
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis of β-lactams: recent examples
Adrian
Saura-Sanmartin
 *a and
Laura
Andreu-Ardil
b
*a and
Laura
Andreu-Ardil
b
aDepartamento de Química Orgánica, Facultad de Química, Universidad de Murcia, 30100 Murcia, Spain. E-mail: adrian.saura@um.es
bFacultad de Veterinaria, Universidad de Murcia, 30100 Murcia, Spain
First published on 6th April 2023
Abstract
The synthesis of β-lactam derivatives is a research topic of great interest due to the biological activity of these molecules. Indeed, there are several antibiotics which include a β-lactam core in their structures, such as penicilins, monobactams, carbacephems and cephamycins. The development of stereoselective approaches to access these molecular architectures turns out to be necessary in order to take advantage of the distinct properties provided by the different stereoisomers. This review covers recent advances towards the stereoselective synthesis of β-lactams, including Staudinger syntheses, cascade reactions, metal-catalyzed syntheses and base-promoted cyclizations. Within these methods, some particularly novel synthetic approaches are highlighted, such as the induction of chirality through bimetallic synergistic catalysis or the transfer of chirality between components in mechanically interlocked molecules. Additionally, a critical opinion on the state of the art of this research field is offered, remarking key points on which the future research should be focused on.
1. Introduction
β-Lactams are organic compounds well known for their biological activity, highlighting their antibiotic properties. Although the discovery of this type of ring arose with the development of penicillin, there are several natural products containing this four-membered ring.1 Thus, β-lactams are privileged scaffolds for the preparation of a vast range of antibiotics, including penicillins, cephalosporins, monobactams and carbacephems, among others. This extensive presence in a wide range of antibiotics has led to an increased interest of several synthetic chemists in the development of methodologies to access this type of compounds.2The traditional approaches to synthesize β-lactams followed the method developed by Hermann Staudinger in 1907, in which an imine reacts with a ketene via a [2 + 2] cycloaddition to afford the target product.3 But the development of new synthetic methodologies has allowed to obtain these bioactive molecules in several ways.4–21
Progress in the preparation of β-lactams ran parallel with the development of stereoselective synthetic methodologies, allowing to access to the different isomers (cis or trans) and also to obtain enantioenriched products.22–26 This review is focused on recent advances towards the stereoselective synthesis of β-lactams, dividing the manuscript into three main sections: (i) metal-free cycloaddition reactions; (ii) transition metal-catalysis; and (iii) base-promoted synthesis. In addition to highlighting selected examples, a critical opinion on the state of the art is provided.
The examples highlighted in this review have been carefully selected from literature published from 2018, when Hosseyni and Jarrahpour published a review on the synthesis of β-lactams.2 For details of the exact chronology of the synthesis of β-lactams and specific synthetic approaches towards their preparation, we refer readers to previously reported reviews.1,9,22,23,25–27
2. Metal-free cycloaddition reactions
Multicomponent cycloaddition reactions have been the most widely employed methods in order to obtain β-lactam derivatives, highlighting the synthesis based on the Staudinger ketene-imine cycloaddition.2,28–33 This section discusses selected recent examples of metal-free stereoselective cycloaddition syntheses of β-lactams.2.1. Ketene-imine cycloaddition reactions
As abovementioned, the ketene-imine cycloaddition has been widely employed in the preparation of β-lactams. A representative example reported by Bhalla and coworkers describes the synthesis of a series of trans-4-(thiophenyl)pyrazolylyl β-lactams 1.34 The synthesis proceeds through a Staudinger cycloaddition reaction of (thiophenyl)pyrazol substituted Schiff's bases 2 and 2-substituted ethanoic acid or acid chloride derivatives 3 (Scheme 1a) in the presence of NEt3 or POCl3/NEt3. The ketene intermediates are generated in situ through the deprotonation of carboxylic acid derivatives 1 (X = OH) by using NEt3, followed by the formation of an active ester with POCl3, which subsequently reacts with NEt3, or by direct reaction with NEt3 if ethanoyl chloride derivatives 1 (X = Cl) are employed. When the reaction was carried out in chloroform, tetrahydrofuran or toluene at reflux, only the trans-β-lactams 1 were obtained in high yield (67–94%). Interestingly, one cis derivative (R1 = acetyl, R2 = p-methoxyphenyl) could be obtained using CH2Cl2 as solvent and setting the temperature from 0 °C to room temperature.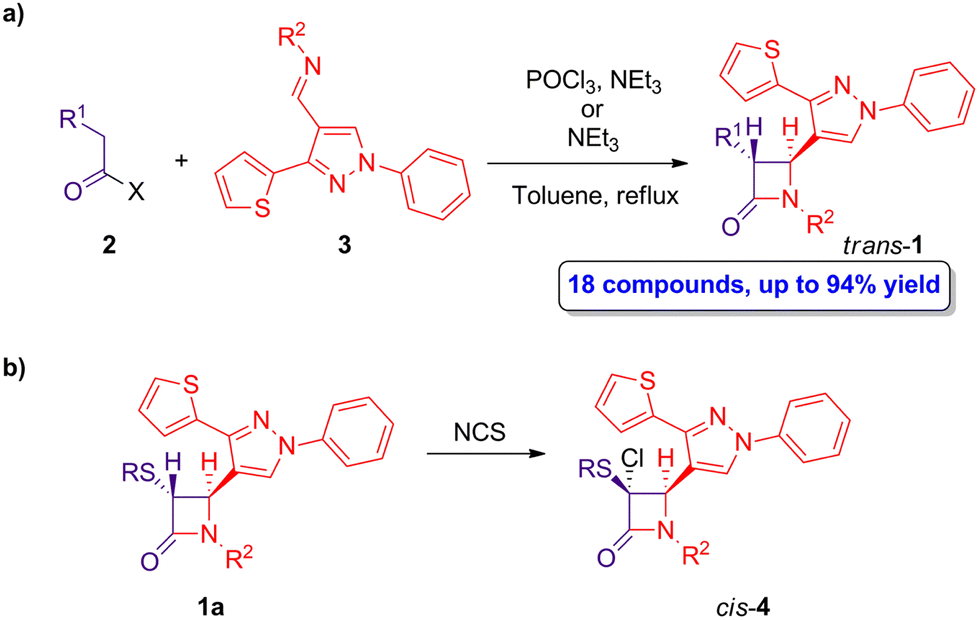 | ||
| Scheme 1 (a) Diastereoselective ketene-imine cycloaddition reaction of 2 and 3 to afford trans-β-lactams 1; and (b) preparation of cis-β-lactams 4. The original results were reported by Bhalla and colleagues.34 | ||
Noteworthy, the treatment of trans-β-lactams 1a having sulfonyl substituents with N-chlorosuccinimide (NCS) afforded cis-β-lactams 4 (Scheme 1b), which can further react through nucleophilic substitution to afford a series of functionalized cis-β-lactams.
The pharmacophore hybridization approach, in which two distinct bioactive functions are combined into a single molecule, was employed by Jarrahpour and coworkers in the synthesis of hybrid molecules combining fused chromenes and β-lactams.35 Both chromenes and β-lactams are well-known for their biological applications, such as the development of antibacterial, antifungal or anticancer products, among others. Through pharmacophore hybridization is possible to improve some parameters, such as therapeutic potential, mode of action and pharmacokinetics. A diastereoselective ketene-imine cycloaddition reaction between 2-(4-formylphenoxy)acetic acid (5) and imine 6 using NEt3 as base and p-toluensulfonyl chloride afforded the target aldehyde-functionalized β-lactams 7 in high yields (up to 88%), showing only cis geometry (Scheme 2a). The subsequent functionalization of β-lactams 7 through a multicomponent reaction with the chromene precursors 8 and 9a,b in the presence of triethylenediamine (DABCO) yielded the chromeno β-lactam hybrids 10a,b (Scheme 2b). Interestingly, anti-inflammatory and anticancer functions of both aldehyde-functionalized β-lactams 7 and chromeno β-lactams 10a,b were evaluated, observing good anti-inflammatory ratios (up to 53.4), whose activity depends on the different substitution at the N1 and C2 atoms of the lactam ring, and an anticancer response in vitro against SW1116 colon cancer cells.
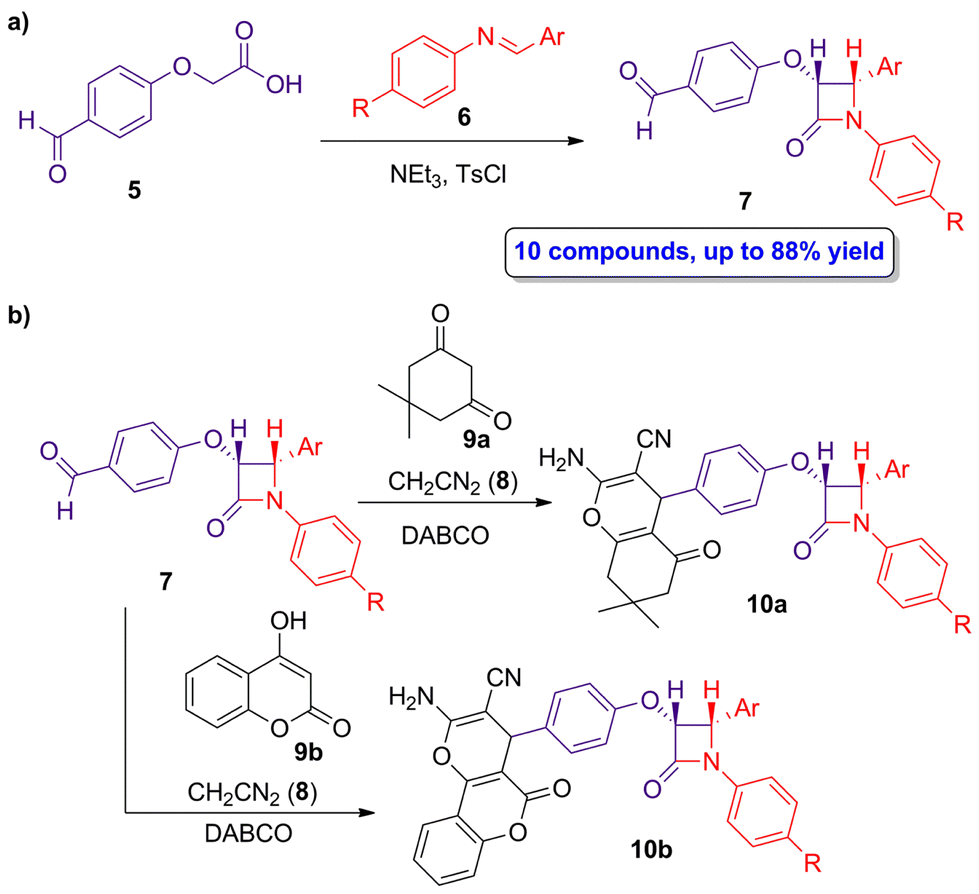 | ||
| Scheme 2 (a) Diastereoselective ketene-imine cycloaddition reaction of 5 and 6 to afford β-lactams 7; and (b) preparation of chromeno β-lactams hybrids 10a,b. The original results were reported by Jarrahpour and colleagues.35 | ||
The Staudinger synthesis was also employed by Sharma and colleagues in the preparation of monocyclic β-lactams having a 1,3,4-thiadiazole motif, which also has important biological properties per se.36 Phenoxyacetyl chloride 11 was employed as source of ketene and thiadiazol-functionalized imines 12 as Schiff bases in the [2 + 2] cycloaddition reaction, which afforded a wide range of a monocyclic β-lactams 13 in high yield (up to 89%) and in a diastereoselective manner (Scheme 3), showing cis geometry as it was determined by the coupling constants of the hydrogens of the lactam ring using 1H-NMR. Interestingly, in vitro assays demonstrated antibacterial activity against the Staphylococcus aureus ATCC 25904 (Gram-positive) and Escherichia coli ATCC 12435 (Gram-negative) pathogens of mostly of the monocyclic cis-β-lactams 13 products, using ciprofloxacin as the reference control. In addition, the presence of electron-withdrawing groups placed at the aryl motif from the imine scaffold increased the antibacterial activity. Due to the stereoselectivity, high yield and easy purification step of this protocol, as well as the vast possibility of functionalization, this synthetic approach paves the way to the obtention of new monocyclic β-lactams having potential applications in medicinal chemistry.
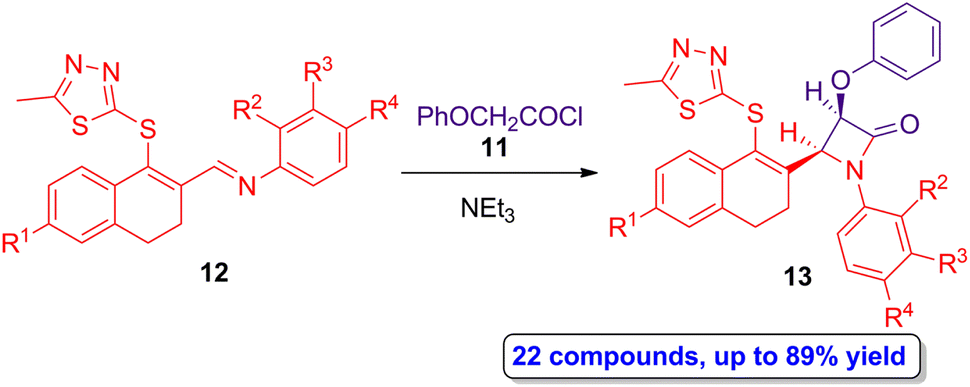 | ||
| Scheme 3 Diastereoselective ketene-imine cycloaddition reaction of 11 and 12 to afford thiadiazol-functionalized monocyclic β-lactams 13. The original results were reported by Sharma and colleagues.36 | ||
The same research group also employed an analogous strategy using acid anhydrides as ketene precursors to obtain a series of monocyclic cis-β-lactams.37
2.2. Mannich lactamization reactions
The Mannich reaction has been successfully employed in the synthesis of several heterocycles.38–42Through an isothiourea-catalyzed Mannich/lactamization cascade reaction, Zhao, Deng and colleagues reported the asymmetric preparation of β-lactam derivatives.43 Bench stable carboxylic acids 14 and isatin-derived ketimines 15 were employed as substrates and the chiral homobenzotetramisole 16 as catalyst in the presence of pivaloyl chloride, affording spirooxindole β-lactams 17 in high yields (up to 98%), showing excellent diastereo- and enantioselectivities (up to 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 dr and >99% ee, respectively). The proposed mechanism starts with the reaction in situ of carboxylic acid 14 and pivaloyl chloride, leading to the formation of acid anhydride intermediate 18 (Scheme 4). The subsequent acylation of 16 affords intermediate 19, whose subsequent deprotonation yields intermediate 20. Then, a Si-face attack Mannich reaction takes places, leading to intermediate 21. A final intramolecular cyclization leads to the obtention of the target enantioenriched spirooxindole β-lactams 17 showing cis geometry.
:6 dr and >99% ee, respectively). The proposed mechanism starts with the reaction in situ of carboxylic acid 14 and pivaloyl chloride, leading to the formation of acid anhydride intermediate 18 (Scheme 4). The subsequent acylation of 16 affords intermediate 19, whose subsequent deprotonation yields intermediate 20. Then, a Si-face attack Mannich reaction takes places, leading to intermediate 21. A final intramolecular cyclization leads to the obtention of the target enantioenriched spirooxindole β-lactams 17 showing cis geometry.
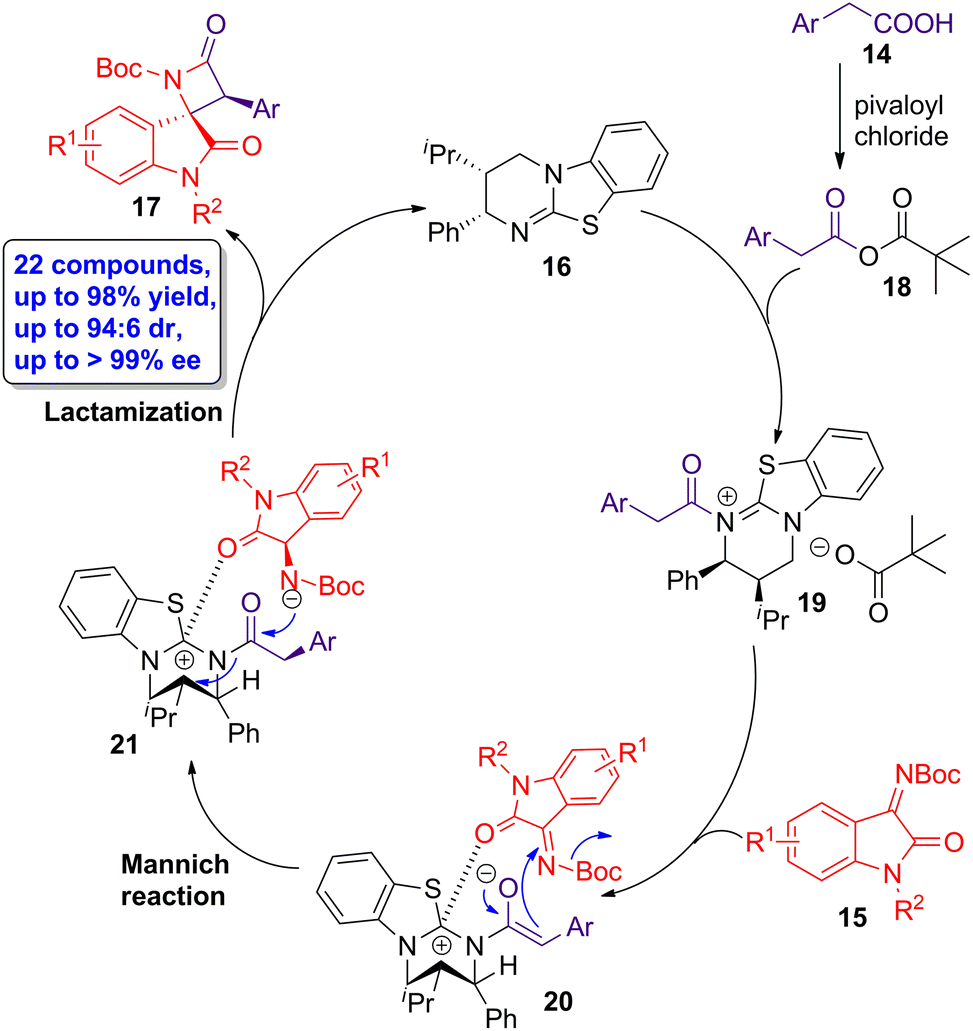 | ||
| Scheme 4 Proposed catalytic cycle for the synthesis of enantioenriched spirooxindole β-lactams 17 from carboxylic acids 14 and isatin-derived ketimines 15. The original results were reported by Zhao, Deng and colleagues.43 | ||
2.3. Ugi and Michael addition cascade reactions
Diversity-oriented synthesis (DOS) through cascade reactions has turned out to be useful approaches towards the synthesis of different nitrogen-containing heterocyclic compounds. Galons, Lu and coworkers employed the DOS approach carrying out an Ugi–Michael addition cascade reaction to synthesize β-lactams 22via the previous formation of Ugi-adduct intermediate 23 (Scheme 5).44 In the model reaction, maleic acid derivative 24, amine 25, isocyanide 26 and nitrogen heterocyclic aldehyde 27 were employed as substrates. In the optimized conditions, 22 was obtained in 80% yield, having a good diastereoselectivity (3.0:1 dr). X-ray analysis and bidimensional NMR spectra allowed to determine that the main diastereoisomer is the lactam having pyridine ring and methyl ester group in cis-conformation. Different substrates were tested, observing a good tolerance for different hydrogen bond acceptor substituents placed at the aldehyde scaffold, thus even being possible to use 2-oxo-2-phenylacetaldehyde and ethyl 2-oxoacetate as aldehyde derivatives. Thus, control over the reaction can be provided by carbonyl groups acting as hydrogen bond acceptors. Interestingly, the use of fumaric acid derivatives, which are the geometric isomers of the maleic ones, also leads to the target lactams but affecting the diastereoselectivity of the Michael addition step, which is the one determining the chemo- and diastereoselectivities according to DFT calculations.
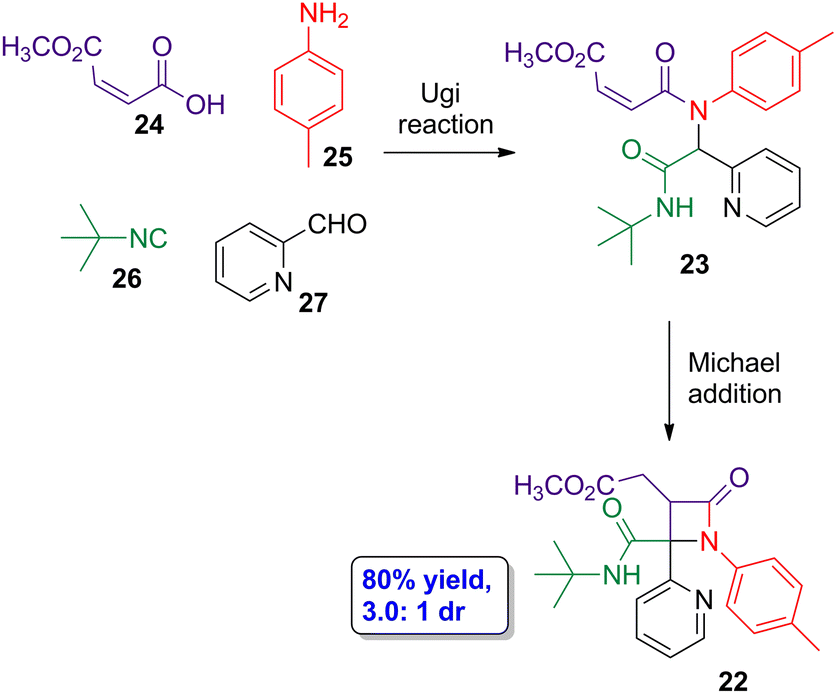 | ||
| Scheme 5 Diastereoselective Ugi-Michael cascade reaction to synthesize β-lactam 22 from carboxylic acid 24, aniline 25, isocyanide 26 and aldehyde 27. The original results were reported by Galons, Lu and colleagues.44 | ||
3. Metal-assisted synthesis
The use of transition-metal-catalysis has turned out to be a suitable strategy to access to a vast range of heterocyclic compounds.45–49 Thus, through metal-assisted synthesis, several β-lactams could be synthesized. This section covers recent selected examples on the use of transition-metals to catalyze the formation of the target lactams.3.1. Palladium-assisted reactions
A palladium-catalyzed enantioselective intramolecular C(sp3)–H amidation of quinoline-functionalized amides 28 was recently employed by He, Liu, Chen and colleagues in the asymmetric synthesis of β-lactams 29.50 The chiral fluorinated BINOL 30 was employed as the ligand, PdCl2(PhCN)2 as the catalyst and Cs2CO3 as the base in the optimized cyclization reaction of 28 in the presence of 2-methoxy-5-chlorophenyl iodide as oxidant (Scheme 6a). A series of β-lactams bearing quinoline-based motifs were synthesized in good to high yields (up to 94%) with high enantioselectivity (up to 94% ee). Interestingly, the removal of the quinoline motifs was feasible by treatment with cerium ammonium nitrate, retaining the enantiomeric excess of the product. The reaction proceeds forming an enantioenriched Pd(II)-palladacycle 31 whose enantioselectivity is controlled by the fluorinated BINOL-based ligand 30 (Scheme 6b). The subsequent oxidative addition of the aryl iodide affords the intermediate 32, which can then experience a ligand exchange with HCO3− leading to palladacycle 33. This cycle can undergo either C–C or C–N reductive elimination. Computational studies allowed to conclude that the use of the nonconventional aryl iodide oxidant leads to high chemoselectivity due to electronic and steric effects, thus favouring the C–N reductive elimination over the C–C one. This synthetic methodology allows to access to a wide range of β-aryl-β-lactams in high yields and excellent enantioselectivities.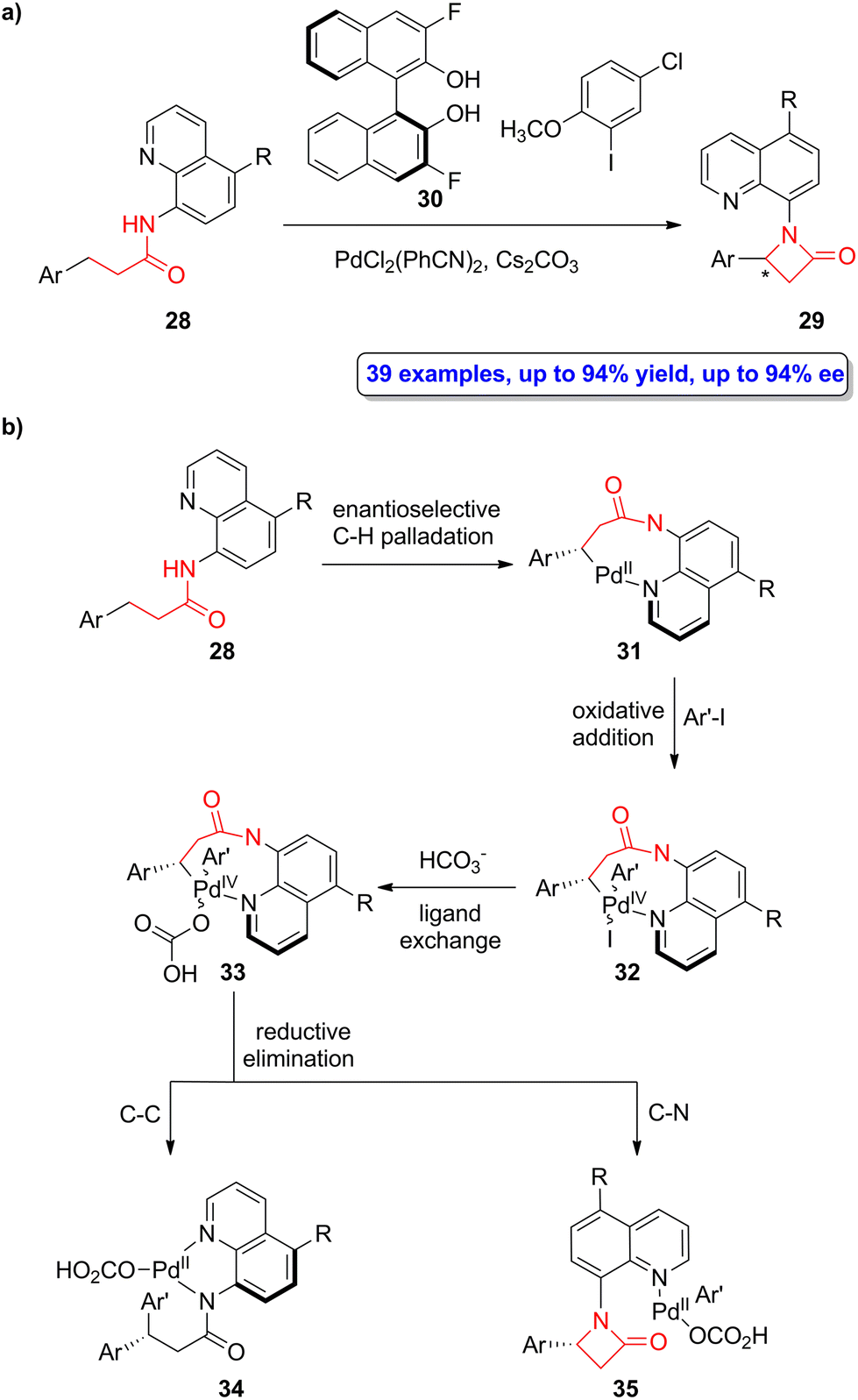 | ||
| Scheme 6 (a) Palladium-catalyzed enantioselective synthesis of β-lactams 29; and (b) proposed mechanism for the formation of β-lactams 29. The original results were reported by He, Liu, Chen and colleagues.50 | ||
Yu and coworkers reported the mono-selective synthesis of a series of β-lactams using a related strategy, the β-C(sp3)–H lactamization of amides having commonly used protecting groups (tosyl, 4-nitrobenzenesulphonyl, 4-cianobenzenesulfonyl, 2-trimethylsilylethanesulfonyl and mesyl) instead of quinoline-based substituents, thus diversifying the range of products that can be obtained through C(sp3)–H lactamization of amides.51
Huang and colleagues reported a novel palladium-catalyzed double C–H bond activation synthetic approach to access β-lactams via carbonylative formal cycloaddition.52 Thus, the authors carried out the reaction of alkylarenes 36 and aldimines 37 in the presence of carbon monoxide, using Pd(CH3CN)2Cl2 as the catalyst, Xantphos as the ligand, di-tert-butyl peroxide (DTBP) as the oxidant and 1,2,2,6,6-pentamethylpiperidine (PMP) as the base (Scheme 7). Through this synthetic approach, a series of trans-β-lactams 38 were synthesized in moderate to high yields (up to 94%). The reaction proceeds through the prior formation of the palladium-ketene complexes 39 formed by the reaction of the radical intermediates 40 and carbon monoxide, which can subsequently react with aldimines 37 to afford the target heterocycles in a diastereoselective manner.
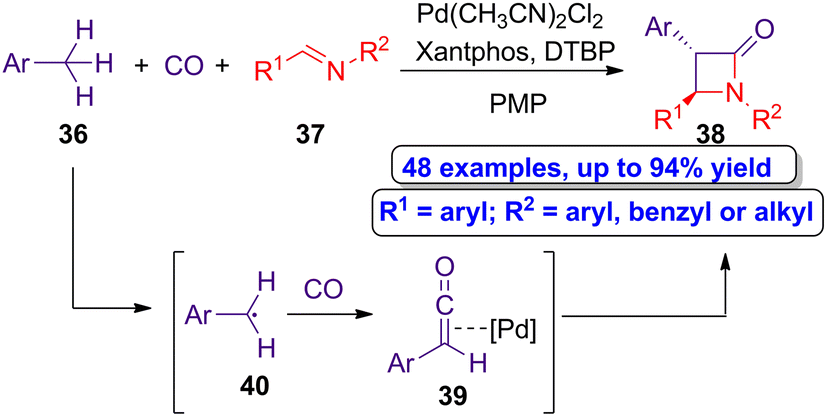 | ||
| Scheme 7 Diastereoselective carbonylative formal cycloaddition of alkylarenes 36 and aldimines 37 to afford trans-β-lactams 38. The original results were reported by Huang and colleagues.52 | ||
3.2. Rhodium-assisted reactions
Sun and coworkers reported the diastereoselective synthesis of highly functionalized β-lactams through a three-component rhodium-organo relay catalysis.53 The reaction of N-hydroxyanilines amphiphilic nucleophiles 41, enynones 42 and diazo compounds 43 affords only furyl-functionalized cis-β-lactams 44, using Rh2(OAc)4 as catalyst and benzoylquinine 45 as organic base (Scheme 8). The target lactams were obtained in high yields (up to 90%) having an excellent diastereoselectivity (dr > 20:1). The reaction proceeds through the formation of an imine derivative from a 2-furyl rhodium carbene, generated in situ with the corresponding enynone 42, and the amphiphilic nucleophile 41. The sequential Wolff rearrangement of diazo 43 to the ketene, and the subsequent ketene-imine cycloaddition leads to the obtention of the four-membered ring lactams. Interestingly, this synthetic methodology is compatible with the functionalization of N-hydroxyamine, enynone and diazo compounds, with both electron-withdrawing or electron-donating groups, not affecting the yield of the reaction, affording the desired compounds in good to high yields and with high stereoselectivity. A mechanistic study allowed to conclude that the diastereoselectivity of the reaction arises during the course of an umpolung Staudinger cyclization, in which a ketene-based enolate acts as the nucleophile and the imine acts as the electrophile, thus leading to the formation of fully substituted β-lactams having exclusively cis geometry.
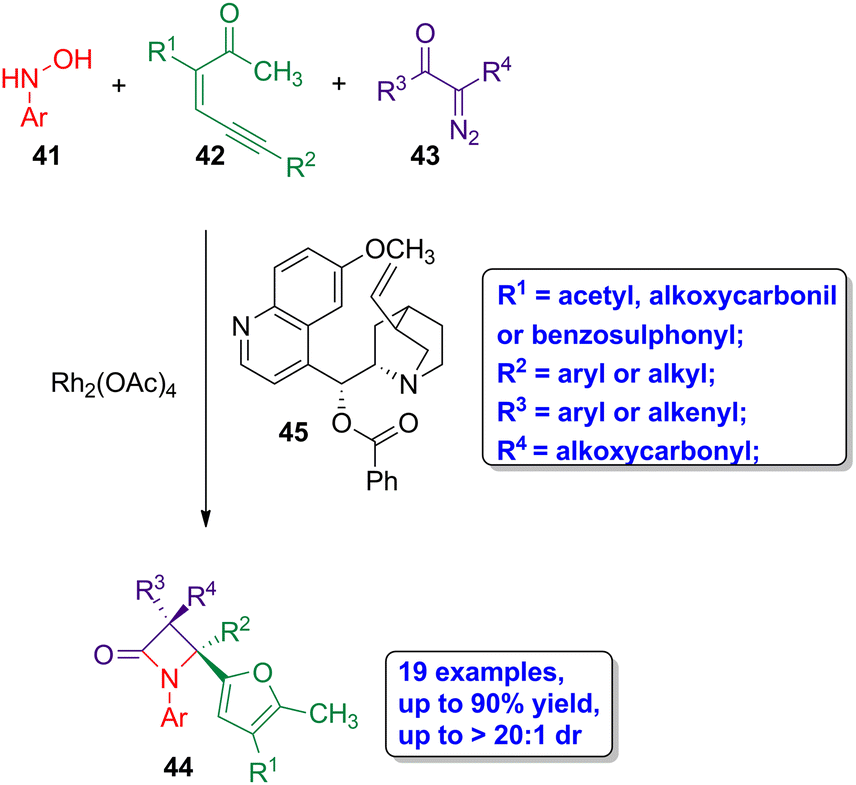 | ||
| Scheme 8 Rhodium-catalyzed diastereoselective synthesis of cis-β-lactams 44 from N-hydroxyanilines 41, enynones 42 and diazo compounds 43. The original results were reported by Sun and colleagues.53 | ||
A rhodium-catalyzed approach was also employed by Novikov and coworkers in the assembly of spirocyclic β-lactams.54 The reaction of the halogenated azirine derivatives 46 with diazocompounds 47 in the presence of Rh2(OAc)4 leads to the formation of azadienes 48 (Scheme 9a). The reaction of these azadienes with diazo-Meldrum's acid 49 using Rh2(Piv)4 as catalyst affords the spiro β-lactams 50 in moderate yields (21% to 59%) and with high stereoselectivity, showing a preferentially formation of the (1RS,4RS) diastereoisomer. A possible mechanism could be the formation of the rhodium carbenoid 51, from diazo-Meldrum's acid 49, which acts as the ketene source (52) after a Wolff rearrangement (Scheme 9b), thus allowing a Staudinger cycloaddition in the final step. Therefore, the four-member ring skeleton of the lactam is assembled via two separate rhodium-catalyzed steps from two distinct diazo derivatives. Interestingly, the formation of the target lactams was also possible using isoxazoles derivatives as starting materials instead of azirines. Noteworthy, this work is the first reported example of the formation of diazo-Meldrum's acid-based iminium-type ylides.
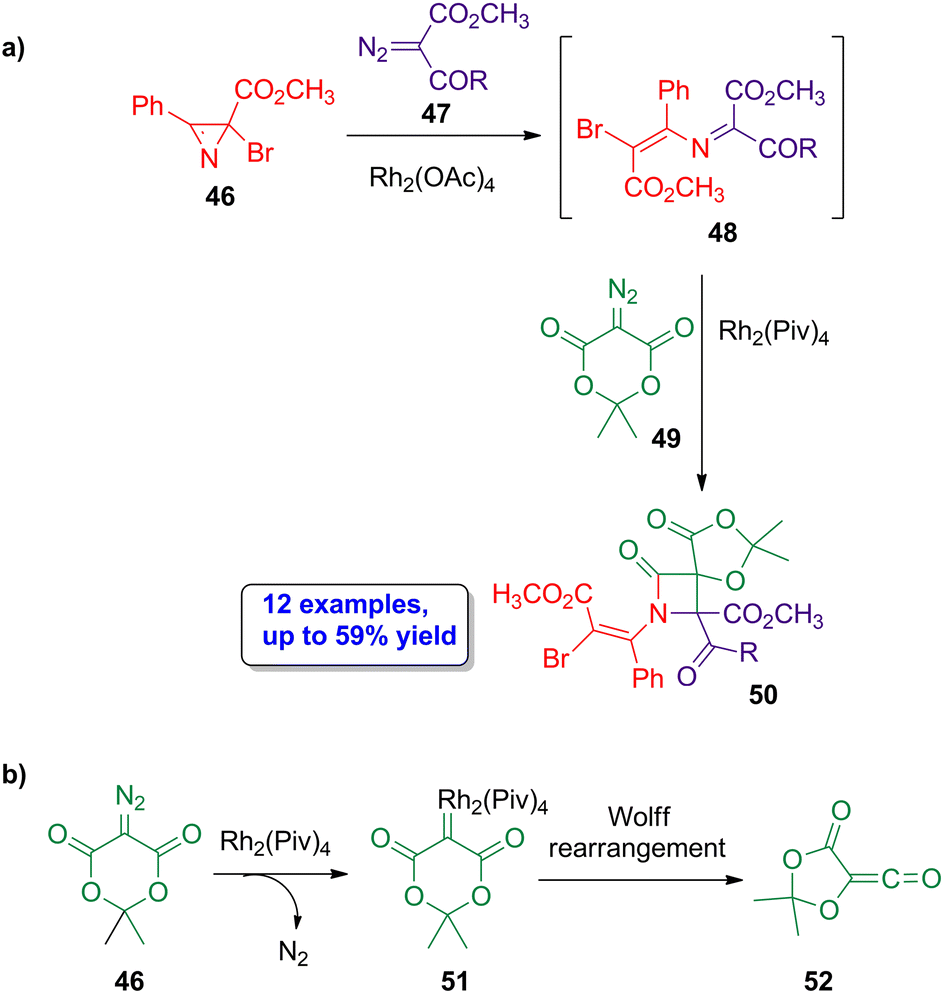 | ||
| Scheme 9 (a) Rhodium-catalyzed diastereoselective synthesis of spirocyclic β-lactams 50 from halogenated azirine derivatives 46, diazocompounds 47 and diazo-Meldrum's acid 49; and (b) possible mechanism pathway for the formation of the ketene 52, involving the generation of the rhodium carbenoid 51. The original results were reported by Novikov and colleagues.54 | ||
3.3. Aluminum-assisted reactions
Lindsay and colleagues reported the formation of a series of chiral β-lactams from enantioenriched cyclopropanone equivalents.55 Noteworthy, the authors carried out a gram scale synthesis which allowed to obtain 1.25 g of a chiral lactam showing >99% enantiomeric excess. This large-scale synthesis started by the three-step formation of the chiral sulfonylcyclopropane 53 from methyl phenyl sulfone (54) and (R)-propylene oxide (55) (Scheme 10). Through this three-step protocol involving treatment with benzenesulfonyl chloride and n-BuLi, the target cyclopropane 53 was obtained in a 66% yield showing excellent enantioselectivity (>99% ee). A subsequent α-hydroxylation protocol afforded the enantiopure cyclopropanone equivalent 56 in 80% yield, retaining the stereospecificity. Then, the cyclopropenone equivalent 56 reacted with N-benzylhydroxylamine chloride (BnNHOH) and NEt3 leading to the formation of the stable hemiaminal intermediate 57, which experiences an in situ rearrangement after addition of Al(OTf) to yield the target β-lactam 58 in 88% yield showing almost enantiopurity. Thus, the stereochemical information is completely transfer from the initial chiral cyclopropane to the prepared β-lactam. The employment of cyclopropanones as highly reactive intermediates leads to envision the use of this methodology to access to a wide variety of chiral heterocycles that can be used in different implementations, such as development of new generations of antibiotics.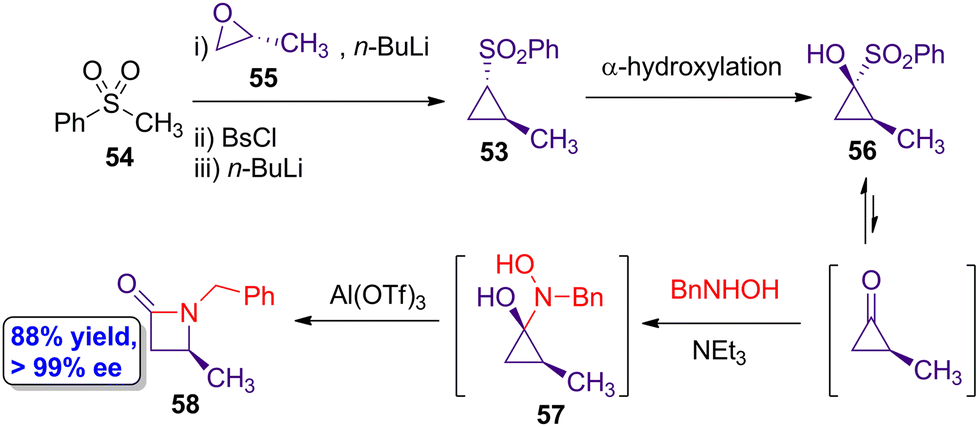 | ||
| Scheme 10 Gram scale synthesis of chiral β-lactam 58 through the formation of the cyclopropenone equivalent 56. The original results were reported by Lindsay and colleagues.55 | ||
3.4. Copper-assisted reactions
Copper-catalyzed Kinugasa/Michael domino reactions were employed by Enders and colleagues in the asymmetric synthesis of a series of spirocyclic β-lactams.56 The reaction of prochiral cyclohexadienone 59 and nitrones 60 using Cu(OTf)2 as catalyst, ligand 61 and NHiBu2 as base (Scheme 11a), leads to the formation of the chiral spirocyclic β-lactams 62 in good yields, showing excellent diastereoselectivity (up to >20:1) and enantioselectivity (up to 97% ee). The proposed catalytic cycle starts with the formation of catalytically active copper(I) species by the in situ reduction of the catalyst (Scheme 11b). These active metallic species react with the prochiral cyclohexanodienone 59 to afford the copper acetylide intermediate 63. A copper-isoxazoline intermediate 64 is then formed through a [3 + 2] dipolar cycloaddition of acetylide 63 and the corresponding nitrone 60. A rearrangement of 64 leads to the formation of the copper β-lactam-based enolate 65, which subsequently experiences a desymmetrized Michael addition, yielding the target spirocyclic β-lactams 62. Interestingly, the scale up of this protocol retains the excellent regio-, chemo-, diastereo- and enantioselectivities of the process. This methodology paves the way to the preparation of spirocyclic β-lactams having four neighbouring stereocenters in a versatile manner, allowing to access to improved bioactive compounds.
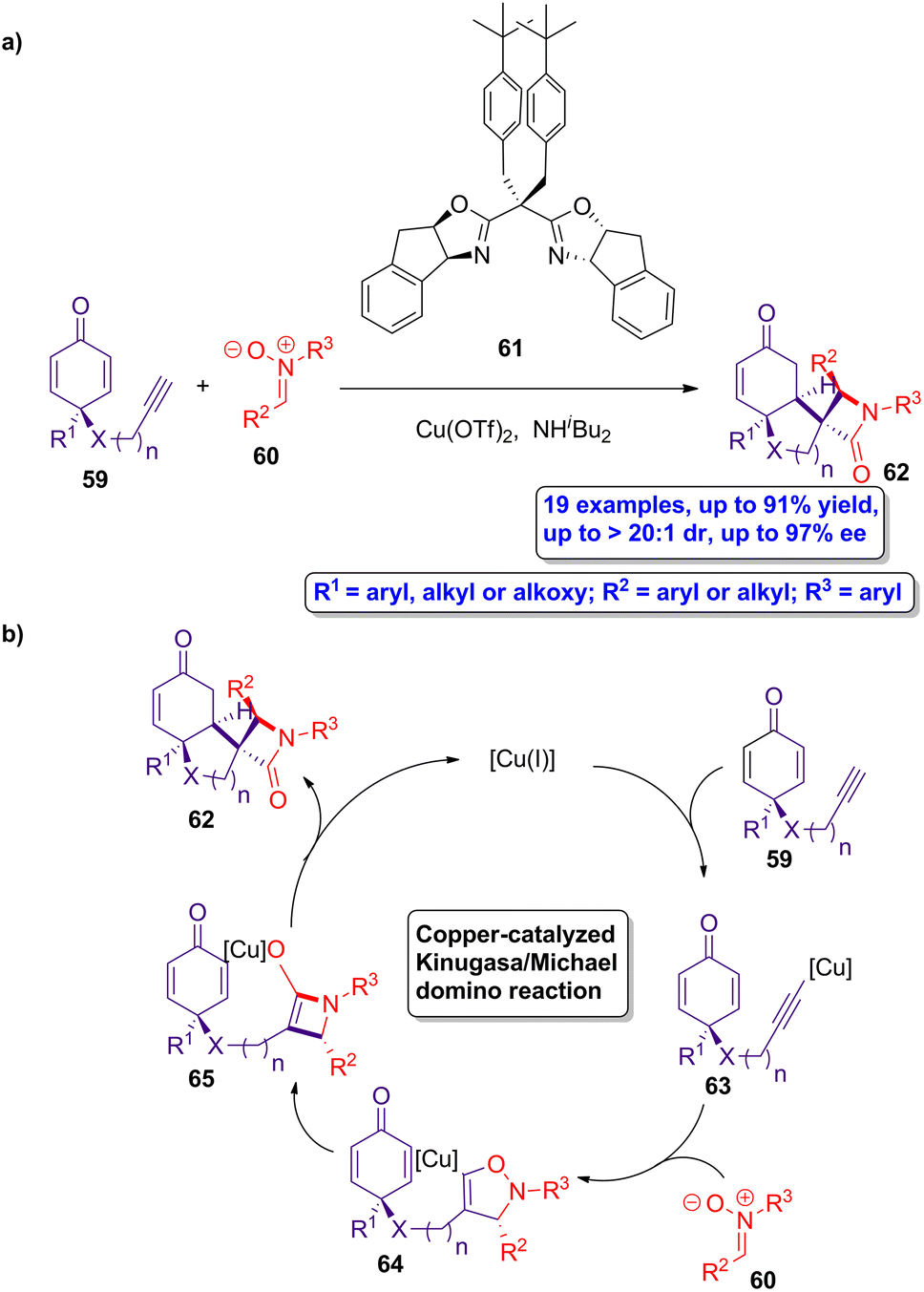 | ||
| Scheme 11 (a) Asymmetric synthesis of spirocyclic β-lactams 62 from cyclohexadienones 59 and nitrones 60; and (b) catalytic cycle of the copper-catalyzed Kinugasa/Michael domino reaction. The original results were reported by Enders and colleagues.56 | ||
Copper(I)-catalyzed asymmetric interrupted Kinugasa reactions were employed by Xu and coworkers in the preparation of chiral β-lactams having sulfur groups in α-position.57 In this protocol, a sulfur-based electrophile is employed to capture the copper(I) four-membered enolate intermediate, thus avoiding the proton transfer process and affording the target sulfur-functionalized lactams. Thus, the reaction of alkynes 66, nitrones 67 and benzenesulfonothioates 68 using Cu(CH3CN)4PF6 as catalyst, ligand 69 and K2CO3 as base (Scheme 12a), afforded chiral sulfur-functionalized β-lactams 70 in excellent enantioselectivity (up to 96.5:3.5 er). An analogous reaction using the disulfur transfer reagent TsSStBu 71 as sulfur source, alkynes 72 and nitrones 73, afforded the disulfur-functionalized β-lactam 74 showing up to 96:4 er (Scheme 12b). Both chiral products (70 and 74) can be easily functionalized to obtain bioactive molecules, turning out to be useful building blocks for the preparation of available products which can be employed in pharmaceutical chemistry, food chemistry and chemical biology.
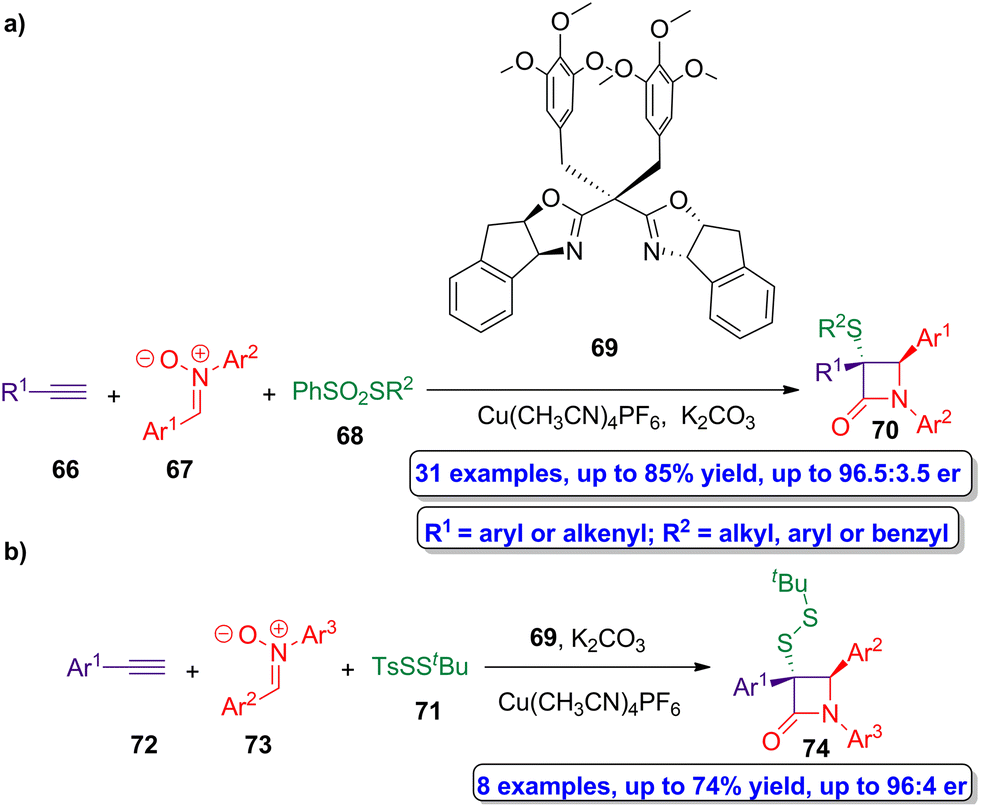 | ||
| Scheme 12 (a) Asymmetric synthesis of β-lactams 70 from alkynes 66, nitrones 67 and benzenesulfonohioates 68; and (b) asymmetric syntesis of β-lactams 74 from alkynes 72, nitrones 73 and disulfur transfer reagent 71. The original results were reported by Xu and colleagues.57 | ||
Cai and coworkers reported a copper-catalyzed interrupted asymmetric Kinugasa reaction of N-(2-iodo-aryl)-propiolamides and nitrones to obtain spirocyclic β-lactams.58 In this example, an intramolecular aryl C–C coupling reaction was employed in order to trap the copper intermediate formed in the Kinugasa reaction.
Gu, Hong, Liu and colleagues reported the asymmetric synthesis of a series of alkyne-functionalized β-lactams 75 through a copper-catalyzed enantioconvergent C(sp3)–C(sp) cross-coupling of tertiary α-bromo functionalized β-lactams 76, which act as tertiary electrophiles, and alkynes 77 (Scheme 13).59 In this synthetic approach, CuTc was employed as the catalyst, compound 78 as the N,N,N-ligand and Cs2CO3 as the base. The target four-membered heterocycles were obtained in high yields (up to 90%) showing excellent enantioselectivities (up to 92% ee). This synthetic strategy allows to access to enantioenriched α-quaternary β-lactams, which turned out to be highly relevant due to the biological significance of this core motif and its underrepresentation in the literature.
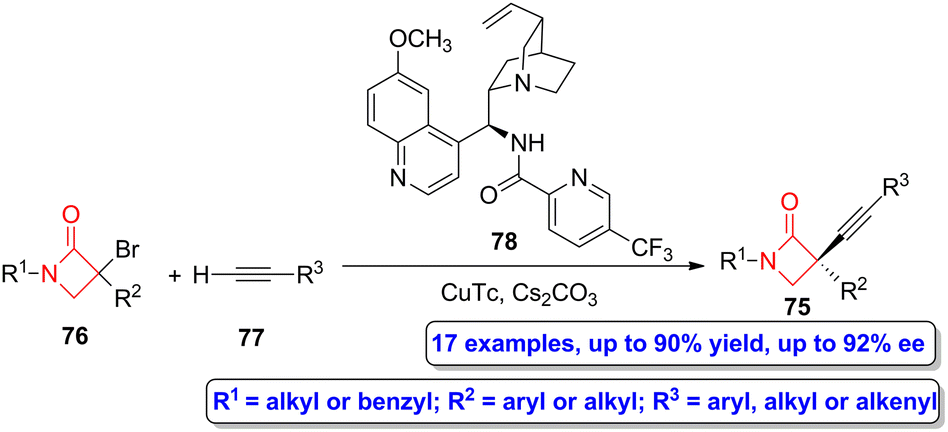 | ||
| Scheme 13 Asymmetric copper-catalyzed enantioconvergent radical C(sp3)–C(sp) synthesis of alkyne-functionalized β-lactams 75 from bromo-functionalized β-lactams 76 and alkynes 77. The original results were reported by Gu, Hong, Liu and colleagues.59 | ||
Recently, a copper-catalyzed enantioconvergent radical C(sp3)–C(sp2) cross coupling reaction was employed by Li, Liu and coworkers to synthesize α-quaternary β-lactams.60 The reaction of bromo-functionalized β-lactams 79 with organoboronate esters 80 using CuBr·S(CH3)2 as the catalyst, compound 81 as the N,N,N-ligand and LiOtBu as the base afforded α-quaternary β-lactams 82 (Scheme 14) in moderate to high yield (up to 92%) and excellent enantioselectivities (up to 99% ee). This synthetic strategy allows to access to a vast range of possible functionalization, covering both alkenyl- and (hetero)arylboronate esters. Interestingly, the authors also tested a sequential cross-coupling followed by hydrogenation, thus being possible to synthesize a α-quaternary-β-lactam through a C(sp3)–C(sp3) cross-coupling reaction.
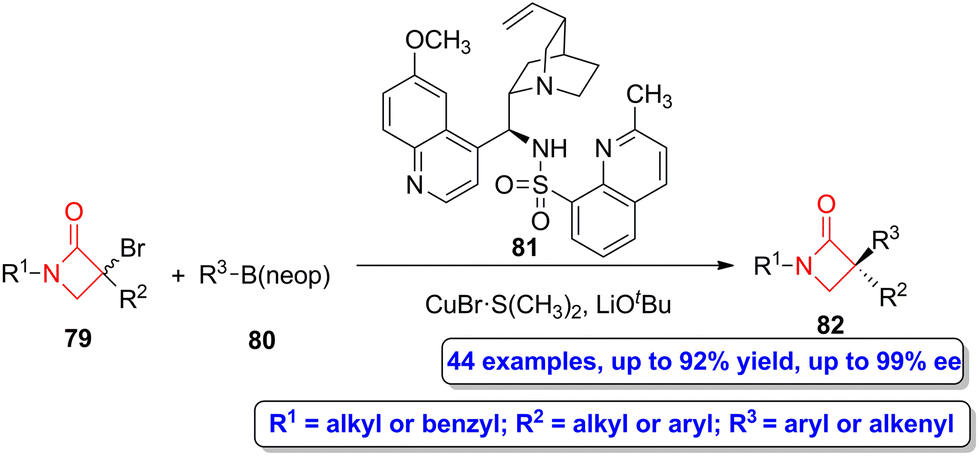 | ||
| Scheme 14 Asymmetric copper-catalyzed enantioconvergent radical C(sp3)–C(sp2) synthesis of α-quaternary β-lactams 82 from bromo-functionalized β-lactams 79 and organoboronate esters 80. The original results were reported by Li, Liu and colleagues.60 | ||
3.4. Bimetallic-assisted reactions
The research group of Xu went a step further and developed a one pot bimetallic catalysis methodology to access to a series of α-quaternary chiral β-lactams.61 The interesting point of this three-component synthetic protocol is the synergy of the asymmetric copper/palladium-catalyzed multicomponent reactions, which allow the modular preparation of the enantioenriched β-lactams by the coupling of transient organometallic species. The authors envision the simultaneously utilization of Cu(CH3CN)4PF6/ligand 69 and Pd2(dba)3/bis[(2-diphenylphosphino)phenyl] ether (DPEPHOS) as combination of ligands and catalysts driving the process. However, in order to accomplish a synergistic bimetallic catalytic cycle, both individual catalytic cycles must be synchronized in order to afford the target product except the two-component Kinugasa adduct. Alkynes 83, nitrones 84 and allylic electrophiles 85 were selected as the components of the reaction, leading to the target chiral allyl-functionalized β-lactams 86 (Scheme 15a). The proposed mechanism of this novel Kinugasa allylic alkylation reaction combines in a synergistic way a copper(I)-catalyzed Kinugasa cycle and a palladium-catalyzed allylic alkylation one (Scheme 15b). Thus, the reaction of active copper(I) species with alkynes 83 leads to the formation of copper(I)-acetylide 87, which then accomplish a cycloaddition reaction with nitrones 84 affording the four-membered copper(I)-based enolate intermediate 88. This enolate intermediate reacts with the corresponding allylic palladium intermediate 89, which is formed from the reaction of palladium catalyst with allylic electrophiles 85 in the palladium-catalyzed cycle. The combination of both organometallic species (88 and 89) allows the formation of palladium-based β-lactam complexes 90, that finally leads to the target α-quaternary chiral β-lactams 86. Noteworthy, these products are formed in good yields (up to 81%) showing excellent enantioselectivity (up to 96.5:3.5 er).
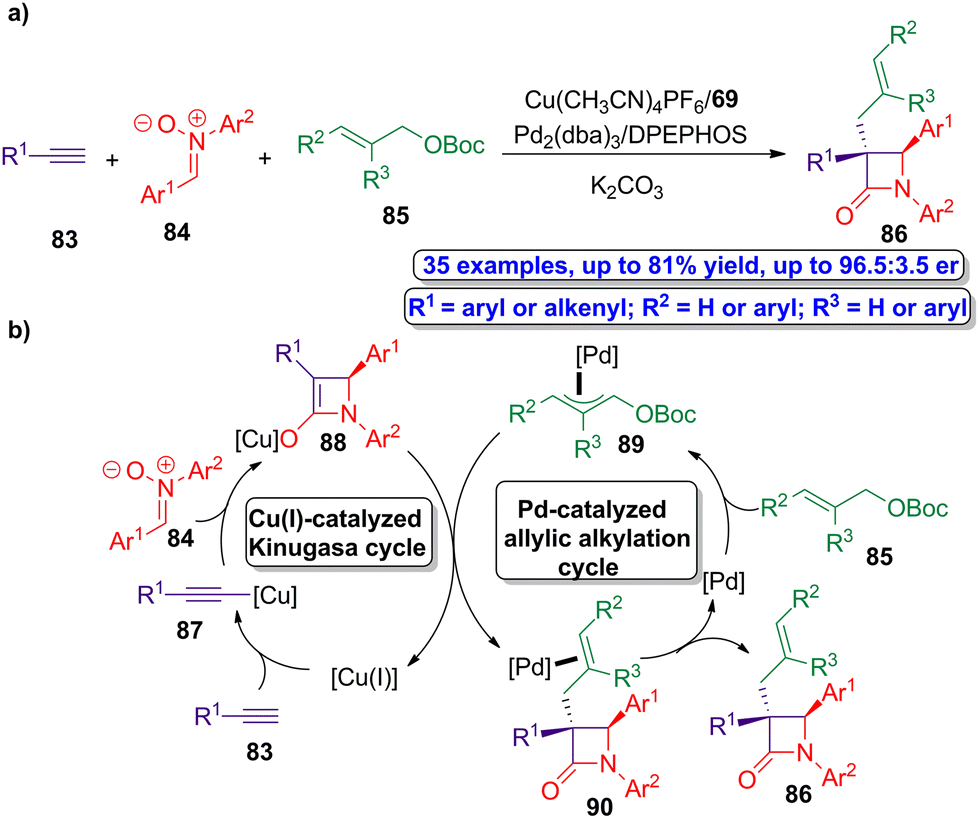 | ||
| Scheme 15 (a) Asymmetric synergistic synthesis of allyl-functionalized β-lactams 86 from alkynes 83, nitrones 84 and allylic electrophiles 85; and (b) proposed synergistic catalytic cycle of the Kinugasa allylic alkylation reaction. The original results were reported by Xu and colleagues.61 | ||
The same research group developed an analogous simultaneous dual bimetallic catalysis using copper- and iridium-based catalysts.62 Through this stereodivergent methodology, chiral β-lactams having contiguous tertiary/quaternary/tertiary stereocenters were synthesized. As in the previous example, a three-component interrupted Kinugasa allylic alkylation reaction was accomplished, selecting alkynes 91, nitrones 92 and allylic electrophiles 93 as the starting materials, and using the combination of Cu(CH3CN)4PF6/ligand 69 and Ir2(cod)2Cl2/ligand 94 as catalysts (Scheme 16). The catalytic cycle is similar to that described in the previous example, but the iridium-catalyzed cycle provides an additional stereocenter in the allyl motif, whose configuration is determined by the absolute configuration of the ligand 94. Thus, when ligand (Sa,S,S)-94 was employed in the synergistic synthetic protocol, β-lactams (3S,4R,5S)-95 were obtained as main products in good yields (up to 75%) showing excellent diastereoselectivity (up to 12:1 dr) and enantioselectivity (up to >99% ee). When the ligand (Ra,R,R)-94 was used, the reaction afforded (3S,4R,5R)-95 as main products showing high diastereoselectivity (up to 1:12 dr) and enantioselectivity (up to >99% ee). This protocol further expands the application of asymmetric synchronized bimetallic-catalyzed cascade reaction in the synthesis of chiral bioactive β-lactams.
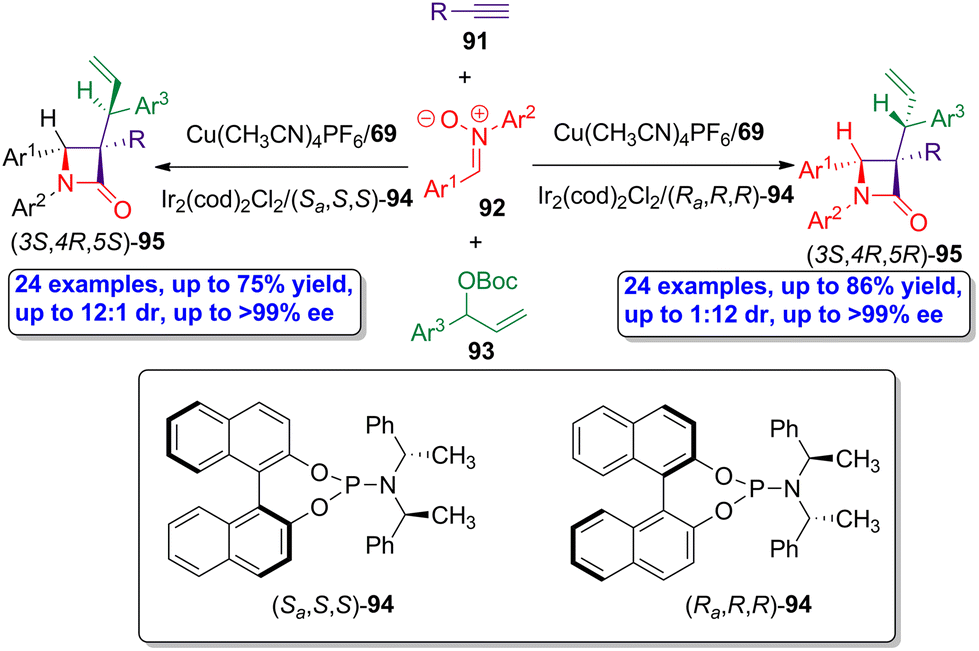 | ||
| Scheme 16 Asymmetric dual copper/iridium-catalyzed synthesis of allyl-functionalized β-lactams 95 having consecutive tertiary/quaternary/tertiary stereocenters, from alkynes 91, nitrones 92 and allylic electrophiles 93. The original results were reported by Xu and colleagues.62 | ||
4. Base-promoted synthesis
Base-promoted cyclization reactions to afford β-lactams are also available synthetic approaches.63–66 This section includes selected recent examples of cyclization reactions promoted by bases in order to prepare β-lactam derivatives.4.1. Potassium tert-butoxide catalyzed reactions
Zeni and coworkers described a stereo- and regioselective base-catalyzed intramolecular cyclization of N-benzyl-N-methyl-propiolamides 96 to form α-methylene-β-lactam compounds 97 employing potassium tert-butoxide as the base and dimethylsulfoxide as the solvent.67 The target lactams were obtained in yields up to 87% observing high selectivity for the 4-exo-dig cyclization over the 5-endo-dig process. The highest yields were observed when aryl motifs having electron-donating substituents in the para position were employed. The authors proposed a mechanism in which potassium tert-butoxide and dimethylsulfoxide first form a dimsyl ion, thus allowing the abstraction of a hydrogen placed at the benzylic position of 96 (Scheme 17). The newly formed carbanion 98 experiences an intramolecular nucleophilic attack to the alkyne functionality in a 4-exo-dig manner, leading to the formation of the heterocyclic intermediate 99. The subsequent protonation reaction leads to the target lactam 97. Noteworthy, these heterocyclic scaffolds can be functionalized through Heck coupling with aryl iodide derivatives, thus paving the way to the incorporation of substituents having also biological activity.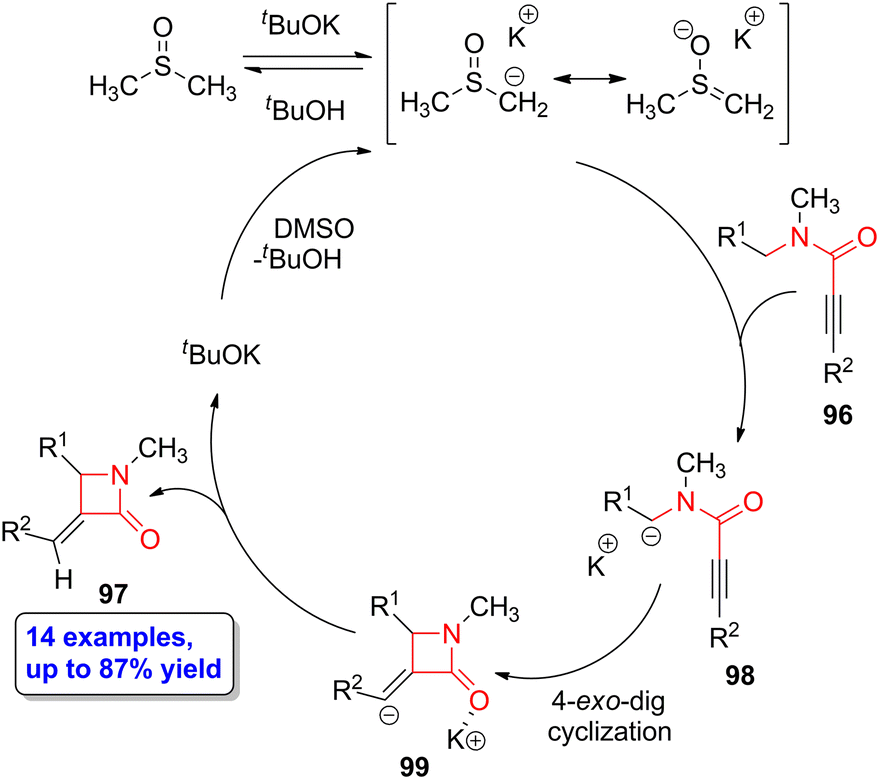 | ||
| Scheme 17 Propose potassium tert-butoxide-catalyzed cycle to obtain β-lactams 97 from N-benzyl-N-methyl-propiolamides 96. The original results were reported by Zeni and colleagues.67 | ||
4.2. Mechanical bond-activated reactions
Beyond the synthetic strategies described above, the use of N-(arylmethyl)fumaramide-based tetralactam [2]rotaxanes in the stereocontrolled synthesis of β-lactams, reported by Berna and colleagues, further expands the possibilities to access to this type of bioactive scaffolds.68–70 This macrocycle-activated synthesis takes place through the base-promoted cyclization of the corresponding interlocked N-(arylmethyl)fumaramide linear counterparts within the confined space provided by the cavity of the macrocycle. The macrocycle is the activating element but also the element that dictates the diastereoselectivity, which leads to the obtention of the trans-adducts as the only products. In order to obtain the noninterlocked β-lactams, a dethreading process is carried out,71 thus allowing the obtention of the target bioactive heterocycles.But in addition to the regio- and diastereoselections, this research group carried out a rational design incorporating stereocenters, either positioned in the linear counterpart72 or in the cyclic one,73 to induce enantioselection in the process.
The incorporation of a chiral α-methyl benzyl group as one of the ends of the fumaramide linear counterpart (100) led to the preparation of enantioenriched β-lactam 101 after carrying out a dethreading process (Scheme 18).72 Although the synthesis can be accomplished as a one pot protocol, it is possible to isolate the enantioenriched interlocked β-lactam 102 after the cesium hydroxide-promoted cyclization. The chiral information that the α-methyl benzyl group provided to the thread is retained throughout the creation of the quaternary stereocenter in the target lactam 101, showing high enantioselectivity (93:7 er) and excellent diastereoselectivity (>99:1 dr).
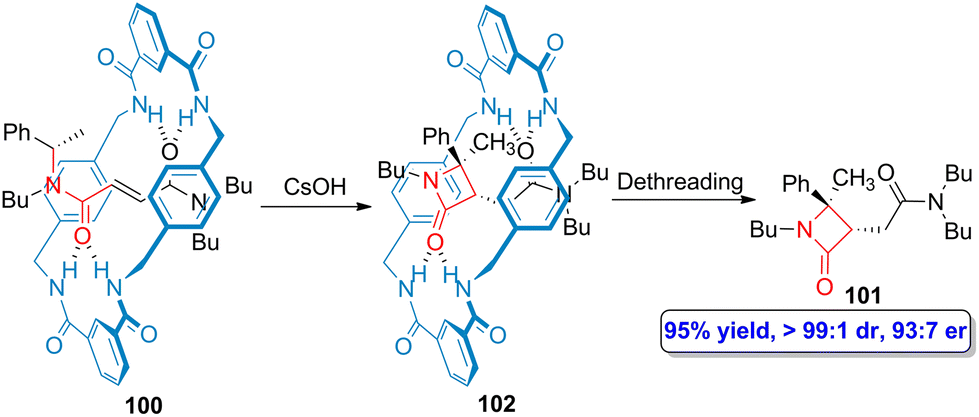 | ||
| Scheme 18 Base-promoted cyclization of N-α-(methylbenzyl)fumaramide-based tetralactam [2]rotaxanes 100 to afford enantioenriched β-lactams 101. The original results were reported by Berna and colleagues.72 | ||
In a more recent example, one methyl group having R configuration was incorporated in the tetralactam macrocycle, thus affording N-(arylmethyl)fumaramide-based rotaxanes 103, which are orientational mechanostereoisomers.73 Through a one pot protocol involving cesium hydroxide-promoted cyclization and the subsequent dethreading process, the enantioenriched β-lactams 104 were obtained in high yield (up to 94%). Interestingly, the lactams (3S,4R)-104 were obtained as the main products (up to 86:14 er) independently of the interlocked mechanical epimer [(R,Rmp)-103 or (R,Smp)-103] employed as starting material (Scheme 19). This result allows to conclude that the (R)-methyl stereocenter placed at the tetralactam macrocycle is the element directing the enantioselectivity of this process activated by the mechanical bond. Thus, the high level of enantioselectivity of this process is a consequence of the chirality transfer from the tetralactam macrocyclic counterpart to the linear one through the mechanical bond induced by the symmetry breaking generated by one small methyl group.
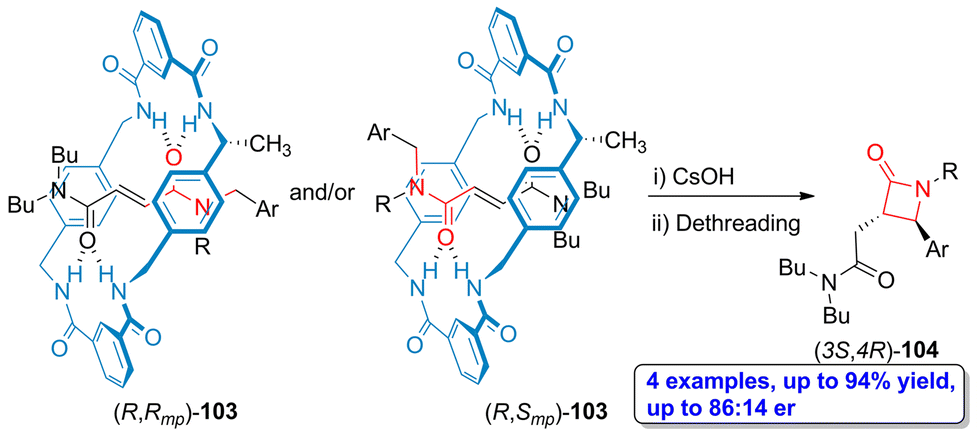 | ||
| Scheme 19 Base-promoted cyclization of N-(arylmethyl)fumaramide-based tetralactam [2]rotaxanes 103 to afford enantioenriched β-lactams 104. The original results were reported by Berna and colleagues.73 | ||
5. Conclusions
The synthesis of β-lactam derivatives is an important area of research due to their relevant bioactive properties. Indeed, there are several widely employed antibiotics having a β-lactam ring as a central core. Due to the major problem of antibiotic resistance,74 the development of novel broad-spectrum antibiotics turns out to be necessary. In this scenario, the stereoselective synthesis of a vast range of β-lactams can contribute to avoiding a future antibiotic resistance crisis. This review article aims to highlight different suitable synthetic protocols to access to these heterocyclic molecules, highlighting selected recent examples.As evidenced throughout the manuscript, the synthesis of β-lactams can be accomplished in diastereo- or/and enantioselective manners using a pool of different reactions, from the “traditional” syntheses to those using transition metal-catalysis or even through the activation of the mechanical bond in rotaxane-based systems. Through these protocols, high stereoselectivity has been obtained towards different functionalized β-lactams, allowing to incorporate several stereocenters. But despite these promising results, there is still a niche for future development. Synthetic efforts towards specific control over the configuration of each individual stereocenters are envisioned as a key future research area in order to synthesize β-lactam compounds having enhanced biological activity.
The industrial scale up is also another milestone to overcome. Although for the utility of large-scale synthetic processes, it is not only required to reproduce yields and stereoselectivity, but also simple purification processes which lead to high purity and in a cost-effective manner. In this direction, the collaboration of academia and industrial sector, especially pharmaceutical industry, will lead to this end.
The privileged properties of β-lactams together with the continuous advances in organic synthesis lead to envision a great future development towards the stereoselective synthesis of these bioactive scaffolds. Thus, the preparation of highly functionalized β-lactams having multiple stereocenters with a precise control over the absolute configuration is anticipated.
Author contributions
Conceptualization, L. A.-A. and A. S.-S.; writing – original draft preparation, L. A.-A. and A.-S. S.; writing – review and editing, A. S.-S.; visualization, L. A.-A. and A. S.-S.; supervision, A. S.-S.; funding acquisition, A. S.-S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union – NextGenerationEU (Margarita Salas postdoctoral grant, Ministerio de Universidades of the Government of Spain). All figures included in this manuscript are original and have been redrawn based on the content of the referenced research articles by using ChemBioDraw® Ultra 12.0 (CambridgeSoft, PerkinElmer Inf.).References
- K. Tahlan and S. E. Jensen, J. Antibiot., 2013, 66, 401–410 CrossRef CAS PubMed.
- S. Hosseyni and A. Jarrahpour, Org. Biomol. Chem., 2018, 16, 6840–6852 RSC.
- H. Staudinger, Justus Liebigs Ann. Chem., 1907, 356, 51–123 CrossRef CAS.
- A. E. Taggi, A. M. Hafez, H. Wack, B. Young, W. J. Drury and T. Lectka, J. Am. Chem. Soc., 2000, 122, 7831–7832 CrossRef CAS.
- S. Schunk and D. Enders, Org. Lett., 2000, 2, 907–910 CrossRef CAS PubMed.
- B. L. Hodous and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 1578–1579 CrossRef CAS PubMed.
- S. France, H. Wack, A. M. Hafez, A. E. Taggi, D. R. Witsil and T. Lectka, Org. Lett., 2002, 4, 1603–1605 CrossRef CAS PubMed.
- C. M. L. Delpiccolo, L. Méndez, M. A. Fraga and E. G. Mata, J. Comb. Chem., 2005, 7, 331–344 CrossRef CAS PubMed.
- C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930–7953 CrossRef CAS PubMed.
- A. Jarrahpour, E. Ebrahimi, V. Sinou, C. Latour and J. M. Brunel, Eur. J. Med. Chem., 2014, 87, 364–371 CrossRef CAS PubMed.
- J. A. Rad, A. Jarrahpour, C. C. Ersanli, Z. Atioğlu, M. Akkurt and E. Turos, Tetrahedron, 2017, 73, 1135–1142 CrossRef.
- L. Chen, L. Zhang, Y. Shao, G. Xu, X. Zhang, S. Tang and J. Sun, Org. Lett., 2019, 21, 4124–4127 CrossRef CAS PubMed.
- A. Moslehi and M. Zarei, New J. Chem., 2019, 43, 12690–12697 RSC.
- R. Heiran, S. Sepehri, A. Jarrahpour, C. Digiorgio, H. Douafer, J. M. Brunel, A. Gholami, E. Riazimontazer and E. Turos, Bioorg. Chem., 2020, 102, 104091 CrossRef CAS PubMed.
- M. K. Mishra, V. N. Singh, S. Muhammad, Z. Aloui, S. Sangeeta, N. Noorussabah, K. Ahmad, M. Choudhary and S. Sharma, J. Mol. Struct., 2020, 1219, 128638 CrossRef CAS.
- M. K. Mishra, V. N. Singh, K. Ahmad and S. Sharma, Mol. Divers., 2021, 25, 2073–2087 CrossRef CAS PubMed.
- V. N. Singh and S. Sharma, New J. Chem., 2021, 45, 19347–19357 RSC.
- Y. Shao, S. Tian, J. Zhu, S. Tang and J. Sun, New J. Chem., 2022, 46, 9989–9993 RSC.
- A. Jarrahpour, Z. Jowkar, Z. Haghighijoo, R. Heiran, J. A. Rad, V. Sinou, F. Rouvier, C. Latour, J. M. Brunel and N. Özdemir, Med. Chem. Res., 2022, 31, 1026–1034 CrossRef CAS.
- M. J. Oddy, D. A. Kusza, R. G. Epton, J. M. Lynam, W. P. Unsworth and W. F. Petersen, Angew. Chem., Int. Ed., 2022, 61, e202213086 CrossRef CAS PubMed.
- Z. Zhuang, S. Liu, J.-T. Cheng, K.-S. Yeung, J. X. Qiao, N. A. Meanwell and J.-Q. Yu, Angew. Chem., Int. Ed., 2022, 61, e202207354 CAS.
- C. Palomo, J. M. Aizpurua, I. Ganboa and M. Oiarbide, Eur. J. Org. Chem., 1999, 3223–3235 CrossRef CAS.
- P. A. Magriotis, Angew. Chem., Int. Ed., 2001, 40, 4377–4379 CrossRef CAS PubMed.
- S. France, A. Weatherwax, A. E. Taggi and T. Lectka, Acc. Chem. Res., 2004, 37, 592–600 CrossRef CAS PubMed.
- Q. Zhang, K. Chen and B.-F. Shi, Synlett, 2014, 1941–1945 CAS.
- K. Dong, L. Qiu and X. Xu, Curr. Org. Chem., 2016, 20, 29–40 CrossRef CAS.
- P. Wang, D. Yang and H. Liu, Chin. J. Org. Chem., 2021, 41, 3448–3458 CrossRef CAS.
- A. Kapoor and J. K. Rajput, J. Heterocycl. Chem., 2021, 58, 2304–2323 CrossRef CAS.
- S. Berry, S. S. Bari, P. Yadav, A. Garg, S. Khullar, S. K. Mandal and A. Bhalla, Synth. Commun., 2020, 50, 2969–2980 CrossRef CAS.
- V. E. Filatov, J. Kuznetsova, L. Petrovskaya, D. Yuzabchuk, V. A. Tafeenko, N. V. Zyk and E. K. Beloglazkina, ACS Omega, 2021, 6, 22740–22751 CrossRef CAS PubMed.
- O. M. Habib, A. S. Mohamed, Y. A. Ibrahim and N. A. Al-Awadi, J. Org. Chem., 2021, 86, 14777–14785 CrossRef CAS PubMed.
- V. E. Filatov, D. A. Iuzabchuk, V. A. Tafeenko, Y. K. Grishin, V. A. Roznyatovsky, D. A. Lukianov, Y. A. Fedotova, M. A. Sukonnikov, D. A. Skvortsov, N. V. Zyk and E. K. Beloglazkina, Int. J. Mol. Sci., 2022, 23, 6666 CrossRef CAS PubMed.
- P. Saini, S. S. Bari, S. Thakur, A. Garg, S. Kumar, S. K. Mandal and A. Bhalla, Synth. Commun., 2022, 52, 1742–1755 CrossRef CAS.
- P. Saini, S. S. Bari, S. C. Sahoo, S. Khullar, S. K. Mandal and A. Bhalla, Tetrahedron, 2019, 75, 4591–4601 CrossRef CAS.
- N. Borazjani, S. Sepehri, M. Behzadi, A. Jarrahpour, J. A. Rad, M. Sasanipour, M. Mohkam, Y. Ghasemi, A. R. Akbarizadeh, C. Digiorgio, J. M. Brunel, M. M. Ghanbari, G. Batta and E. Turos, Eur. J. Med. Chem., 2019, 179, 389–403 CrossRef CAS PubMed.
- M. K. Mishra, S. Sharma, K. Ahmad and V. N. Singh, ChemistrySelect, 2020, 5, 3784–3788 CrossRef CAS.
- V. N. Singh and S. Sharma, J. Heterocycl. Chem., 2021, 58, 2163–2173 CrossRef CAS.
- K. A. Frolow, V. V. Dotsenko, S. G. Krivokolysko and V. P. Litvinov, Russ. Chem. Bull., 2005, 54, 2226–2228 CrossRef.
- S.-T. Ruan, J.-M. Luo, Y. Du and P.-Q. Huang, Org. Lett., 2011, 13, 4938–4941 CrossRef CAS PubMed.
- D. Leonte, L. C. Bencze, C. Paizs, F. D. Irimie and V. Zaharia, Molecules, 2015, 20, 12300–12313 CrossRef CAS PubMed.
- S. K. Singh, N. Chandna and N. Jain, Org. Lett., 2017, 19, 1322–1325 CrossRef CAS PubMed.
- J. L. Galman, I. Slabu, F. Parmeggiani and N. J. Turner, Chem. Commun., 2018, 54, 11316–11319 RSC.
- J.-H. Jin, J. Zhao, W.-L. Yang and W.-P. Deng, Adv. Synth. Catal., 2019, 361, 1592–1596 CrossRef CAS.
- X. Gao, C. Shan, Z. Chen, Y. Liu, X. Zhao, A. Zhang, P. Yu, H. Galons, Y. Lan and K. Lu, Org. Biomol. Chem., 2018, 16, 6096–6105 RSC.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS PubMed.
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed.
- J. D. Neuhaus and M. C. Willis, Org. Biomol. Chem., 2016, 14, 4986–5000 RSC.
- A. Baccalini, G. Faita, G. Zanoni and D. Maiti, Chem. – Eur. J., 2020, 26, 9749–9783 CrossRef CAS PubMed.
- M. Choury, A. B. Lopes, G. Blond and M. Gulea, Molecules, 2020, 25, 3147 CrossRef CAS PubMed.
- H.-R. Tong, W. Zheng, X. Lv, G. He, P. Liu and G. Chen, ACS Catal., 2020, 10, 114–120 CrossRef CAS.
- Z. Zhuang, S. Liu, J.-T. Cheng, K.-S. Yeung, J. X. Qiao, N. A. Meanwell and J.-Q. Yu, Angew. Chem., Int. Ed., 2022, 61, e202207354 CAS.
- Y. Ding, J. Wu and H. Huang, J. Am. Chem. Soc., 2023, 145, 4982–4988 CrossRef CAS PubMed.
- L. Chen, K. Wang, Y. Shao and J. Sun, Org. Lett., 2019, 21, 3804–3807 CrossRef CAS PubMed.
- A. A. Golubev, I. A. Smetanin, A. V. Agafonova, N. V. Rostovskii, A. F. Khlebnikov, G. L. Starova and M. S. Novikov, Org. Biomol. Chem., 2019, 17, 6821–6830 RSC.
- C. M. Poteat, Y. Jang, M. Jung, J. D. Johnson, R. G. Williams and V. N. G. Lindsay, Angew. Chem., Int. Ed., 2020, 59, 18655–18661 CrossRef CAS PubMed.
- T. Shu, L. Zhao, S. Li, X.-Y. Chen, C. von Essen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 10985–10988 CrossRef CAS PubMed.
- J. Qi, F. Wei, S. Huang, C.-H. Tung and Z. Xu, Angew. Chem., Int. Ed., 2021, 60, 4561–4565 CrossRef CAS PubMed.
- X. Zhong, M. Huang, H. Xiong, Y. Liang, W. Zhou and Q. Cai, Angew. Chem., Int. Ed., 2022, 61, e202208323 CAS.
- F.-L. Wang, C.-J. Yang, J.-R. Liu, N.-Y. Yang, X.-Y. Dong, R.-Q. Jiang, X.-Y. Chang, Z.-L. Li, G.-X. Xu, D.-L. Yuan, Y.-S. Zhang, Q.-S. Gu, X. Hong and X.-Y. Liu, Nat. Chem., 2022, 14, 949–957 CrossRef CAS PubMed.
- F.-L. Wang, L. Liu, C.-J. Yang, C. Luan, J. Yang, J.-J. Chen, Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202214709 CAS.
- J. Qi, F. Wei, C.-H. Tung and Z. Xu, Angew. Chem., Int. Ed., 2021, 60, 13814–13818 CrossRef CAS PubMed.
- J. Qi, T. Song, Z. Yang, S. Sun, C.-H. Tung and Z. Xu, ACS Catal., 2023, 13, 2555–2564 CrossRef CAS.
- G. Gerona-Navarro, M. T. García-López and R. González-Muñiz, J. Org. Chem., 2002, 67, 3953–3956 CrossRef CAS PubMed.
- P. Pérez-Faginas, F. O'Reilly, A. O'Byrne, C. García-Aparicio, M. Martín-Martínez, M. J. Pérez de Vega, M. T. García-López and R. González-Muñiz, Org. Lett., 2007, 9, 1593–1596 CrossRef PubMed.
- L. Zhang, L. Ma, H. Zhou, J. Yao, X. Li and G. Qiu, Org. Lett., 2018, 20, 2407–2411 CrossRef CAS PubMed.
- N. Riemer, M. Riemer, M. Krüger, G. J. Clarkson, M. Shipman and B. Schmidt, J. Org. Chem., 2021, 86, 8786–8796 CrossRef CAS PubMed.
- A. R. Claus, T. A. C. Goulart, D. F. Back and G. Zeni, Eur. J. Org. Chem., 2021, 2180–2187 CrossRef CAS.
- A. Martinez-Cuezva, C. Lopez-Leonardo, D. Bautista, M. Alajarin and J. Berna, J. Am. Chem. Soc., 2016, 138, 8726–8729 CrossRef CAS PubMed.
- A. Martinez-Cuezva, C. Lopez-Leonardo, M. Alajarin and J. Berna, Synlett, 2019, 893–902 CAS.
- A. Martinez-Cuezva, A. Pastor, M. Marin-Luna, C. Diaz-Marin, D. Bautista, M. Alajarin and J. Berna, Chem. Sci., 2021, 12, 747–756 RSC.
- A. Saura-Sanmartin, Eur. J. Org. Chem., 2023, 26, e202201512 CrossRef CAS.
- A. Martinez-Cuezva, D. Bautista, M. Alajarin and J. Berna, Angew. Chem., Int. Ed., 2018, 57, 6563–6567 CrossRef CAS PubMed.
- C. Lopez-Leonardo, A. Saura-Sanmartin, M. Marin-Luna, M. Alajarin, A. Martinez-Cuezva and J. Berna, Angew. Chem., Int. Ed., 2022, 61, e202209904 CrossRef CAS PubMed.
- G. L. French, Int. J. Antimicrob. Agents, 2010, 36, S3–S7 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |