
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complementary strategies for synthesis of sulfinamides from sulfur-based feedstock†
Miloš
Jabczun
,
Vladimír
Nosek
and
Jiří
Míšek
 *
*
Department of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030/8, 12843 Prague 2, Czech Republic. E-mail: misek@natur.cuni.cz
First published on 17th March 2023
Abstract
We describe a straightforward one-pot reductive protocol for the synthesis of sulfinamides from sulfonyl chlorides. This method enables the preparation of sulfinamides with a broad range of functional groups. Furthermore, we have expanded a known oxidative pathway to sulfinamides starting from thiols. These methods together provide a general strategy for the synthesis of sulfinamides from common sulfur-based feedstock that is available with large structural and functional group diversity.
Introduction
Sulfinamides have become a valuable chiral auxiliary in asymmetric organic synthesis, and the efficient chirality transfer has also been utilized in the catalytic format.1–3 However, sulfinamides are also very useful intermediates in the preparation of sulfoximines and sulfonimidamides that are gaining popularity in medicinal chemistry.4–17 The development of efficient synthetic methods for this kind of hexavalent sulfur compound will enable further exploration of this underexplored chemotype. Sulfoximines and sulfonimidamides offer a unique 3D scaffold for the display of three different substituents around the central tetrahedral sulfur atom (Fig. 1). In order to fully utilize these compounds in drug discovery, a mild and general method that allows a straightforward installation of a diverse set of substituents with a variety of functional groups is essential. Sulfinamides provide a key intermediate with C- and N-substituents as places for diversification. | ||
| Fig. 1 Structures of underexplored sulfur-based chemotypes. | ||
Sulfinamides are commonly synthesized from sulfinic acids by conversion to chloride and subsequent reaction with an amine (Scheme 1).18–20 DCC and EDC reagents can also be used for activation of sulfinic acids.21,22 However, sulfinic acids are not widely available due to the limited stability caused by possible disproportionation.23–25 Another way of making sulfinamides is from starting sulfinate or thiolsulfinate esters, which can undergo aminolysis with amines or metal amides.26–29 These starting materials are commonly prepared from, again, sulfinic acids or disulfides.30–32 Thiols can be converted to sulfinamides in a one-pot format using SO2Cl2 and acetic acid to obtain sulfinyl chlorides that react with the corresponding amine.33 Copper/palladium-catalysed oxidative transformation of thiols to sulfinamides has also been reported.34,35 Alternatively, metal-catalyzed transsulfinamidation can be utilized for diversification of the sulfinamide pool.36,37 An elegant approach to sulfinamide synthesis was recently reported by Willis et al. The authors utilized sulfur dioxide surrogate DABSO in reaction with organomagnesium/lithium compounds to generate a sulfinate that is further converted to sulfinamide.38 Similarly, arylboron coumpounds have been reported as a starting material for one-pot reaction leading to sulfinamides.39,40 An interesting approach was developed by Harmata et al. by utilization of sulfonyl chlorides as a starting material.41In situ reduction of sulfonyl chloride by PPh3 provides an intermediate that reacts directly with amines to provide sulfinamides. This method was further improved by using a MOF catalyst.42 However, the method suffers from limited substrate scope as only certain aromatic sulfonyl chlorides afford the desired sulfinamides in a reasonable yield. Sulfonyl chlorides, however, represent a common chemical feedstock, and thousands of sulfonyl chlorides with a variety of substitution patterns and functional groups are now commercially available. Therefore, they represent a good starting material for the diversification of sulfinamides at the C-substituent. Sulfonyl chlorides can be reduced to sulfinate salt, most commonly by zinc powder or sodium sulfite.43–46
 | ||
| Scheme 1 Selected one-pot protocols for the synthesis of sulfinamides. | ||
We wanted to explore if sulfonyl chlorides can be utilized for the straightforward, efficient, and general synthesis of sulfinamides. Thiols represent another sulfur-based feedstock that is available with diverse structural and functional group features and can be converted to sulfinyl chloride through above mentioned oxidation with SO2Cl2 and acetic acid and further to sulfinamides.33 Our goal was to explore this oxidative approach as a complementary strategy to the reductive pathway with sulfonyl chlorides and to broaden the structural and functional group diversity of the corresponding sulfinamides. Ultimately, we wanted to show that sulfinamides with structurally diverse C-substituents can be converted to sulfonimidamides and thus allow preparation of libraries of compounds from the broadly available feedstock.
Here, we report on two methods that use optimized reductive and oxidative conditions for the preparation of sulfinamides from sulfonyl chlorides and thiols. The direct protocols use a one-pot format without any isolation of intermediates and allow efficient synthesis of sulfinamides with a broad spectrum of functional groups.
Results and discussion
Our reductive synthetic strategy relies on the fact that sulfinate salts can be easily converted to sulfinyl chlorides that readily react with amines forming sulfinamides. This reaction sequence can be performed in a one-pot protocol. Therefore, a key step is the efficient reduction of sulfonyl chloride to a sulfinate salt. This reduction is usually carried out using sodium sulfite or zinc powder in an aqueous environment. Despite these reductions can form the desired sulfinates with high yields, the aqueous conditions prevent performing the entire desired reaction sequence in situ as the subsequent generation of sulfinyl chloride would not be compatible. The sulfinates can be isolated; however, our goal was to devise a direct method without any isolation of intermediates. Moreover, some zinc sulfinates are known to crystalize as hydrates, which could also complicate the preparation of chlorides.45 Therefore, we turn to a zinc-based non-aqueous reduction of sulfonyl chlorides. Toluenesulfonyl chloride (TsCl) and benzylamine were used for a model reaction. Reduction of TsCl with a zinc powder in organic solvents was not very efficient, however in agreement with the literature, we observed that the addition of 1.5 equiv. of DMF in chlorinated solvents significantly improved the yield of the desired sulfinate.47 Therefore, these conditions have been utilized for the reaction sequence. TsCl was reduced with zinc powder, the resulting mixture was filtered, rotavaped and the residues dissolved in THF and reacted with SOCl2. Subsequent addition of benzylamine (1.5 equiv.) with Et3N (1.5 equiv.) provided the desired sulfinamide. Optimization of the individual steps of the sequence resulted in conditions providing sulfinamide 1a in 80% isolated yield (Table 1).| a Reaction conditions: RSO2Cl (0.81 mmol, 1 equiv.), DMF (1.22 mmol, 1.5 equiv.), Zn powder (0.89 mmol, 1.1 equiv.), DCM, reflux, 1 h, then SOCl2 (1.63 mmol, 2.0 equiv.) THF, −40 °C, 45 min followed by Et3N (1.22 mmol, 1.5 equiv.) and benzylamine (1.22 mmol, 1.5 equiv.), rt, on. |
|---|

|
Having the optimized procedure in hands, we proceeded to determine the scope of the synthetic method with respect to the starting sulfonyl chlorides. Satisfyingly, the method is mild enough to tolerate a broad range of functional groups. In general, both (het)aryl and alkyl sulfonyl chlorides were suitable substrates with moderate to good yields (Table 1). Importantly, functional groups that can be attached to sulfinamides at the C-substituent include secondary amide (1d), cyano group (1b), ester (1g), ketone (1k) and alkyl chloride (1l). Thiophene-containing sulfonyl chlorides can be converted to sulfinamides (1f and 1g) as well. Also, sterically hindered mesityl sulfonyl chloride was a good substrate providing the desired product 1i in 71% yield. Out of the tested functional groups, the nitro group turned out to be incompatible with the method as it gets partially reduced under the reaction conditions. Also, N-heterocyclic substrates provided a complex mixture of products.
Next, we turned out attention to the oxidative pathway starting from thiols. Conversion of thiols to sulfinyl chlorides and subsequently to sulfinamides was described previously on a small number of substrates.33 We wanted to explore the substrate scope in comparison to our reductive pathway. One-pot conversion of thiols to sulfinyl chlorides by SO2Cl2 and acetic acid and further to sulfinamides was compatible with a variety of aryl and alkyl substituted substrates with functional groups including ester, nitrile, alkyl chloride, secondary amide as well as a nitro group (Table 2). Interestingly, the original report on the preparation of sulfinyl chlorides from thiols states that secondary amides are not compatible with this method. In our hands, sulfinamide 1d with a secondary amide was obtained in a good yield (70%). In contrast to the reductive pathway, sulfur-containing heterocycles were not compatible substrates due to the oxidative damage of the heterocycle. Also, it has been previously observed that this method is not suitable for the preparation of sulfinamides from thiols containing N-heterocycles. Our experiments with pyrimidine-2-thiol indeed failed to produce the corresponding sulfinamide using the standard conditions. However, the detailed investigation of the reaction indicated that the major product of the reaction is sulfenamide instead of sulfinamide. This product was difficult to isolate in a pure form. Therefore, we decided to oxidize this product in situ to obtain the desired sulfinamide. Delightfully, the careful addition of 1 equiv. of mCPBA at the end of the reaction led to the isolation of sulfinamide 1q in 66% yield. Using the same modification, pyridine-containing sulfinamide 1r was also obtained in 58% yield. Given the importance of N-heterocycles in drug discovery, this modification further broadens the scope of useful products available.

In the following experiments, we showed that in addition to benzylamine, sulfinamides can also be successfully produced with other primary, secondary and (het)aryl amines, which is in agreement with a previous report (Table 3).38 Various primary and secondary amines provided sulfinamides in good yields including sterically demanding tert-butylamine. Yields of the products with (het)aryl amines are moderate, however it should be noted that they represent unoptimized reaction conditions. It is not unreasonable to expect that reaction condition optimization, for instance by reversing the order of addition of reagents, could increase the yields, as double sulfufinylation of the nucleophilic nitrogen can be supressed.
| a Reaction conditions: sulfonyl chloride or thiol was converted to sulfinyl chloride by method A or B and reacted with the corresponding amine. For more details see ESI.† |
|---|
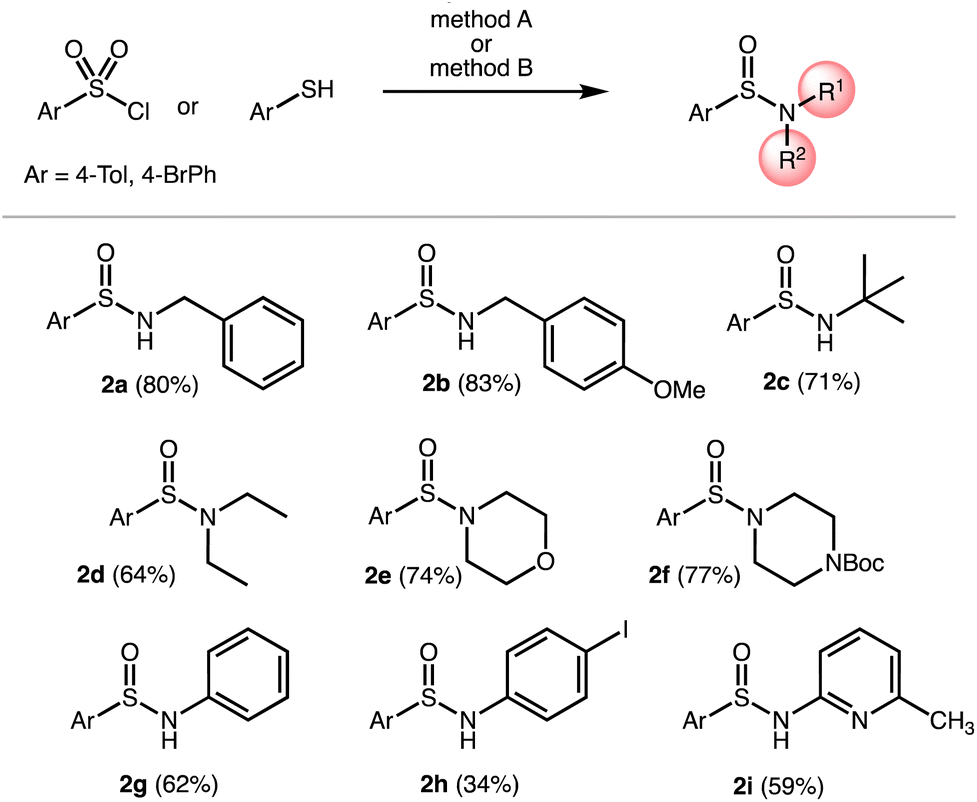
|
In order to show the utility of the method, we have further converted the sulfinamides to sulfonimidamides.38,48 Reaction of sulfinamides with 0.4 equiv. of trichloroisocyanuric acid (TCCA) and subsequent addition of morpholine (1.5 equiv.) provided sulfonimidamides 3a–3i in good yields (Table 4). This experiment demonstrated that the two complementary methods for the preparation of sulfinamides allow to introduce a set of C-substituents with high structural and functional group diversity, and this diversity can be transferred to the corresponding sulfonimidamides.
| a Reaction conditions: sulfinamide (0.16 mmol, 1 equiv.), TCCA (0.06 mmol, 0.4 equiv.), MeCN, rt, 30 min then Et3N (0.24 mmol, 1.5 equiv.) and morpholine (0.24 mmol, 1.5 equiv.), rt, 60 min. |
|---|

|
Conclusions
In conclusion, we have developed a new straightforward one-pot protocol for the preparation of sulfinamides from sulfonyl chlorides. The method tolerates a broad range of structural features and functional groups. Also, we have tested a complementary oxidative approach to sulfinamides starting from thiols. We have shown that the substrate scope for this known method is broader than previously thought and developed a modification for the preparation of N-heterocyclic sulfinamides. In a broader perspective, when compared to other methods, the reductive pathway enables products with oxidation-sensitive groups (e.g., thiophenes) that are incompatible with oxidative approaches starting from thiols. Also, diverse functional groups (secondary amides, ketones, esters, etc.) can be installed in the sulfinamide products, which would be difficult with approaches starting with organometallic reagents. Furthermore, these sulfinamides with various functional groups can be efficiently converted to the corresponding sulfonimidamides. Given the structural diversity of commercially available sulfonyl chlorides and thiols, these two complementary methods are set for broad C-substituent diversification of sulfinamides and sulfonimidamides for discovery chemistry.Author contributions
M. J. and V. N. designed and performed the experiments and analysed the data. J. M. designed the experiments, analysed the data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support of the Czech Science Foundation (GACR 20-26798S).References
- J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS.
- S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed.
- P. Dinér, A. Sadhukhan and B. Blomkvist, ChemCatChem, 2014, 6, 3063–3066 CrossRef.
- Y. Aota, T. Kano and K. Maruoka, Angew. Chem., Int. Ed., 2019, 58, 17661–17665 CrossRef CAS PubMed.
- Y. Aota, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2019, 141, 19263–19268 CrossRef CAS PubMed.
- S. Greed, O. Symes and J. A. Bull, Chem. Commun., 2022, 58, 5387–5390 RSC.
- T. Q. Davies, A. Hall and M. C. Willis, Angew. Chem., Int. Ed., 2017, 56, 14937–14941 CrossRef CAS PubMed.
- P. Mäder and L. Kattner, J. Med. Chem., 2020, 63, 14243–14275 CrossRef PubMed.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed.
- Y. Han, K. Xing, J. Zhang, T. Tong, Y. Shi, H. Cao, H. Yu, Y. Zhang, D. Liu and L. Zhao, Eur. J. Med. Chem., 2021, 209, 112885 CrossRef CAS PubMed.
- G. C. Nandi and P. I. Arvidsson, Adv. Synth. Catal., 2018, 360, 2976–3001 CrossRef CAS.
- T. Q. Davies, M. J. Tilby, J. Ren, N. A. Parker, D. Skolc, A. Hall, F. Duarte and M. C. Willis, J. Am. Chem. Soc., 2020, 142, 15445–15453 CrossRef CAS PubMed.
- M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed.
- U. Lücking, Chem. – Eur. J., 2022, 28, e202201993 Search PubMed.
- M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Chem. – Eur. J., 2021, 27, 17293–17321 CrossRef CAS PubMed.
- M. Andresini, S. Carret, L. Degennaro, F. Ciriaco, J.-F. Poisson and R. Luisi, Chem. – Eur. J., 2022, 28, e202202066 CrossRef CAS PubMed.
- Y. Tu, D. Zhang, P. Shi, C. Wang, D. Ma and C. Bolm, Org. Biomol. Chem., 2021, 19, 8096–8101 RSC.
- C. K. Savile and R. J. Kazlauskas, Adv. Synth. Catal., 2006, 348, 1183–1192 CrossRef CAS.
- M. Asada, M. Iwahashi, T. Obitsu, A. Kinoshita, Y. Nakai, T. Onoda, T. Nagase, M. Tanaka, Y. Yamaura, H. Takizawa, K. Yoshikawa, K. Sato, M. Narita, S. Ohuchida, H. Nakai and M. Toda, Bioorg. Med. Chem., 2010, 18, 1641–1658 CrossRef CAS.
- N.-Q. Shao, Y.-H. Chen, C. Li and D.-H. Wang, Org. Lett., 2020, 22, 7141–7146 CrossRef CAS PubMed.
- M. Furukawa and T. Okawara, Synthesis, 1976, 339–340 CrossRef CAS.
- S. H. Gafur, S. L. Waggoner, E. Jacobsen, C. G. Hamaker and S. R. Hitchcock, Synthesis, 2018, 50, 4855–4866 CrossRef CAS.
- W. E. Truce and F. E. Roberts Jr., J. Org. Chem., 1963, 28, 593–594 CrossRef CAS.
- J. Kice and K. Bowers, J. Am. Chem. Soc., 1962, 84, 605–610 CrossRef CAS.
- The Chemistry of Sulphinic Acids, Esters and Derivatives, ed. S. Patai, Wiley, Chichester; New York, 1st edn, 1990 Search PubMed.
- J. L. García Ruano, A. Parra, F. Yuste and V. Mastranzo, Synthesis, 2008, 311–319 CrossRef.
- J. L. García Ruano, A. Parra, L. Marzo, F. Yuste and V. M. Mastranzo, Tetrahedron, 2011, 67, 2905–2910 CrossRef.
- J. L. García Ruano, J. Alemán, M. B. Cid and A. Parra, Org. Lett., 2005, 7, 179–182 CrossRef PubMed.
- G. Liu, D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1997, 119, 9913–9914 CrossRef CAS.
- C. Mioskowski and G. Solladie, Tetrahedron, 1980, 36, 227–236 CrossRef CAS.
- C. Worch, I. Atodiresei, G. Raabe and C. Bolm, Chem. – Eur. J., 2010, 16, 677–683 CrossRef CAS PubMed.
- P. Brownbridge and I. C. Jowett, Synthesis, 1988, 252–254 CrossRef CAS.
- J.-H. Youn and R. Herrmann, Synthesis, 1987, 72–73 CrossRef CAS.
- N. Taniguchi, Eur. J. Org. Chem., 2016, 2157–2162 CrossRef CAS.
- N. Taniguchi, Eur. J. Org. Chem., 2010, 2010, 2670–2673 CrossRef.
- H. Yu, Z. Li and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 15602–15605 CrossRef CAS PubMed.
- D. Wen, Q. Zheng, C. Wang and T. Tu, Org. Lett., 2021, 23, 3718–3723 CrossRef CAS PubMed.
- P. K. T. Lo, G. A. Oliver and M. C. Willis, J. Org. Chem., 2020, 85, 5753–5760 CrossRef CAS.
- Q. Wang, X.-Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2016, 55, 10811–10815 CrossRef CAS PubMed.
- P. K. T. Lo and M. C. Willis, J. Am. Chem. Soc., 2021, 143, 15576–15581 CrossRef CAS PubMed.
- M. Harmata, P. Zheng, C. Huang, M. G. Gomes, W. Ying, K.-O. Ranyanil, G. Balan and N. L. Calkins, J. Org. Chem., 2007, 72, 683–685 CrossRef CAS PubMed.
- R. Chakravarti, A. Mano, H. Iwai, S. S. Aldeyab, R. P. Kumar, M. L. Kantam and A. Vinu, Chem. – Eur. J., 2011, 17, 6673–6682 CrossRef CAS PubMed.
- F. C. Whitmore and F. H. Hamilton, Org. Synth., 1922, 2, 89–90 CrossRef.
- N. Chumachenko and P. Sampson, Tetrahedron, 2006, 62, 4540–4548 CrossRef CAS.
- F. O'Hara, R. D. Baxter, A. G. O'Brien, M. R. Collins, J. A. Dixon, Y. Fujiwara, Y. Ishihara and P. S. Baran, Nat. Protoc., 2013, 8, 1042–1047 CrossRef PubMed.
- M. Kulka, J. Am. Chem. Soc., 1950, 72, 1215–1218 CrossRef CAS.
- H. Uchiro and S. Kobayashi, Tetrahedron Lett., 1999, 40, 3179–3182 CrossRef CAS.
- C. R. Johnson and A. Wambsgans, J. Org. Chem., 1979, 44, 2278–2280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00050h |
| This journal is © The Royal Society of Chemistry 2023 |