
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective oxidation of phenoxathiin-based thiacalix[4]arenes – stereomutation of sulfoxide groups†
N.
Broftová
a,
T.
Landovský
a,
H.
Dvořáková
 b,
V.
Eigner
c,
M.
Krupička
a and
P.
Lhoták
*a
b,
V.
Eigner
c,
M.
Krupička
a and
P.
Lhoták
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague (UCTP), Technická 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz
bLaboratory of NMR Spectroscopy, UCTP, Technická 5, 166 28 Prague 6, Czech Republic
cDepartment of Solid State Chemistry, UCTP, Technická 5, 166 28 Prague 6, Czech Republic
First published on 15th May 2023
Abstract
A phenoxathiin-based macrocycle represents an inherently chiral building block, well accessible in two steps from the starting thiacalix[4]arene. The oxidized derivatives bearing one sulfoxide group and three sulfonyl groups were found to exhibit unexpected stereochemical preferences of the sulfoxide group during transformations. The sulfoxide moiety is always pointing out of the cavity (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O out), while the opposite (SO in) configuration was never obtained by direct oxidation. In order to achieve full oxidation to sulfone, the configuration of the sulfoxide group must first be changed by a photochemical inversion before the final oxidation occurs. The phenomenon of stereomutation of the sulfoxide group in the thiacalixarene series was studied using a combination of experimental (NMR and single crystal X-ray analysis) and theoretical (DFT) approaches.
O out), while the opposite (SO in) configuration was never obtained by direct oxidation. In order to achieve full oxidation to sulfone, the configuration of the sulfoxide group must first be changed by a photochemical inversion before the final oxidation occurs. The phenomenon of stereomutation of the sulfoxide group in the thiacalixarene series was studied using a combination of experimental (NMR and single crystal X-ray analysis) and theoretical (DFT) approaches.
Introduction
After their discovery more than two decades ago,1 thiacalixarenes quickly became an integral part of the calixarene family.2 The introduction of sulfur instead of classical methylene bridges makes these macrocycles very interesting for various applications in supramolecular chemistry. On the one hand, these derivatives retain some typical properties of classical calixarenes, such as the existence of four basic conformations/atropisomers called cone, partial cone, 1,2- and 1,3-alternates.2 On the other hand, the presence of heteroatoms brings about some new properties that are not possible with the parent systems. Such new features include altered conformational preferences, significantly different complexation properties, and last but not least, new methods of chemical derivatization of the basic skeleton.3Although the formation of spirodienone derivatives4 has long been known in the classical calixarene series and is used for the synthesis of some unusual derivatisation patterns,5 the application of this approach to thiacalixarenes led to the discovery of a completely new type of chemistry. As shown in Scheme 1, the starting thiacalix[4]arene 1 can be smoothly transformed into spirodienone 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ,6 which is under acidic conditions unexpectedly rearranged into phenoxathiin-based macrocycle 3 in a very high yield.7 Due to the presence of the condensed heterocyclic moiety, compound 3 represents a highly rigidified inherently chiral system with potential application in the design of chiral receptors. During our attempts at the shaping of this skeleton (formation of various conformations) and the oxidation of sulfur bridges, we found8 that sulfur bridges can be oxidised to yield sulfone 4, which was alkylated to form intermediate 5.
,6 which is under acidic conditions unexpectedly rearranged into phenoxathiin-based macrocycle 3 in a very high yield.7 Due to the presence of the condensed heterocyclic moiety, compound 3 represents a highly rigidified inherently chiral system with potential application in the design of chiral receptors. During our attempts at the shaping of this skeleton (formation of various conformations) and the oxidation of sulfur bridges, we found8 that sulfur bridges can be oxidised to yield sulfone 4, which was alkylated to form intermediate 5.
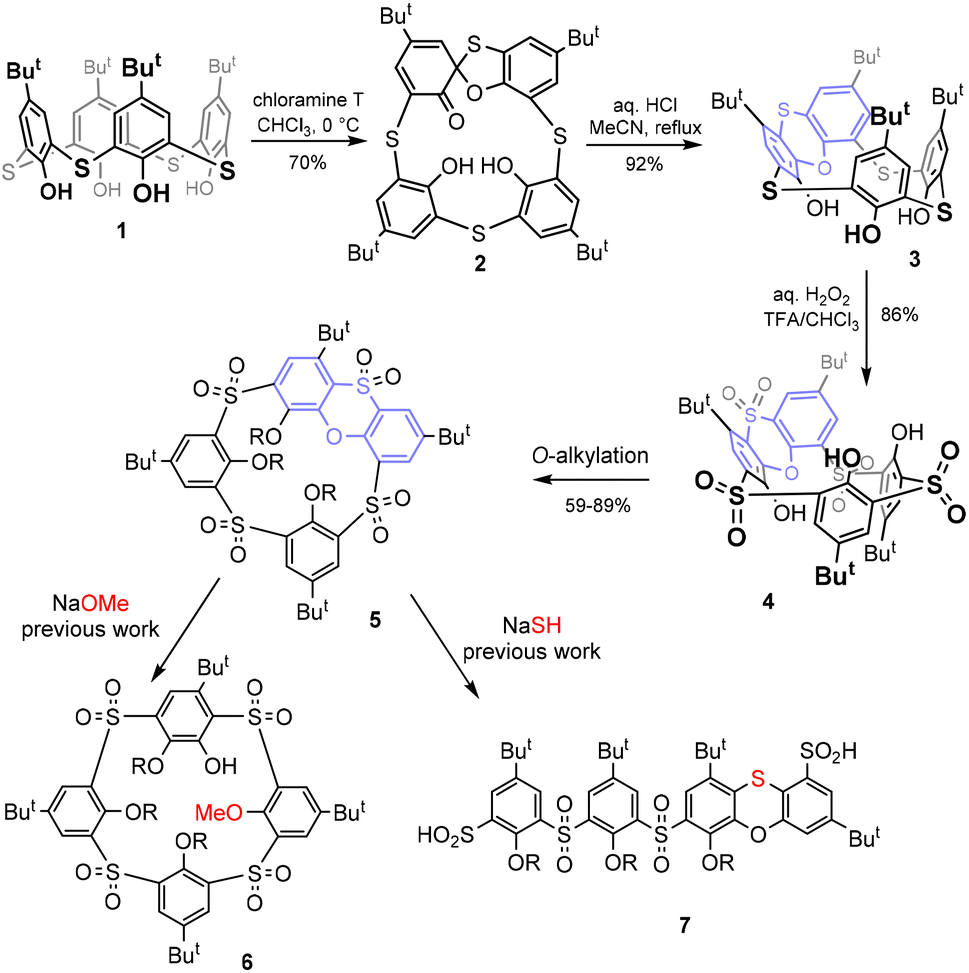 | ||
| Scheme 1 Synthesis of phenoxathiin-based thiacalixarene and its further transformations. | ||
Recently, rigid calix[4]arenes9 with high internal strain have been recognized as surprisingly reactive systems enabling unusual transformations.10 Similarly, it was found that compound 5 can be used for further unexpected reactions. In the presence of alkoxides, it is subject to a nucleophilic attack11 leading to a hitherto unexplored type of macrocycle 6, or it undergoes an unprecedented cascade of nucleophilic cleavage/rearrangements (with SH−) to form linear oligomers 7 (Scheme 1).12
In this paper, we focused on some other interesting aspects of oxidised compound 5 and related systems concerning their unusual stereochemical preferences during the oxidation of sulfur bridges. We hope that a better understanding of this phenomenon can be used in further derivatization of these systems and contribute to the application of these compounds in supramolecular chemistry.
Results and discussion
Thiacalixarene 1 was oxidized using the known procedure7b (chloramine T/CHCl3-MeOH) to obtain the corresponding spirodienone derivatives 2 in 74% yield (Scheme 1). The acid-catalysed rearrangement then led to the isolation of 3 in 92% yield. During the oxidation of 3 using the literature conditions (excess of 30% aq. H2O2 in TFA/CHCl3),8 we noticed that the outcome of the reaction can vary from case to case. The careful inspection of the reaction conditions revealed that the ratio of products 4 and 8 depends not only on the reaction time and temperature (as already observed) but is also unexpectedly dependent on the presence or absence of light. Indeed, the reaction carried out at rt in the dark (flask wrapped with aluminum foil) gave compound 8 as the only product in 98% yield (Scheme 2). On the other hand, the same reaction conditions under light led to a mixture of both compounds 4 and 8. Increasing the temperature (reflux) and prolonging the reaction time (3 days) finally led to the isolation of compound 4 in 86% yield. Interestingly, an attempt at further oxidation of 8 with fresh oxidation agents (in the dark) did not lead to any reaction while the same reaction under light provided 4 in a good yield.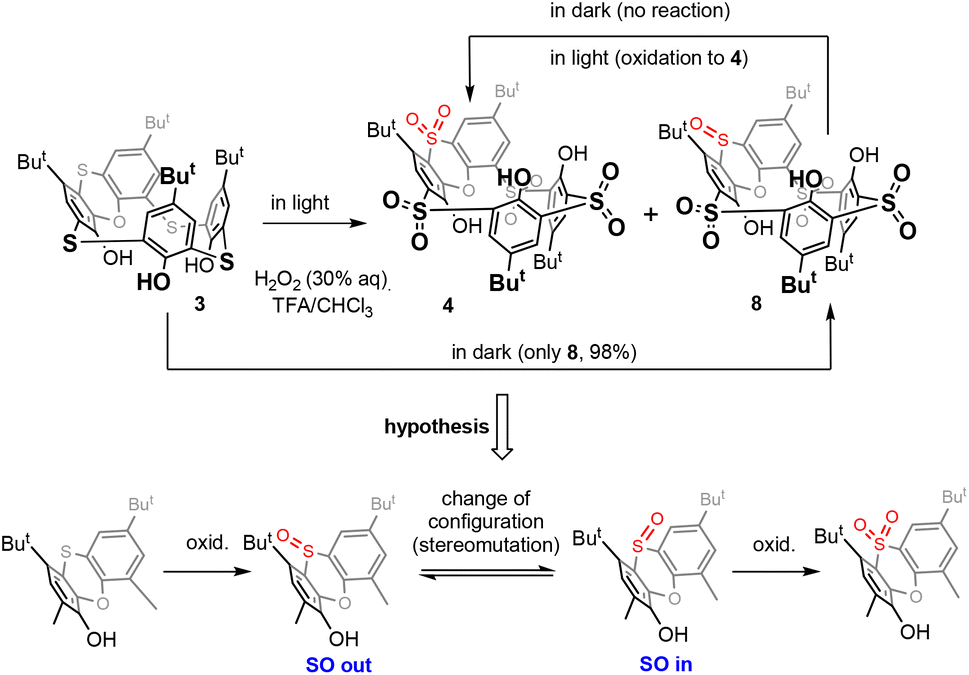 | ||
| Scheme 2 Oxidation of phenoxathiin-based thiacalixarenes. | ||
Based on the above results, we can therefore postulate the following unexpected assumption: for the oxidation of the sulfoxide group in compound 8, a change of the configuration to the opposite stereoisomer is first required, and only then can the oxidation occur by itself (Scheme 2).
The stereochemistry of the sulfoxide group in compound 8 was already assigned by single crystal X-ray measurement8 which revealed its “out of cavity” configuration (Scheme 2). The fact that the reaction stopped spontaneously at the 3 × SO2, 1 × SO stage (compound 8) indicates that the access of the oxidizing agent from inside the cavity is greatly disadvantaged. Even to such an extent that a change in configuration is required first for oxidation (to compound 4) to occur at all.
This is reminiscent of our earlier observation13 that the oxidation of sulfur to sulfoxide in thiacalix[4]arenes in the cone conformation always occurs on the side opposite to the lower rim of calixarene.14 In other words, the oxidizing agent always approaches from the side of the upper rim, which is probably sterically more accessible (Fig. 1). In this case, however, the selectivity is not so pronounced and after the oxidation of all bridges to sulfoxide, further oxidation to sulfone normally follows.
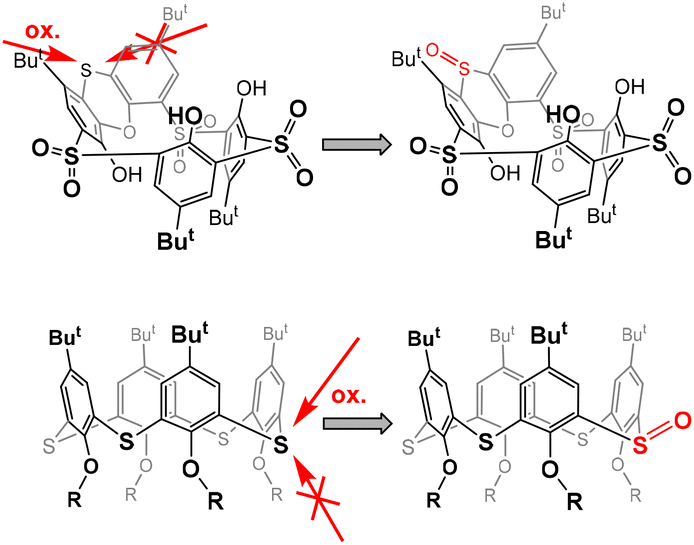 | ||
| Fig. 1 Preferred approaches for the oxidizing agent during oxidation to sulfoxide. | ||
Since the non-alkylated derivatives 4 and 8 are difficult to handle (they form long tails in thin-layer chromatography), we decided to study the last oxidation step on alkylated derivatives, which are easily chromatographically separable. Compound 8 was alkylated using an excess of Cs2CO3 as a base and alkyl iodide (MeI, EtI, and PrI) as an alkylating agent in acetone. The corresponding peralkylated compounds 9a–c were isolated in good yields (Scheme 3).
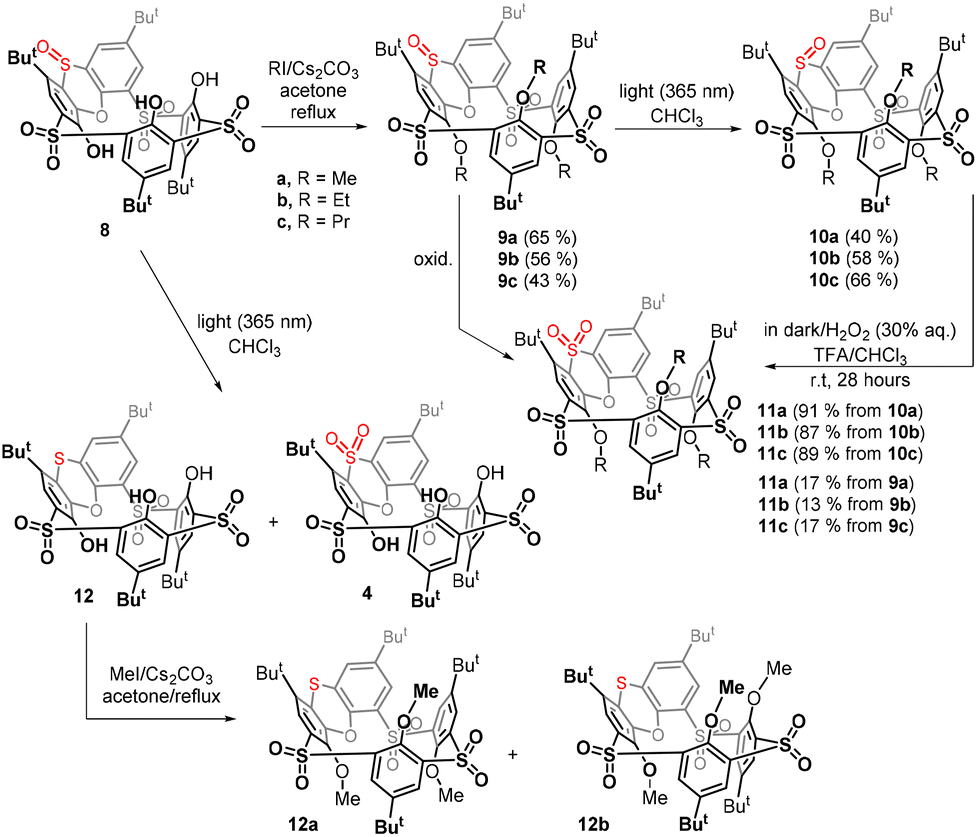 | ||
| Scheme 3 Stereomutation of the sulfoxide group in phenoxathiin-based thiacalixarenes and subsequent oxidation. | ||
In order to study the possible photochemical stereomutation15 of the sulfoxide moiety, we first chose an appropriate wavelength of light. The UV/vis spectrum of the starting sulfoxide 9a in CHCl3 (see the ESI Fig. 43†) showed two maxima at 295 nm (ε295 = 9.02 × 103 l mol−1 cm−1) and 328 nm (ε328 = 7.77 × 103 l mol−1 cm−1), and the maxima for 9b and 9c were almost identical (ESI Fig. 44 and 45†). The sample of 9a was irradiated with light (commercially available LED with a maximum at a wavelength of 311 nm, which was close to the measured maximum of the compound). The reaction was carried out at room temperature and quenched after one day of irradiation. However, we found that under these conditions, the molecule was degraded to some unidentified products and the expected thiacalixarene derivatives were not found in the reaction mixture.16
To avoid harsh conditions, we used light with a maximum at 365 nm (commercially available LED). After 24 h, derivative 10a was isolated from the reaction mixture in 40% yield, together with 31% of the unreacted starting material 9a (Scheme 3). A similar reaction of 9b gave product 10b with the opposite configuration in 58% yield (11% of 9b was isolated), while a propoxy derivative provided 10c in 66% yield (14% of 9c was isolated).
The structure of 10a was confirmed by HRMS ESI+ analysis, which showed a peak at m/z = 895.2283, which was in very good agreement with the value of the [M + Na]+ cation (895.2285) predicted for 10a. The number of peaks and the splitting patterns of the 1H NMR spectrum (CDCl3, 400 MHz, 298 K) of 10a are the same as those of the starting compound. However, some differences are clearly visible here, such as the shift of the singlet of the MeO group from 3.55 (9a) to 3.17 (10a) or the change in the chemical shift of the tert-Bu group from 0.73 (9a) to 0.97 (10a). In both cases, these are groups that come into close proximity to the SO function facing the cavity of compound 10a.
The unequivocal evidence of the inversion of sulfoxide stereochemistry was obtained by single crystal X-ray analysis. Slow crystallization from a CHCl3–EtOH mixture provided 10a as a solvate with ethanol in the partial cone conformation (monoclinic system, space group P21/c). As shown in Fig. 2a, the sulfur atom of the phenoxathiin system is tilted into the cavity, and the sulfoxide group points toward the neighbouring But and MeO groups (see above for changes in the NMR spectrum). The inversion of configuration is best highlighted by a comparison with the starting compound 9a (Fig. 2b). At the same time, the sulfoxide group is responsible for trapping ethanol, which is bound through a hydrogen bond (SO⋯H–O = 1.977 Å).
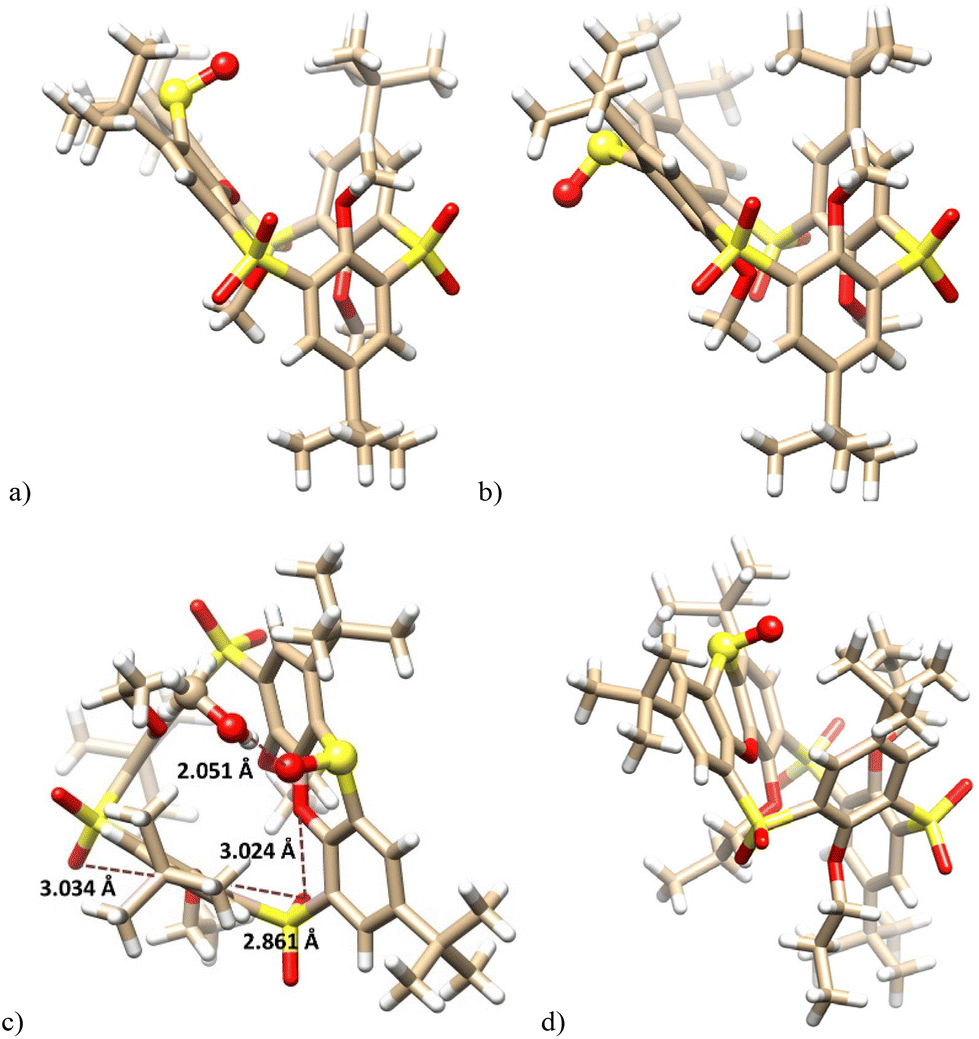 | ||
| Fig. 2 X-ray structure of: (a) 10a – side view (EtOH not shown for better clarity); (b) the starting 9a – side view (ref. 8a); (c) 10b – side view; (d) 10c – side view, (sulfoxide group shown as balls). | ||
Compound 10b crystallized in a triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , adopting the partial cone conformation. The solvent molecule (CH3OH) is held by HB interactions with the sulfoxide group (SO⋯H–O = 2.051 Å), resembling thus the solvate of 10a (Fig. 2c). The arrangement is further stabilised by intramolecular close contacts between the sulfonyl oxygen atoms and the ether oxygen atoms of the calixarene skeleton (3.034 Å, 2.861 Å and 3.024 Å). These distances are shorter than the corresponding sum of the van der Waals radii (ca.3.04 Å),17 indicating attractive interactions between the oxygen atoms which can be classified as chalcogen bonding.18 The same features can be also found within the above 10a·EtOH complex. Compared to Me or Et derivatives, the introduction of propyl groups had only a minimal effect on the basic geometrical parameters of the system; see the structure of 10c (Fig. 2d).
, adopting the partial cone conformation. The solvent molecule (CH3OH) is held by HB interactions with the sulfoxide group (SO⋯H–O = 2.051 Å), resembling thus the solvate of 10a (Fig. 2c). The arrangement is further stabilised by intramolecular close contacts between the sulfonyl oxygen atoms and the ether oxygen atoms of the calixarene skeleton (3.034 Å, 2.861 Å and 3.024 Å). These distances are shorter than the corresponding sum of the van der Waals radii (ca.3.04 Å),17 indicating attractive interactions between the oxygen atoms which can be classified as chalcogen bonding.18 The same features can be also found within the above 10a·EtOH complex. Compared to Me or Et derivatives, the introduction of propyl groups had only a minimal effect on the basic geometrical parameters of the system; see the structure of 10c (Fig. 2d).
Having in hand the corresponding stereoisomers 9a–c and 10a–c possessing the out and in configuration of the sulfoxide group, we were interested in how their subsequent oxidation would proceed. The oxidation of 9a to tetrasulfone 11a carried out in the dark under the standard conditions (30% aq. H2O2/TFA/CHCl3, room temp) showed only 17% conversion of the starting compound after 28 h. Similar results were also obtained for ethyl and propyl derivatives 9b and 9c, where we observed 13% and 17% conversion (1H NMR), respectively (Scheme 3).
In sharp contrast, oxidations of derivatives 10a–c with the reversed configuration proceeded smoothly in all cases with 100% conversion, and the respective sulfones 11a–c were isolated by preparative TLC in 91%, 87%, and 89% yields, respectively. The above result confirms our assumptions that the oxidation of the “in” configuration is much easier than that of the “out” configuration.
In order to confirm these conclusions also for non-alkylated derivatives, compound 8 was irradiated in the same way (light 365 nm, CHCl3, rt) as was done for compounds 9a–c to achieve the stereomutation of the sulfoxide group.
In addition to the unreacted starting compound 8, the crude reaction mixture showed the formation of a small amount of sulfone 4, identified by a comparison of the NMR signals with an authentic sample, while the main product 12 remained unknown (Scheme 3). Since the chromatographic separation of non-alkylated derivatives is very difficult, both products were converted by a reaction with methyl iodide (MeI, K2CO3/acetone/reflux) to the corresponding methoxy derivatives and then separated by preparative TLC on silica gel. The formation of sulfone 4 was confirmed by its alkylated product 11a. Interestingly, the alkylation of 12 led to a mixture of two products 12a and 12b possessing the same molecular mass but different 1H NMR spectra, which indicated the formation of two different conformers. Surprisingly, the HRMS ESI+ analysis of the methoxy derivative 12a showed a molecular peak at m/z = 879.2335, which was in perfect agreement with the [M + Na]+ cation (879.2336) predicted for a compound lacking one oxygen atom. Thus, the main product is not sulfoxide with altered stereochemistry, but compounds 12a and 12b possessing a sulfide bridge.
While methylation of tetrasulfone 4 or trisulfone monosulfoxide 8 leads to a single conformation (partial cone), the formation of two conformers 12a and 12b suggests that the conformational preferences of the sulfur-bridged derivative 12 are significantly altered. To reveal the possible mobility of alkylated derivatives 12a and 12b, the VT 1H NMR studies of both products were carried out. While heating of 12a (500 MHz, CDCl2–CDCl2) did not lead to any change of the spectrum (see the ESI†), compound 12b assigned as the 1,2-alternate was gradually transformed into 12a at 110 °C (see Fig. 4). This suggests that (i) the sulfur bridge in 12 presents less steric hindrance than sulfone 4 or sulfoxide 8, where a similar change is not possible; (ii) the partial cone conformer 12a represents a thermodynamically more stable conformation than 12b. This was further confirmed by heating a solution of compound 12b in CDCl2–CDCl2 overnight at 120 °C, which led to complete conversion to compound 12a.
The final proof of the structure was obtained by the single crystal X-ray analysis of 12a. This revealed that compound 12a crystallized as a solvate with cyclohexane (12a·C6H12) in a monoclinic system, space group P21/c. As shown in Fig. 3a, the molecule adopts the partial cone conformation and the missing oxygen atom was removed in the phenoxathiin moiety, thus forming a sulfide bridge. The conformation was stabilised by an array of intramolecular close contacts between the oxygen atoms from SO2 and OR moieties (2.890 Å, 2.883 Å, 2.869 Å and 3.039 Å), which can be classified as chalcogen bonds (Fig. 3b).
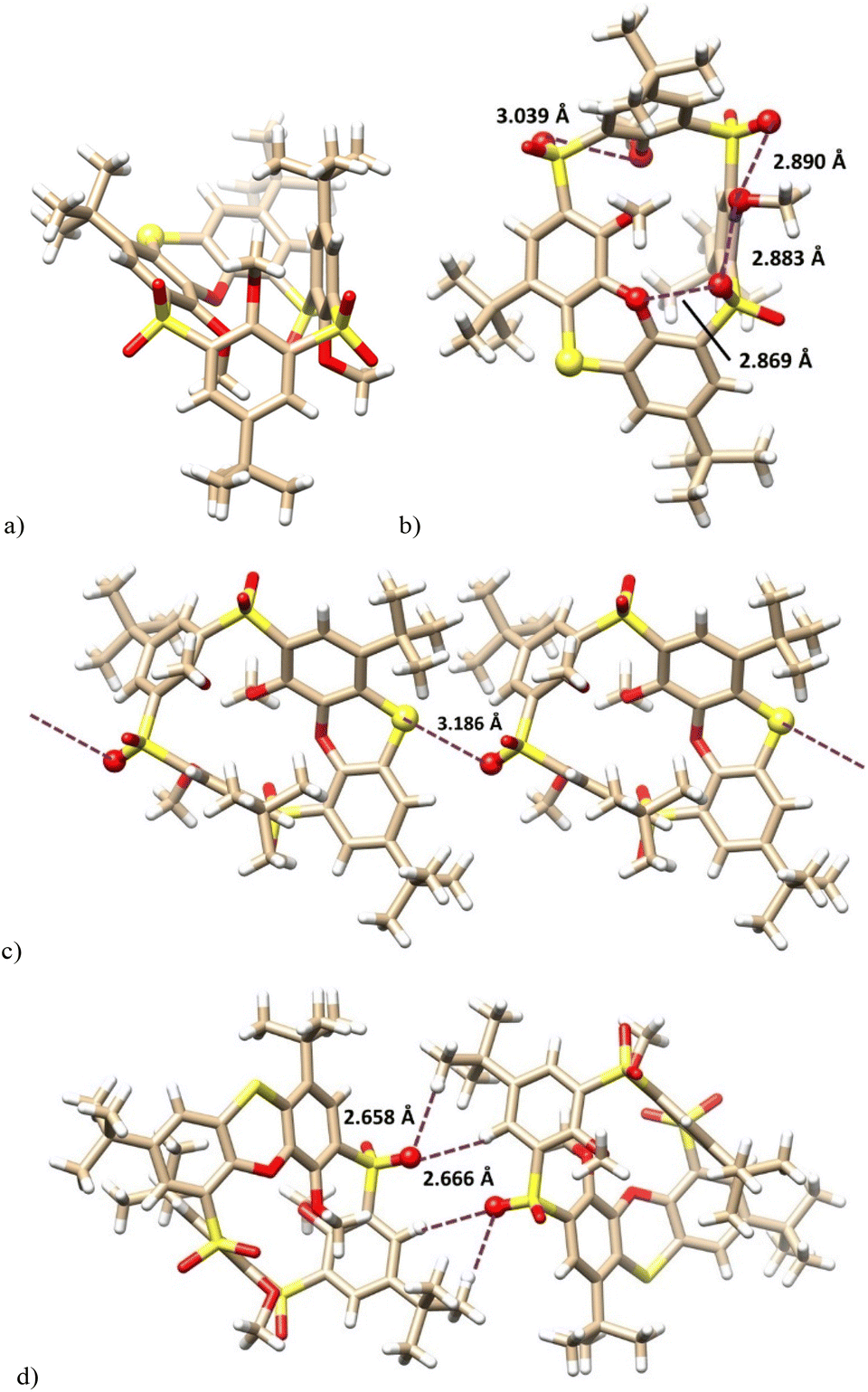 | ||
| Fig. 3 X-ray structure of: (a) 12a — side view; (b) 12a — bottom view of the same indicating the close contacts between the oxygen atoms; (c) the chalcogen bonds in the infinite chain; (d) non-classical hydrogen bonds between the chains. | ||
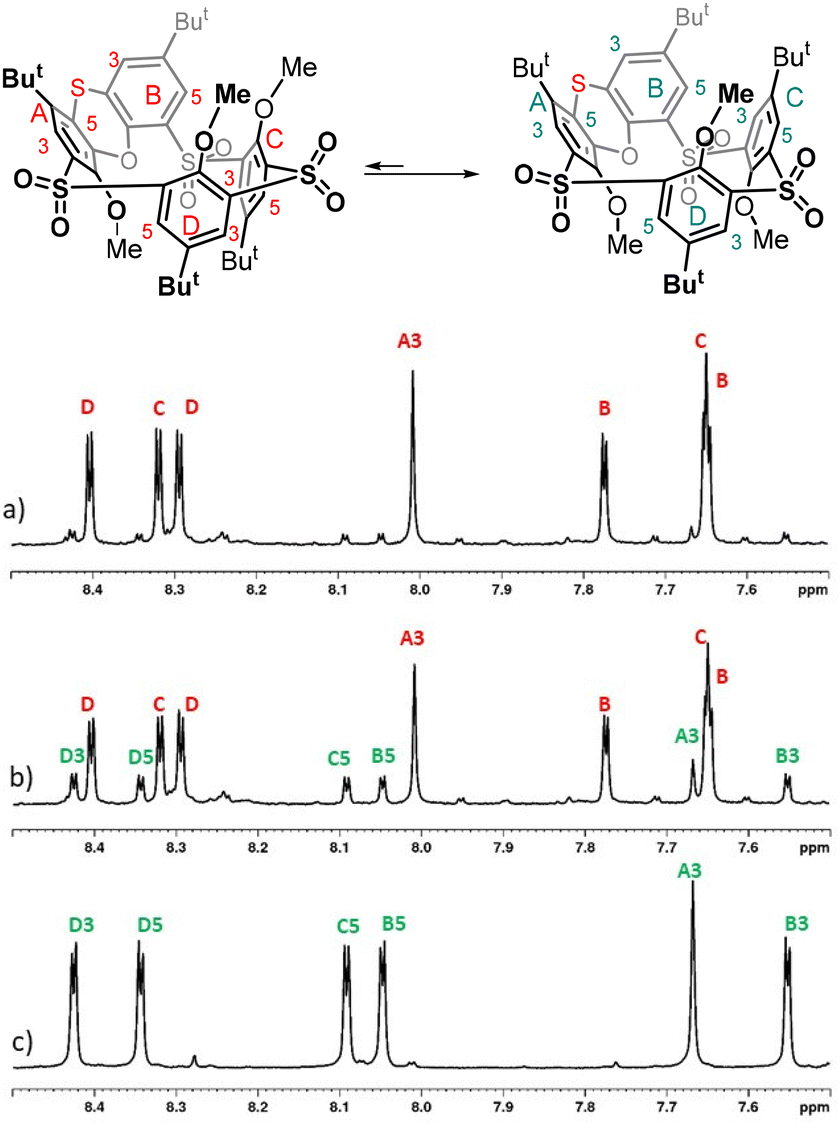 | ||
| Fig. 4 1H NMR spectra of: (a) 12b before heating (sample contained a small amount of isomer 12a due to non-ideal preparative TLC separation of products; (b) 12b after heating up to 110 °C (peaks corresponding to 12a visibly increased); (c) 12a. | ||
The crystal packing of 12a (Fig. 3c) was characterised by the infinite chains of calixarenes held together by the intermolecular chalcogen bonds between the sulfide S atoms and one of the SO bonds of the opposite sulfone bridge. An S⋯OS distance of 3.186 Å was reasonably shorter than the corresponding sum of vdW radii (3.32 Å).17 The individual chains are interconnected (see Fig. 3d) by non-classical hydrogen bonds between SO groups and hydrogens in tert-butyl groups (SO⋯H = 2.658 Å) and on the aromatic subunits (SO⋯Harom = 2.666 Å).
The formation of both reduced and oxidised compounds during the attempted stereomutation of 8 was rather unexpected. To avoid possible confusion about the oxygen source for oxidation, the reaction was performed under argon in carefully degassed chloroform, but the result was the same. It indicates that the sulfoxide SO bond in 8 was cleaved during the reaction, yielding 12. Such reactions (deoxygenations) are known from the chemistry of aromatic sulfoxides,19 and the resulting highly reactive oxygen species20 usually react with the environment (solvent) to form oxidized products. In our case, the only oxidizable molecule in the system (besides chloroform) is the starting sulfoxide 8, so the formation of a small amount of by-product 4 seems to be a more or less logical consequence.
To further support this idea, we repeated the reaction under identical conditions but with the addition of diphenyl sulfide as the presumed oxygen acceptor. After 11 hours of irradiation (364 nm) and subsequent methylation of the crude reaction mixture with MeI, the 1H NMR analysis showed three calixarene compounds 12a, 12b and 9a in 14%, 7% and 79% yields. The isomers 12a and 12b were obtained after column chromatography in 12% and 5% yields.
In any case, compound 12 represents the only example of its kind, where there is an unoxidized sulfur bridge together with the sulfone bridges. To the best of our knowledge, a similar type of compound has not yet been described for thiacalixarenes, because the oxidation of sulfur atoms always proceeds in such a way that sulfones begin to form only after all the sulfur bridges have been oxidized to sulfoxides.
Important parts of the reaction pathway were modelled using DFT. The detailed sequence of the multiple oxidation steps is beyond the scope of this research, hence we assumed that the sulphur bridges are fully oxidized before phenoxathiine oxidation takes place. Conformational screening of all structures was performed using CREST, and 30 lowest-lying conformers were optimized at the B3LYP/def2-SVP level with D3BJ corrections. The energy of the most favourable conformer is reported. The reactivity is expected to be guided by steric repulsion, which is usually well described at the DFT level, but due to a wide range of functional groups present in the molecule, final DLPNO-CCSD(T)/def2-QZVPP single point energy evaluation was performed. The relative energies at the DFT level differ quite substantially from CCSD(T) and both values are reported, with the DFT value (in italics).
From the thermodynamic point of view, the structures of 9a and 10a, possessing the out and in configuration of the sulfoxide group are close in energy, with the isomer 9a (out) being preferred by 3.3 (0.5) kcal mol−1. The reaction energy of the second oxidation step from sulfoxide 9a to sulfone 11a is −68 (−51) kcal mol−1, significantly more favoured than the first step, sulfide 12a to sulfoxide (9a or 10a), which is −41 (−28) kcal mol−1.
The kinetics of the oxidation steps was examined with respect to the approach of the oxidant, modelled using trifluoroperoxoacetic acid (TFPA). Both transition states leading to 9a and 10a were found, and the attack from outside was preferred by about 2.1 (−1.7) kcal mol−1. The second oxidation step, from sulfoxide to sulfone, proceeds from above the molecule, rather across the calixarene ring. The activation energy of the oxidation of 9a is 1.1 (−0.3) kcal mol−1 higher than that of the 10a isomer. The whole reaction sequence is shown in Fig. 5.
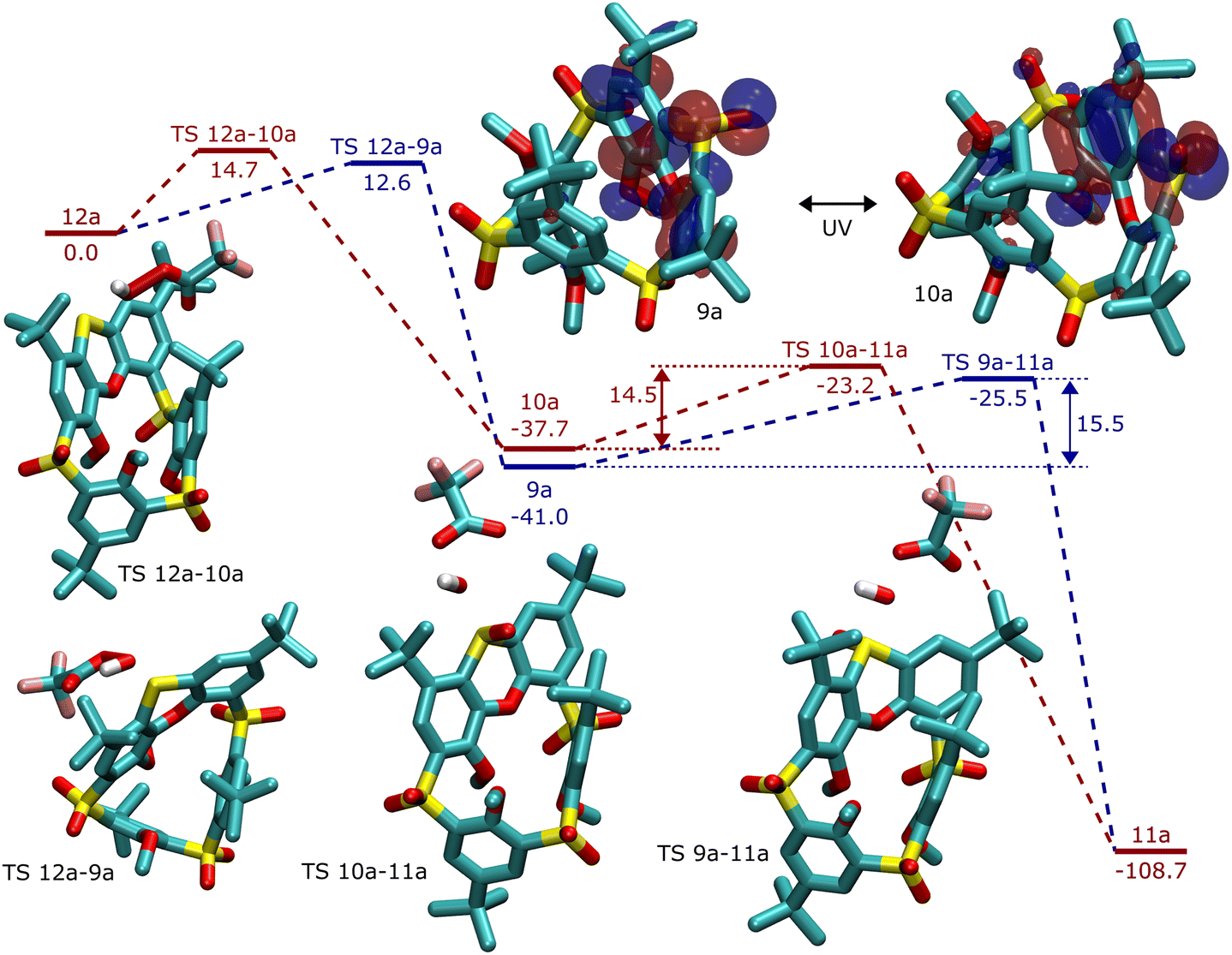 | ||
| Fig. 5 Reaction profile for stepwise oxidation of sulfide 12a to sulfone 11a through two different (in–out) sulfoxides, 9a and 10a. Relative energies were calculated at the CCSD(T) level. In the insets, the structures of transition states are shown, together with the HOMO orbitals (at the DFT level) of sulfoxides 9a and 10a. | ||
The difference in activation energy for the isomers agrees with the experimental results of the first oxidation step. In the second step, the calculated energy difference favors the oxidation of 10a, with SO pointing inwards, which is impossible without the isomerization of sulfoxide. The intuitive explanation using the argument of the steric hindrance of the calixarene ring is probably justified in this case. In addition, the extent of HOMO delocalization to aromatic rings is smaller in 10a, making the lone pair on sulphur more reactive in the SO in isomer.
The insufficiency of a computational model may come into play. Nevertheless, steric interactions are usually well described at the applied DFT-D3 level of theory, providing good geometries of intermediates and transition states. The role of solvent is not expected to be very important, namely, when comparing two very similar stereoisomers. The steric effects are presumably not the only contribution, for example in Fig. 5, phenoxathiin-oxide is deformed inwards during the oxidation of 10a, contributing to the increased activation energy.
Conclusion
Phenoxathiin-based thiacalixarene is well accessible in two steps starting from thiacalix[4]arene and represents an inherently chiral building block. The oxidised forms of this macrocycle bearing 3 sulfonyl and 1 sulfoxide groups were found to exhibit unexpected stereochemical preferences during its transformation to a fully oxidised macrocycle (4 × SO2). The sulfoxide group, which is part of the phenoxathiin moiety, is always pointing out of the cavity (SO out), while the opposite (SO in) configuration has never been obtained by direct oxidation. In order to achieve full oxidation, the configuration of the sulfoxide group must first be changed by a photochemical reaction before the final oxidation step occurs. The phenomenon of stereomutation of the sulfoxide group in this thiacalix[4]arene series was studied using a combination of experimental (NMR and single crystal X-ray analysis) and theoretical (DFT) approaches.
Experimental section
General experimental procedures
All chemicals were purchased from commercial sources and were used without further purification. Acetone was dried and distilled using conventional methods and acetonitrile was dried using a column solvent purification system, PureSolv MD7 (Inert). All samples were dried in a desiccator over P2O5 under vacuum (1 Torr) for 8 hours. Melting points were measured on Heiztisch Mikroskop Polytherm A (Wagner & Munz) and they were not corrected. 1H, 13C, COSY, HMQC and HMBC, and NOE spectra were measured on Bruker AvanceIII 600 operating at 600.13 MHz for 1H and 150.92 MHz for 13C, Bruker Avance III 500 MHz operating at 500.13 MHz for 1H and 125.77 MHz for 13C or Agilent 400-MR DDR2 operating at 400 MHz for 1H. Chemical shifts are given in δ-units (ppm) and were referenced to TMS. IR spectra were measured on a Nicolet 6700 (Thermo-Nicolet) FTIR spectrometer connected with a diamond ATR attachment GladiATR (PIKE) and DTGS detector. The measurement parameters were 4000–400 cm−1 spectral range, 4 cm−1 resolution, 64 spectral accumulations and Happ-Genzel apodization. ESI HRMS spectra were measured on a Q-TOF (Micromass) spectrometer. UV-Vis spectra were acquired on a dual beam UV-Vis spectrophotometer Varian CARY 50 CONC (Varian). UV spectra were recorded in the range of 200–800 nm with a step of 4 nm. The sample concentration was 1 × 10−3 mol L−1. The purity of the substances and courses of the reactions were monitored by thin layer chromatography (TLC) using silica gel 60 F254 on aluminum-backed sheets (Merck) and analyzed at 254 and 365 nm. Column chromatography was performed on silica gel 60 with a particle size of 0.063–0.200 mm (Merck). Radial chromatography was carried out on Chromatotron (Harrison Research) connected with Lab Pump RHSY2 (Fluid Metering). Self-prepared glass discs were covered using silica gel 60 PF254 containing CaSO4 (Merck). Preparative TLC was carried out on self-prepared glass plates (25 × 25 cm) covered by silica gel 60 PF254 containing CaSO4 (Merck).Synthetic procedures
Data for 4 were in accordance with the previously published data.8
Data for 8 were in accordance with the previously published data.8 Compounds 9a–c were prepared according to the published procedure.8
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 111.2 mg, 40%).
Data for 10a. M.p. > 250 °C. 1H NMR (400 MHz, CDCl3, 298 K): 8.52 (d, 1H, Ar–H, J = 2.5 Hz), 8.50 (d, 1H, Ar–H, J = 2.3 Hz), 8.41 (d, 1H, Ar–H, J = 2.5), 8.20 (d, 1H, Ar–H, J = 2.5), 8.15 (d, 1H, Ar–H, J = 2.3 Hz), 7.90 (s, 1H), 7.03 (d, 1H, Ar–H, J = 2.5 Hz), 4.14 (s, 3H, O–CH3), 3.78 (s, 3H, O–CH3), 3.17 (s, 3H, O–CH3), 1.65 (s, 9H, tBu–H), 1.50 (s, 9H, tBu–H), 1.47 (s, 9H, tBu–H), 0.97 (s, 9H, tBu–H). 13C NMR (126 MHz, CDCl3, 298 K): 155.61, 155.34, 148.97, 148.11, 147.38, 147.08, 143.47, 141.69, 140.76, 138.33, 136.71, 136.39, 135.70, 134.49, 132.87, 132.61, 132.01, 131.60, 130.66, 130.57, 130.33, 127.24, 124.55, 118.66, 77.30, 77.04, 76.79, 66.55, 65.35, 63.31, 32.81, 31.24, 31.22, 30.25. IR (ATR) ν (cm−1): 2957, 2870, 1478, 1423, 1316, 1133. HRMS (ESI+) (C43H52O11S4) m/z calc: 895.2285 [M + Na]+, found: 895.2283 [M + Na]+.
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 117.3 mg, 58%).
Data for 10b. M.p. > 250 °C. 1H NMR (500 MHz, CDCl3, 298 K): 8.45 (d, 1H, Ar–H, J = 2.3 Hz), 8.40 (d, 1H, Ar–H, J = 2.5 Hz), 8.33 (d, 1H, Ar–H, J = 2.5 Hz), 8.12 (d, 1H, Ar–H, J = 2.5 Hz), 8.07 (d, 1H, Ar–H, J = 2.3 Hz), 7.90 (s, 1H, Ar–H), 6.92 (d, 1H, Ar–H, J = 2.4 Hz), 4.41–4.32 (m, 1H, O–CH2), 4.28–4.13 (m, 2H, O–CH2), 4.10–4.01 (m, 1H, O–CH2), 3.95–3.86 (m, 1H, O–CH2), 3.36–3.27 (m, 1H, O–CH2), 1.59 (s, 9H, tBu–H), 1.43 (s, 9H, tBu–H), 1.41 (s, 9H, tBu–H), 1.37 (t, 3H, O–CH3, J = 6.9 Hz,), 0.92 (s, 9H, tBu–H), 0.82 (t, 3H, O–CH3, J = 7.0 Hz), 0.48 (t, 3H, O–CH3, J = 6.9 Hz).13C NMR (126 MHz, CDCl3, 298 K): 154.82, 152.84, 148.76, 147.59, 146.64, 146.41, 144.62, 142.26, 141.04, 137.55, 137.41, 136.36, 135.90, 134.85, 132.28, 132.06, 131.61, 131.26, 131.04, 130.94, 130.42, 127.62, 124.44, 119.49, 77.31, 77.06, 76.80, 76.58, 73.74, 72.29, 37.89, 35.50, 35.44, 34.71, 32.93, 31.29, 31.25, 30.32, 29.73, 15.52, 14.45, 14.26. IR (ATR) ν (cm−1): 2960, 2870, 1456, 1434, 1315, 1133. HRMS (ESI+) (C46H58O11S4) m/z calc: 937.2754 [M + Na]+, found: 937.2752 [M + Na]+.
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 73.7 mg, 66%).
Data for 10c. M.p. > 250 °C. 1H NMR (400 MHz, CDCl3, 298 K): 1H NMR (CDCl3, 400 MHz, 298 K) δ (ppm): 8.51 (d, 1H, Ar–H, J = 2.3 Hz), 8.45 (d, 1H, Ar–H, J = 2.5 Hz), 8.37 (d, 1H, Ar–H, J = 2.5 Hz), 8.18 (d, 1H, Ar–H, J = 2.5 Hz), 8.12 (d, 1H, Ar–H, J = 2.3 Hz), 7.94 (s, 1H, Ar–H), 7.03 (d, 1H, Ar–H, J = 2.5 Hz), 4.30–4.22 (m, 1H, O–CH2), 4.22–4.17 (m, 1H, O–CH2), 4.17–4.11 (m, 1H, O–CH2), 4.05–3.96 (m, 1H, O–CH2), 3.96–3.87 (, 1H, O–CH2), 3.24–3.13 (m, 1H, O–CH2), 1.97–1.83 (m, 2H, –CH2), 1.63 (s, 9H, tBu–H), 1.47 (s, 9H, tBu–H), 1.46 (s, 9H, tBu–H), 1.39–1.23 (m, 2H, –CH2), 1.21–1.03 (m, 2H, –CH2), 0.99 (t, 3H, –CH3, J = 7.4 Hz), 0.98 (s, 9H, tBu–H), 0.60 (t, 3H, –CH3, J = 7.5 Hz), 0.28 (t, 3H, –CH3, J = 7.5 Hz).13C NMR (126 MHz, CDCl3, 298 K): 155.02, 153.43, 148.70, 147.34, 146.59, 146.06, 144.55, 142.25, 140.97, 137.38, 137.37, 136.23, 135.65, 134.85, 132.33, 132.04, 131.76, 131.52, 131.51, 131.06, 130.25, 127.60, 124.42, 119.55, 82.55, 79.89, 78.14, 77.25, 77.00, 76.74, 37.72, 35.45, 35.35, 34.65, 32.83, 31.21, 31.15, 30.25, 22.97, 22.31, 21.84, 10.09, 9.81, 9.01. HRMS (ESI+) (C49H64O11S4) m/z calc: 979.3224 [M + Na]+, found: 979.3224 [M + Na]+.
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 23.2 mg, 91%).
Data for 11a were in accordance with the previously published data.8
Compound 11a was obtained in 17% yield under identical reaction conditions using derivative 9a as the starting compound.
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 22.6 mg, 89%).
Data for 11b were in accordance with the previously published data.8
Compound 11b was obtained in 13% yield under identical reaction conditions using derivative 9b as the starting compound.
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 22.1 mg, 87%).
Data for 11c were in accordance with the previously published data.8
Compound 11c was obtained in 17% yield under identical reaction conditions using derivative 9c as the starting compound.
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 4.3 mg, 17%).
Data for 11a were in accordance with the previously published data.8
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 3.3 mg, 13%).
Data for 11b were in accordance with the previously published data.8
:cyclohexane 1:10 v/v) as a racemic mixture (white solid, 4.3 mg, 17%).
Data for 11c were in accordance with the previously published data.8
The residue was suspended in dry acetonitrile (10 mL) and caesium carbonate (800 mg, 2.45 mmol) was added to the mixture. The suspension was stirred for 30 minutes. After that, methyl iodide (0.18 mL, 2.9 mmol) was added and the reaction mixture was heated under reflux overnight. Then, the solvent was removed under reduced pressure. The residue was dissolved in chloroform (10 mL) and water (10 mL). The layers were separated and the aqueous phase was extracted with chloroform (3 × 20 mL). Combined organic layers were dried over MgSO4. The solvent was evaporated and the residue was purified by silica gel chromatography (eluent ethyl acetate:cyclohexane 1:6 v/v) to give compound 12a (12 mg, 12%) and compound 12b (5 mg, 5%) as white solids together with 55 mg of compound 11a (from unreacted 8).
X-ray measurements
, a = 13.7722 (5) Å, b = 14.5337 (5) Å, c = 25.6391 (9) Å, α = 79.646 (2)°, β = 88.001 (2)°, γ = 78.359 (2)°, Z = 4, V = 4944.5 (3) Å3, Dc = 1.280 g.cm−3, μ(Cu-Kα) = 2.25 mm−1, crystal dimensions of 0.42 × 0.27 × 0.07 mm. Data were collected at 180 (2) K on a Bruker D8 Venture Photon CMOS diffractometer with Incoatec microfocus sealed tube Cu-Kα radiation. Data reduction, scaling and absorption correction were performed in Apex4.25 The structure was solved by charge flipping methods22 and anisotropically refined by full matrix least squares on F squared using CRYSTALS23 to final value R = 0.075 and wR = 0.191 using 18737 independent reflections (θmax = 70.4°), 1232 parameters and 66 restraints. The hydrogen atoms bonded to carbon atoms were placed in calculated positions refined with riding constraints, while hydrogen atoms bonded to oxygen were refined using soft restraints, after which they were refined using riding constraints. The disordered functional group positions were found in different electron density maps and refined with restrained geometry. MCE24 was used for the visualization of electron density maps. The occupancy of disordered functional groups was constrained to full. The structure has been deposited into the Cambridge Structural Database under number CCDC 2227441.†
, a = 12.4947 (5) Å, b = 12.7653 (6) Å, c = 19.6418 (8) Å, α = 92.4959 (17)°, β = 90.0704 (16)°, γ = 112.6404 (15)°, Z = 2, V = 2888.1 (2) Å3, Dc = 1.311 g cm−3, μ(Mo-Kα) = 0.38 mm−1, crystal dimensions of 0.53 × 0.34 × 0.23 mm. Data were collected at 180 (2) K on a Bruker D8 Venture Photon CMOS diffractometer with Incoatec microfocus sealed tube Mo-Kα radiation. Data reduction, scaling and absorption correction were performed in Apex4.25 The structure was solved by charge flipping methods22 and anisotropically refined by full matrix least squares on F squared using CRYSTALS23 to the final value R = 0.057 and wR = 0.169 using 12169 independent reflections (θmax = 26.7°), 736 parameters and 89 restrains. The hydrogen atoms bonded to carbon atoms were placed in calculated positions refined with riding constraints, while hydrogen atoms bonded to oxygen were refined using soft restraints after which they were refined using riding constraints. The disordered functional group positions were found in different electron density maps and refined with restrained geometry. MCE24 was used for the visualization of electron density maps. The occupancy of disordered functional groups was constrained to full. The structure has been deposited into the Cambridge Structural Database under number CCDC 2227442.†
, a = 14.1073 (2) Å, b = 17.6934 (5) Å, c = 19.5984 (4) Å, α = 70.921 (2)°, β = 89.7620 (14)°, γ = 83.5928 (17)°, Z = 4, V = 4591.58 (3) Å3, Dc = 1.286 g cm−3, μ(Cu-Kα) = 2.39 mm−1, crystal dimensions of 0.42 × 0.29 × 0.07 mm. Data were collected at 95 (2) K on a Rigaku OD Supernova using an Atlas S2 CCD detector with Cu-Kα radiation from a microfocus sealed X-ray tube. Data reduction, scaling and absorption correction were performed in CrysAlis PRO.26 The structure was solved by charge flipping methods22 and anisotropically refined by full matrix least squares on F squared using CRYSTALS23 to the final value R = 0.051 and wR = 0.136 using 18212 independent reflections (θmax = 74.6°), 1167 parameters and 116 restraints. The hydrogen atoms were placed in calculated positions refined with riding constraints. The disordered functional group positions were found in different electron density maps and refined with restrained geometry. MCE24 was used for the visualization of electron density maps. The occupancy of disordered functional groups was constrained to full. Despite significant efforts, a suitable solvent disorder model could not be found; therefore, the solvent contribution to the structure factors was accounted for using PLATON squeeze,27 resulting in a total solvent void volume of 374.6 Å3. The structure has been deposited into the Cambridge Structural Database under number CCDC 2227443.†
265 independent reflections (θmax = 26.4°), 710 parameters and 255 restraints. The hydrogen atoms were placed in calculated positions refined with riding constraints. The disordered functional group positions were found in different electron density maps and refined with restrained geometry. MCE24 was used for the visualization of electron density maps. The occupancy of disordered functional groups was constrained to full. The structure has been deposited into Cambridge Structural Database under number CCDC 2227444.†
Calculations
The conformational screening was performed in CREST version 2.11,28 using default settings. The DFT geometry optimizations were performed in ORCA 5.0.3,29 using B3LYP30 functional with a def2-SVP basis set31 employing the RIJCOSX approximation,32 with a def2/J auxiliary basis set33 and D3BJ corrections.34Final single point energies were obtained at DLPNO-CCSD(T) with a def2-QZVPP31 basis set, def2/J auxiliary basis set33 and def-QZVPP/C auxiliary basis set for CC,35 using the RIJCOSX approximation.32
Geometries of the most favourable isomers of all stationary points at the DFT level are reported in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Czech Science Foundation (Grant 20-08667S).References
- H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971 CrossRef CAS.
- For selected books on calixarenes and their applications, see: (a) Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M. X. Wang, Springer, Cham, Switzerland, 2016 Search PubMed; (b) C. D. Gutsche, Calixarenes: An Introduction, Royal Society of Chemistry, Cambridge, U.K., 2nd edn, 2008 Search PubMed; (c) Calixarenes in the Nanoworld, ed. J. Vicens, J. Harrowfield and L. Backlouti, Springer, Dordrecht, The Netherlands, 2007 Search PubMed; (d) Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer, Dordrecht, The Netherlands, 2001 Search PubMed; (e) Calixarenes in Action, ed. L. Mandolini and R. Ungaro, Imperial College Press, London, 2000 Search PubMed.
- For recent reviews on thiacalixarenes, see: (a) R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824 RSC; (b) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS PubMed; (c) P. Lhotak, Eur. J. Org. Chem., 2004, 1675 CrossRef CAS.
- (a) A. M. Litwak and S. E. Biali, J. Org. Chem., 1992, 57, 1945 CrossRef; (b) S. E. Biali, Synlett, 2003, 1 CrossRef CAS; (c) A. M. Litwak, F. Grynszpan, O. Aleksiuk, S. Cohen and S. E. Biali, J. Org. Chem., 1993, 58, 393 CrossRef CAS.
- For selected examples of a spirodienone route to the derivatization of classical calixarenes, see: (a) C. Gaeta, F. Troisi, C. Talotta, T. Pierro and P. Neri, J. Org. Chem., 2012, 77, 3634 CrossRef CAS PubMed; (b) F. Troisi, T. Pierro, C. Gaeta and P. Neri, Tetrahedron Lett., 2009, 50, 4416 CrossRef CAS; (c) S. Simaan and S. E. Biali, J. Org. Chem., 2004, 69, 95 CrossRef CAS PubMed; (d) S. Simaan, K. Agbaria and S. E. Biali, J. Org. Chem., 2002, 67, 6136 CrossRef CAS PubMed; (e) K. Agbaria and S. E. Biali, J. Am. Chem. Soc., 2001, 123, 12495 CrossRef CAS PubMed; (f) K. Agbaria, J. Wöhnert and S. E. Biali, J. Org. Chem., 2001, 66, 7059 CrossRef CAS PubMed.
- N. Morohashi, M. Kojima, A. Suzuki and Y. Ohba, Heterocycl. Commun., 2005, 11, 249 CAS.
- (a) K. Polivkova, M. Simanova, J. Budka, P. Curinova, I. Cisarova and P. Lhotak, Tetrahedron Lett., 2009, 50, 6347 CrossRef CAS; (b) L. Vrzal, M. Kratochvilova-Simanova, T. Landovsky, K. Polivkova, J. Budka, H. Dvorakova and P. Lhotak, Org. Biomol. Chem., 2015, 13, 9610 RSC.
- (a) T. Landovsky, H. Dvorakova, V. Eigner, M. Babor, M. Krupicka, M. Kohout and P. Lhotak, New J. Chem., 2018, 42, 20074–20086 RSC; (b) T. Landovsky, M. Tichotova, L. Vrzal, J. Budka, V. Eigner, H. Dvorakova and P. Lhotak, Tetrahedron, 2018, 74, 902–907 CrossRef CAS.
- (a) K. Flidrova, P. Slavik, V. Eigner, H. Dvorakova and P. Lhotak, Chem. Commun., 2013, 49, 6749 RSC; (b) P. Slavik, V. Eigner and P. Lhotak, CrystEngComm, 2016, 18, 4964 RSC.
- (a) P. Slavik, H. Dvorakova, M. Krupicka and P. Lhotak, Org. Biomol. Chem., 2018, 16, 838 RSC; (b) P. Slavik, M. Krupicka, V. Eigner, L. Vrzal, H. Dvorakova and P. Lhotak, J. Org. Chem., 2019, 84, 4229 CrossRef CAS PubMed.
- T. Landovsky, V. Eigner, M. Babor, M. Tichotova, H. Dvorakova and P. Lhotak, Chem. Commun., 2020, 56, 78–81 RSC.
- T. Landovsky, M. Babor, J. Cejka, V. Eigner, H. Dvorakova, M. Krupicka and P. Lhotak, Org. Biomol. Chem., 2021, 19, 8075–8085 RSC.
- J. Miksatko, V. Eigner, H. Dvorakova and P. Lhotak, Tetrahedron Lett., 2016, 57, 3781–3784 CrossRef CAS.
- Similar observations regarding exo- and endo- stereoisomers of calixarenes and an exo/endo attack with respect to the calixarene cavity were also discussed in: F. Troisi, T. Pierro, C. Gaeta and P. Neri, Org. Lett., 2009, 11, 697–700 CrossRef CAS PubMed.
- For selected example papers on photochemical stereomutation of the sulfoxide group, see: (a) K. Mislow, M. Axelrod, D. R. Rayner, H. Gotthardt, L. M. Coyne and G. S. Hammond, J. Am. Chem. Soc., 1965, 87, 4958–4959 CrossRef CAS; (b) W. S. Jenks, D. D. Gregory, Y. Guo, W. Lee and T. Tetzlaff, Mol. Supramol. Photochem., 1997, 1, 1–56 CAS; (c) W. S. Jenks, W. Lee and D. Shutters, J. Phys. Chem., 1994, 98, 2282–2289 CrossRef CAS; (d) W. Lee and W. S. Jenks, J. Org. Chem., 2001, 66, 474–480 CrossRef CAS PubMed.
- This is in contrast to the only paper devoted to the photoisomerization of the sulfoxide group in thiacalix[4]arenes, where the authors successfully used 283 nm light. On the other hand, they used much shorter reaction times (4 min), see: R. Cacciapaglia, S. Di Stefano, O. Lanzalunga, L. Maugeri and M. Mazzonna, Eur. J. Org. Chem., 2012, 1426–1430 CrossRef CAS.
- (a) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS; (b) R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS; (c) M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- (a) L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed; (b) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed; (c) W. Wang, B. Ji and Y. Zhang, J. Phys. Chem. A, 2009, 113, 8132–8135 CrossRef CAS PubMed; (d) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- For examples of deoxygenation of sulfoxides, see: (a) J. Drabowicz, T. Numata and S. Oae, Org. Prep. Proced. Int., 1977, 9, 63–83 CrossRef CAS; (b) G. M. Gurria and G. H. Posner, J. Org. Chem., 1973, 38, 2419–2420 CrossRef CAS; (c) L. Shiri and M. Kazemi, Res. Chem. Intermed., 2017, 43, 6007–6041 CrossRef CAS.
- For the nature of highly reactive oxygen species formed, see: (a) D. D. Gregory, Z. Wan and W. S. Jenks, J. Am. Chem. Soc., 1997, 119, 94–102 CrossRef CAS; (b) Z. Wan and W. S. Jenks, J. Am. Chem. Soc., 1995, 117, 2667–2668 CrossRef CAS.
- Bruker, APEX3, SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2015 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- J. Rohlicek and M. Husak, J. Appl. Crystallogr., 2007, 40, 600 CrossRef CAS.
- Bruker, APEX4, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- Rigaku OD, CrysAlis PRO, Rigaku Oxford Diffraction Ltd, Yarnton, Oxfordshire, England, 2020 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc., 2007, 117(4), 587–597 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral characterization of new compounds, temperature-dependent NMR spectra of 12a and 12b, and DFT calculations. CCDC 2227440–2227444. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00530e |
| This journal is © The Royal Society of Chemistry 2023 |