Bioluminescence, photophysical, computational and molecular docking studies of fully conformationally restricted enamine infraluciferin†
Received
15th February 2023
, Accepted 10th March 2023
First published on 13th March 2023
Abstract
A new rationally designed fully rotationally restricted luciferin has been synthesised. This synthetic luciferin, based upon the structure of infraluciferin, has two intramolecular H-bonds to reduce degrees of freedom, an amine group to enhance ICT process, and an alkenyl group to increase π-conjugation. In the spectroscopic measurements and computational calculations, enamine luciferin showed more red-shifted absorption and fluorescence emission than LH2 and iLH2. With PpyWT luciferase enamine luciferin gave bioluminescence at 564 nm which is similar to LH2 at 561 nm. Further investigation by docking studies revealed that the emission wavelength of enamine luciferin might be attributed to the unwanted twisted structure caused by Asp531 within the enzyme. With mutant luciferase FlucRed, the major emission peak was shifted to 606 nm, a distinct shoulder above 700 nm, and 21% of its spectrum located in the nIR range.
Introduction
Non-invasive bioluminescence imaging (BLI) with the luciferin-luciferase assay is one of the most popular methods for the non-invasive visualisation of biological process including detection, localisation and monitoring of in vivo and in vitro events.1–3 Unlike fluorescence-based imaging, bioluminescence imaging provides visible photons without an external excitation source which negates noise and interference from unnecessary light scattering. As a result, bioluminescent probes have very low background signals, in comparison to fluorescence probes, resulting in high signal to noise ratios. Based on this unique property, bioluminescent probes can be used for highly sensitive and accurate bioimaging.4 The most well-studied bioluminescence system consists of firefly luciferin, also called D-luciferin (1, LH2), and firefly luciferase (Fluc) pairs. In the presence of oxygen, Mg2+ ions and adenosine triphosphate (ATP), D-luciferin is catalytically oxidised by its luciferase to generate an oxidised form oxyluciferin 2 in its excited state. The excited state ketone species relaxes to its ground state and emits a quantised photon of light (Scheme 1).5 The emission wavelength ranges from 530 nm to ∼630 nm depending on subtle differences in luciferase mutants and pH.6 The bioluminescence quantum yield (Øblu) of D-luciferin is very high, determined to be 0.41 by total-photon-flux measurement.7 However, the wavelength of light emitted by D-luciferin is readily absorbed by melanin, haemoglobin and tissue, which compromises the resolution of BLI in mammals. To improve tissue penetration and obtain better resolution, red-shifted emissions, especially in the near Infra-red (nIR) range (λblu = 650 nm to 800 nm), is desirable. There have been many efforts towards this goal, historically by the development of mutant luciferases and more recently by the synthesis of D-luciferin analogues. A number of mutant luciferases have been reported8–11 that allow D-luciferin to express different wavelengths of emitted light and shifting the emission wavelength above 600 nm. However, there is a limitation to this approach due to the inherent structure of D-luciferin allowing red shifting only so far. The synthesis of D-luciferin analogues has given bioluminescent molecules with far greater red shifted emission (vide infra), but due to their often-compromised stability, the development of rapid and efficient synthetic methods is slow and challenging. In addition, the random modification of the D-luciferin skeleton can result in the loss of bioluminescence activity with different luciferase variants.12–15 Therefore, the synthesis of novel D-luciferin analogues which are compatible with luciferases is important for the development of new red shifted bioluminescent probes for application in deep tissue imaging.
 |
| | Scheme 1 Adenylation and oxidation of D-luciferin to give firefly bioluminescence. | |
Over the past few decades many synthetic luciferin analogues that exhibit red shifted light emission have been designed and synthesised (Fig. 1). White, who was a pioneer in bioluminescence research, demonstrated that the 6-hydroxyl group of D-luciferin could be replaced by a primary amine. The introduction of the more electron donating amino group in place of the naturally occurring hydroxyl group resulted in amino luciferin 3 giving red-shifted light emission (λblu = 593 nm), in part due to increasing intramolecular charge transfer. Unfortunately, 3 gave a much reduced light output compared to D-luciferin of about 10%.16 Developing this concept further, Miller17,18 synthesised a series of more electron donating alkyl and cyclic amino substituted luciferins which exhibited emission wavelengths above 600 nm. Among these analogues, a pair of Cycluc10 4 and mutant luciferase R218K had the longest wavelength at 648 nm which was close to nIR. Although matching the analogue with a luciferase mutant to give the longest wavelength of emission, this cyclic analogue was 2–3 orders of magnitude less bright than D-luciferin. In 2012, Conley reported amino selenium luciferin 5 which gave bioluminescence emission at 600 nm, but its quantum yield was about 74% compared to amino 3 due to the heavy atom effect.19 In 2013, Maki and co-workers developed non-benzothiazole extended luciferin 6 bearing two double bonds between the aromatic ring and the thiazoline.20 With wild type luciferase, luciferin 6 showed nIR emission at 675 nm. Inspired by Maki's work, in 2014 we reported the synthesis of infra luciferin (7, iLH2), a π-extended version of D-luciferin, which has a bridging alkenyl group between the benzothiazole and thiazoline ring systems of D-luciferin.21 This π-extended luciferin provided nIR emission up to 730 nm with mutant CBR2.11 As the structure retained the 6-hydroxyl group, it was also shown to be sensitive to colour modulation. Later this was capitalised upon by demonstrating nIR dual bioluminescence imaging in mouse models of cancer. However, like other D-luciferin analogues, iLH2 suffered from reduced light output (∼1000 times less bright than D-LH2) and lower bioactivity with respect to D-luciferin.22 In 2018, Hall reported the installation of an extra aromatic ring fused to the benzothiazole moiety to increase the π-conjugation of the natural system. With mutant luciferase CBR2, NH2-NpLH28 showed nIR emission at 730 nm which was shown to be useful for deep tissue bioluminescence imaging and tomography. In vivo studies of NH2-NpLH28 demonstrated greater cell membrane permeability in mice brain cells.23 However, the intensity of light output was again weak (∼5000–500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold less than D-luciferin). In an attempt to increase light output by reducing radiationless decay from freely rotating bonds, we have synthesised rigid benzimidazole infra-luciferin analogue 9 that has reduced degrees of freedom around one of the linking alkene groups single bonds due to a covalent linker. With mutant luciferase x11, luciferin 9 had nIR bioluminescence at 714 nm with 5 times higher photon flux than its rotationally un-restricted form 10 at 561 nm. These results indicated that rigidification benefited longer emission wavelength and stronger light output. However, luciferin 9 was still 10 times less bright than iLH2, and ∼1000 weaker than D-LH2 with mutant ×11.24 More recently, Yao and co-workers described a partially restricted luciferin (11, PHOH-Luc) in which one of the linking groups single bonds was rotationally restricted by an intramolecular H-bond.25 According to the fluorescence study, luciferin 11 gave a longer fluorescence wavelength at 569 nm than its rotationally un-restricted form Ph-Luc 12 at 445 nm suggesting that the interlocking H-bond provided a more planar π-conjugated system in its excited state. Additionally, density functional theory (DFT) calculation of luciferin 9 indicated that nIR bioluminescence emission could potentially reach 850 nm. However, in the bioluminescence assay, luciferins 9 and 10 were “dark” with wild type luciferase. Further studies with a complimentary designed luciferase showed light emission for PhOH-Luc 9 at λblu = 608 nm and additionally gave a broad spectrum in which about 31% of the emission was in the nIR region. In this paper we report the synthesis, photophysical and bioluminescence properties of our own H-bonded rotation restricted π-extended infra-luciferin derivative that effectively reduces the degrees of freedom around both of the linking groups single bonds and report its photophysical features.
000 fold less than D-luciferin). In an attempt to increase light output by reducing radiationless decay from freely rotating bonds, we have synthesised rigid benzimidazole infra-luciferin analogue 9 that has reduced degrees of freedom around one of the linking alkene groups single bonds due to a covalent linker. With mutant luciferase x11, luciferin 9 had nIR bioluminescence at 714 nm with 5 times higher photon flux than its rotationally un-restricted form 10 at 561 nm. These results indicated that rigidification benefited longer emission wavelength and stronger light output. However, luciferin 9 was still 10 times less bright than iLH2, and ∼1000 weaker than D-LH2 with mutant ×11.24 More recently, Yao and co-workers described a partially restricted luciferin (11, PHOH-Luc) in which one of the linking groups single bonds was rotationally restricted by an intramolecular H-bond.25 According to the fluorescence study, luciferin 11 gave a longer fluorescence wavelength at 569 nm than its rotationally un-restricted form Ph-Luc 12 at 445 nm suggesting that the interlocking H-bond provided a more planar π-conjugated system in its excited state. Additionally, density functional theory (DFT) calculation of luciferin 9 indicated that nIR bioluminescence emission could potentially reach 850 nm. However, in the bioluminescence assay, luciferins 9 and 10 were “dark” with wild type luciferase. Further studies with a complimentary designed luciferase showed light emission for PhOH-Luc 9 at λblu = 608 nm and additionally gave a broad spectrum in which about 31% of the emission was in the nIR region. In this paper we report the synthesis, photophysical and bioluminescence properties of our own H-bonded rotation restricted π-extended infra-luciferin derivative that effectively reduces the degrees of freedom around both of the linking groups single bonds and report its photophysical features.
 |
| | Fig. 1 Representative synthetic D-luciferin analogues with their bioluminescence wavelengths. | |
Results and discussion
Design and synthesis
By considering the previously discussed modifications of D-Luciferin analogues (stronger intramolecular charge transfer, extended π-system, rigidification), we set out to design and synthesise a fully rotationally restricted D-luciferin analogue, enamine luciferin 13 (Fig. 2), based on the structure of iLH2.21 To the best of our knowledge, there is no example of a fully rotationally restricted D-luciferin analogue. We postulated that full rigidification can be achieved by two intramolecular hydrogen bonds between an enamine -NH2 group and the two nitrogen atoms in each of the benzothiazole and thiazoline rings. This design maintains the alkene linker between the two heterocycles, the extended π-system, which we have shown has a dramatic effect on the red shifting of bioluminescence emission. The two hydrogen bonds reduce the rotational degrees of freedom around each of the σ-bonds linking the alkene spacer to the heterocyclic rings, rigidifying the system. In addition, 13 possesses an additional electron donating NH2 substituent in comparison to infraluciferin (7) which may provide better intramolecular charge transfer for longer wavelength emission. The introduction of this conformational restraint causes the sulfur atoms of the enamine-infraluciferin analogue 13 to sit in the cis position. In contrast, the crystal structures of D-luciferin and iLH2 transition state mimics in the luciferase enzyme showed that the conformer where the two sulfur atoms adopt a trans relationship is the most likely emissive conformation.22,26–28 We were therefore interested in investigating the bioluminescence emission of the fully rotationally restricted enamine-infraluciferin (13). To avoid any unwanted epimerisation of the carboxylic acid, the racemic form of luciferin 13 was synthesised for comparison to D-LH21 and racemic iLH27.
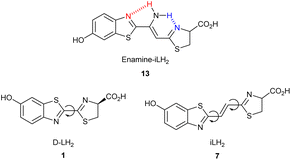 |
| | Fig. 2 The structures of racemic luciferin 13, D-LH21 and iLH27. | |
The synthesis began with the protection of the phenol group of known benzothiazole 1729 as its triisopropylsilyl ether 18 in quantitative yield (Scheme 2). Treatment of nitrile 18 with anionic acetonitrile solution provided enamine 19via a Thorpe reaction30,31 in 83% yield. The triisopropylsilyl protecting group was cleaved under mild conditions to give the nitrile precursor 20 in quantitative yield. The final step was biomimetic formation of the thiazoline by treatment with cysteine, but this proved to be a challenging step. Standard reaction12 of nitrile 20, racemic cysteine hydrochloride (1.1 eq.), and K2CO3 (1.1 eq.) did not provide the desired product. Therefore, solvent and temperature screens were attempted to find suitable conditions (Table S1†). It was found that cyclization could be achieved with anhydrous methanol as solvent and heating the reaction at 80 °C overnight to give luciferin 13 in an isolated yield of 20%.
 |
| | Scheme 2 The synthesis of novel Enamine-iLH2 13. | |
Spectroscopic properties
The spectroscopic data of Enamine-iLH2 (13) was collected alongside that of D-luciferin (1) and infraluciferin (7) for comparison purposes. Absorption spectra were measured in dilute solutions of anhydrous DMSO at 298 K (Fig. 3).
 |
| | Fig. 3 The normalised absorption and emission spectra of D-luciferin and its analogues in DMSO. | |
The absorption wavelengths of the luciferins in DMSO were in the order Enamine-iLH213 (λabs = 360 nm) > iLH27 (λabs = 348 nm) > D-LH21 (λabs = 318 nm) (Table 1). The extinction coefficients (ε) of each luciferin were determined as LH21 (79200 M−1cm−1) > Enamine-iLH213 (76000 M−1cm−1) > iLH27 (66500 M−1cm−1). To achieve the most intense emission spectra the maximum absorption wavelength was used to excite these fluorophores. The emission spectra showed the same trend of absorption, Enamine-iLH213 (λem = 496 nm) > iLH27 (λem = 470 nm) > LH21 (λem = 446 nm). The shortest wavelength of LH21 can be ascribed to the absence of the π-extending alkenyl linker group. Although luciferins iLH27 and Enamine-iLH213 bear the same π-extended system, Enamine-iLH213 gave a longer emission wavelength than iLH27 by 26 nm with a distinct blue-shift shoulder. Based on the excited state intramolecular proton transfer (ESIPT) mechanism,32–35 the enamine 13 might contribute to the shoulder emission ranging from 400 to 470 nm, and the longer wavelength emission at 496 nm might be from the tautomer imine 21 form (Fig. 4). Additionally, enamine-iLH213 has a more planar structure rigidified by the intramolecular H-bonds, improving its conjugation over less rigid 7 in their excited states, which would promote a more red-shifted emission of 13.25 The band gaps (E0−0) of each luciferin were estimated from the intersection of the normalised absorption and emission spectra (λopt).36–38 The data suggested that Enamine-iLH213 has the narrowest band gap (3.01 eV) which ascends to iLH27 (3.08 eV) and LH21 (3.22 eV). From these spectroscopic characteristics, it was expected that 13 could generate an interesting bioluminescence result.
 |
| | Fig. 4 Plausible enamine imine tautomerization via ESIPT process.32,33 | |
Table 1 Spectroscopic properties of luciferins
| Luciferins |
λ
abs (nm) |
ε (M−1 cm−1) |
λ
em (nm) |
E
0−0 (eV) |
| Absorption and fluorescence emission were recorded in anhydrous DMSO solutions at 298 K. The concentrations for absorption and fluorescence were 10−5 M. Band gap E0−0 was measured from the intersection of the absorption and emission spectra, E = 1240/λopt. |
| LH21 |
318 |
79200 |
446 |
3.22 |
| iLH27 |
348 |
66500 |
470 |
3.08 |
| Enamine-iLH213 |
360 |
76000 |
496 |
3.01 |
Computational studies
Computational modelling was applied to investigate whether the intramolecular hydrogen bonds could rigidify luciferin 13. Four possible conformations were examined using QChem5.4,39 fully-restricted 13, partially restricted 13a and 13b, and flexible 13c (Fig. 5). The geometry optimizations of each conformation were calculated using DFT with the ωB97X-D3 functional and the def-SVP basis set with PCM method in DMSO to obtain the lowest energy. For the conformers that have H-bond interactions, the distances of the enamine hydrogen bond donor and the benzothiazole acceptor was approximately 2.3 Å. The H-bond interaction between thiazole and the enamine group was 2.1 Å. The computational results also showed that the fully-restricted 13 had the lowest energy along with the smallest torsion angles (θ1 = −0.5° and θ2 = −0.9°), due to the two bridged H-bond interactions. In contrast 13c, which has no intermolecular H-bonding, had the highest energy owing to its highly-twisted conformation disabling conjugation. When compared to coplanar structure 13, the relative energies calculated by the degree of rigidification via hydrogen bond(s) showed that 13a–c have higher energies at ∼15 kJ mol−1, ∼44 kJ mol−1, ∼49 kJ mol−1, respectively. The computational calculation and the spectroscopic observation (Fig. 3) suggested enamine 13 possesses a planar conformation, restricted by intramolecular H-bonds and is the most stable conformation in the solvent DMSO.
 |
| | Fig. 5 Calculated hydrogen bond distances and dihedral angles of possible hydrogen bonded conformers. | |
Fully-restricted luciferin 13 had the most red-shifted photo-emission among the three luciferins (1, 7, 13) from our spectroscopic studies. Therefore, we expected that their oxidised forms (22, 23, and 24) (Fig. 6) would show a corresponding trend with respect to their absorption and emission spectra. Oxy luciferins are notoriously unstable40 so to test this hypothesis, computer-aided calculations were employed to estimate these compounds’ basic electronic transition and spectroscopic properties. The method of choice to describe excitation and emission energies was TDA-TDDFT with the ωB97-X-D3 functional and the def2-SV(P) basis set. For the simulation of the H2O solvent, the absorption vertical energies were modified with the (ptSS+LR)-PCM method,41 as this method was able to account for the dynamical response of the fast component of the solvent polarisation (ptSS) and excitonic coupling and dispersion effects (LR). For the excited state optimizations and emission energies the LR-PCM method was used, with the assumption that the solvent molecules have time to reorient in response to changes in the solute's geometry. The ΔEabs (eV) values of oxy luciferins exhibited a slightly different trend to those measured for the parent luciferins; 22 (2.30 eV), 23 (2.84 eV), and 24 (2.39) (Table 2). This demonstrated that 22 had the smallest excitation energy. From ΔEem (eV) and the dipole moment difference Δμge, 22 has the smallest energy gap and largest change of the Δμge (1.80 eV/2.90 Debye), when compared to 23 (2.44 eV/1.25 Debye) and 24 (2.03 eV/1.69 Debye). These results suggested that anion 22 has the best ICT in its excited state and is better able to generate more red-shifted emission.42
 |
| | Fig. 6 Deprotonated Oxidized forms of luciferins. | |
Table 2 Summarised of calculated results
| Compounds |
ΔEabs (eV) |
ΔEem (eV) |
μ
g (Debye) |
μ
e (Debye) |
Δμge |
| ΔEabs: vertical absorption energy; ΔEem: vertical emission energy; μg (Debye): dipole moment in the ground state; μe (Debye): dipole moment in the excited state; Δμge: Dipole moment change between ground state and excited state. |
| Oxy enamine iLH222 |
2.30 |
1.79 |
16.99 |
14.09 |
2.90 |
| Oxy LH223 |
2.84 |
2.44 |
9.84 |
8.59 |
1.25 |
| Oxy iLH224 |
2.39 |
2.03 |
11.44 |
9.75 |
1.69 |
The frontier orbitals of oxyluciferins 22–24 were visualised through the Avogadro platform (Fig. 7).43 The HOMOs and LUMOs demonstrated that the electron densities were well delocalized on the backbones, which implies that these oxyluciferins have highly conjugated skeletons. Moreover, these oxyluciferins with their push–pull skeleton showed strong ICT characteristics. The HOMOs are mainly located on the hydroxyl benzothiazole moiety and the LUMOs on the thiazolone segment. The route of ICT is from the election rich phenolate group to the electron deficient carbonyl group. It is worth noting that the -NH2 group on 22 also acts as a second electron donor and contributes electrons to the electron acceptor via ICT.
 |
| | Fig. 7 Selected frontier orbitals of the oxy luciferins 22, 23, 24 (isovalue = 0.007 e3·Bohr−1). | |
Bioluminescence
The bioluminescence results of luciferins D-LH21, iLH27 and Enamine-iLH213 were examined with two luciferases Photinus pyralis firefly luciferase (PpyWT) and its mutant FlucRed (a mammalian codon optimized P. pyralis Luc variant containing 16 mutations)22 at pH = 7.4 at 25 °C (Table 3). With D-LH2, enzymes exhibited their expected wavelengths at 561 nm (PpyWT) and 604 nm (FlucRed). The iLH2 displayed nIR wavelengths at 705 nm (PpyWT) and 707 nm (FlucRed), respectively. The new analogue Enamine-iLH213 showed surprisingly similar emission wavelengths to D-LH2 at 564 nm (PpyWT) and 606 nm (FlucRed).
Table 3 Bioluminescence emission maxima and relative activities of Luc/substrate pairs
| Luciferase/substrate |
Specific activity |
BL, 25 °C (λmax ± 2 nm) |
| Specific activities were obtained from assays at pH 7.4 and 23 °C by monitoring and integrating signal intensity for 2 min using purified enzyme (2.5 μg), 50 μM substrate, 1 mM ATP, and 3 mM MgSO4. The background light emission expressed in specific activity terms with PpyLuc (<0.0008) and with FlucRed (<0.0005) without substrates added. Data are expressed as the mean ± standard deviation and are reported relative to the Luc2/LH2 value, defined as 100 and were corrected for the spectral response of the CCD detector. Data determined from BL emission spectra measured were obtained as described in the ESI.† Bandwidths at full width at half-maximum values are given in parentheses. |
| PpyWT/no added |
<0.0008 |
–– |
| FlucRed/no added |
<0.0005 |
–– |
| PpyWT/LH21 |
100 ± 4 |
561 (86) |
| FlucRed/LH21 |
47.11 ± 2.6 |
601 (90) |
| PpyWT/iLH27 |
0.07 ± 0.001 |
705 (102) |
| FlucRed/iLH27 |
0.14 ± 0.02 |
707 (90) |
| PpyWT/enamine-iLH213 |
0.004 ± 0.002 |
564 (104) |
| FlucRed/enamine-iLH213 |
0.001 ± 0.003 |
606 (108) |
The normalised bioluminescence emission spectra were depicted in Fig. 8. With enzyme PpyWT, 1 and 13 have very similar spectra which nearly overlap. With FlucRed, Enamine-iLH213 displayed a broad spectrum with 21% of the spectrum located in the nIR region. The distinct shoulder >700 nm is suggestive of a species capable of nIR emission similar to that of iLH2 with FlucRed.
 |
| | Fig. 8 Normalised bioluminescence emission spectra of luciferins with luciferases (a) PpyWT and (b) mutant luciferase FlucRed. | |
Docking study
The results of the bioluminescence data with PpyWT for the enamine-iLH213 showed no shift in the wavelength of emission compared to D-LH21 despite possessing extended conjugation. This result was inconsistent with the fluorescence data and computational studies for enamine-iLH213. Due to these conflicting but interesting results, computational molecular docking between PpyLuc luciferase and enamine-iLH213 was examined to interrogate the subtle intermolecular interactions within the micro-environment of the active site which might affect the bioluminescence wavelength. Such an analysis may also inform the rational design of luciferin/luciferase pairs.
The X-ray cocrystal structures of the active site of PpyLuc luciferase with a luciferin inhibitor LH2-5′-O-[(N-dehydroluciferyl)-sulfamoyl]-adenosine 25 (LH2-DLSA)24 and an infraluciferin inhibitor 5′-O-[(N-dehydroinfraluciferyl)-sulfamoyl]-adenosine 26 (iLH2-DLSA)22 have previously been determined (Fig. 9). Therefore, adenylated enamine-iLH2-DLSA 27 is a reasonable ligand candidate to use in molecular docking calculations. Preliminary docking calculations were performed using AutoDock Vina44 and visualized by Chimera,45,46 screening the potential binding modes of adenylated 27 with PpyLuc luciferase.
 |
| | Fig. 9 The structures of LH2-DLSA 25, iLH2-DLSA 26, and enamine-iLH2-DLSA 27. | |
The calculations suggested nine possible conformations readily fit into the PpyLuc active site (Table S2†). The conformation of enamine-iLH2-DLSA 27 with the lowest affinity energy was chosen to further investigate the H-bond interactions with PpyLuc. From the cocrystals of LH2-DLSA 25 (Fig. S1†)26 and iLH2-DLSA 26 (Fig. S2†)22 it can be seen that the thiazoline and the benzothiazole rings are planar with respect to each other within the enzyme pocket, allowing π-overlap between them. Unlike LH2-DLSA 25 and iLH2-DLSA 26, the heterocyclic rings of enamine-iLH2-DLSA 27 appear twisted far from planarity with each other with a torsion angle (θ3 = −60 °C) (Fig. 10). This unwanted twist can be attributed to the potential interactions among Asp531 with the primary amine (3.96 Å), Lys529 with the nitrogen atom of the thiazoline (5.04 Å), and the intramolecular H-bond interactions between the benzothiazole with the two hydroxyl groups of the adenosine (4.03 Å, 4.33 Å). As a result, the generation of longer emission wavelength was unfavoured due to the poor conjugation. Previous work has shown that Lys529 acts as a hydrogen bond donor to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the amide which determines the adenylate reaction and is crucial for its bioactivity.47 From the cocrystal structures (Fig. S1 and S2†) and our docking prediction (Fig. 10), all the luciferins occupy the active site and bioluminescence is observed, although enamine-iLH2 exhibited the lowest specific activity (Table 3). The X-ray cocrystal structures of LH2-DLSA 25 and iLH2-DLSA 26 showed that the distances between Lys529 to the oxygen atom of the CO were measured at 1.94 Å, and 2.41 Å, respectively. However, enamine-iLH2-DLSA 27 had a longer distance (4.74 Å) than the other two luciferins in the docking model. The weaker interaction between enamine-iLH2-DLSA 27 with Lys529 may account for the lower bioactivity of enamine-iLH213.
O group of the amide which determines the adenylate reaction and is crucial for its bioactivity.47 From the cocrystal structures (Fig. S1 and S2†) and our docking prediction (Fig. 10), all the luciferins occupy the active site and bioluminescence is observed, although enamine-iLH2 exhibited the lowest specific activity (Table 3). The X-ray cocrystal structures of LH2-DLSA 25 and iLH2-DLSA 26 showed that the distances between Lys529 to the oxygen atom of the CO were measured at 1.94 Å, and 2.41 Å, respectively. However, enamine-iLH2-DLSA 27 had a longer distance (4.74 Å) than the other two luciferins in the docking model. The weaker interaction between enamine-iLH2-DLSA 27 with Lys529 may account for the lower bioactivity of enamine-iLH213.
 |
| | Fig. 10 Preliminary docking study of enamine-iLH2-DLSA 27 within PpyLuc. | |
Conclusions
The first fully conformationally restricted enamine-iLH213 has been synthesized and subjected to bioluminescence and spectroscopic measurements, and computational and molecular docking calculations. Fluorescence measurements and DFT calculations support full conformational restriction in the liquid phase and demonstrated that enamine-iLH213 had the smallest band gap and gave the longest fluorescence wavelength in comparison to LH21 and iLH27. However, the maximum bioluminescence peak of enamine-iLH213 was observed at 564 nm, which is not in line with the emission expected for extended conjugation luciferin substrates. Docking study calculations suggest that the incorporation of the enamine group, although potentially providing planarity and ICT character to the bioluminescent emitter, encourages disadvantageous hydrogen bonding in the micro-environment of the luciferase active site, resulting in the bound structure being significantly non-planar and therefore unable to take advantage of an extended π-conjugation to red shift the emission wavelength. Further study with mutant enzyme FlucRed showed that the bioluminescence emission peak could be shifted to 606 nm with a distinct shoulder above 700 nm. This result implies that enamine-iLH2 can generate nIR photons by mutation of native luciferase. With a suitable mutant luciferase, the enamine luciferin 13 should be able to emit more red-shifted bioluminescence emission than iLH27.
Experimental
General experimental details - please see ESI†
6-(Triisopropylsilyloxy)benzo[d]thiazole-2-carbonitrile (18).
To a solution of 2-cyano-6-hydroxybenzothiazole (17)29 (0.20 g, 1.1 mmol), imidazole (0.12 g, 1.8 mmol), and DMF (20 mL) was added TIPSCl (0.32 mL, 1.5 mmol) at rt, and the solution stirred for 24 h. The solution was extracted with EtOAc (3 × 20 mL), the organic layers combined, dried (MgSO4), and filtered. After the volatile materials were removed in vacuo, the crude product was purified by column chromatography (silica 80% hexane:20% EtOAc) to give 18 as a brown oil (0.38 g, 99%). Rf = 0.63 (80% hexane:20% EtOAc); IR υmax (solution in CDCl3): 2940 (C–H), 2863 (C–H), 2225 (CN), 1594 (CC), cm−1; 1H NMR (400 MHz, CDCl3) δ 8.04 (1H, d, J = 8.0, ArH), 7.36 (1H, d, J = 4.0, ArH), 7.19 (1H, dd, J = 8.0, 4.0, ArH), 1.31 (3H, sept, J = 8.0, OSiCH(CH3)2, 1.12 (18H, d, J = 8.0, OSiCH(CH3)2). 13C NMR (150 MHz, CDCl3) δ 157.4 (ArC), 147.2 (ArC), 137.3 (ArCH), 133.8 (ArCH), 125.9 (ArC), 122.5 (ArC), 113.4 (ArC), 110.8 (ArCH), 18.0 (OSiCH), 12.8 (OSiCH(CH3)2); HRMS (ESI+) Calcd for C17H25N2OSSi [M + H]+ 333.1457, found 333.1443.
(Z)-3-Amino-3-(6-(triisopropylsilyloxy)benzo[d]thiazol-2-yl)acrylonitrile (19).
To a solution of acetonitrile (0.070 mL, 1.2 mmol) in an anhydrous THF (1 mL) was added n-BuLi (0.76 mL, 1.6 M, 1.2 mmol) at −78 °C and stirred at this temperature for 1 h. This solution was then added dropwise to a solution of 18 (0.25 g, 0.75 mmol) in anhydrous THF (2 mL) at −78 °C under N2. The mixture was stirred at −78 °C for 1 h and quenched with water. The mixture was extracted with EtOAc (3 × 20 mL), the organic layers were combined, dried (MgSO4), and filtered. After the volatile materials were removed in vacuo, the crude product was purified by column chromatography (silica 80% hexane:20% EtOAc) to give 19 (0.25 g, 83%) as a pale-yellow solid. m.p. 177 °C; Rf = 0.45 (80% hexane:20% EtOAc); IR υmax (solution in CDCl3): 3458.7 (NH2), 3321 (NH2), 2939 (C–H), 2930 (C–H), 2887 (C–H), 2194 (CN), 1618 (CC) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.87 (1H, d, J = 8.0, ArH), 7.31 (1H, d, J = 4.0, ArH), 7.08 (1H, dd, J = 8.0, 4.0, ArH), 5.63 (2H, s, NH2), 4.68 (1H, s, CCH–CN), 1.30 (3H, sept, J = 8.0, OSiCH(CH3)2, 1.12 (18H, d, J = 8.0, OSiCH(CH3)2). 13C NMR (150 MHz, CDCl3) δ 158.6 (ArCOSi), 155.8 (CNH2), 152.6 (ArC), 147.7 (ArC), 136.6 (ArC), 124.7 (ArCH), 121.2 (ArCH), 118.3 (ArCN), 111.4 (ArCH), 66.3 (CC–CN), 18.0 (OSiCH(CH3)2), 12.8 (OSiCH(CH3)2); HRMS (ESI+) Calcd for C19H28N3OSSi [M + H]+ 374.1722, found 374.1731.
(Z)-3-Amino-3-(6-hydroxybenzo[d]thiazol-2-yl)acrylonitrile (20).
To a solution of 19 (0.20 g, 0.70 mmol), and anhydrous pyridine (20 mL) was added TBAF (2.5 mL, 1.0 M, 2.1 mmol) at 0 °C, and the reaction was stirred for 2 h. After the volatile materials were removed in vacuo, the crude product was purified by column chromatography (silica 50% hexane:50% EtOAc) to give 20 (0.15 g, 95%) as a pale-yellow solid. m.p. 169 °C (dec.); Rf = 0.2 (80% hexane:20% EtOAc); IR υmax (solution in MeOH): 3448.9 (NH2), 3369.9 (OH), 2202.7 (CN), 1635.2 (CC), 1592.9 (N–H) cm−1; 1H NMR (600 MHz, CD3OD) δ 7.86 (1H, d, J = 8.0, ArH), 7.30 (1H, s, ArH), 7.04 (1H, d, J = 8.0, ArH), 4.70 (1H, s, CCH–CN). 13C NMR (150 MHz, CD3OD) δ 159.7 (ArCOH), 158.6 (CNH2), 155.0 (ArC), 148.0 (ArC), 138.2 (ArC), 125.6 (ArCH) 119.6 (ArCN), 118.0 (ArCH), 107.3 (ArCH), 63.9 (CC–CN); HRMS (ESI+) Calcd for C10H8N3OS [M + H]+ 218.0413, found 218.0388.
Enamine-iLH213.
A solution of 20 (10 mg, 50 μmol), D,L-cysteine (6.0 mg, 50 μmol), and anhydrous K2CO3 (6.2 mg, 50 μmol) in anhydrous MeOH (1 mL) were stirred at 80 °C for 24 h under N2. After the volatile materials were removed in vacuo, the crude product was purified by reverse phase C18 HPLC with gradient elution (5% Methanol:95% H2O followed by 95% Methanol:5% H2O) to give Enamine-iLH213 (3.0 mg, 20%) as a pale yellow solid. m.p. 141.6 °C (dec.); IR υmax (solution in CDCl3): 3413 (COOH), 2953 (C–H), 2863 (C–H), 1618 (CO, CN, CC) cm−1; 1H NMR (700 MHz, CD3OD) δ 7.79 (1H, d, J = 8.0, ArH), 7.25 (1H, s, ArH), 6.98 (1H, d, J = 8.0, ArH), 5.45 (1H, s, NH2–CCH), 4.98 (1H, app t, J = 8.0, CH(COOH)), 3.44 (2H, dd, J = 8.0, 4.0, CH2S). 13C NMR (175 MHz, CD3OD) δ 179.4 (COOH), 166.8 (CCH–NCS), 161.9 (NCS), 159.1 (ArCOH), 147.7 (ArC), 146.2 (NH2–CCH), 137.8 (ArC), 124.8 (ArCH). 117.9 (ArCH), 107.4 (ArCH), 91.1 (NH2–CCH), 83.5 (CHCOOH), 36.5 (CH2); HRMS (ESI+) Calcd for C13H12N3O3S [M + H]+ 322.0333, found 322.0320.
Bioluminescence (BL) emission spectra11
BL was initiated by mixing equal volumes (0.25 mL) of a solution of 50 mM Tricine pH 7.4 containing 2 mM ATP and 6 mM MgSO4 with a solution of assay buffer containing 12.5 μg of enzyme and 0.1 mM of either LH2 or iLH2 in a quartz cuvette. All solutions were pre-warmed to 25 °C. The final concentrations of the mixture (0.5 mL) in 50 mM Tricine pH 7.4 were 0.4 μM enzyme, 50 μM of the indicated analog, 1 mM ATP, and 3 mM MgSO4. Emission spectra were acquired at 25 °C after a 1 min delay with a Horiba Jobin–Yvon iHR 320 imaging spectrometer equipped with a liquid N2 cooled CCD detector. Data were collected over the wavelength range 450 nm to 925 nm, with the excitation source turned off and the emission slit width set to 10 nm, and were corrected for the spectral response of the detector using a correction curve provided by the manufacturer. Assay pH values were confirmed before and after spectra were obtained. In order to obtain sufficient signal strength with BL reactions containing Enamine-iLH2, it was necessary to use 6-fold more enzyme and a slit width of 50 nm.
Specific activities
All assays were performed in triplicate in white 96-well microtiter plates containing 2.5 μg of purified enzyme and 50 μL of 0.1 mM analogue in 50 mM Tricine pH 7.4. BL was initiated by the automated injection of 50 μL of 50 mM Tricine pH 7.4 containing 2 mM ATP, and 6 mM MgSO4. Signals were monitored over 2 min using a Synergy™ 2 microplate luminometer (BioTek, Winooski, VT). Data were integrated and corrected for the spectral response of the Hamamatsu R928 PMT detector.
Author contributions
JCA designed and supervised the project. C-HC performed the chemical synthesis. DF and BRB determined the photophysical and bioluminescence data and interpreted the results. C-HC, SG and PF peformed the computational calculations and docking studies. All authors contributed to writing the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Leverhulme Trust RPG-2019-360 for funding and Dr K. Karu for mass spectra. Mr Aziz Khan (UCL undergraduate project dissertation Chemistry, 2015) is thanked for preliminary studies.
References
- J. A. Prescher and C. H. Contag, Curr. Opin. Chem. Biol., 2010, 14, 80–89 CrossRef CAS PubMed.
- C. H. Contag and M. H. Bachmann, Annu. Rev. Biomed. Eng., 2002, 4, 235–260 CrossRef CAS PubMed.
- S. Y. Teow, K. Liew, M. F. C. Mat, M. Marzuki, N. A. Aziz, T. L. Chu, M. Ahmad and A. S. B. Khoo, BMC Biotechnol., 2019, 19, 1–11 CrossRef PubMed.
- M. A. Paley and J. A. Prescher, MedChemComm, 2014, 5, 255–267 RSC.
- B. R. Pule, C. E. Behney, T. L. Southworth, D. M. Fontaine, A. M. Gulick, D. J. Vinyard and G. W. Brudvig, J. Am. Chem. Soc., 2015, 137, 7592–7595 CrossRef PubMed.
- H. H. Seliger and W. D. McElroy, Proc. Natl. Acad. Sci. U. S. A., 1964, 52, 75–81 CrossRef CAS PubMed.
- Y. Ando, K. Niwa, N. Yamada, T. Enomoto, T. Irie, H. Kubota, Y. Ohmiya and H. Akiyama, Nat. Photonics, 2008, 2, 44–47 CrossRef CAS.
- B. R. Branchini, T. L. Southworth, N. F. Khattak, E. Michelini and A. Roda, Anal. Biochem., 2005, 345, 140–148 CrossRef CAS PubMed.
- K. V. Wood, Y. A. Lam, H. H. Seliger and W. D. McElroy, Science, 1989, 244, 700–702 CrossRef CAS PubMed.
- V. R. Viviani, Cell. Mol. Life Sci., 2002, 59, 1833–1850 CrossRef CAS PubMed.
- B. R. Branchini, D. M. Fontaine, D. Kohrt, B. P. Huta, A. R. Racela, B. R. Fort, T. L. Southworth and A. Roda, Int. J. Mol. Sci., 2022, 23, 2451 CrossRef CAS PubMed.
- D. C. McCutcheon, M. A. Paley, R. C. Steinhardt and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 7604–7607 CrossRef CAS PubMed.
- S. Ioka, T. Saitoh, S. Iwano, K. Suzuki, S. A. Maki, A. Miyawaki, M. Imoto and S. Nishiyama, Chem. – Eur. J., 2016, 22, 9330–9337 CrossRef CAS PubMed.
- D. K. Sharma, S. T. Adams, K. L. Liebmann, A. Choi and S. C. Moller, Org. Lett., 2019, 21, 1641–1644 CrossRef CAS PubMed.
- Y. Ikeda, T. Saitoh, K. Niwa, T. Nakajima, N. Kitada, S. A. Maki, M. Sato, D. Citterio, S. Nishiyama and K. Suzuki, Chem. Commun., 2018, 54, 1774–1777 RSC.
- E. H. White, E. Rapaport, H. H. Seliger and T. A. Hopkins, Bioorg. Chem., 1971, 1, 92–122 CrossRef CAS.
- G. R. Reddy, W. C. Thompson and S. C. Miller, J. Am. Chem. Soc., 2010, 132, 13586–13587 CrossRef CAS PubMed.
- D. M. Mofford, G. R. Reddy and S. C. Miller, J. Am. Chem. Soc., 2014, 136, 13277–13282 CrossRef CAS PubMed.
- N. R. Conley, A. Dragulescu-Andrasi, J. Rao and W. E. Moerner, Angew. Chem., Int. Ed., 2012, 51, 3350–3353 CrossRef CAS PubMed.
- S. Iwano, R. Obata, C. Miura, M. Kiyama, K. Hama, M. Nakamura, Y. Amano, S. Kojima, T. Hirano, S. Maki and H. Niwa, Tetrahedron, 2013, 69, 3847–3856 CrossRef CAS.
- A.
P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, Angew. Chem., Int. Ed., 2014, 53, 13059–13063 CrossRef CAS PubMed.
- C. L. Stowe, T. A. Burley, H. Allan, M. Vinci, G. Kramer-Marek, D. M. Ciobota, G. N. Parkinson, T. L. Southworth, G. Agliardi, A. Hotblack, M. F. Lythgoe, B. R. Branchini, T. L. Kalber, J. C. Anderson and M. A. Pule, eLife, 2019, 8, 1–22 CrossRef PubMed.
- M. P. Hall, C. C. Woodroofe, M. G. Wood, I. Que, M. V. Root, Y. Ridwan, C. Shi, T. A. Kirkland, L. P. Encell, K. V. Wood, C. Löwik and L. Mezzanotte, Nat. Commun., 2018, 9, 132–143 CrossRef PubMed.
- J. C. Anderson, C. H. Chang, A. P. Jathoul and A. J. Syed, Tetrahedron, 2019, 75, 347–356 CrossRef CAS.
- Z. Yao, B. S. Zhang, R. C. Steinhardt, J. H. Mills and J. A. Prescher, J. Am. Chem. Soc., 2020, 142, 14080–14089 CrossRef CAS PubMed.
- J. A. Sundlov, D. M. Fontaine, T. L. Southworth, B. R. Branchini and A. M. Gulick, Biochemistry, 2012, 51, 6493–6495 CrossRef CAS PubMed.
- T. Nakatsu, S. Ichiyama, J. Hiratake, A. Saldanha, N. Kobashi, K. Sakata and H. Kato, Nature, 2006, 440, 372–376 CrossRef CAS PubMed.
- D. Dennis and R. H. J. Stanford, Acta Crystallogr., 1973, B29, 1053–1058 CrossRef.
- D. C. McCutcheon, W. B. Porterfield and J. A. Prescher, Org. Biomol. Chem., 2015, 13, 2117–2121 RSC.
- M. V. Gool, S. A. A. De Diego, O. Delgado, A. A. Trabanco, F. Jourdan, G. J. Macdonald, M. Somers and L. V. Donck, ChemMedChem, 2017, 12, 905–912 CrossRef PubMed.
- H. Baron, F. G. P. Remfry and J. F. Thorpe, J. Chem. Soc. Trans., 1904, 85, 1726–1761 RSC.
- C.-L. Chen, Y.-T. Chen, A. P. Demchenko and P.-T. Chou, Nat. Rev. Chem., 2018, 2, 131–143 CrossRef CAS.
- H.-W. Tseng, J.-Q. Liu, Y.-A. Chen, C.-M. Chao, K.-M. Liu, C.-L. Chen, T.-C. Lin, C.-H. Hung, Y.-L. Chou, T.-C. Lin, T.-L. Wang and P.-T. Chou, J. Phys. Chem. Lett., 2015, 6, 1477–1486 CrossRef CAS PubMed.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- N. Suzuki, K. Suda, D. Yokogawa, H. Kitoh-Nishioka, S. Irle, A. Ando, L. M. G. Abegão, K. Kamada, A. Fukazawa and S. Yamaguchi, Chem. Sci., 2018, 9, 2666–2673 RSC.
- R. Y.-Y. Lin, F. L. Wu, C. H. Chang, H. H. Chou, T. M. Chuang, T. C. Chu, C. Y. Hsu, P. W. Chen, K. C. Ho, Y. H. Lo and J. T. Lin, J. Mater. Chem. A, 2014, 2, 3092–3101 RSC.
- R. Y.-Y. Lin, H. W. Lin, Y. S. Yen, C. H. Chang, H. H. Chou, P. W. Chen, C. Y. Hsu, Y. C. Chen, J. T. Lin and K. C. Ho, Energy Environ. Sci., 2013, 6, 2477–2486 RSC.
- C. H. Chang, Y. C. Chen, C. Y. Hsu, H. H. Chou and J. T. Lin, Org. Lett., 2012, 14, 4726–4729 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr., H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. V. Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- O. V. Maltsev, N. K. Nath, P. Naumov and L. Hintermann, Angew. Chem., Int. Ed., 2014, 53, 847–850 CrossRef CAS PubMed.
- J. M. Mewes, Z. Q. You, M. Wormit, T. Kriesche, J. M. Herbert and A. Dreuw, J. Phys. Chem. A, 2015, 119, 5446–5464 CrossRef CAS PubMed.
- B. Carsten, J. M. Szarko, H. J. Son, W. Wang, L. Lu, F. He, B. S. Rolczynski, S. J. Lou, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2011, 133, 20468–20475 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Chem. Inf., 2012, 4, 17 CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- J. E. Chen, C. C. Huang and T. E. Ferrin, Bioinformatics, 2015, 31, 1484–1486 CrossRef CAS PubMed.
- B. B. Branchini, R. A. Magyar, M. H. Murtiashaw and N. C. Portier, Biochemistry, 2001, 40, 2410–2418 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental, optimising reaction conditions for 13, docking studies method and copies of 1H and 13C NMR, and IR spectra of new compounds. See DOI: https://doi.org/10.1039/d3ob00247k |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Danielle M.
Fontaine
c,
Panagiotis
Fikas
a,
Bruce R.
Branchini
c and
James C.
Anderson
b,
Danielle M.
Fontaine
c,
Panagiotis
Fikas
a,
Bruce R.
Branchini
c and
James C.
Anderson











