methyl-9-mesitylacridinium (Mes-Acr+) as the photocatalyst in 1,4 dioxane under blue LEDs (Scheme 11).32
 | ||
| Scheme 11 Acridinium photoredox catalyzed trifluoromethylation of vinyl azides with the Langlois reagent. | ||
In this protocol, alkyl, aryl, and heteroaryl-substituted vinyl azides 20 worked well under the optimized conditions to afford the desired trifluoromethylated product 23. Based on mechanistic studies, the authors proposed that the Langlois reagent 22 was oxidized by the photoexcited *Mes-Acr+, generating the ˙CF3 radical intermediate 11Avia a SET process. This intermediate 11A upon radical addition to vinyl azide 20 gives the iminyl radical intermediate 11B. Next, intermediate 11B upon SET/protonation gives access to the imine intermediate 11C. Eventually, intermediate 11C upon hydrolysis affords the desired product 23.
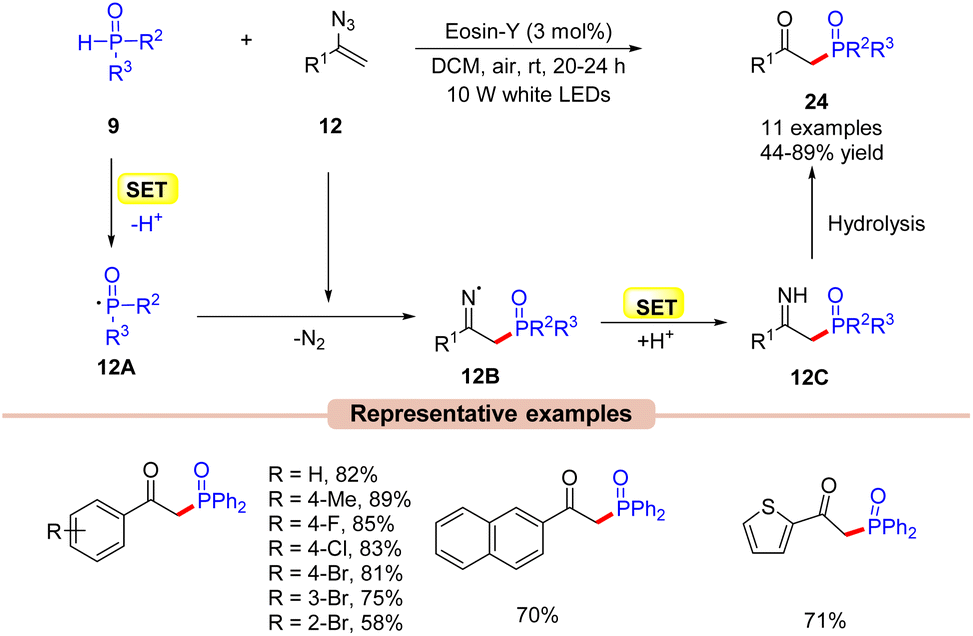
A similar setup can also be applied to the synthesis of phosphorous-containing organic compounds such as β-keto phosphine oxides. Kim and co-workers in 2020, explored a visible light-induced Eosin-Y catalyzed phosphorylation of vinyl azides 12 with phosphine oxides 9 to afford β-keto phosphine oxides 24 (Scheme 12).33 In this protocol, Eosin-Y was utilized as an organophotoredox catalyst in DCM under the irradiation of white LEDs (10 W) as a light source. Various substituents on vinyl azides 12 reacted efficiently to give the desired products 24 in 44–89% yields. Notably, the alkyl-substituted vinyl azide also delivered the required product in a 44% yield. In addition, this methodology was also extended to the synthesis of naphthyl- and heteroaryl-substituted β-keto phosphines in good yields. The authors also demonstrated that this reaction could occur at the gram scale level. As shown in Scheme 12, the authors proposed that phosphine oxide 9 is oxidized by the photoexcited catalyst *EY via a SET process, followed by deprotonation to give the key phosphonyl radical intermediate 12A. Thereafter, this intermediate 12A adds to vinyl azide 12 to give the iminyl radical intermediate 12B, which upon SET/protonation gives intermediate 12C. Lastly, the hydrolysis of intermediate 12C led to the β-keto phosphine oxide product 24. Unfortunately, despite mild reaction conditions and use of a metal-free photocatalyst, the reaction was marred by a limited substrate scope.
 | ||
| Scheme 12 Eosin-Y catalyzed phosphorylation of vinyl azides. | ||
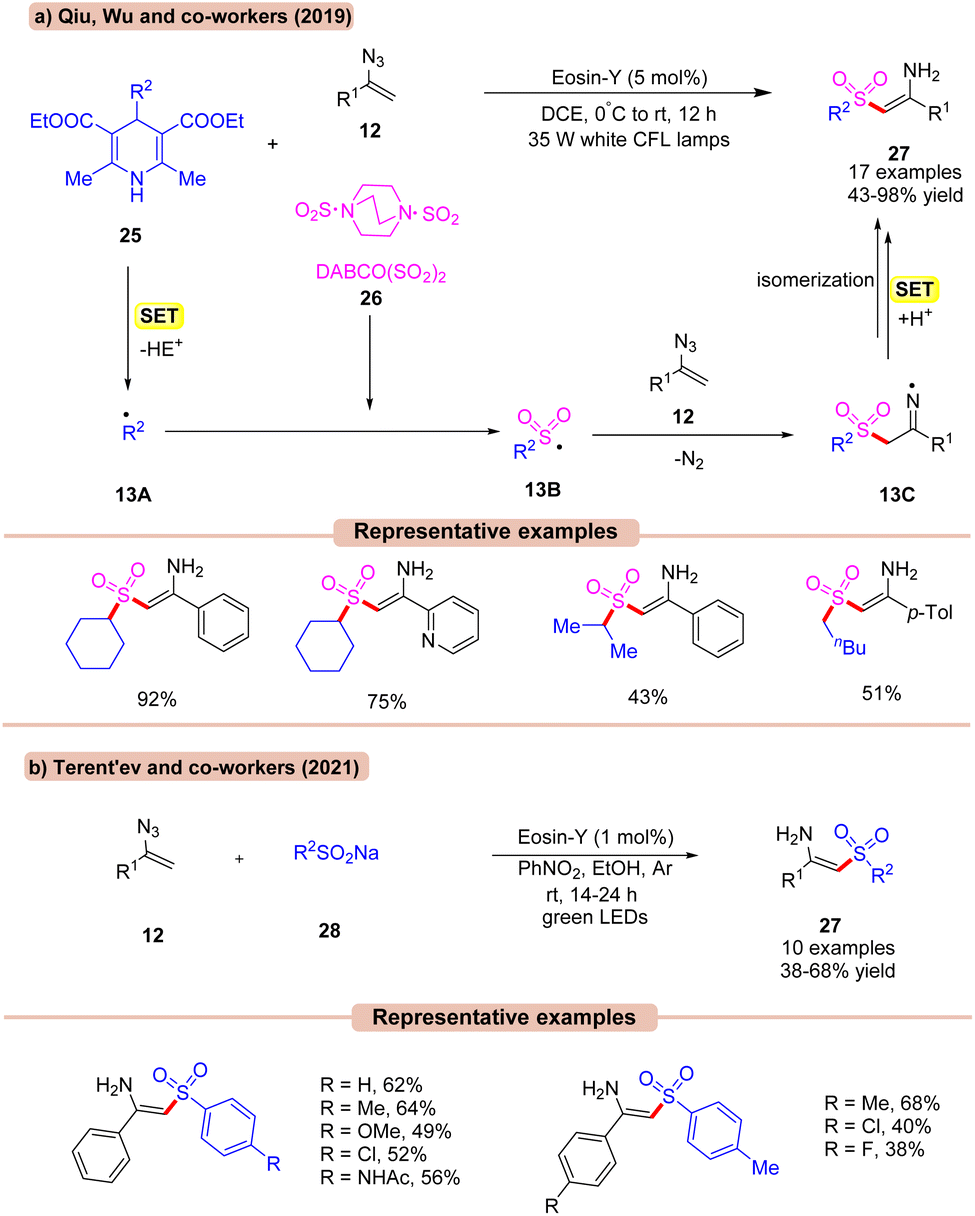
In 2019, Qiu, Wu, and co-workers reported a visible light Eosin-Y catalyzed multicomponent (MCR) reaction using vinyl azides 12, with substituted Hantzsch esters 25 as an alkyl radical reservoir and DABCO(SO2)226 as an SO2 source in DCE (Scheme 13a).34a In this methodology, a diverse range of N-unsubstituted enaminosulfones 27 were synthesized. Various substituents, including chloro and bromo groups attached to the phenyl ring of vinyl azides 12, led to the corresponding products 27 in moderate to good yields. Interestingly, the ortho-substituent on the phenyl ring of vinyl azides 12 also reacted smoothly. In addition, both primary and secondary (cyclic and acyclic) alkyl groups on the Hantzsch ester 25 worked well. The addition of TEMPO resulted in only a trace amount of the desired product, indicating that a radical process might be involved. Based on several control experiments, the authors proposed that the Hantzsch ester 25 was oxidized by the photoexcited catalyst *EY via SET to produce the alkyl radical 13A. This resulting radical intermediate is trapped by DABCO·(SO2)226 to give the alkyl sulfonyl radical intermediate 13B. This sulfonyl radical 13B reacts with vinyl azide 12 to generate the N-centered radical intermediate 13C. Next, intermediate 13C undergoes a SET/protonation, followed by isomerization to yield the final product 27 in 43–98% yields.
 | ||
| Scheme 13 Eosin-Y catalyzed synthesis of N-unsubstituted enaminosulfones from vinyl azides. | ||
In continuation with the previous report (Scheme 13a), Terent'ev and co-workers, in 2021, developed a similar method to synthesize N-unsubstituted enaminosulfones 26 in moderate yields using sodium sulfinates 28, Eosin-Y, and PhNO2 in EtOH under visible light irradiation (Scheme 13b).34b In this protocol, nitrobenzene was utilized as an electron shuttle in ethanol solvent. Despite a relatively narrow substrate scope, this strategy opens a potential route for the synthesis of N-unsubstituted enaminosulfones 27.
Amongst the two, the strategy by Qiu, Wu, and co-workers (Scheme 13a) displays a higher efficiency in terms of a wider substrate scope and higher reaction yield.
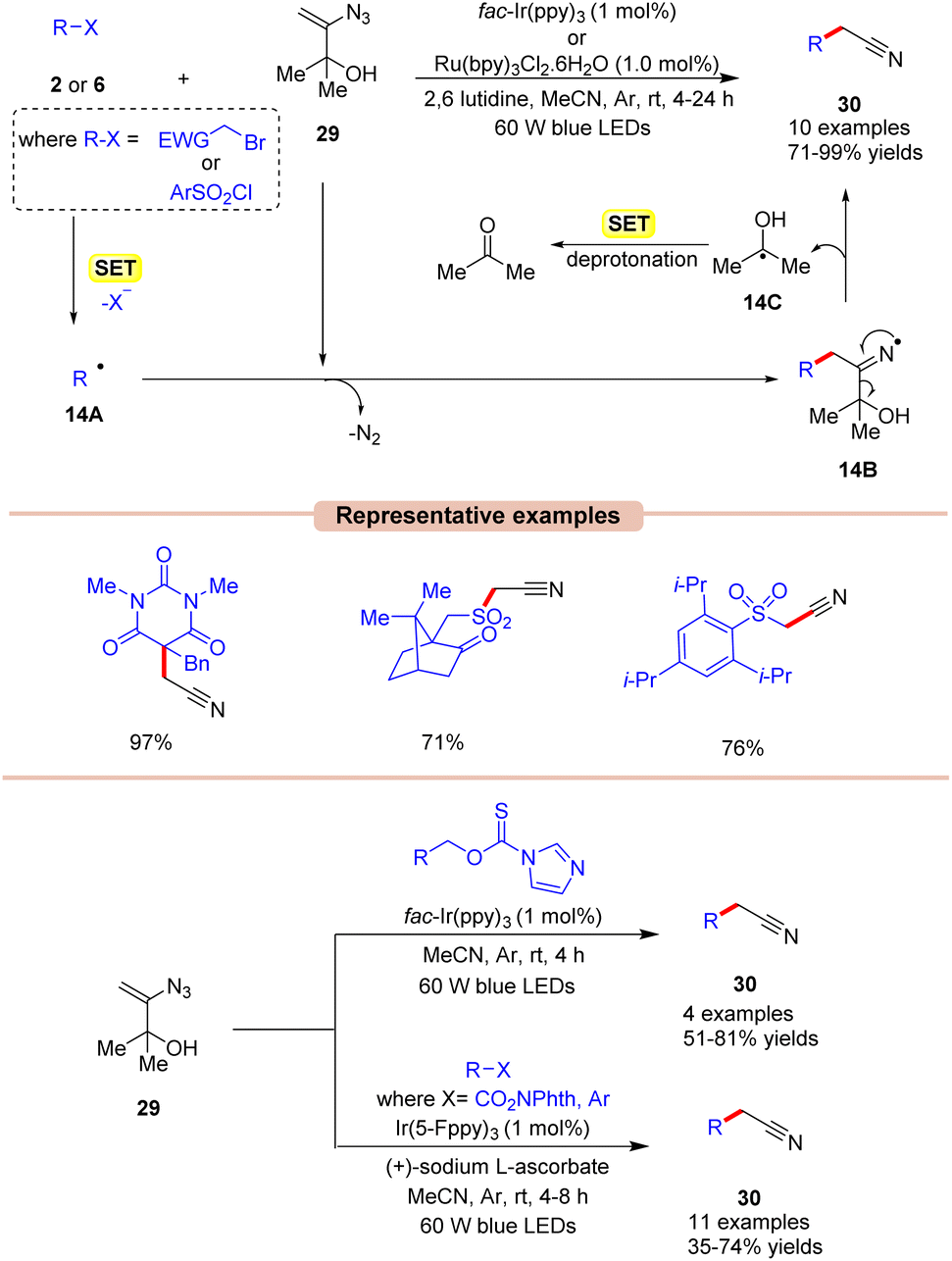
Nitriles are crucial functional groups having a wide range of biological activities, making them synthetic candidates of immense importance.35 In this framework, in 2019, Donald and co-workers described a cost-effective and operationally simple strategy for radical cyanomethylation via vinyl azides 29 in the presence of visible light illumination (Scheme 14).36 In this protocol, various vinyl azides 29 were explored by employing either fac-Ir(ppy)3 or Ru(bpy)3Cl2 as a photocatalyst, with 2,6 lutidine as a base in MeCN at room temperature. In this strategy, the base played a crucial role in neutralizing the accumulation of HBr and maintaining the pH of the reaction. The radical trapping experiment with the TEMPO reagent entirely suppressed the reaction, suggesting that a radical pathway might be involved in this protocol. As shown in Scheme 14, the authors proposed that 2-bromoacetophenone 2 is reduced by the excited photocatalyst via SET to give the alkyl radical 14A. This is followed by the radical addition of 14A to the double bond of vinyl azide 29, giving the iminyl radical intermediate 14B with the expulsion of dinitrogen. This intermediate 14B undergoes α-C–C cleavage to afford the desired nitrile product 27 along with intermediate 14C. Thereafter, the generated intermediate 14C upon SET gives ketone as the side product. This strategy was also applicable to the late-stage functionalization of various pharmaceutical drugs to afford the corresponding nitriles.
 | ||
| Scheme 14 Photoredox catalyzed radical cyanomethylation of vinyl azides. | ||
Under mild conditions, this strategy provides access to synthetically versatile cyanomethyl groups without the need for a strong base or external cyanide source.
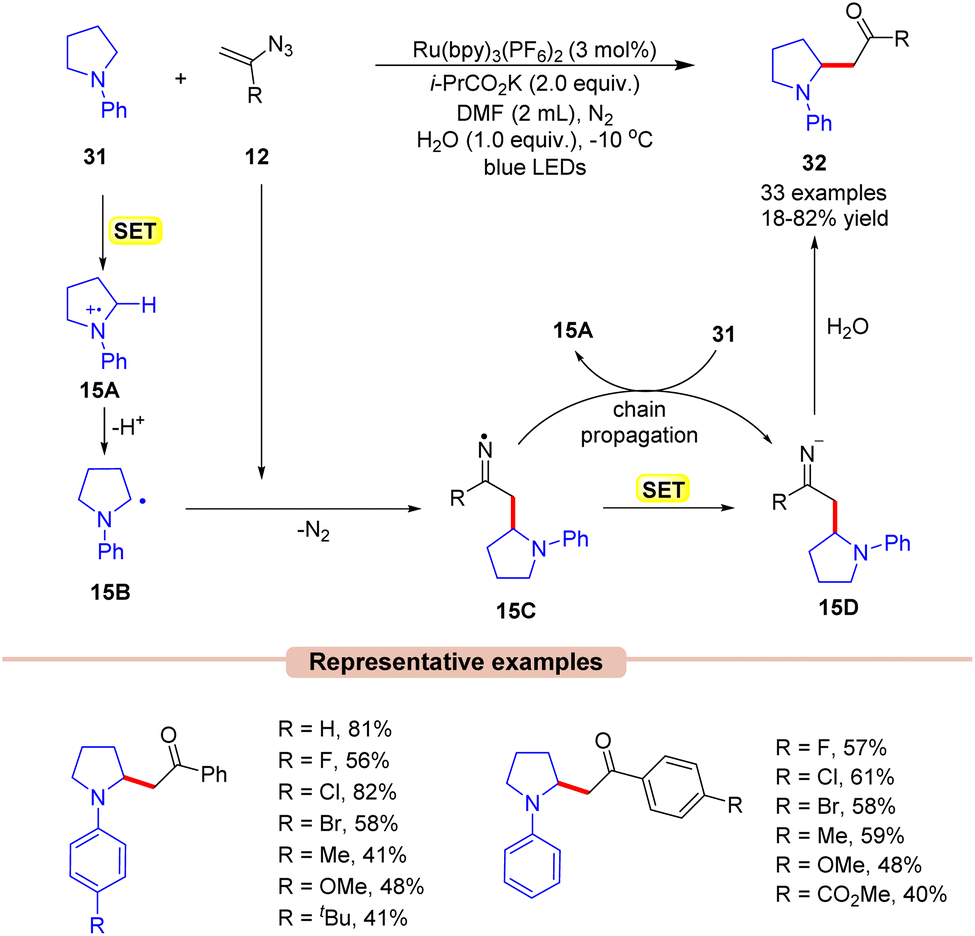
β-Amino ketones are one of the key building blocks present in many pharmacophores and natural products and act as an essential intermediate for the synthesis of crucial N-containing molecules such as β-amino acids, β-amino alcohols, and β-lactams.37 In 2019, Xu and co-workers developed a new, simple, and straightforward approach toward the synthesis of potent β-amino ketone derivatives 32via the reaction of vinyl azides 12 with tertiary arylamines 31 (Scheme 15).38 The authors employed Ru(bpy)3(PF6)2 as a photocatalyst with i-PrCO2K as a base in DMF under visible light irradiation. In this protocol, the addition of H2O to the reaction mixture also enhanced the reaction yields. Both electron-donating, as well as electron-withdrawing groups on vinyl azides 12 were found to be compatible with the reaction conditions. A variety of cyclic (5-membered, 6-membered, and 7-membered) and acyclic N-aryl amines 31 reacted smoothly to access the desired products 32. Unfortunately, the ortho-substituted aryl groups in N-aryl pyrrolidine did not afford the desired product due to steric hindrance.
 | ||
| Scheme 15 Ru-photoredox catalyzed α-alkylation of cyclic tertiary arylamines. | ||
A quantum yield of ϕ = 4.83 indicated that the reaction proceeded through a radical chain propagation. As represented in Scheme 15, under visible light irradiation, the excited photocatalyst *Ru(II) is reduced by amine 31 to produce the α-amino radical 15Bvia SET/deprotonation. This intermediate 15B adds to vinyl azide 12, leading to the iminyl radical intermediate 15C. Thereafter, the resulting radical intermediate 15C undergoes SET with the reduced photocatalyst, followed by hydrolysis, leading to the corresponding β-amino ketone product 32. The resulting intermediate 15C can further interact with 31, generating the radical intermediate 15A, thus continuing the radical chain process.
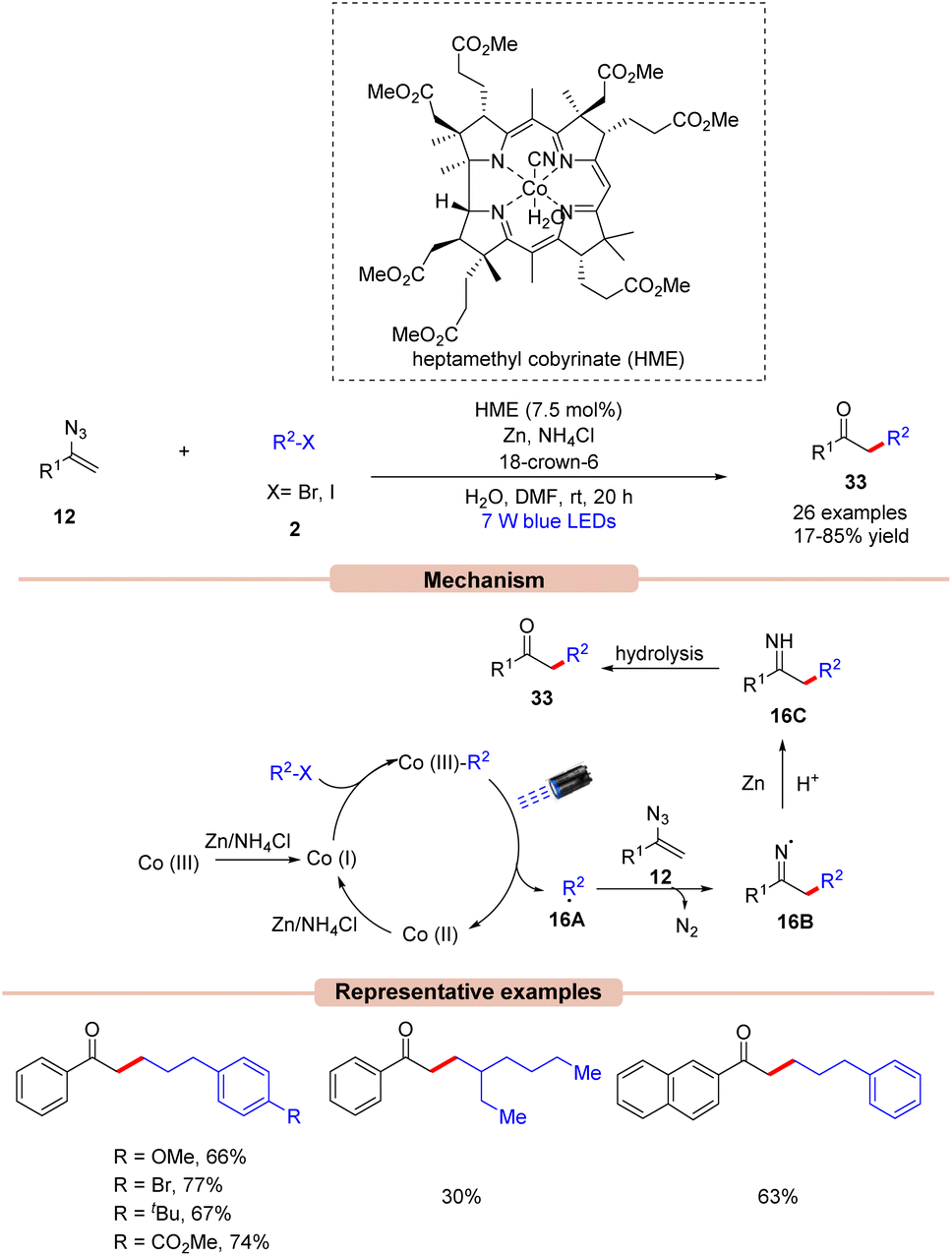
Impressively, Gryko and co-workers in 2021, demonstrated a visible light-induced vitamin B12-catalyzed synthesis of unsymmetrical ketones 33 from vinyl azides 12 and alkyl halides 2 (Scheme 16).39 Alkyl bromide 2 having electron-rich and electron-poor groups on the phenyl ring reacted well. Unfortunately, secondary bromides did not undergo the reaction probably due to steric hindrance. In addition, alkyl chloride and tosylate were also not found to be suitable coupling partners with vinyl azide. Mechanistically, HME [Co(III)] is promoted to its super nucleophilic form Co(I) in the presence of Zn/NH4Cl. Thereafter, Co(I) species reacts with alkyl bromides 2 forming alkyl cobalamin, which would generate the radical intermediate 16A upon visible light irradiation. The generated radical species 16A reacts with vinyl azide 12 to form the N-radical intermediate 16B. Finally, 16B is reduced by Zn, followed by protonation to give intermediate 16C. This is followed by hydrolysis to afford the unsymmetrical ketone derivatives 33 in 17–85% yields.
 | ||
| Scheme 16 Vitamin B12-catalyzed synthesis of unsymmetrical ketones from vinyl azides. | ||
3. Formation of 2H-azirine intermediate based reactions
The manifold reactivity of vinyl azides with the synthetic appeal of energy transfer catalysis offers a powerful strategy to achieve novel molecular structures. Majorly, the reactions involving vinyl azide via the EnT pathway form the key 2H-azirine intermediate. The rate of EnTs is dependent on the triplet excited state energy of the substrates and the excited photocatalyst. Kinetic studies and calculations revealed that the vinyl azides possess an excited-state triplet energy of 26–30 kcal mol−1, which may vary with the extended conjugation and electronic nature of the substituents on vinyl azides.2a This section highlights the visible light-mediated applications of vinyl azides via the 2H-azirine intermediate in organic synthesis.3.1. Formation of cyclic compounds
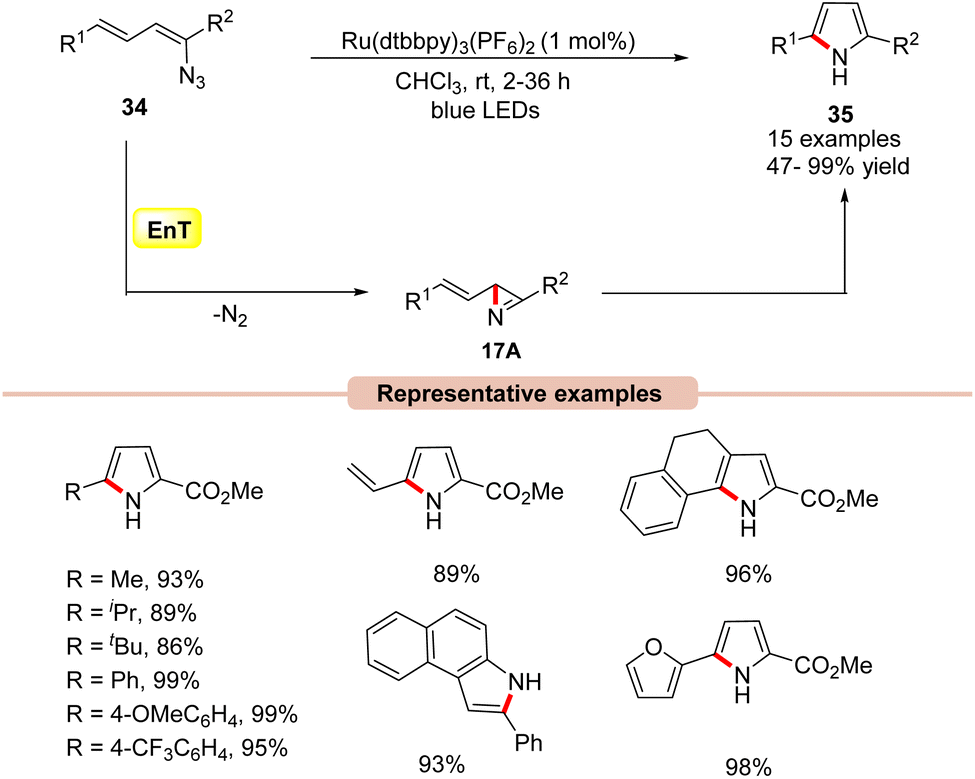
Pyrrole derivatives are omnipresent in many natural products, pharmaceuticals, biologically active molecules, and materials science.40 Several effective strategies such as the Knorr, Paal–Knorr, and Hantzsch synthesis, and 1,3-dipolar cycloaddition reactions are well known to synthesize pyrrole derivatives.41In this framework, the construction of functionalized pyrrole derivatives 35 was established by Yoon and co-workers in 2014, under visible light irradiation via intramolecular cyclization of dienyl azides 34 (Scheme 17).42 The reaction parameters revealed that Ru(dtbbpy)3(PF6)2 as a photocatalyst in CHCl3 under the irradiation of blue LEDs at room temperature was best suited for this protocol, affording the desired pyrrole products 35 in high yields. A range of dienyl azides 34 bearing aliphatic and aromatic functionalities smoothly underwent intramolecular cyclization to access the corresponding substituted pyrroles 35 in 47–99% yields. Notably, aryl azides were also well tolerated under these reaction protocols, affording the desired products in 75–93% yields. Experimental studies suggested the involvement of an EnT process due to the well-matched energy of the triplet excited vinyl azide 34 (ET = 45.4 kcal mol−1) and the photoexcited catalyst *Ru(II) (ET = 46 kcal mol−1). Trapping experiments with cyclopentadiene under control conditions resulted in a hetero-Diels–Alder cycloadduct product; this proved the involvement of the azirine intermediate 17A in the reaction mechanism. Based on these experimental outcomes, the authors postulated that the 2H-azirine intermediate 17A is formed under visible light irradiation, which then undergoes a formal intramolecular [3 + 2] cycloaddition to afford the desired pyrrole product 35.
 | ||
| Scheme 17 Visible light-mediated intramolecular cycloaddition of vinyl azides for the synthesis of pyrroles. | ||
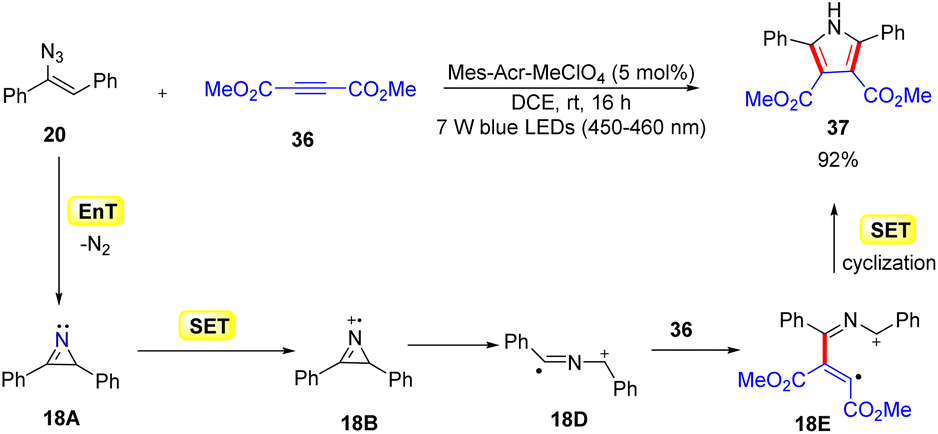
In 2014, Lu, Xiao, and co-workers disclosed another mild and efficient method for the construction of pyrroles 37via a [3 + 2] cycloaddition of activated alkyne 36 with the resulting 2H-azirines by using an organophotocatalyst (Scheme 18).43 The use of vinyl azides 20 and alkynes 36 as the starting materials with Mes-Acr+ as the organophotocatalyst resulted in the concise synthesis of functionalized pyrroles 37. During optimization, it was observed that the Ru and Ir-complexes were inefficient in catalyzing this transformation. The authors proposed that the reaction proceeded through a dual EnT and redox pathway. In this manner, the protocol represented an unusual photocascade reaction, which combines the EnT and SET processes. The reaction was initiated by the photosensitization of vinyl azide 20via an energy transfer (EnT) process with excited *Mes-Acr+, generating the 2H-azirine intermediate 18A. Next, intermediate 18A undergoes an SET event with the excited catalyst *Mes-Acr+, generating the 2H-azirine cation radical 18B. Subsequently, the radical cation 18B generates another radical cation 18Dvia ring-opening. Thereafter, the radical addition of 18D to the electron-deficient alkyne 36 forms intermediate 18E, which undergoes an SET process followed by intramolecular cyclization to form the required polysubstituted pyrrole derivatives 37.
 | ||
| Scheme 18 Dual EnT/SET photoredox catalyzed synthesis of pyrrole. | ||
Despite the narrow substrate scope, this strategy opened a potential route for the synthesis of the substituted pyrrole with excellent yields.
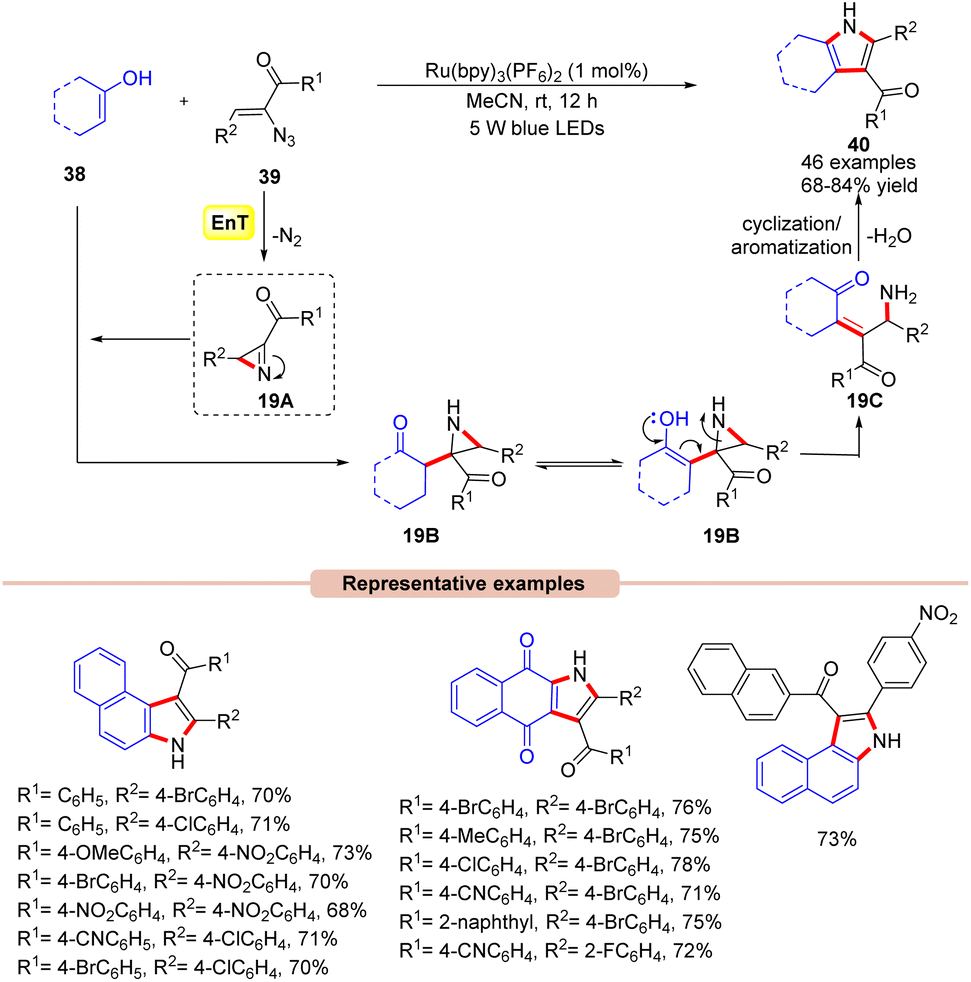
Impressively, another method for the synthesis of 2,3-fused pyrrole derivatives 40 was demonstrated by Maurya and co-workers in 2019 via a visible light-mediated intermolecular cyclization reaction between α-azido chalcones 39 with either 2-hydroxy-1,4-naphthoquinone or 1/2-naphthol 38 derivatives (Scheme 19).44 In this method, the authors utilized Ru(bpy)3(PF6)2 as a photocatalyst in MeCN to obtain the corresponding desired product 40 in 68–84% yields at room temperature with N2 and H2O as the only by-products. In this protocol, α-azido chalcones 39 bearing both electron-deficient and electron-rich functionalities on the phenyl ring reacted efficiently with a wide range of substituted 2-hydroxy-1,4-naphthoquinone or 1/2-naphthol 38 derivatives. Unfortunately, the reaction remained inert with 2,4-dimethylphenol, phenol, and resorcinol as coupling partners with α-azido chalcones 39 due to their propensity towards the unfavourable dearomatization/rearomatization step. In contrast, α-azido chalcones 39 with heteroaromatic rings were unable to give the target product. Moreover, the reaction could also be scaled up to the gram scale with a good yield (70%). Detailed spectroscopic studies confirmed the formation of the key 2H-azirine intermediate 19A. It is revealed that the nucleophilic attack of the enolic carbon of 38 on the reactive 2H-azirine intermediate 19A generates the aziridine intermediate 19B. The enolization of 19B followed by ring opening leads to intermediate 19C. Finally, intermediate 19C undergoes intramolecular cyclization/aromatization to yield the 2,3-fused pyrrole product 40.
 | ||
| Scheme 19 Ru-photocatalyzed synthesis of 2,3 fused pyrroles from α-azido chalcones. | ||
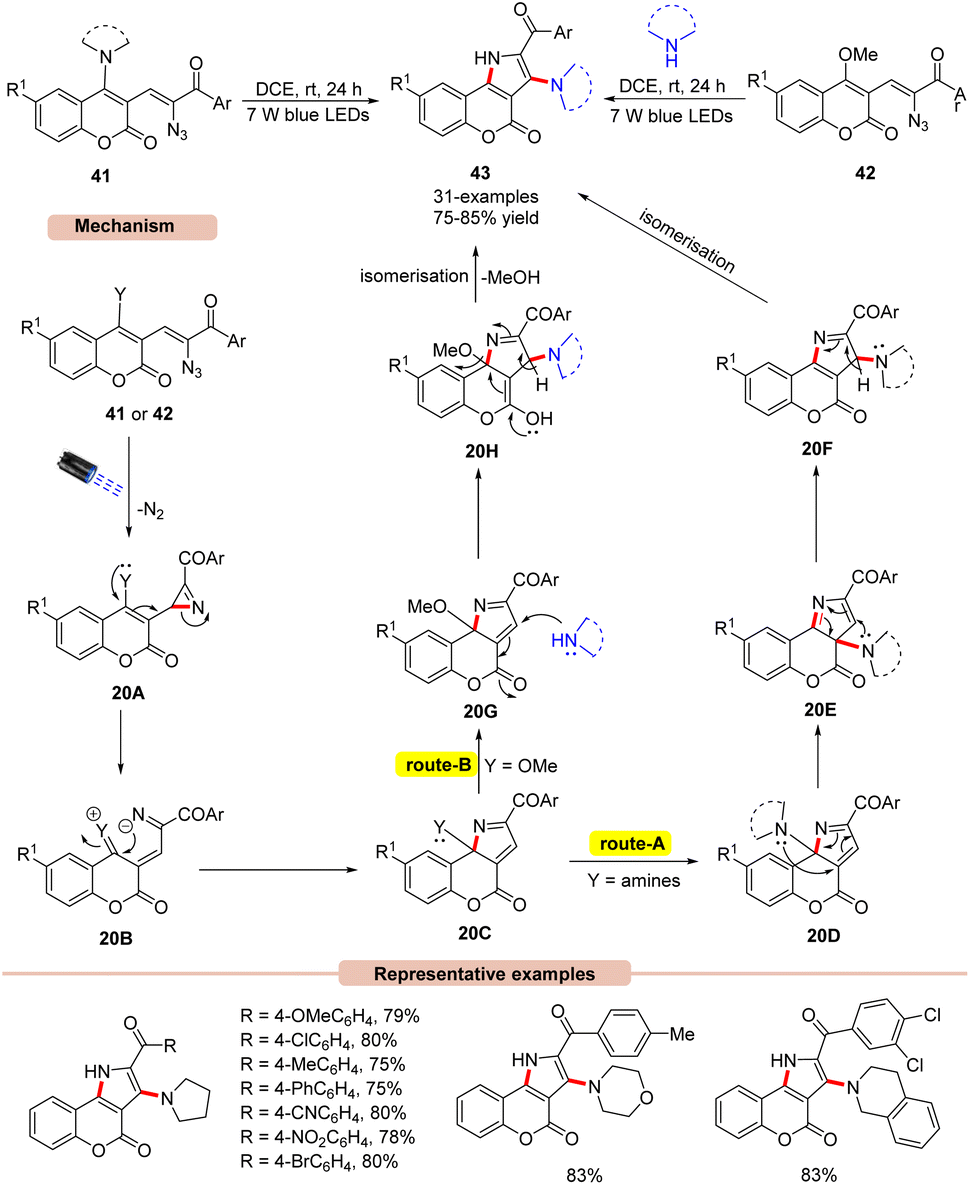
Later, the same group disclosed a unique photocatalyst-free visible light-assisted synthesis of 2,3-fused pyrroles 43 using α-keto vinyl azides 41 or 42 (Scheme 20).45 In the reaction protocol, it was observed that vinyl azides 41 bearing 1,2,3,4-tetrahydroisoquinoline, diethylamine, morpholine, and piperidine units were found compatible under the given reaction conditions, affording the desired pyrrole product 43 in 80–85% yields. Unfortunately, vinyl azides bearing primary and secondary amino groups failed to undergo the photodecomposition reaction. Impressively, methoxy-substituted vinyl azides 42 as starting materials reacted well with secondary amines under the reaction protocol to afford the required pyrrole product 43 in 75–84% yields. UV-visible absorption studies of vinyl azides 41 and 42 revealed a strong absorption of light due to the extended conjugation in their structure, thereby avoiding the use of photocatalysts in the reaction medium. Control experiments supported the formation of the key 2H-azirine intermediate 20A as well as the involvement of the 1,3-amino intramolecular migration. As illustrated in Scheme 20, in the presence of visible light irradiation, vinyl azide 41 or 42 is initially excited to its excited triplet state, which gives the 2H-azirine intermediate 20Avia the release of N2. The resulting intermediate 20A undergoes subsequent ring opening followed by intramolecular cyclization to give 20C. In route-A, the amine group of intermediate 20D undergoes two successive [1,5]-sigmatropic shifts to afford intermediate 20F, which upon isomerization yields the desired product 43. Similarly, route-B involves the nucleophilic attack of the secondary amine on intermediate 20G, which yields intermediate 20H. Lastly, the isomerization of 20H delivers the final pyrrole product 43. The striking features of this strategy include harvesting visible light energy without any external photocatalyst, compared to the previous methods (Schemes 17–19).
 | ||
| Scheme 20 Visible light-mediated synthesis of substituted pyrroles. | ||
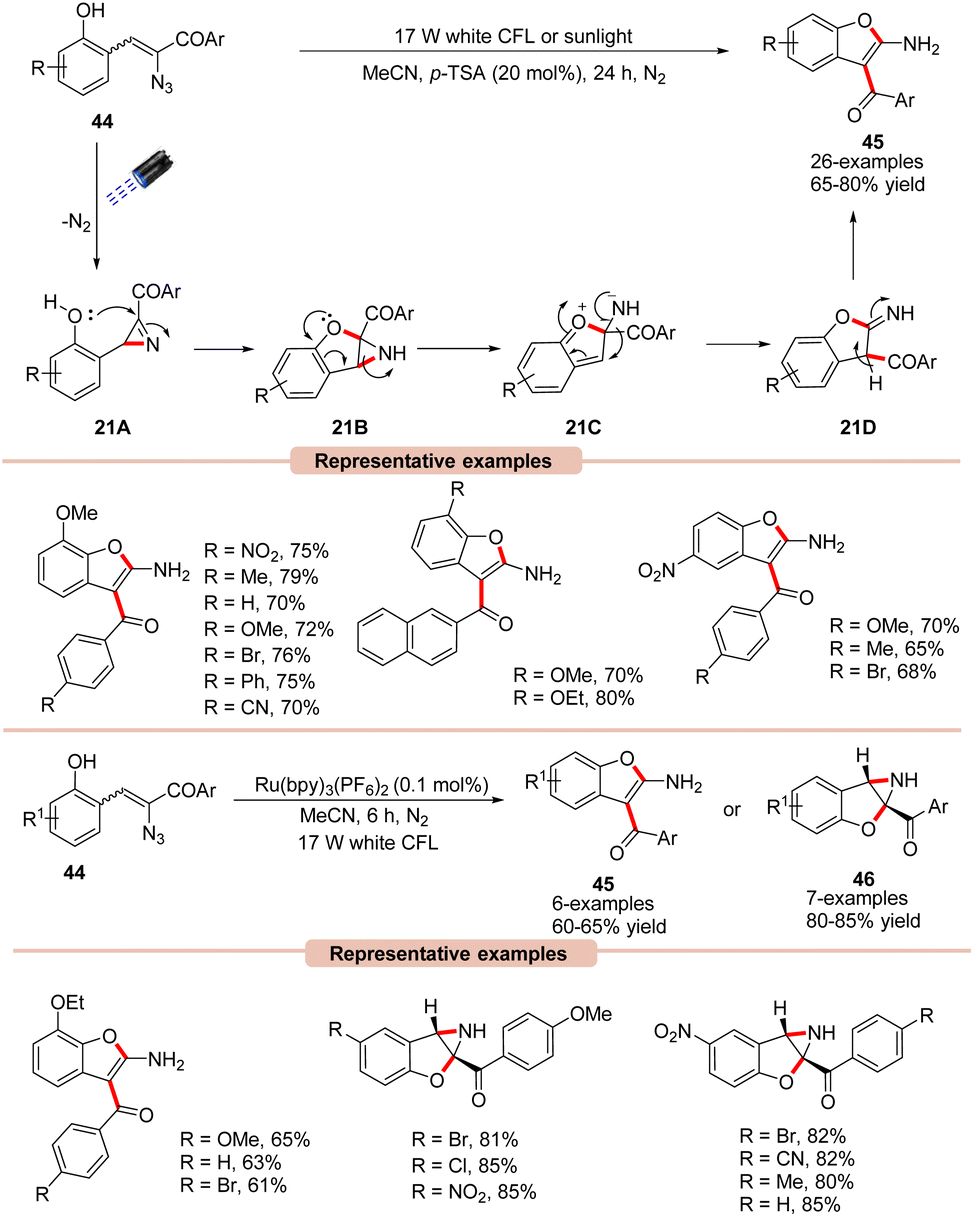
Benzofurans are important scaffolds in medicinal chemistry. Therefore, considerable research has gone into developing efficient synthetic methods for the construction of substituted benzofurans.46 In this regard, Maurya and co-workers reported a green and efficient photocatalyst-free synthesis of benzofuran derivatives 45via an intramolecular cyclization α-azido chalcones 44 (Scheme 21).47 This protocol takes advantage of the extended conjugation of α-azido chalcone derivatives 44, allowing it to act as a photosensitizer without the utilization of a photocatalyst, making it more sustainable, practical, and green in nature. UV-visible absorption studies further revealed that α-azido chalcones 44 exhibited an absorption maxima at 378 nm, with strong absorption of light in the 300–430 nm range. The controlled parameters suggested the involvement of an intramolecular 1,2-acyl migration. Moreover, Brønsted acid (p-TSA) was found to be very crucial for this transformation, facilitating the ring-opening of 2H-azirine via its interaction with the non-bonding electrons of the N-atom. Various α-azido chalcones 44 were well tolerated under the optimized conditions to afford the desired 2-aminobenzofuran product 45 in 65–80% yields.
 | ||
| Scheme 21 Visible light-assisted synthesis of 2-aminobenzofuran from α-azido chalcones. | ||
Impressively, this reaction could also be performed under irradiation with sunlight. As outlined in Scheme 21, the 2H-azirine intermediate 21A is generated from the α-azido chalcone 44 under irradiation with visible light. The resulting intermediate 21A undergoes intramolecular cyclization by the nucleophilic attack of the OH group, which leads to the aziridine intermediate 21B. This aziridine 21B undergoes ring-opening accompanied by a 1,2-acyl migration to yield intermediate 21D. Lastly, the isomerization of 21D leads to the desired product 45. Besides, it was observed that Ru(bpy)3(PF6)2 could also efficiently catalyze this reaction, affording the desired 2-aminobenzofuran product 45 in 60–65% yields, while α-azido chalcones 44 bearing electron-withdrawing functionalities lead to aziridine products 46 due to ring deactivation, preventing 1,2-acyl migration and isomerization.
2-Aminothiazoles are common building blocks found in natural products, pharmaceuticals, and agrochemicals and have remarkable biological activities including anticancer, anticonvulsant, antiviral, antidiabetic, and antituberculosis activities.48
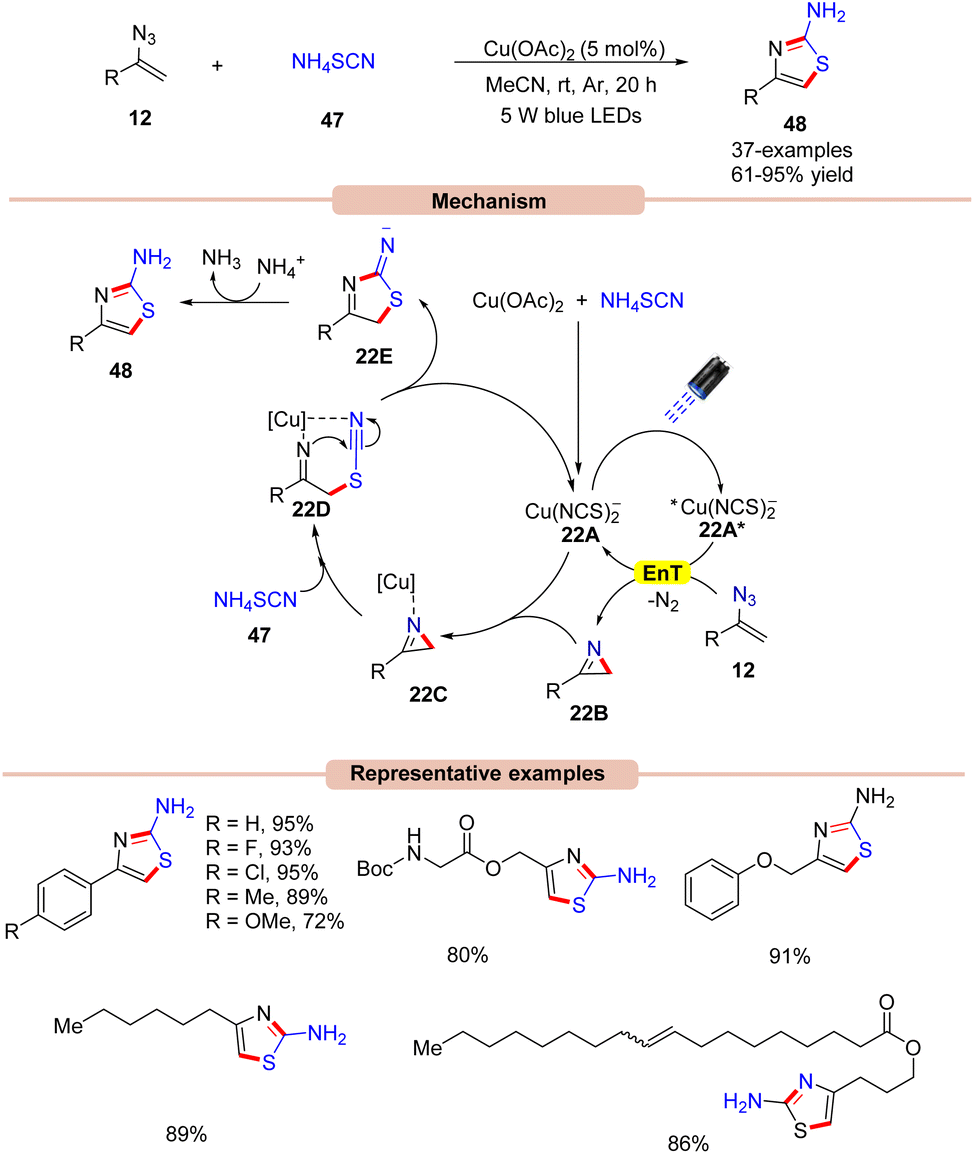
In 2017, Liu and co-workers developed a dual Cu-catalyzed synthesis of 4-alkyl/aryl-2-aminothiazoles 48 from ammonium thiocyanate 47 and vinyl azides 12 (Scheme 22).49
 | ||
| Scheme 22 Photo-mediated Cu-catalyzed synthesis of 4-alkyl/aryl-2-aminothiazoles. | ||
The control experiments indicated that the 2H-azirine 22B was the key intermediate in the reaction protocol and is formed via a Cu-photoredox catalyzed EnT process with vinyl azide 12. Interestingly, the in situ-formed copper species [Cu(NCS)2−] acts as a photocatalyst in this dual catalytic cycle. Cu(OAc)2 was found to be the most compatible out of various copper catalysts screened. UV-visible absorption studies of the mixture of Cu(OAc)2 and NH4SCN reveal an intense absorption band in the 350–560 nm region, which indicated an in situ formation of the photocatalyst. Based on these mechanistic findings, the authors proposed a mechanism, as shown in Scheme 22. The reaction is initiated by the photoexcitation of the copper catalyst 22A to give the excited state complex 22A*, which transfers its energy to vinyl azides 12 to produce the 2H-azirine intermediate 22B with the elimination of N2. Next, the resulting intermediate 2H-azirine 22B coordinates with the copper catalyst 22A to give the chelated intermediate 22C. Later, intermediate 22C upon nucleophilic attack by the thiocyanide nucleophile is converted into intermediate 22Dvia ring-opening. Finally, intramolecular cyclization of 22D releases intermediate 22E along with the regenerated copper catalyst 22A. Lastly, proton abstraction, followed by tautomerization forms the desired 2-aminothiazole derivatives 48. High yields, low catalytic loading, and an operationally simple strategy were the key advantages of this protocol.
Functionalized aziridines are the smallest saturated aza-heterocycles found in many natural biologically active compounds and are valuable precursors for the synthesis of diverse N-containing heterocyclic compounds.50
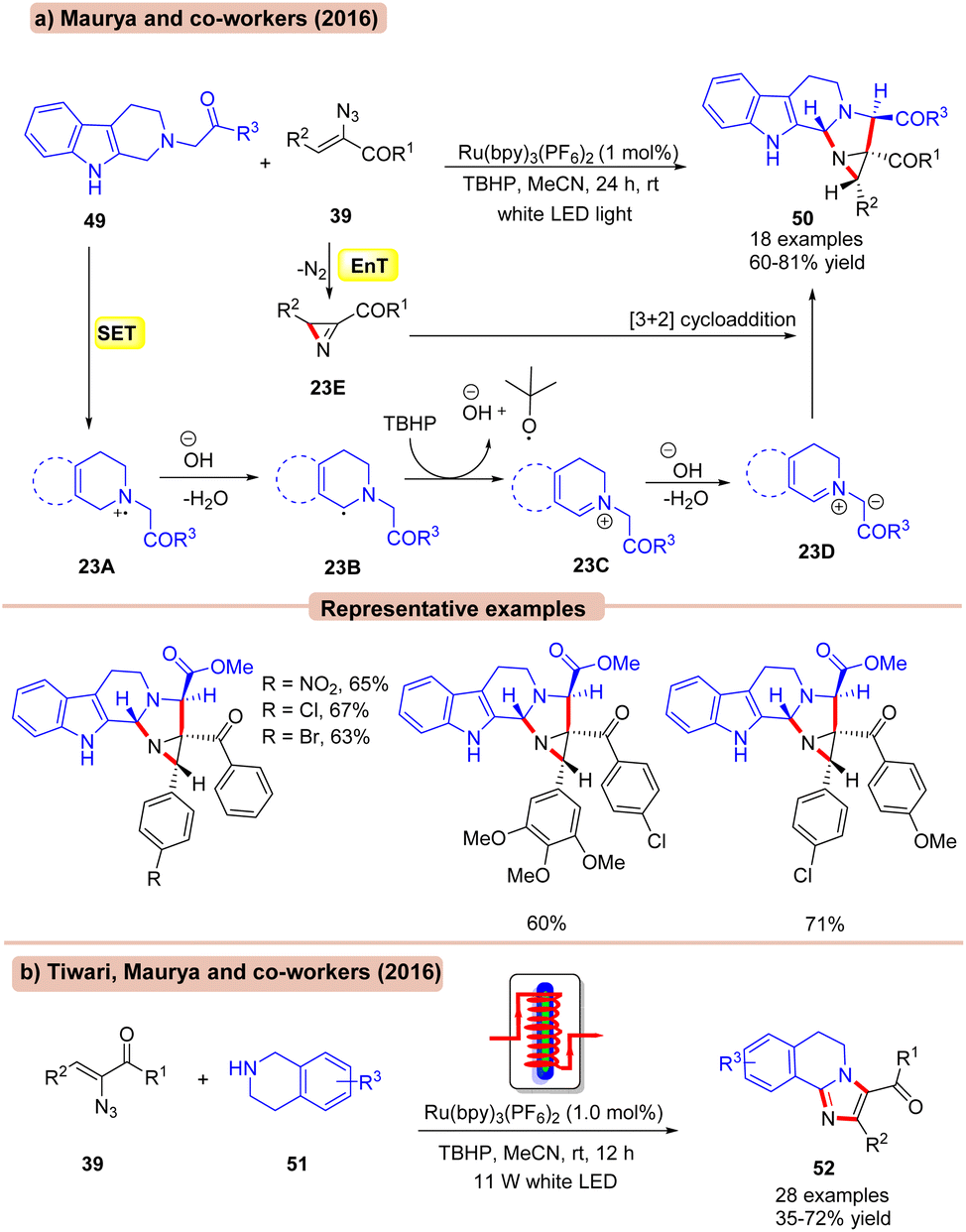
In this context, in 2016, Maurya and co-workers reported a dual EnT/SET Ru-photoredox catalyzed coupling of 1,2,3,4-tetrahydro-β-carbolines 49 with α-keto vinyl azides 39 for the synthesis of functionalized aziridines 50 using tert-butyl hydroperoxide (TBHP) as an oxidant in MeCN (Scheme 23a).51a This protocol exhibits a broad substrate scope and functional group compatibility by successfully employing a variety of ester and ketone-containing β-carboline substrates 49. Unfortunately, 1,2,3,4-tetrahydroisoquinoline remained an inactive coupling partner in this conversion. Also, heteroaromatic and aliphatic vinyl azides failed to deliver the target products. Mechanistically, 1,2,3,4-tetrahydro-β-carbolines 49 is oxidized by the photoexcited *Ru(II) via SET to generate the aminyl radical cation 23A, which upon deprotonation leads to the α-amino radical species 23B. Later, the radical intermediate 23B is further oxidized by TBHP to give the iminium ion 23C, accompanied by deprotonation to form the key 1,3-dipolar species 23D. This resulting intermediate 23D undergoes [3 + 2] cycloaddition with the azirine intermediate 23E, which was initially formed by the photosensitization of the α-azido chalcone 39 resulting in the functionalized aziridine product 50.
 | ||
| Scheme 23 Ru-photoredox catalyzed coupling of secondary amines with vinyl azides. | ||
In continuation, Tiwari, Maurya and co-workers disclosed the synthesis of structurally diverse imidazoles 52 by the interaction of tetrahydroisoquinolines 51 and α-keto vinyl azides 39 (Scheme 23b).51b In this strategy, Ru(bpy)3(PF6)2 was employed as a photocatalyst with TBHP as an oxidant in MeCN using a flow microreactor at room temperature as the ideal reaction conditions. Other photocatalysts such as Eosin-Y, Rose Bengal, and Ru(bpy)3(Cl)2 were ineffective in catalyzing this reaction. A broad range of vinyl azide 39 bearing substituted aryl and hetero(aryl) functionalities reacted efficiently with tetrahydroisoquinolines 51, affording the desired imidazole products 52 in 53–72% yields. Besides, the reaction was also compatible with acyclic secondary amines such as dibenzyl amine, N-benzyl-p-anisidine, and N-benzyl aniline, affording the targeted products 52 in 35–58% yields. The desired product 52 is formed via a mechanistic pathway similar to that discussed above.
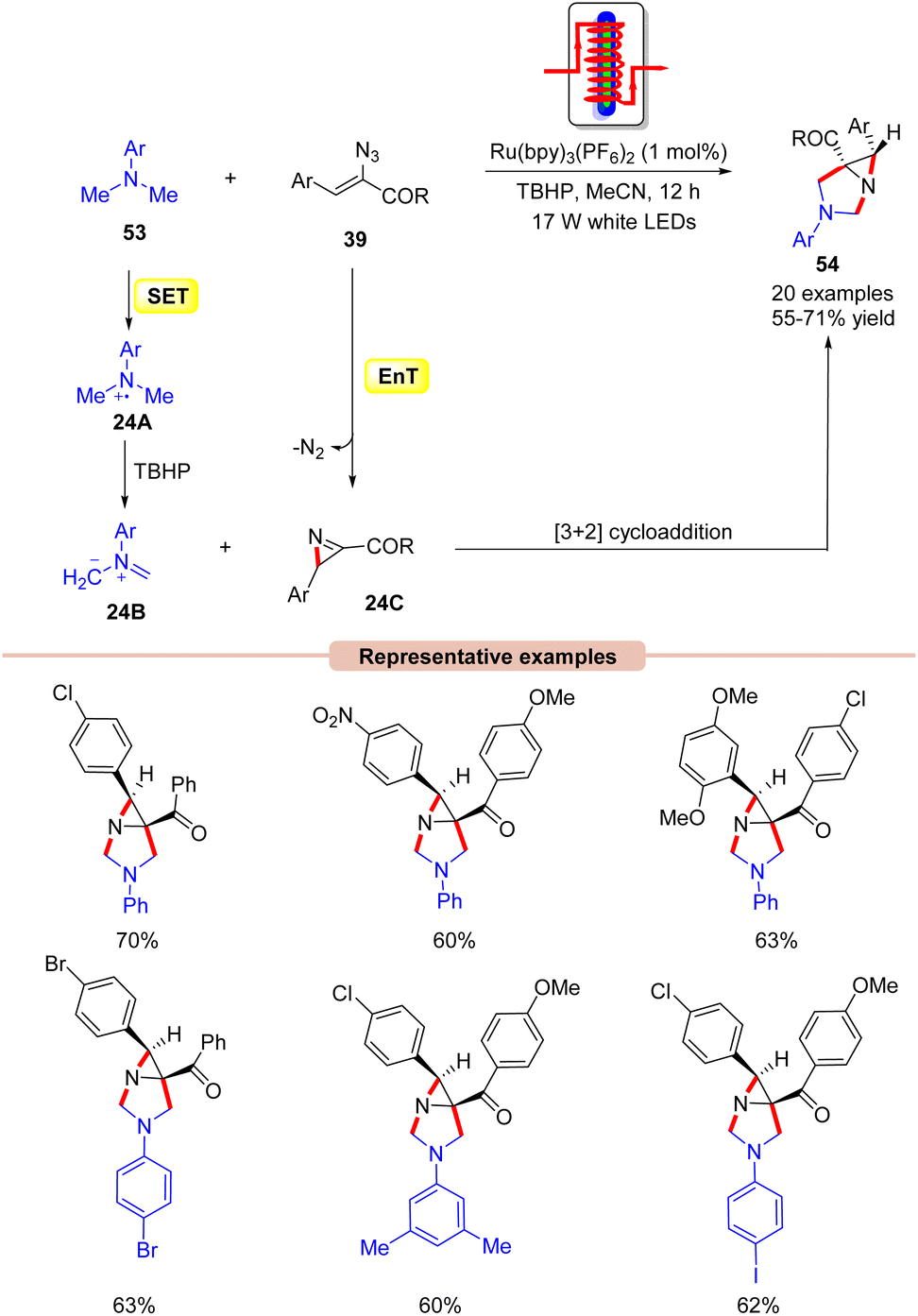
Interestingly, Maurya and co-workers in 2017, extended their work to the diastereoselective synthesis of functionalized aziridine 54 by the coupling reaction of N,N-dimethylanilines 53 with α-azido chalcones 39 (Scheme 24).52 In this strategy, Ru(bpy)3(PF6)2 functions as a dual-energy and electron-transfer photocatalyst, efficiently catalyzing the cycloaddition reaction in the presence of TBHP as an oxidant under the irradiation of 17 W white LEDs. Other photocatalysts such as Ir-complex, Rose Bengal, and Eosin-Y were also screened but were found to be ineffective for this transformation. It is worth noting that two sp3 C–H bonds were activated at once in this reaction. Moreover, vinyl azides 39 with electron-withdrawing and electron-rich substituents on the aromatic ring worked efficiently, affording the desired products 54 in 55–71% yield. In contrast, heteroaromatic and aliphatic substituents on vinyl azides remained inert as in the previous case. As shown in Scheme 24, the authors revealed that the photoexcited catalyst *Ru(II) is reduced by N,N-dimethylaniline 53via a SET process, generating the amine radical cation intermediate 24A, which is oxidized by TBHP, generating the non-stabilized azomethine ylide 24B. At the same time, the photoexcited catalyst *Ru(II) can transfer its energy to vinyl azides 39, generating an excited vinyl azide, which decomposes to yield the 2H-azirine intermediate 24C. Lastly, the azomethine ylide 24B undergoes a [3 + 2] cycloaddition reaction with the 2H-azirine intermediate 24C to give the final product 54.
 | ||
| Scheme 24 Ru-photoredox catalyzed synthesis of functionalized aziridines. | ||
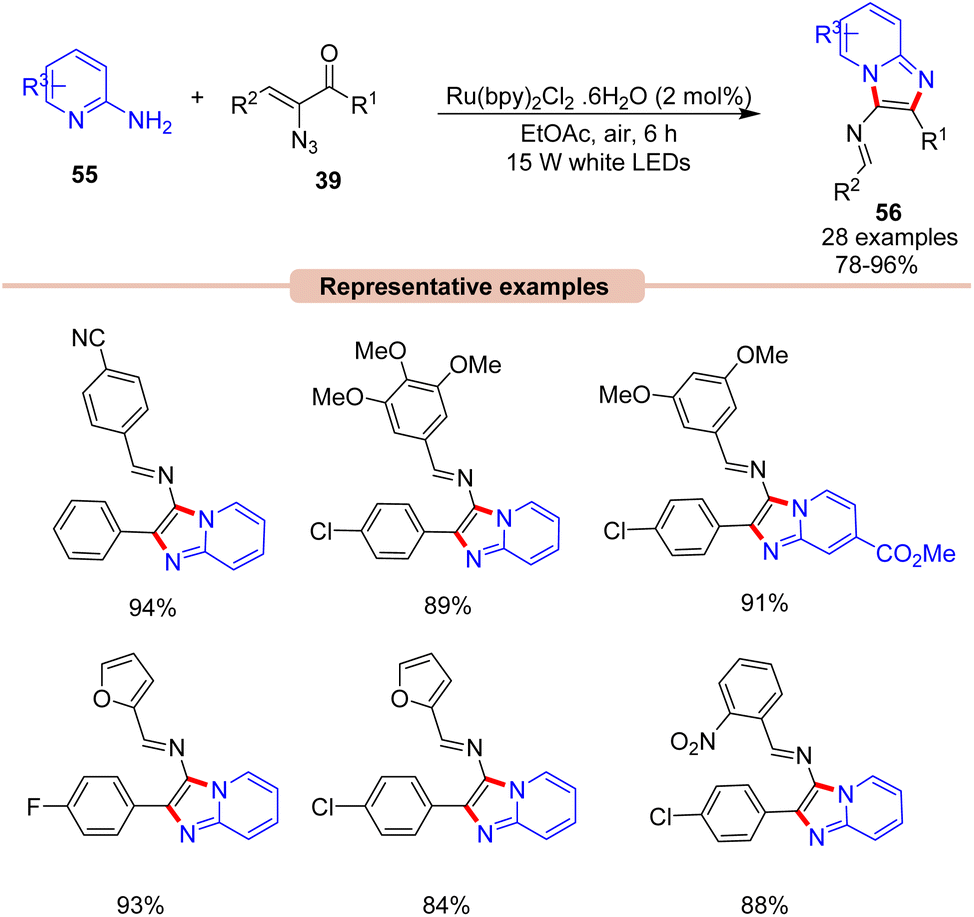
Later that year, Kamal, Maurya, and co-workers, reported a visible light-induced coupling of 2-aminopyridines 55 with α-ketovinyl azides 39 for the direct construction of imidazo[1,2-a]pyridines 56 (Scheme 25).53 This one-pot protocol comprised the Ru(bpy)3(PF6)2 photocatalyst in EtOAc under the irradiation of white LEDs as the ideal reaction condition to afford the corresponding imidazopyridine product 56. This reaction displayed a wide substrate scope, α-keto vinyl azides 39 bearing alkyl, aryl, and heterocyclic substituents with OMe, CN, Cl, Br, and NO2 functionalities were well tolerated, providing the desired product 56 in 78–96% yields. Control experiments supported the formation of a 2H-azirine intermediate.
 | ||
| Scheme 25 Ru-photoredox catalyzed synthesis of imidazopyridine derivatives. | ||
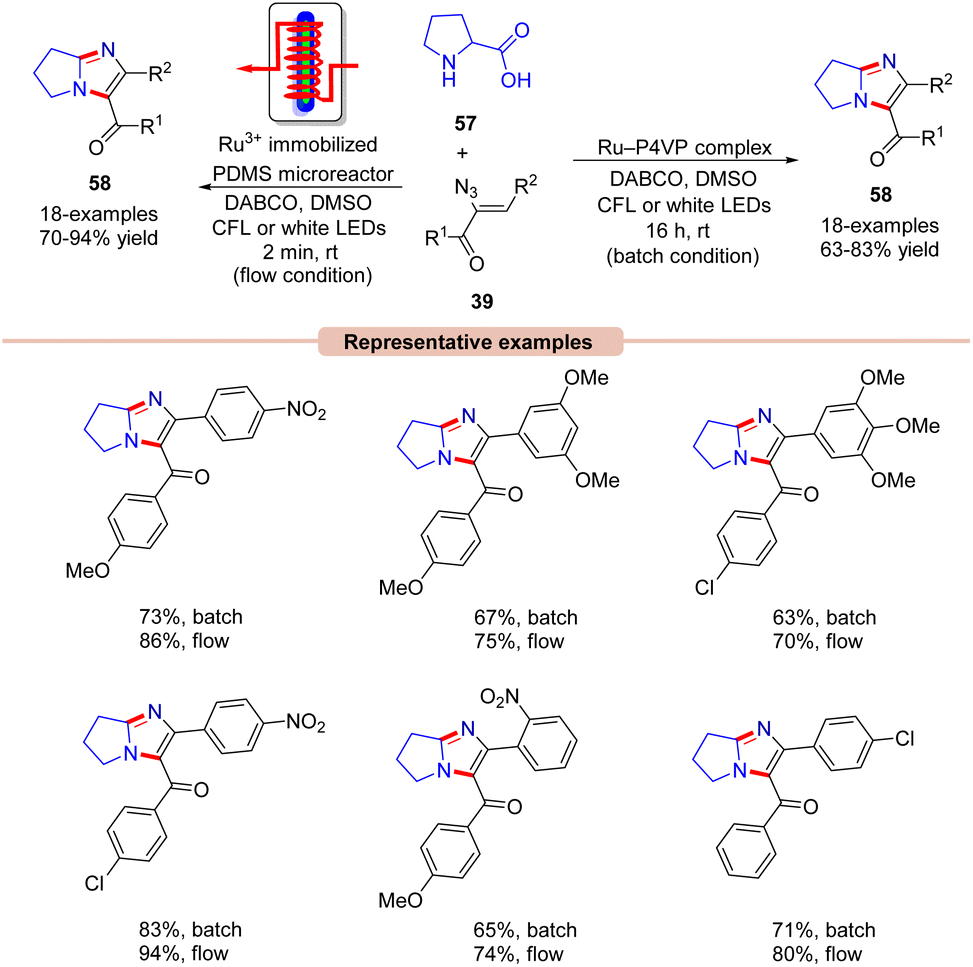
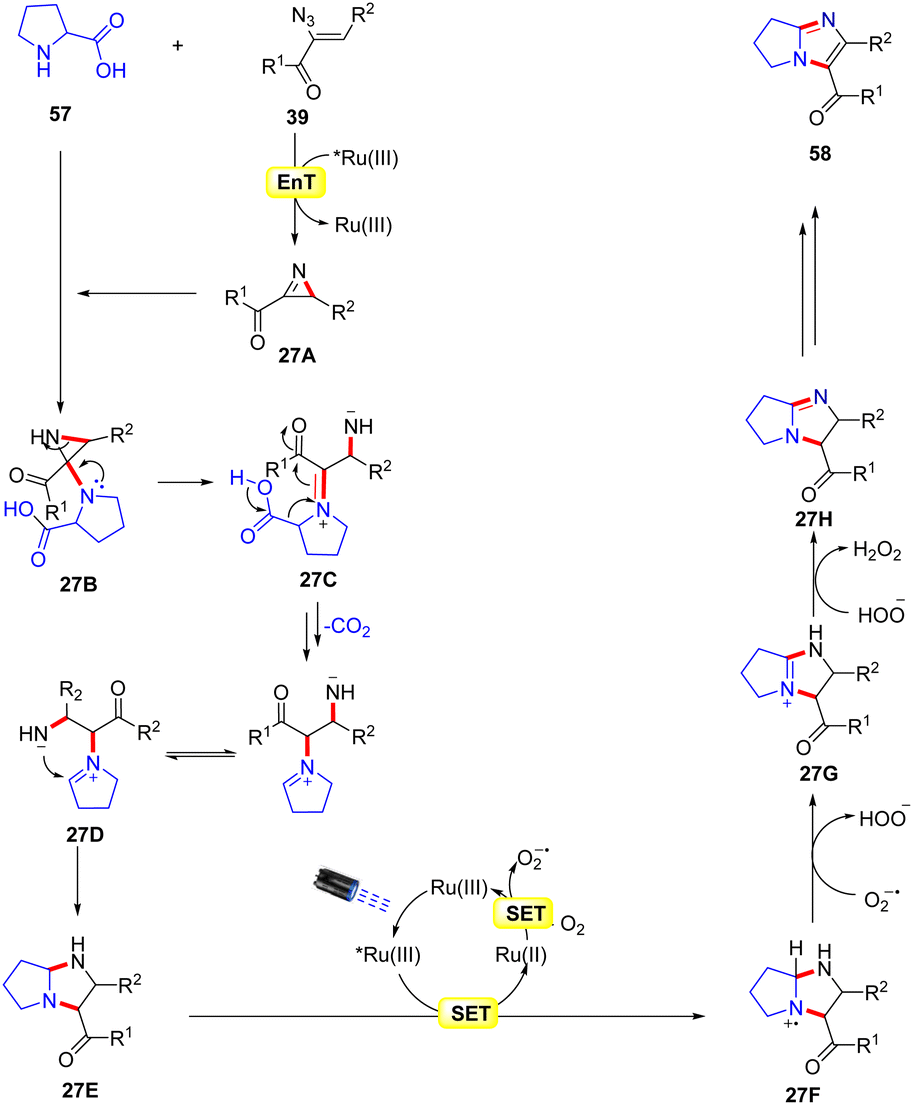
A similar method was proposed by Adiyala, Kim, and co-workers in 2020 for the construction of fused imidazole derivatives 58via decarboxylative cyclization of α-azido chalcones 39 with L-proline 57 using a continuous-flow microreactor (Scheme 26).54 Initially, the authors employed a polyvinyl pyridine (P4VP) complex of ruthenium(III) chloride as an efficient photocatalyst under batch and flow conditions, which was prepared with ease and found to be stable as it could be recycled up to six times. Other photocatalysts such as Ru(bpy)3Cl2·6H2O, Ru(bpy)3PF6, and Ir(ppy)6 were also reactive in catalyzing the reaction, affording the desired product 58 in comparable yields. A wide range of α-azido chalcones 39 reacted well with L-proline 57 to give the desired imidazole product 58 in 63–83% yields. Notably, other amino acids such as glycine, alanine, leucine, tryptophan, and isoleucine did not respond to the reaction and α-azido chalcones 39 bearing substituted aliphatic and heteroaromatic functionalities remained inactive under the optimized conditions. The authors further modified the strategy using an Ru(III) immobilized PDMS microreactor, affording the target products in 70–94% yields under 2 min of reaction time. Experimental investigations revealed the formation of the key 2H-azirine intermediate 27Avia an EnT process. Moreover, a radical pathway was suggested to be involved in the radical trapping experiment. Based on these experimental findings, the authors put forward a mechanism, as illustrated in Scheme 27. Mechanistically, the photo-excited catalyst *Ru(III) undergoes an EnT process with α-azido chalcone 39 to give the 2H-azirine intermediate 27A, which reacts with L-proline 57, producing intermediate 27B.
 | ||
| Scheme 26 Ru-complex synthesis of imidazoles from vinyl azides and α-amino acids. | ||
 | ||
| Scheme 27 Ru-complex synthesis of imidazoles from vinyl azides and α-amino acids. | ||
This intermediate 27B undergoes a ring opening reaction followed by decarboxylation to afford the zwitterion 27D. This resulting intermediate 27D triggers an intramolecular cyclization, leading to the hexahydro-1H-pyrrole[1,2-a]imidazole 27E, which is oxidized by the excited photocatalyst *Ru(III) via a SET process, resulting in the amine radical cation 27F and the reduced catalyst Ru(II). The resulting catalyst Ru(II) was re-oxidized by O2, generating the ground state photocatalyst Ru(III) and superoxide radical (O2˙−), which abstracts a proton from the radical cation 27F to deliver the amine cation 27G. Next, another proton is abstracted from intermediate 27G by HOO− resulting in intermediate 27H. Moreover, a similar SET oxidation and deprotonation cycle affords the desired imidazole product 58. The key advantage of this protocol is the formation of two new C–N bonds with the elimination of CO2 and N2 as by-products.
Pyrazines are a well-known class of nitrogen heterocycles with a wide range of applications in treating neurological and obesity disorders. Owing to their wide range of applications, considerable attention has been paid towards the synthesis of pyrazine derivatives.55
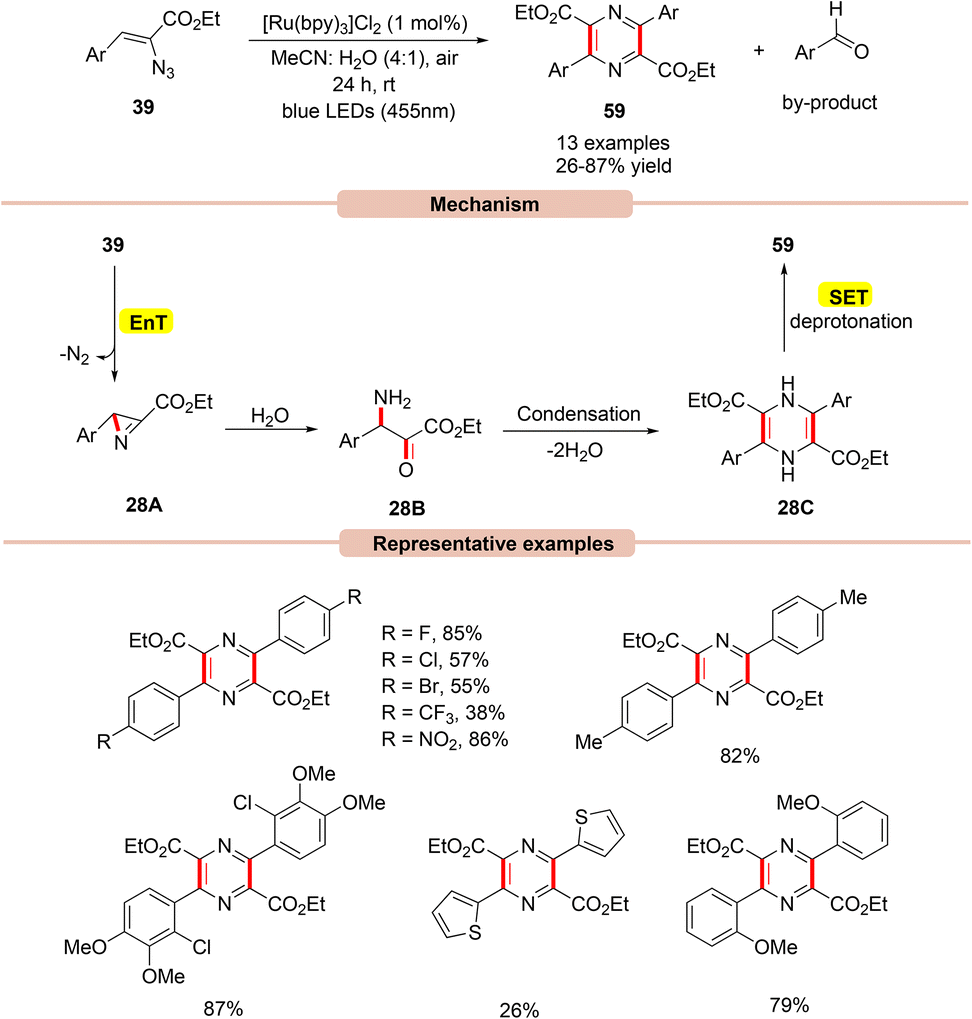
In this context, in 2017, Reiser and co-workers developed a simple and efficient approach to access multi-substituted pyrazines 59via a dual-energy and electron transfer strategy in the presence of visible light irradiation (Scheme 28).56
 | ||
| Scheme 28 Ru-photoredox catalyzed formation of pyrazines from vinyl azides via azirine intermediate. | ||
This strategy utilized Ru(bpy)3Cl2 as a photocatalyst with atmospheric oxygen as the terminal oxidant and blue LEDs (455 nm) as the light source. In this process, both electron-rich and electron-deficient groups at the ortho-, meta-, and para-positions on the aryl groups of vinyl azides 39 were well tolerated. Notably, vinyl azides bearing carbonyl groups instead of ester groups did not respond to the reaction. The authors proposed that initially, the photoexcited *Ru(II) catalyst transfers its energy to vinyl azide 39, which gets converted into a triplet excited state followed by the extrusion of dinitrogen to give the highly reactive 2H-azirine intermediate 28A. This 2H-azirine 28A gives dihydropyrazine 28Cvia a ring-opening by the interaction with H2O, followed by a condensation reaction. Next, SET/deprotonation of the dihydropyrazine species 28C gives access to the desired aromatized product 59. Moreover, a small amount of the aromatic aldehyde by-product was obtained at the end of the reaction.
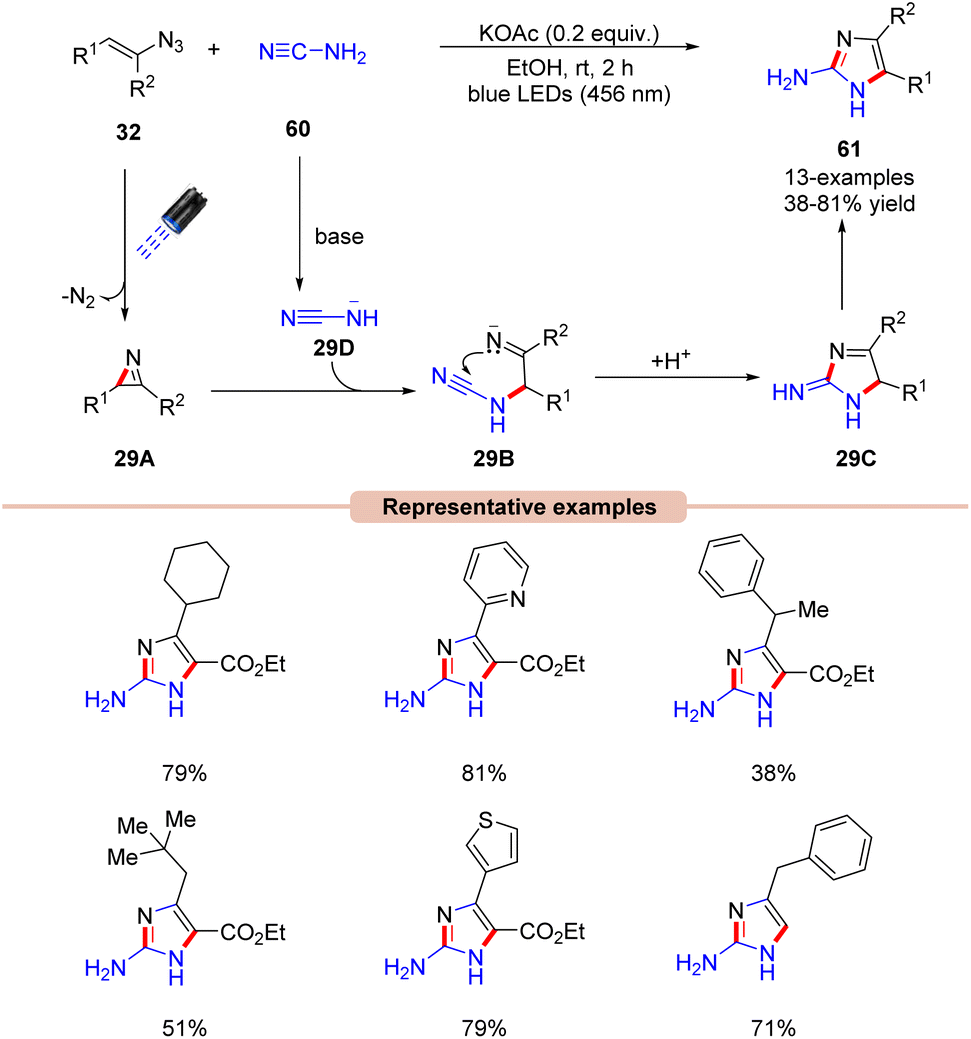
Handlon and co-workers in 2019 investigated the photoinduced synthesis of 2-aminoimidazoles 61via the sequential annulation of vinyl azides 32 with cyanamides 60 under visible light irradiation at ambient temperature (Scheme 29).57 Initial investigations revealed that the reaction can be carried out without an external photocatalyst under the irradiation of blue light (456 nm) and KOAc base in ethanol for 2 h. The reaction scope comprised of cyclic, sterically hindered, and α-branched aliphatic and aromatic vinyl azides 32, affording the desired products 61 in 38–81% yields. Product formation was further supported by X-ray crystal studies.
 | ||
| Scheme 29 Visible light-mediated synthesis of 2-aminoimidazoles from vinyl azides and cyanamides. | ||
The plausible mechanism of this protocol is illustrated in Scheme 29. The azirine intermediate 29A is generated upon the photodecomposition of vinyl azide 32 under visible light irradiation. Consequently, the addition of the base deprotonated cyanamide anion 29D to the 2H-azirine intermediate 29A affords the iminyl anion intermediate 29B, which undergoes an exo-trig cyclization to deliver the cyclic intermediate 29C. Finally, intermediate 29C undergoes tautomerization to form the 2-aminoimidazole product 61.
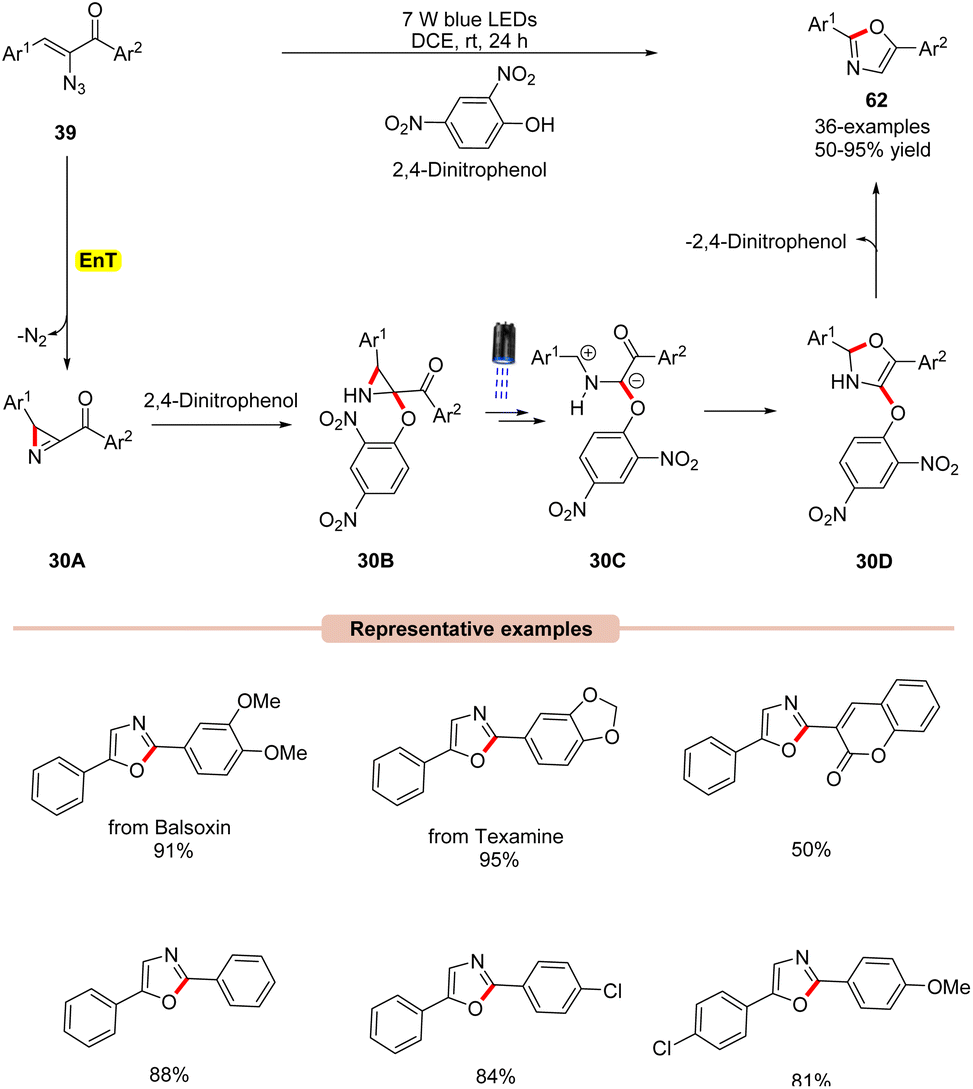
Thereafter, a 2,4-dinitrophenol (2,4-DNP) mediated synthesis of 2,5-diaryloxazoles 62 from α-azidochalcones 39 was reported by Maurya and co-workers in 2022, under visible light irradiation (Scheme 30).58 The substrate scope revealed that a library of substituted α-azidochalcones 39 reacted well under the optimized conditions and underwent photoannulation to give the desired 2,5-diaryloxazole products 62 in 50–95% yields. The robustness of this reaction was further demonstrated in the synthesis of balsoxin and texamine compounds bearing an oxazole core in good yields. Mechanistic studies concluded that 2,4-DNP functions as a photocatalyst as well as an additive. Moreover, NMR-studies at different reaction times under control conditions confirmed the involvement of the key 2H-azirine intermediate. Based on these studies, it is proposed that 2,4-dinitrophenol (2,4-DNT) undergoes EnT process with α-azidochalcones 39, which generates the key 2H-azirine intermediate 30A. Next, 2,4-DNT interacts with intermediate 30A to give intermediate 30B. Furthermore, in the presence of visible light, intermediate 30B is excited to its excited state, followed by C–C bond cleavage to form intermediate 30C. Later, 30C undergoes intramolecular cyclization to give intermediate 30D, accompanied by the subsequent release of 2,4-dinitrophenol to give the final product 62.
 | ||
| Scheme 30 Visible light 2,4-dinitrophenol assisted synthesis of 2,5-diaryloxazoles from α-azidochalcones. | ||
3.2. Formation of acyclic compounds
In 2019, Maurya and co-workers disclosed an efficient photocatalyst-free visible light mediated decomposition of vinyl azides 63 to substituted (E)-stilbenes 64 in moderate to good yields (Scheme 31).59 In addition, the cyclic by-product 65 was also formed in minor amounts in all the cases. Out of various reaction conditions screened, a catalyst-free photo-decomposition of vinyl azides 63 was found best suited for this protocol. Various solvents were screened, but acetonitrile (MeCN) was found to display high efficiency. UV-visible absorption studies reveal that vinyl azides 63 showed strong absorption maxima at 326 and 380 nm, which can absorb light and undergo denitrogenative photodecomposition. It was suggested that the reaction was initiated with the photodecomposition of vinyl azide 63 under visible light irradiation, affording 2H-azirine 31A with the extrusion of nitrogen. This 2H-azirine 31A may undergo ring-opening assisted by the neighboring amine group, leading to the formation of intermediate 31B, which may be converted into E-stilbene 64via the 1,2 acyl migration of 31B′ followed by enolization. In addition, the by-product indole 65 is also formed via the annulation of intermediate 31B followed by a [1,5] hydrogen atom transfer to give the side-product 65, which was also separated from the reaction mixture.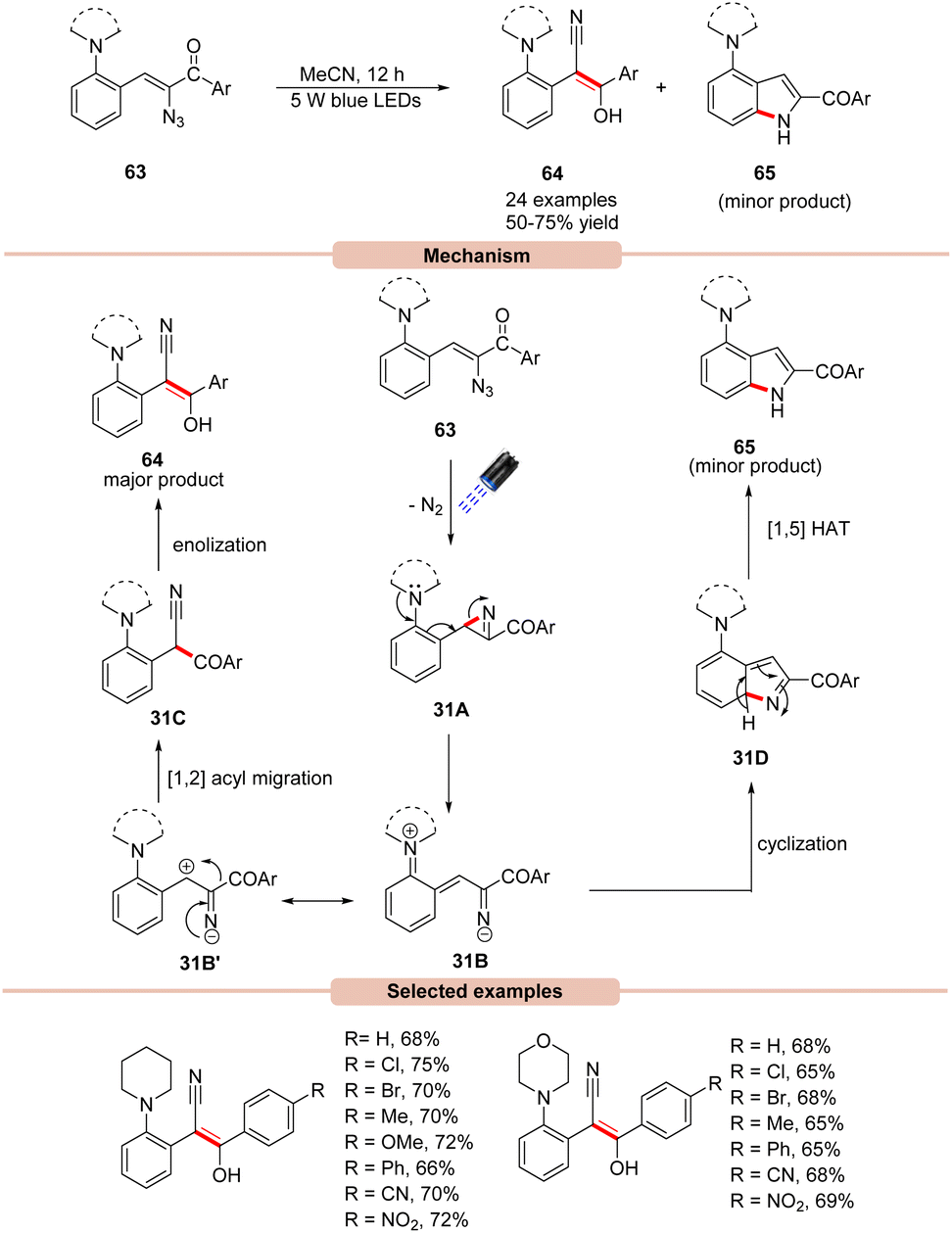 | ||
| Scheme 31 Synthesis of (E)-stilbene derivatives from the photo-decomposition of vinyl azides. | ||
In this reaction, both electron-rich and electron-deficient substituents on vinyl azides 63 were found to be compatible to afford various (E)-stilbene derivatives 64. The key advantage of this method is that it does not require any external photocatalyst for its successful execution.
4. Conclusions
In summary, this review has covered the synthetic applications of vinyl azides in organic synthesis under visible light-induced photoredox conditions, which include (i) iminyl radical formation and (ii) 2H-azirine intermediate formation based reactions. Vinyl azides have been proven to be versatile synthetic intermediates for constructing various complex N-heterocycles and other functional groups. Due to their wide range of synthetic applications, these methodologies will help promote continued interest in the visible light-induced photoredox-catalyzed organic transformation of vinyl azides. The synthetic utility of vinyl azides has been widely explored mainly because of the high intrinsic reactivity driven by the excellent leaving group N2. The mild conditions also enable vinyl azides with various coupling partners to synthesize molecular architectures that are otherwise difficult to access via conventional methods. The transformation of vinyl azides involving different reaction intermediates has to be carried out with the addition of transition metal photoredox catalysts and organic dyes or without any catalyst. However, many opportunities and challenges remain, such as: (1) more efforts are needed to address the enantioselectivity in these visible light-induced reactions; (2) the exploration of cheap and ecologically benign photocatalysts that have comparable redox potentials with the substrates is needed; (3) photocatalyst free radical formation via the EDA (electron-donor–acceptor) complex as well as visible light-mediated homolysis strategies would undoubtedly be a worthy goal and a great opportunity for researchers to explore; (4) new radical precursors such as aryl halides, diaryliodonium salts, aryl diazonium salts, benzenesulfinic acids, aryazosulfones, pyridinium salts, etc. should be explored for vinyl azides under visible light conditions; and (5) there are few reports on the oxidative coupling of radical intermediates with vinyl azides under visible light photocatalysis, which needs further study. Overall, it is believed that vinyl azide chemistry will significantly expand in the near future.4.1. Safety warnings while handling vinyl azides
Vinyl azides are highly energetic molecules that exhibit potentially explosive properties. They are shock-sensitive and can readily decompose when exposed to a small amount of energy from an external source (light, pressure, heat, etc.). Vinyl azides should be handled with care and should not be overheated while concentrating on the rotary evaporator and physical handling of the isolated products. Generally, the explosion risk of organic azides increases as the fraction of nitrogen in the molecular mass increases. In order to use organic azides safely, Smith's rules must be followed. (a) (NC + NO)/NN ≥ (N = number of atoms).7a,60 (b) The number of nitrogen atoms must be (NN) and must not exceed that of carbon (NC). Any synthesized azides should be stored in tightly closed containers at low temperatures and in the dark.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from UCOST (UCS & T/R & D-35/20-21), Government of Uttarakhand, and SERB (CRG/2022/002691), India, is gratefully acknowledged. B. S. and J. T. thank CSIR for the Senior Research Fellowship. R. I. P. thanks UGC for the Senior Research Fellowship.References
- (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed; (b) Organic Azides, Syntheses and Applications, ed. S. Bräse and K. Banert, Wiley, Chichester, 2010, pp. 115–166 Search PubMed; (c) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC; (d) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831–3846 RSC; (e) S. Chiba, Synlett, 2012, 21–44 CrossRef CAS; (f) E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368 CrossRef CAS; (g) P. Sivaguru, Y. Ning and X. Bi, Chem. Rev., 2021, 121, 4253–4307 CrossRef CAS PubMed.
- (a) G. L′abbé, Angew. Chem., Int. Ed. Engl., 1975, 14, 775–782 CrossRef; (b) S. Chiba, Chimia, 2012, 66, 377–381 CrossRef CAS PubMed; (c) N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 12169–12171 CrossRef CAS PubMed; (d) H. Ni, G. Zhang and Y. Yu, Curr. Org. Chem., 2015, 19, 776–789 CrossRef CAS.
- (a) S. Cludius-Brandt, L. Kupracz and A. Kirschning, Beilstein J. Org. Chem., 2013, 9, 1745–1750 CrossRef PubMed; (b) B. Hu, Z. Wang, N. Ai, J. Zheng, X. H. Liu, S. Shang and Z. Wang, Org. Lett., 2011, 13, 6362–6365 CrossRef CAS PubMed; (c) Y.-F. Wang, K. K. Toh, S. Chiba and K. Narasaka, Org. Lett., 2008, 10, 5019 CrossRef CAS PubMed; (d) E. P. J. Ng, Y.-F. Wang and S. Chiba, Synlett, 2011, 783 CAS; (e) F.-L. Zhang, Y.-F. Wang, G. H. Lonca, X. Zhu and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 4390 CrossRef CAS PubMed; (f) F.-L. Zhang, X. Zhu and S. Chiba, Org. Lett., 2015, 17, 3138 CrossRef CAS PubMed; (g) A. Hassner, E. S. Ferdinandi and R. J. Isbister, J. Am. Chem. Soc., 1970, 92, 1672–1675 CrossRef CAS; (h) F. L. Zhang, Y. F. Wang, G. H. Lonca, X. Zhu and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 4390–4394 CrossRef CAS PubMed.
- S. Cao, Q. Ji, H. Li, M. Pang, H. Yuan, J. Zhang and X. Bi, J. Am. Chem. Soc., 2020, 142(15), 7083–7091 CrossRef CAS PubMed.
- Z. Liu, P. Liao and X. Bi, Org. Lett., 2014, 16, 3668–3671 CrossRef CAS PubMed.
- F. W. Fowler, A. Hassner and L. A. Levy, J. Am. Chem. Soc., 1967, 89, 2077–2082 CrossRef CAS.
- (a) B. Hu and S. G. Dimagno, Org. Biomol. Chem., 2015, 13, 3844–3855 RSC; (b) F. Gholami, F. Yousefnejad, B. Larijani and M. Mahdavi, RSC Adv., 2023, 13, 990–1018 RSC; (c) Y. Jun, J. Xiaoyue, H. Shugui and W. Jing, Chin. J. Org. Chem., 2018, 38(4), 791–801 CrossRef.
- H. Hayashi, A. Kaga and S. Chiba, J. Org. Chem., 2017, 82, 11981–11989 CrossRef CAS PubMed.
- J. Fu, G. Zanoni, E. A. Anderson and X. Bi, Chem. Soc. Rev., 2017, 46, 7208–7228 RSC.
- (a) G. Büchi, C. G. Inman and E. S. Lipinsky, J. Am. Chem. Soc., 1954, 76, 4327–4331 CrossRef; (b) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed; (c) E. E. Coyle and M. Oelgemöller, Photochem. Photobiol. Sci., 2008, 7, 1313–1322 CrossRef CAS; (d) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (e) M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (c) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed; (d) Y. Wu, D. Kim and T. S. Teets, Synlett, 2021, 33, 1154–1179 Search PubMed; (e) C. R. J. Stephenson, T. Yoon and D. W. C. Macmillan, Visible light photocatalysis in organic chemistry, Wiley-VCH, Germany, 2018 CrossRef; (f) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed; (g) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (h) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed; (i) D. Ravelli, S. Protti and M. Fognoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (j) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (k) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (l) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121(10), 5741–5829 CrossRef CAS PubMed; (m) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (n) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (o) T. Constantin, F. Julia and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed; (p) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Chem. Rev., 2022, 122, 2695–2751 CrossRef CAS PubMed; (q) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed; (r) S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (s) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed; (t) R. I. Patel, J. Singh and A. Sharma, ChemCatChem, 2022, 14, e202200260 CrossRef CAS; (u) J. Singh, R. I. Patel and A. Sharma, Adv. Synth. Catal., 2022, 364, 2289 CrossRef CAS.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (c) N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed; (d) A. Tlili and S. Lakhdar, Angew. Chem., 2021, 133, 19678–19701 CrossRef; (e) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS PubMed; (f) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC; (g) R. I. Patel, A. Sharma, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 1694–1718 RSC; (h) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC; (i) S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC; (j) A. Bosveli, T. Montagnon, D. Kalaitzakis and G. Vassilikogiannakis, Org. Biomol. Chem., 2021, 19, 3303–3317 RSC; (k) N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed; (l) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (m) X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed; (n) M. A. Miranda and H. Garcia, Chem. Rev., 1994, 94, 1063–1089 CrossRef CAS; (o) P. P. Singh and V. Srivastava, Org. Biomol. Chem., 2021, 19, 313–321 RSC.
- (a) D. W. Manley and J. C. Walton, Beilstein J. Org. Chem., 2015, 11, 1570 CrossRef CAS PubMed; (b) S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4, 456 CrossRef CAS; (c) D. Franchi and Z. Amara, ACS Sustainable Chem. Eng., 2020, 8, 15405 CrossRef CAS.
- (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed; (b) C. G. S. Lima, T. D. M. Lima, M. Duarte, I. D. Jurbery and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS; (c) Y. Q. Yuan, S. Majumder, M. H. Yang and S. R. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (d) P. Garra, J. P. Fouassier, S. Lakhdar, Y. Yagci and J. Lalevée, Prog. Polym. Sci., 2020, 107, 101277 CrossRef CAS; (e) B. Saxena, R. I. Patel and A. Sharma, Adv. Synth. Catal., 2023, 365, 1538–1564 CrossRef CAS.
- (a) A. Suzuki, M. Tabata and M. Ueda, Tetrahedron Lett., 1975, 16, 2195–2198 CrossRef; (b) A. F. Bamford, M. D. Cook and B. P. Roberts, Tetrahedron Lett., 1983, 24, 3779–3782 CrossRef CAS.
- (a) N. Thirupathi, F. Wei, C. H. Tung and Z. Xu, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed; (b) Z. Zhong, Z. Xiao, X. Liu, W. Cao and X. Feng, Chem. Sci., 2020, 11, 11492–11497 RSC; (c) A. Chakrabarty and S. Mukherjee, Angew. Chem., Int. Ed., 2022, 61, e202115821 ( Angew. Chem. , 2022 , 134 , e202115821 ) CrossRef CAS PubMed.
- (a) Y. F. Wang, G. H. Lonca and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 1067–1071 CrossRef CAS PubMed; (b) Y.-F. Wang, G. H. Lonca, M. L. Runigo and S. Chiba, Org. Lett., 2014, 16, 4272–4275 CrossRef CAS PubMed; (c) J. C. Yang, J. J. Zhang and L. N. Guo, Org. Biomol. Chem., 2016, 14, 9806–9813 RSC.
- K. Banert, M. Chityala, M. Hagedorn, H. Beckers, T. Stüker, S. Riedel, T. Rüffer and H. Lang, Angew. Chem., Int. Ed., 2017, 56, 9582–9586 CrossRef CAS PubMed.
- (a) E. C. Taylor and G. G. Spence, Chem. Commun., 1968, 1037–1038 RSC; (b) O. B. Abdel-Halim, T. Morikawa, S. Ando, H. Matsuda and M. Yoshikawa, J. Nat. Prod., 2004, 67, 1119–1124 CrossRef CAS PubMed; (c) I. Kock, D. Heber, M. Weide, U. Wolschendorf and B. Clement, J. Med. Chem., 2005, 48, 2772–2777 CrossRef CAS PubMed; (d) P. H. Bernardo, K. F. Wan, T. Sivaraman, J. Xu, F. K. Moore, A. W. Hung, H. Y. K. Mok, V. C. Yu and C. L. L. Chai, J. Med. Chem., 2008, 51, 6699–6710 CrossRef CAS PubMed; (e) T. Nakanishi and M. Suzuki, Org. Lett., 1999, 1, 985–988 CrossRef CAS PubMed; (f) T. Nakanishi, M. Suzuki, A. Saimoto and T. Kabasawa, J. Nat. Prod., 1999, 62, 864–867 CrossRef CAS PubMed.
- (a) R. Yanada, K. Hashimoto, R. Tokizane, Y. Miwa, H. Minami, K. Yanada, M. Ishikura and Y. Takemoto, J. Org. Chem., 2008, 73, 5135–5138 CrossRef CAS PubMed; (b) D. A. Candito and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 6713–6716 CrossRef CAS PubMed; (c) L. R. Donaldson, D. Haigh and A. N. Hulme, Tetrahedron, 2008, 64, 4468–4477 CrossRef CAS; (d) T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572–577 CrossRef CAS PubMed; (e) L. Zhang, G. Y. Ang and S. Chiba, Org. Lett., 2010, 12, 3682–3685 CrossRef CAS PubMed.
- X. Sun and S. Yu, Chem. Commun., 2016, 52, 10898–10901 RSC.
- (a) M. C. Carreão, Chem. Rev., 1995, 95, 1717–1760 CrossRef; (b) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC; (c) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- (a) L.-L. Mao, D.-G. Zheng, X.-H. Zhu, A.-X. Zhou and S.-D. Yang, Org. Chem. Front., 2018, 5, 232–236 RSC; (b) L.-L. Mao, L.-X. Quan, X.-H. Zhu, C.-B. Ji, A.-X. Zhou, F. Chen and D.-G. Zheng, Synlett, 2019, 955–960 CAS.
- Y. Li, Y. Zhu and S.-D. Yang, Org. Chem. Front., 2018, 5, 822–826 RSC.
- J.-C. Yang, J.-Y. Zhang, J.-J. Zhang, X.-H. Duan and L.-N. Guo, J. Org. Chem., 2018, 83, 1598–1605 CrossRef CAS PubMed.
- M.-J. Jiao, Q. Hu, X.-Q. Hu and P.-F. Xu, J. Org. Chem., 2021, 86, 17156–17163 CrossRef CAS PubMed.
- (a) P. Sridhar, M. Alagumuthu, S. Arumugam and S. R. Reddy, RSC Adv., 2016, 6, 64460–64468 RSC; (b) K. Devi, Y. Asmat, M. Agrawal, S. Sharma and J. Dwivedi, J. Chem. Sci., 2013, 125, 1093–1101 CrossRef CAS; (c) M. R. E. Aly, M. M. Ibrahim, A. M. Okael and Y. A. M. H. Gherbawy, Russ. J. Bioorg. Chem., 2014, 40, 214–227 CrossRef CAS.
- Q. Wang, J. Huang and L. Zhou, Adv. Synth. Catal., 2015, 357, 2479–2484 CrossRef CAS.
- X. Huang, X. Li, X. Xie, K. Harms, R. Riedel and E. Meggers, Nat. Commun., 2017, 8, 2245 CrossRef PubMed.
- S. Sarkar, A. De, S. Santra, I. A. Khalymbadzha, G. V. Zyryanov and A. Majee, Eur. J. Org. Chem., 2022, e202200503 CAS.
- (a) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937–1945 CrossRef CAS PubMed; (b) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284–2294 CrossRef CAS PubMed; (c) M. Li, H. Zheng, X.-S. Xue and J.-P. Cheng, Tetrahedron Lett., 2018, 59, 1278–1285 CrossRef CAS.
- H.-T. Qin, S.-W. Wu, J.-L. Liu and F. Liu, Chem. Commun., 2017, 53, 1696–1699 RSC.
- H. I. Jung and D. Y. Kim, Synth. Commun., 2019, 50, 380–387 CrossRef.
- (a) X. Wang, H. Li, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 2062–2065 RSC; (b) O. M. Mulina, A. I. Ilovaisky, T. Opatz and A. O. Terent'ev, Tetrahedron Lett., 2021, 64, 152737 CrossRef CAS.
- (a) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed; (b) J. Wang and H. Liu, Chin. J. Org. Chem., 2012, 32, 1643 CrossRef CAS; (c) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed; (d) G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068 CrossRef CAS; (e) T. Najam, S. S. A. Shah, K. Mehmood, A. U. Din, S. Rizwan, M. Ashfaq, S. Shaheen and A. Waseem, Inorg. Chim. Acta, 2018, 469, 408 CrossRef CAS; (f) J. T. Yu, F. Teng and J. Cheng, Adv. Synth. Catal., 2017, 359, 26 CrossRef CAS; (g) W. B. Wu, J. S. Yu and J. Zhou, ACS Catal., 2020, 10, 7668 CrossRef CAS; (h) R. I. Patel, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 3166–3200 RSC.
- J. R. Donald and S. L. Berrell, Chem. Sci., 2019, 10, 5832–5836 RSC.
- (a) S. Abele and D. Seebach, Eur. J. Org. Chem., 2000, 1–15 CrossRef CAS; (b) E. Hagiwara, A. Fujii and M. Sodeoka, J. Am. Chem. Soc., 1998, 120, 2474–2475 CrossRef CAS; (c) W. J. Drury, D. Ferraris, C. Cox, B. Young and T. Leckta, J. Am. Chem. Soc., 1998, 120, 11006–11007 CrossRef CAS; (d) H. Wang, J. F. Yan, X. L. Song, L. Fan, J. Xu, G. M. Zhou, L. Jiang and D. C. Yang, Bioorg. Med. Chem., 2012, 20, 2119–2130 CrossRef CAS PubMed.
- J.-T. Xu, G.-Q. Xu, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2019, 84, 14760–14769 CrossRef CAS PubMed.
- K. R. Dworakowski, S. Pisarek, S. Hassan and D. Gryko, Org. Lett., 2021, 23, 9068–9072 CrossRef CAS PubMed.
- (a) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801–1839 RSC; (b) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed; (c) T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831–1861 CrossRef CAS PubMed; (d) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- (a) L. Zhang, J. Zhang, J. Ma, D. J. Cheng and B. Tan, J. Am. Chem. Soc., 2017, 139, 1714–1717 CrossRef CAS PubMed; (b) Y. Liu, H. Hu, X. Wang, S. Zhi, Y. Kan and C. Wang, J. Org. Chem., 2017, 82, 4194–4202 CrossRef CAS PubMed.
- E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793–797 CrossRef CAS PubMed.
- J. Xuan, X.-D. Xia, T.-T. Zeng, Z.-J. Feng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 1–5 CrossRef PubMed.
- S. Borra, D. Chandrasekhar, U. D. Newar and R. A. Maurya, J. Org. Chem., 2019, 84, 1042–1052 CrossRef CAS PubMed.
- S. Borra, L. Borkotoky, U. D. Newar, B. Das and R. A. Maurya, Adv. Synth. Catal., 2020, 362, 3364–3368 CrossRef CAS.
- (a) S. Bhargava and D. Rathore, Lett. Org. Chem., 2017, 14, 381–402 CrossRef CAS; (b) N. Iqbal, N. Iqbal, D. Maiti and E. J. Cho, Angew. Chem., 2019, 131, 15955–15959 CrossRef; (c) U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669–12673 CrossRef CAS PubMed.
- S. Borra, D. Chandrasekhar, S. Khound and R. A. Maurya, Org. Lett., 2017, 19, 5364–5367 CrossRef CAS PubMed.
- (a) M. A. Metwally, E. A. Latif, F. A. Amer and G. Kaupp, J. Sulfur Chem., 2004, 25, 63–85 CrossRef CAS; (b) A. Meissner, H. Boshoff, M. Vasan, B. P. Duckworth, C. E. Barry and C. C. Aldrich, Bioorg. Med. Chem., 2013, 21, 6385–6397 CrossRef CAS PubMed.
- W.-L. Lei, T. Wang, K.-W. Feng, L.-Z. Wu and Q. Liu, ACS Catal., 2017, 7, 7941–7945 CrossRef CAS.
- (a) F. M. D. Ismail, D. O. Levitsky and V. M. Dembitsky, Eur. J. Med. Chem., 2009, 44, 3373–3387 CrossRef CAS PubMed; (b) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed; (c) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC.
- (a) D. Chandrasekhar, S. Borra, J. B. Nanubolu and R. A. Maurya, Org. Lett., 2016, 18, 2974–2977 CrossRef CAS PubMed; (b) D. K. Tiwari, R. A. Maurya and J. B. Nanubolu, Chem. – Eur. J., 2016, 22, 526–530 CrossRef CAS PubMed.
- S. Borra, D. Chandrasekhar, S. Adhikary, S. Rasala, S. Gokulnath and R. A. Maurya, J. Org. Chem., 2017, 82, 2249–2256 CrossRef CAS PubMed.
- P. R. Adiyala, K. N. V. Sastry, J. Kovvuri, A. Nagarajan, V. G. Reddy, I. B. Sayeed, V. L. Nayak, R. A. Maurya and A. Kamal, ChemistrySelect, 2017, 2, 8158–8161 CrossRef CAS.
- P. R. Adiyala, S. Jang, N. K. Vishwakarma, Y.-H. Hwang and D.-P. Kim, Green Chem., 2020, 22, 1565–1571 RSC.
- (a) L. E. Seitz, W. J. Suling and R. C. Reynolds, J. Med. Chem., 2002, 45, 5604–5606 CrossRef CAS PubMed; (b) S. A. Raw, C. D. Wilfred and R. J. K. Taylor, Chem. Commun., 2003, 2286–2287 RSC.
- A. Hossain, S. K. Pagire and O. Reiser, Synlett, 2017, 28, 1707–1714 CrossRef CAS.
- L. Man, R. C. B. Copley and A. L. Handlon, Org. Biomol. Chem., 2019, 17, 6566–6569 RSC.
- U. D. Newar, S. Borra and R. A. Maurya, Org. Lett., 2022, 24, 4454–4458 CrossRef CAS PubMed.
- S. Borra, L. Borkotoky, U. D. Newar, A. Kalwar, B. Dasc and R. A. Maurya, Org. Biomol. Chem., 2019, 17, 5971–5981 RSC.
- P. A. S. Smith, Open-chain nitrogen compounds, Benjamin, New York, 1966, vol. 2, p. 211 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |