
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzophenone as a cheap and effective photosensitizer for the photocatalytic synthesis of dimethyl cubane-1,4-dicarboxylate†‡
Callum
Prentice
ab,
Alice E.
Martin
 a,
James
Morrison
c,
Andrew D.
Smith
*b and
Eli
Zysman-Colman
*a
a,
James
Morrison
c,
Andrew D.
Smith
*b and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM, School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk
bEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, Fife, Scotland KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
cPharmaceutical Sciences, IMED Biotech Unit, Astra Zeneca, Macclesfield, SK10 2NA, UK
First published on 20th February 2023
Abstract
The key intramolecular [2 + 2] photochemical cycloaddition step in the synthesis of dimethyl cubane-1,4-dicarboxylate is performed with substoichiometric amounts of the photosensitizer benzophenone. The reaction proceeds via a Dexter energy transfer process between the triplet excited state benzophenone and a well-known cubane precursor diene. The use of the cheap and widely available benzophenone as the photosensitizer enables lower energy light to be used than the traditional photochemical process.
Cubane was first theorized to exist by Thorpe and Beesley1 over 100 years ago and consists of eight nominally sp3-hybridized carbon atoms bonded together to produce a highly strained cubic structure.2 Cubane is not only interesting because of its peculiarity as a hydrocarbon Platonic solid, but it is also a useful bioisostere for the benzene ring due to its similar size and shape (Scheme 1A).3–5 Therefore the 1,4-substitution pattern of a cubane molecule could replace a para-substituted phenyl ring in a molecule and potentially offer improved properties including a departure from “flat-land”.6 Furthermore, there are other uses for cubane and its derivatives such as its ability to rearrange into cunenes,7 its utilization in explosives8 and its incorporation into polymers.9
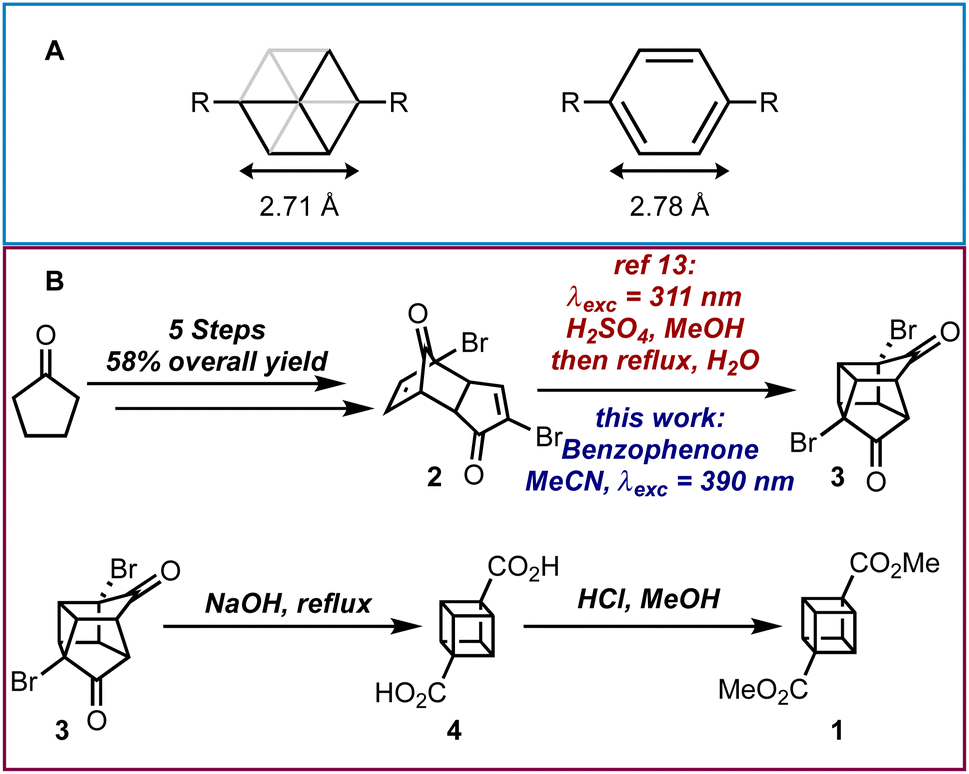 | ||
| Scheme 1 (A) Body diagonals of cubane and benzene. (B) Overall synthesis for dimethyl cubane-1,4-dicarboxylate. | ||
The first successful synthesis of cubane was accomplished by Eaton and Cole10 to give dimethyl cubane-1,4-dicarboxylate 1 in 8 steps with a 12% overall yield from cyclopentenone. Subsequent routes by Chapman11 and Tsanaktsidis12 allowed for cyclopentanone to be used as starting material at comparable synthetic efficiency and overall yield. Further optimizations of Tsanaktsidis's route by Linclau and co-workers allowed for the synthesis of decagram quantities of 1 in overall yields of between 33–40%.13 Despite extensive work, current synthetic routes to 1 rely upon the synthesis of key building block dibromo-dione 2 from cyclopentanone in 5 steps (Scheme 1B). Dione 2 can then undergo an intramolecular photochemical [2 + 2] cycloaddition, which is promoted by the use of either a high-powered Hg lamp or a UV-B lamp (λexc = 311 nm) under acidic conditions and has been successfully scaled-up using continuous flow photochemistry.13 After being refluxed in water to hydrolyse any methyl ketals, 3 can then be transformed into the cubane scaffold using a Favorskii rearrangement to give cubane-1,4-dicarboxylic acid 4, which is then esterified and isolated as 1.
While these synthetic routes have been refined over decades and can now be scaled-up to efficiently produce large quantities of 1, the key [2 + 2] cycloaddition step still requires the use of UV-B lamps and specialist glassware. With this in mind, we set out to identify and optimize alternative conditions that would allow for lower energy light to be used. Two different strategies to achieve this were considered: the use of Lewis acid coordination to 2 to bathochromically shift the absorption into the visible region where it could be directly photoexcited, or the use of an appropriate photosensitizer to generate the same excited state intermediate required but via Dexter energy transfer (DET) instead of direct photoexcitation.
Initial exploration focused on the use of Lewis acids to shift the absorption spectra of 2 closer to the visible region. This has been demonstrated previously for simpler α,β-unsaturated ketones by Bach and coworkers14 using oxazaborolidine-based catalysts for enantioselective reactions and boron trichloride in the racemic version, which achieved bathochromic shifts of nearly 70 nm. To test if this was possible for 2, UV-vis absorption spectra were obtained in the presence and absence of BCl3 (Fig. 1). Without BCl3, 2 shows a band with a peak maximum, λabs, of 250 nm. Pleasingly, in the presence of BCl3 a new band appears at λabs of 300 nm and with a tail into the visible region (Fig. S1†). However, attempts at direct photoexcitation of 2 in the presence of boron trichloride with a 370 nm LED showed no reactivity and only returned unreacted starting material. Therefore, even though the Lewis acid does shift the absorption of 2 as hypothesized, this strategy did not prove productive, and investigation turned to the use of a photocatalytic DET reaction.
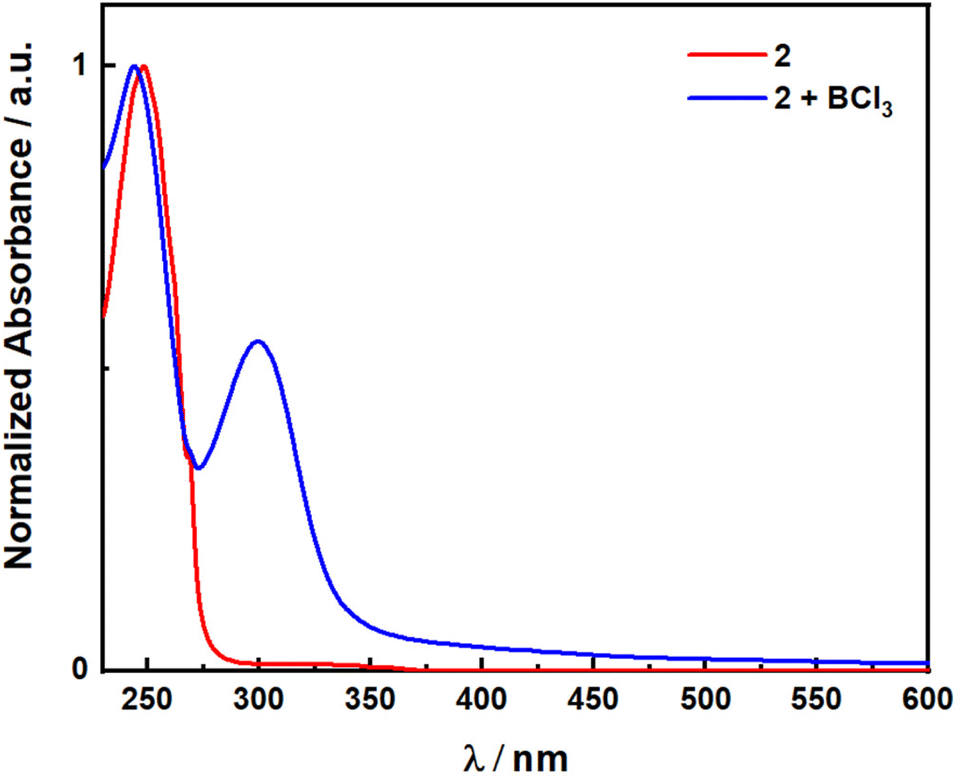 | ||
| Fig. 1 Absorption spectrum of 2 with and without BCl3. | ||
A DET reaction is possible if there is spectral overlap between the S0 → T1 absorption spectrum of the acceptor and the T1 → S0 emission spectrum of the donor.15 However, such absorption spectra are not easily obtained, so instead the triplet energies (ET) of the donor and acceptor are often used as a rough predictive guide. If ET(donor) > ET(acceptor) then the DET is predicted to occur. Density functional theory (DFT) calculations were therefore employed to predict the ET of 2. A small validation study was undertaken to determine the best combination of functional and basis set to accurately model the triplet energy of the system by cross-comparison with the experimentally determined ET. Cyclohexenone was chosen as the test molecule as it has a similar structure to 2 and has an experimentally determined triplet energy (ET = 62.4 kcal mol−1).16 The most accurate methodology used was at the BLYP/6-31G(d,p) level of theory, which predicted an ET of 62.5 kcal mol−1 (Table 1, entry 1). Other functionals tested included B3LYP and PBE0 but they significantly overestimated the ET (Table 1, entries 2 and 3). The ET of 2 was thus calculated at the BLYP/6-31G(d,p) level to be 67.3 kcal mol−1 (Table 1, entry 4). While this value makes it a more challenging substrate than cyclohexenone to engage in DET, we hypothesized that the commonly used and commercially available photosensitizer, benzophenone (ET = 69.0 kcal mol−1),17 could promote the DET and subsequent cyclisation of 2 to 3.
Based on these calculations, a solution of 2 and 25 mol% of benzophenone in MeCN was irradiated with a 390 nm LED over 24 h (Table 2, entry 1). Pleasingly, partial conversion to the desired product (3) was observed using our standard photoreactor (Fig. S1 and S2‡). Increasing the catalyst loading gave increased product conversion over the same period but fell short of complete conversion to 3 (Table 2, entries 2 and 3). To achieve full conversion, an alternative reaction set-up that involved direct irradiation rather than the use of a photoreactor (see ESI‡) was employed that provided more intense irradiation (Table 2, entries 4–6). At this point, control reactions were carried out to confirm the necessity of benzophenone. When using the photoreactor in the absence of benzophenone, no conversion to the desired product was observed, which is consistent with our previous attempts with BCl3 (Table 2, entry 7). However, upon direct irradiation, a modest conversion to 3 was observed in the absence of benzophenone (Table 2, entry 8). This suggests that if the light is sufficiently intense, then photoirradiation at 390 nm should promote the [2 + 2] cycloaddition via direct excitation of 2, albeit much less efficiently. To obtain useful quantities of 3 the reaction was scaled up to a 0.5 mmol scale. Similar results were observed, with full conversion to 3 requiring 0.5 equivalents of benzophenone (Table 2, entries 9–11). Further increase in scale to a 1.0 mmol reaction could also be achieved using the same conditions and an NMR yield of 93% was observed (Table 2, entry 12).
|
|
|||
|---|---|---|---|
| Entry | Amount of 2/mmol | Catalyst loading/mol% | Conversionb/% |
| a Conditions: 2, benzophenone, MeCN (0.1 M), N2, LED (λexc = 390 nm), rt. b From crude 1H NMR. c Reaction performed in a photoreactor (see Fig. S1‡). d 1H NMR yield using 1,3,5-trimethoxybenzene as the internal standard. | |||
| 1c | 0.1 | 25 | 66 |
| 2c | 0.1 | 50 | 73 |
| 3c | 0.1 | 100 | 95 |
| 4 | 0.1 | 25 | 93 |
| 5 | 0.1 | 50 | 100 |
| 6 | 0.1 | 100 | 100 |
| 7c | 0.1 | 0 | 0 |
| 8 | 0.1 | 0 | 28 |
| 9 | 0.5 | 25 | 87 |
| 10 | 0.5 | 50 | 100 |
| 11 | 0.5 | 100 | 100 |
| 12 | 1.0 | 50 | 100 (93)d |
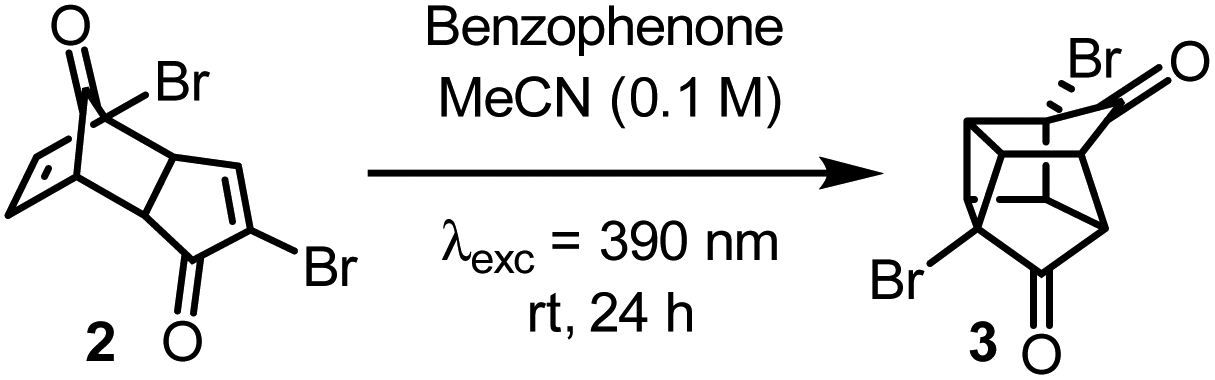
With optimized conditions for the synthesis of 3 in hand (Table 2, entry 12), the synthesis of dimethyl cubane-1,4-dicarboxylate was then completed (Scheme 2). Linclau and co-workers13 reported that crude 3 could only be subjected to the Favorskii rearrangement conditions after being refluxed in water to hydrolyse any methyl ketals present. As our method avoids the formation of any methyl ketals, crude 3 could undergo the desired Favorskii rearrangement without the water hydrolysis step to give crude 4. To allow for facile purification, 4 was then converted to the methyl ester using an acidic methanol solution to give the final compound 1 in 20% yield over three steps from 2. Although this represents a lower overall yield than the 54% achieved by Linclau and coworkers,13 considering the high conversion and NMR yield for the formation of 3 through [2 + 2] cycloaddition we hypothesise this is due to difficulties in precipitating 4 on a much smaller scale.
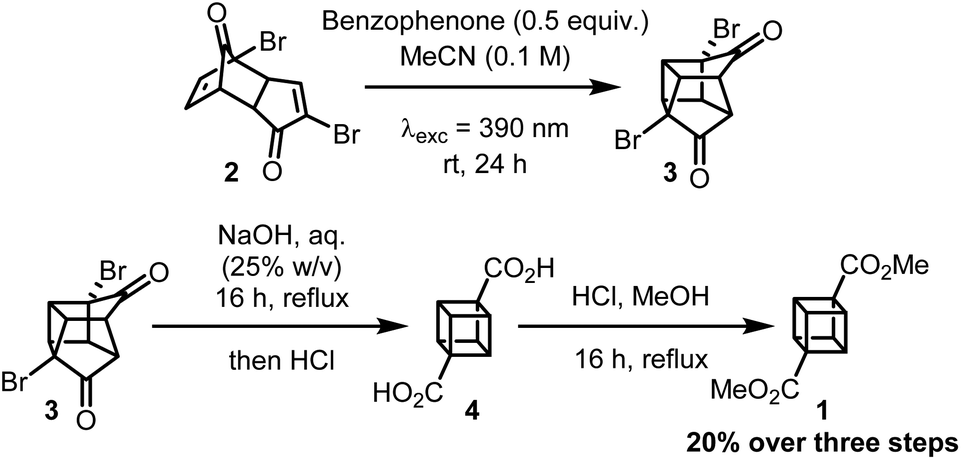 | ||
| Scheme 2 Synthesis of 1. | ||
Having completed the synthesis of 1, our attention turned to the photocatalytic functionalization of cubane using cubane carboxylic acid 5 as a convenient cubyl radical precursor. First, 5 was synthesised via hydrolysis of 1 in 50% yield, with 44% of the unreacted diester being recovered (Scheme 3A). Then, 5 was subjected to a photocatalytic decarboxylation procedure known to work efficiently with other tertiary carboxylic acids such as 1-adamantylcarboxyclic acid;18 however, only trace amounts of the desired product, 6, was observed (Scheme 3B and Fig. S15‡). Other methodologies19,20 were also applied to this reaction but did not achieve any detectable conversion to 6.
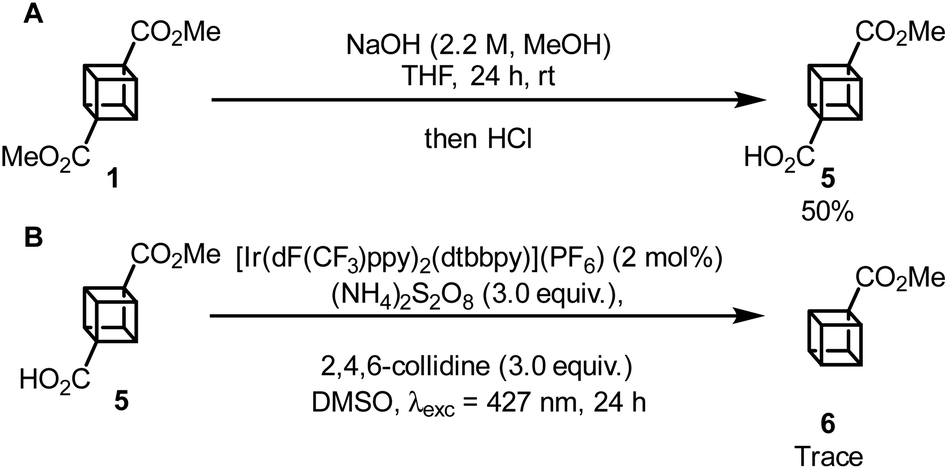 | ||
| Scheme 3 (A) Hydrolysis of cubane. (B) Attempted photocatalytic hydrodecarboxylation of cubane. | ||
In summary, we have developed a synthetic route to dimethyl cubane-1,4-dicarboxylate where the key photochemical [2 + 2] cycloaddition is catalysed by the cheap and widely available photosensitizer benzophenone to give near quantitative yields of the desired product 3 from dione 2. This allowed for the use of much lower energy light (λexc = 390 nm) than previous direct excitation methods that required a high-power Hg lamp or UV-B irradiation. Issues involving scale-up should be remedied using flow chemistry to allow for the photocatalysed [2 + 2] cycloaddition to be performed on a much larger scale. Finally, attempts at photocatalytic decarboxylation of 5 were attempted using conditions known to work for tertiary carboxylic acids; however, these were unsuccessful, with further work towards the photocatalytic functionalization of cubane derivatives ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank AstraZeneca and the University of St Andrews for funding (CASE studentship C.P.).References
- R. M. Beesley and J. F. Thorpe, Proc. Chem. Soc., London, 1913, 29, 343–365 RSC.
- P. E. Eaton, Angew. Chem., Int. Ed. Engl., 1992, 31, 1421–1436 CrossRef.
- B. A. Chalmers, H. Xing, S. Houston, C. Clark, S. Ghassabian, A. Kuo, B. Cao, A. Reitsma, C.-E. P. Murray, J. E. Stok, G. M. Boyle, C. J. Pierce, S. W. Littler, D. A. Winkler, P. V. Bernhardt, C. Pasay, J. J. De Voss, J. McCarthy, P. G. Parsons, G. H. Walter, M. T. Smith, H. M. Cooper, S. K. Nilsson, J. Tsanaktsidis, G. P. Savage and C. M. Williams, Angew. Chem., Int. Ed., 2016, 55, 3580–3585 CrossRef CAS PubMed.
- T. Yildirim, P. M. Gehring, D. A. Neumann, P. E. Eaton and T. Emrick, Carbon, 1998, 36, 809–815 CrossRef CAS.
- E. G. Cox, Rev. Mod. Phys., 1958, 30, 159–162 CrossRef CAS.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- H. Takebe and S. Matsubara, Eur. J. Org. Chem., 2022, e202200567 CAS.
- P. E. Eaton, M.-X. Zhang, R. Gilardi, N. Gelber, S. Iyer and R. Surapaneni, Propellants, Explos., Pyrotech., 2002, 27, 1–6 CrossRef CAS.
- P. E. Eaton, K. Pramod, T. Emrick and R. Gilardi, J. Am. Chem. Soc., 1999, 121, 4111–4123 CrossRef CAS.
- P. E. Eaton and T. W. Cole, J. Am. Chem. Soc., 1964, 86, 962–964 CrossRef CAS.
- N. B. Chapman, J. M. Key and K. J. Toyne, J. Org. Chem., 1970, 35, 3860–3867 CrossRef CAS.
- M. Bliese and J. Tsanaktsidis, Aust. J. Chem., 1997, 50, 189–192 CrossRef CAS.
- D. E. Collin, E. H. Jackman, N. Jouandon, W. Sun, M. E. Light, D. C. Harrowven and B. Linclau, Synthesis, 2021, 53, 1307–1314 CrossRef CAS.
- R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- E. Y. Y. Lam, D. Valentine and G. S. Hammond, J. Am. Chem. Soc., 1967, 89, 3482–3487 CrossRef CAS.
- F. Bayrakçeken, Spectrochim. Acta, Part A, 2008, 71, 603–608 CrossRef PubMed.
- E. B. McLean, D. T. Mooney, D. J. Burns and A.-L. Lee, Org. Lett., 2022, 24, 686–691 CrossRef CAS PubMed.
- J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed.
- C. Cassani, G. Bergonzini and C.-J. Wallentin, Org. Lett., 2014, 16, 4228–4231 CrossRef CAS PubMed.
Footnotes |
| † The research data supporting this publication can be accessed at https://doi.org/10.17630/e66ca579-375d-40f1-85fc-caaec4985476 |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00231d |
| This journal is © The Royal Society of Chemistry 2023 |