
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of radioiodinated estrogens for diagnosis and therapy of male urogenital tumours†
Feodor
Braun
a,
Marcel
Jaschinski
b,
Philipp
Täger
a,
Verena
Marmann
a,
Melanie von
Brandenstein
c,
Barbara
Köditz
c,
Thomas
Fischer
a,
Sergio
Muñoz-Vázquez
a,
Beate
Zimmermanns
a,
Markus
Dietlein
a,
Ferdinand
Sudbrock
a,
Phillip
Krapf
a,
Dietmar
Fischer
d,
Axel
Heidenreich
c,
Alexander
Drzezga
a,
Stefan
Kirsch
 b,
Markus
Pietsch
d and
Klaus
Schomäcker
*a
b,
Markus
Pietsch
d and
Klaus
Schomäcker
*a
aDepartment of Nuclear Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, 50937, Cologne, Germany. E-mail: klaus.schomaecker@uni-koeln.de
bOrganic Chemistry, Bergische Universität Wuppertal, 42119 Wuppertal, Germany
cClinic and Polyclinic for Urology, Faculty of Medicine and University Hospital Cologne, University of Cologne, 50937 Cologne, Germany
dInstitutes I & II of Pharmacology, Center of Pharmacology, Faculty of Medicine and University Hospital Cologne, University of Cologne, 50931 Cologne, Germany
First published on 20th March 2023
Abstract
The preparation of 24 estrogens, their estrogen receptor (ER) affinity and studies of radioiodinated estrogen binding to ER-positive male bladder tumor cells (HTB9) are described. The estrogens with the highest affinity were selected using fluorescence anisotropy assays. A 2,2,2-trifluoroethyl group at the 11β-position caused particularly promising affinity. (Radio)iodination was performed on the 17α-vinyl group. Binding studies on HTB9 cells revealed picomolar affinities of radioconjugates 19 and 31, indicating promising ability for targeting of urogenital tumors.
It has long been recognized that the internalization and intranuclear localization of radioactively labelled estrogen analogues make them promising candidates for receptor-mediated therapy and diagnosis.1–3 There was initial concern that introducing radionuclides into the steroid backbone would require chemical modifications that lower estrogen affinity. However, certain modifications in the 11β-position are well known to increase estrogen analogs’ affinity significantly.1 Various trials, introducing chemical groups such as ethyl or methoxy groups into the 11β-position, confirmed the affinity-raising effect of a 11β-modification.4 In this work, radioactive iodine isotopes were coupled in the 17α-position without significant loss of affinity compared to the native estrogen. The radioisotopes of iodine have a wide range of possible applications. Iodine-123, I-124, I-125, and I-131 have the necessary physical properties for diagnostics and therapy. As a positron emitter, I-124 can be applied in diagnostic procedures, such as positron emission tomography (PET).5 Auger electrons are emitted from I-123 or I-125 at ultra-short ranges (within the dimensions of a cell nucleus) and offer the advantage of high specific cytotoxicity combined with relatively few side effects.6,7 Iodine-131 is a beta emitter that has proven its worth in radiotherapy over decades.8 For a selective coupling of iodine isotopes, radioiododestannylation is the standard method.9,10 This requires the introduction of a domain capable of coupling through prestannylated linkers. It, therefore, includes modifications on 17α- and 11β-positions via Grignard reactions with alkyl groups, such as ethyl- and ethinyl moieties. In particular, the latter provides the ideal scaffold for further derivatization, such as Sonogashira coupling or hydrostannylation reaction, to prepare rapidly accessible arylstannylated compounds.
Affinity measurements using fluorescence anisotropy assays (FAA) were performed on the synthesized estrogen derivatives to determine the influence of structural modifications at the 17α- and 11β-position on the affinity of the estrogen ligands for ERα and ERβ.11–14
It remains to be investigated whether introducing a terminally perfluorinated ethyl group in the 11β-position leads to a further increase in affinity. Further experiments were intended to find suitable linkers for prestannylation without a significant loss of affinity. Various prestannylated and non-radioactive iodine-coupled estrogen ligands were tested for affinity by FAA.
The most promising candidates were then radioiodinated and examined in male bladder tumour cells (HTB9) concerning cellular uptake. The unique feature of using radioestrogens in ER+ tumours in men is that the ubiquitously occurring estrogen receptor expression is significantly lower than in women.15,16 This also applies to the concentration of estrogens circulating in blood.17 In this way, it is possible to target ER+ tumours more precisely in men with less extratumoural accumulation of radioactivity.
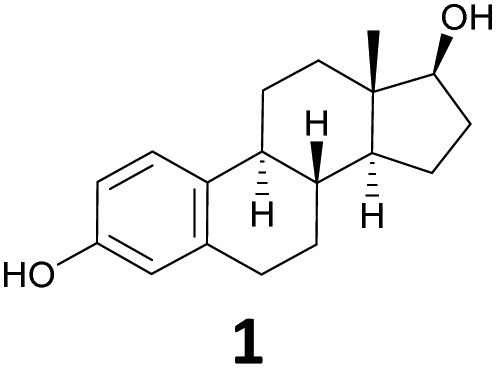
The starting material for chemical modifications was the native 17β-estradiol 1 (Fig. 1). After benzyl protection of both alcohol groups, selective oxidation at positions 9 and 11 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) produced steroid 3 in excellent yields. A subsequent hydroboration/oxidation sequence permitted access to the region- and stereoselective alcohol 4 in an almost quantitative yield and reproducible way. Ketone formation under Swern oxidation conditions yielded steroid 5, which using a Grignard reaction with ethylmagnesium bromide and dry cerium(III) chloride, allowed access to the 11α-ethyl group.18 Deoxygenation and deprotection gave the desired 11β-ethyl steroid 7 in 98% yield in two steps. Finally, using reaction conditions of the Oppenauer reaction followed by the Grignard reaction with ethynylmagnesium bromide under the same conditions as before resulted in steroid 10. The 11β-ethyl-17α-ethinylestradiol (10) was thus successfully synthesised with an overall yield of 48% in 10 steps.
 | ||
| Fig. 1 Synthesis of 11β-ethyl-17α-ethinylestradiol (10). | ||
Further modifications were made to the 17α-alkyne by the introduction of different linkers. The structures of the compounds are listed in (Table S1 ESI†). Cold iodination was performed by selective iododestannylation.9,19 The chemical characterisation was performed via NMR and mass spectrometry (chemical synthesis part, ESI†).
Subsequent work with the synthesised 11β-ethyl-17α-ethinylestradiol (10) included hydrostannylation to the (E)-stannane 18 followed by iodination to the corresponding vinyl iodide 19 (Fig. 2).
 | ||
| Fig. 2 Synthesis of vinyl iodine 19. | ||
Compound 31 (Fig. 3) was synthesised starting from steroid 5 under Peterson olefination conditions.20 The olefin 20 was isolated in 87% yield in two steps via the formation of the alcohol as an intermediate and subsequent cleavage in the acidic milieu in acetone. The desired stereocentre was constructed through a hydroboration/oxidation sequence, leading to the primary alcohol 21.21 Subsequent oxidation according to Swern reaction conditions afforded the desired aldehyde 22 in an excellent yield. The use of (trifluoromethyl)trimethylsilane and catalytic amounts of cesium fluoride resulted in the trifluoride, spectroscopically detected via19F-NMR.22 After trimethylsilyl ether (TMS) deprotection, alcohol 23 was generated in 78% yield in two steps. Deoxygenation to compound 25 was achieved by preparing the thiocarbonate 24 using O-phenylchlorothionoformate followed by Barton's McCombie deoxygenation.23,24 After ether cleavage and regioselective tert-butyldimethylsilyl ether (TBS) protection of the phenolic hydroxy group, the alcohol in 17β-position was oxidised using 2-iodoxybenzoic acid (IBX) in dimethylformamide (DMF) to form the corresponding ketone 28. In a two-step synthesis procedure, the alkyne was introduced in an addition reaction with TMS acetylene and N-butyllithium (n-BuLi) and then underwent TBS deprotection to steroid 29. Finally, the desired trifluoroethylated steroid 31 was successfully obtained after hydrostannylation and iodination in yields of 61% and 79%, respectively. The outcome of these 17 steps was that the crucial trifluoroethyl group was installed successfully in the 11β-position, and the desired steroid 31 was synthesized with an overall yield of 4.2%.
 | ||
| Fig. 3 Synthesis of compound 31. | ||
Fluorescence anisotropy assays25,26 were used to preselect non-radioactive estrogens. In this process, an ER protein is added to a fluorescent estrogen probe with low fluorescence anisotropy (Fluormone™ EL Red), forming a probe/ER protein complex with high fluorescence anisotropy. The various non-radioactive estrogen ligands to be tested for their affinities are capable of displacing the probe from the ER to different degrees. This leads to a decrease in fluorescence anisotropy (Fig. S1, ESI†), which allows the half-maximal inhibitory concentration (IC50) to be determined and the relative binding affinity (RBA) to be calculated.11–14 If the affinity of the estrogen ligands to be tested is significantly higher than that of the fluorescent probe, as it is for estrogens 8,4,2719,28 and 31, the IC50 can no longer be measured precisely.29,30 Such compounds are characterised by very steep dose–response curves in the FAA with IC50 values at the “tight binding limit imposed by the assay conditions”.30
In contrast, previously reported RBAs of <100%, such as estrogen 17,31 could be reproduced in our experimental setting. The method is, therefore, only suitable for excluding test ligands with comparatively low affinity from further investigations. Promising ligands have IC50 values and relative binding affinities close to those achievable with native 17β-estradiol (1) (Table 1). However, the real values of RBA can be orders of magnitude higher. For a prime example, estrogen 8, other authors have published RBAs of up to 3000% (obtained by radioligand binding assays).4,27 Comparing the RBAs of the iodinated ligands 17 and 19, the difference is not too large. However, other authors have reported a 10-fold increase in affinity due to ethylation in the 11β-position.27,28,31 The results and conclusions mentioned here represent only a portion of our extensive investigations into relationships between structure and ER affinity using FAA. To keep the communication thread running, a comprehensive overview of the IC50 values and RBAs of all tested estrogens (Table S1†) and a further discussion of the results (results and discussion S1†) can be found in the ESI† (FAA part).
| Structure | ERα![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IC50 (nM) :IC50 (nM) |
ERα:RBA (%) |
ERβ:IC50 (nM) |
ERβ:RBA (%) |
|---|---|---|---|---|
| a Reported IC50 values of 1 in PolarScreen™ Competitor Assay with Fluormone™ EL Red: ERα, 5.9–16 nM; ERβ, 20.8–23 nM.25,26,32–34 b Reported RBAs: 8, 1000, 3000% (rat uterine ERα);4,2717, 62% (rat uterine ERα);3119, 890% (lamb uterine ERα).28 c N.d., not determined. The synthesis of compound 42 is described in the ESI.† | ||||
|
11.6 ± 1.2a (n = 32) | 100 | 39.5 ± 0.7a (n = 4) | 100 |

|
16.0 ± 1.2 (n = 2) | 72.5b | N.d.c | N.d.c |

|
15.6 ± 3.3 (n = 3) | 74.4b | N.d.c | N.d.c |

|
14.5 ± 1.9 (n = 3) | 80.0b | 37.6 ± 4.2 (n = 3) | 105 |

|
12.9 ± 0.5 (n = 3) | 89.9 | 26.8 ± 3.0 (n = 3) | 147 |

|
38.8 ± 3.4 (n = 3) | 29.9 | 97.7 ± 12.8 (n = 4) | 40.4 |
The most promising candidates for the FAA were estrogens 19 and 31. Next, the corresponding I-131- and I-123-labelled analogues of these two estrogens were prepared via iododestannylation of prestannylated precursors (18 and 30). The radioestrogens were purified by analytical HPLC (Fig. 4). Radioiodination was performed with sufficient radiochemical yields (>80%) and radiochemical purities (>99%), with a molar activity of 24.3 MBq/nmol for I-131 and 8770.7 MBq/nmol for I-123 labelled compounds. The obtained radioligands remained stable in PBS for 24 hr.
 | ||
| Fig. 4 Representative HPLC tracks of I-131-labelled estrogen 19. Because of the short half-life of I-123, I-131 was used for stability testing. | ||
The radioactively labelled estrogens 19 and 31 should be investigated on male urogenital carcinoma cells with the highest possible estrogen receptor expression. For this purpose, western blot examined various available cell lines regarding the expression of ERα and ERβ, leading to the identification of HTB9 cells as those with a comparatively moderate ER expression density (Fig. 5). The expression of ERα and ERβ was detectable in HTB9,35 TCam2,36,37 PC3,38,39 DU14540 cells, BPH41 and LNCap,36,39 cells. In all prostate cell lines used, both receptors were detectable, which corresponds with the literature.42,43 Since the overexpression of estrogens receptors (alpha or beta) seems to be of importance, especially in high-grade bladder cancer, we predominantly focused on HTB-9 cells.44
 | ||
| Fig. 5 Western blot analysis of the appearance of ERα and ERβ in the cell lines HTB9,35 Tcam2,36,37 BPH,42 LNCap,36,39 PC3,38 and DU145.40 GAPDH was used as a housekeeper for neutralization. Weak signals were detectable in all cell lines for ERβ. For calculation of the relative percentage, GAPDH signal density was measured with ImageJ program and used for neutralization. The corresponding signal of the GAPDH line was set as 100% and compared with the density of the ER alpha and ER beta. | ||
HTB9 cells were then titrated with various concentrations of the radioiodinated ligands 19 (0-339.5 pM) and 31 (0-346.6 pM), respectively. After an incubation time of 24 h, the radioactive medium was removed, and the cells were washed and lysed. The radioactivity of the lysates was measured in a borehole measuring station, and the dissociation constant, KD, and the maximum ER saturation, Bmax, were determined. The results for the specific binding of the I-123-labelled analogs of estrogens 19 and 31 are summarised in Fig. 6. In addition, a linearisation was carried out using a Scatchard plot. As a result, the following KD values could be determined for the I-123 labelled ligands: I-123-19: (63 ± 25) pM and I-123-31: (40 ± 4) pM. The maximal specific saturation, Bmax, was obtained as the amount of specifically accumulated radioactivity per well with 106 cells (19.46 KBq for compound 19 and 40.17KBq for compound 31). From this, 1375–2840 of ER per cell were calculated.
 | ||
| Fig. 6 Saturation assays and Scatchard plots of specific ER binding of I-123 labelled products 19 (top) and 31 (bottom). Values of KD were 63 ± 25 pM for compound 19 and 40 ± 4 pM for compound 31. Maximum specific saturation of activity per cell (Bmax) was 19.46–40.17 KBq, corresponding to 2.2–4.6 fmol and 1375–2840 ER per cell. | ||
The KD and Bmax could only be determined with I-123-labelled variants of 19 and 31. The results of the cell studies with the respective I-131-containing derivatives showed an unacceptably high degree of scatter of the measured values. We suspect this was caused by the insufficient molar activity of the radioisotope (manufacturer's information: 24.3 MBq/nmol). Taking into account a Bmax for 106 cells of 2.2–4.6 fmol, this would result in an activity of only appr. 60 Bq. Such activities correspond to the background radiation in normal radionuclide laboratories and can hardly be measured exactly.
In the trials presented here, it was possible to develop and test promising candidates for the ER-mediated transport of radioligands into tumour cells. This is important not only for imaging urogenital carcinomas in men but also for new theragnostic approaches. ER ligands labelled with radionuclides bind to the ER protein in the cytoplasm with high affinity. The estrogen/ER complex is activated and transported into the cell nucleus. There, the complex reacts with transcription-activating regions of the DNA and causes a change in the transcription of specific mRNA and, thus, in protein synthesis.45 As a result, ER ligands can serve as vehicle molecules for radionuclides and deposit ionising radiation in the most radiation-sensitive part of the cell, the DNA. The selective enrichment near the cell nucleus of emitters with the Auger effect, such as I-123 or I-125, leads to cell death due to irreparable DNA double-strand breaks, even with a few radioactive decays per cell.46,47 In addition, extranuclear or extracellular iodine isotopes with the Auger effect are much less toxic, as their radiation effect is limited to the vicinity of the cell nucleus.
Compounds 19 and 31 appear to be auspicious parent compounds for labelling of ER ligands with radionuclides. Further improvement of their tumour targeting properties will be attempted shortly.
Author contributions
Feodor Braun: formal analysis, investigation, writing – original draft preparation, writing – review and editing; Marcel Jaschinski: investigation, writing – review and editing; Philipp Täger: funding acquisition; Verena Marmann: investigation; Melanie von Brandenstein: investigation, supervision, writing – review and editing; Barbara Köditz: investigation, writing – review and editing; Thomas Fischer: investigation, writing – review and editing; Sergio Muñoz-Vázquez: investigation, writing – review and editing; Beate Zimmermanns: data curation, investigation; Markus Dietlein: writing – review and editing; Ferdinand Sudbrock: formal analysis, writing – review and editing; Phillip Krapf: writing – review and editing; Dietmar Fischer: resources, writing – review and editing; Axel Heidenreich: resources, writing – review and editing; Alexander Drzezga: resources, writing – review and editing; Stefan Kirsch: resources, supervision, writing – review and editing; Markus Pietsch: formal analysis, methodology, supervision, writing – original draft preparation, writing – review and editing; Klaus Schomäcker: conceptualization, formal analysis, project administration, supervision, writing – original draft preparation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Köln Fortune Program (Faculty of Medicine, University of Cologne) (389/2021).References
- J. A. Katzenellenbogen, Nucl. Med. Biol., 2021, 92, 24–37 CrossRef CAS PubMed.
- K. Schomäcker, T. Fischer, B. Böttcher, S. A. Mraheel, A. Scharl, B. Gabruk-Szostak, K. Scheidhauer, U. J. Göhring, B. Meller-Rehbein, S. K. Shukla and H. Schicha, Nuklearmedizin, 1998, 37, 134–140 CrossRef.
- T. Fischer, K. Schomacker and H. Schicha, Int. J. Radiat. Biol., 2008, 84, 1112–1122 CrossRef CAS PubMed.
- E. Napolitano, R. Fiaschi, K. E. Carlson and J. A. Katzenellenbogen, J. Med. Chem., 1995, 38, 429–434 CrossRef CAS PubMed.
- H. Kertész, M. Conti, V. Panin, J. Cabello, D. Bharkhada, T. Beyer, L. Papp, W. Jentzen, J. Cal-Gonzalez, J. L. Herraiz, A. López-Montes and I. Rausch, EJNMMI Phys., 2022, 9, 56 CrossRef PubMed.
- R. W. Howell, Int. J. Radiat. Biol., 2020, 1–26 Search PubMed.
- T. Fischer, F. Sudbrock, E. Pomplun, R. Kriehuber, J. Winkler, M. Matzkies, A. Dellweg, M. Dietlein, S. Arnhold, H.-D. Royer, H. Schicha, J. Hescheler and K. Schomäcker, Int. J. Radiat. Biol., 2012, 88, 961–971 CrossRef CAS PubMed.
- S. St James, B. Bednarz, S. Benedict, J. C. Buchsbaum, Y. Dewaraja, E. Frey, R. Hobbs, J. Grudzinski, E. Roncali, G. Sgouros, J. Capala and Y. Xiao, Int. J. Radiat. Oncol., Biol., Phys., 2021, 109, 891–901 CrossRef PubMed.
- T. Taldone, D. Zatorska, S. O. Ochiana, P. Smith-Jones, J. Koziorowski, M. P. Dunphy, P. Zanzonico, A. Bolaender, J. S. Lewis, S. M. Larson, G. Chiosis and N. V. K. Pillarsetty, J. Labelled Compd. Radiopharm., 2016, 59, 129–132 CrossRef CAS PubMed.
- Z. Tu, J. Xu, L. A. Jones, S. Li, D. Zeng, M.-P. Kung, H. F. Kung and R. H. Mach, Appl. Radiat. Isot., 2010, 68, 2268–2273 CrossRef CAS PubMed.
- R. Bolger, T. E. Wiese, K. Ervin, S. Nestich and W. Checovich, Environ. Health Perspect., 1998, 106, 551–557 CrossRef CAS PubMed.
- G. N. Nikov, N. E. Hopkins, S. Boue and W. L. Alworth, Environ. Health Perspect., 2000, 108, 867–872 CrossRef CAS PubMed.
- G. N. Nikov, M. Eshete, R. V. Rajnarayanan and W. L. Alworth, J. Endocrinol., 2001, 170, 137–145 CAS.
- G. J. Parker, T. L. Law, F. J. Lenoch and R. E. Bolger, J. Biomol. Screening, 2000, 5, 77–88 CrossRef CAS PubMed.
- P. S. Cooke, M. K. Nanjappa, C. Ko, G. S. Prins and R. A. Hess, Physiol. Rev., 2017, 97, 995–1043 CrossRef PubMed.
- L. K. Davis, A. L. Pierce, N. Hiramatsu, C. V. Sullivan, T. Hirano and E. G. Grau, Gen. Comp. Endocrinol., 2008, 156, 544–551 CrossRef CAS PubMed.
- A. Kratz, M. Ferraro, P. M. Sluss and K. B. Lewandrowski, N. Engl. J. Med., 2004, 351, 1548–1563 CrossRef CAS PubMed.
- T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392–4398 CrossRef CAS.
- R. N. Hanson, E. Napolitano and R. Fiaschi, J. Med. Chem., 1998, 41, 4686–4692 CrossRef CAS PubMed.
- D. J. Peterson, J. Org. Chem., 1968, 33, 780–784 CrossRef CAS.
- D. C. Labaree, J.-X. Zhang, H. A. Harris, C. O'Connor, T. Y. Reynolds and R. B. Hochberg, J. Med. Chem., 2003, 46, 1886–1904 CrossRef CAS PubMed.
- C.-C. Tseng, G. Baillie, G. Donvito, M. A. Mustafa, S. E. Juola, C. Zanato, C. Massarenti, S. Dall'Angelo, W. T. A. Harrison, A. H. Lichtman, R. A. Ross, M. Zanda and I. R. Greig, J. Med. Chem., 2019, 62, 5049–5062 CrossRef CAS PubMed.
- D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC.
- D. Crich and L. Quintero, Chem. Rev., 1989, 89, 1413–1432 CrossRef CAS.
- PolarScreen™ ER Alpha Competitor Assay, Red, MAN0009428 RevA.0, accessed on 18 January 2022, 3.48 pm.
- PolarScreen™ ER Beta Competitor Assay, Red, MAN0008944 RevA.0, accessed on 18 January 2022, 3.50 pm.
- R. Tedesco, J. A. Katzenellenbogen and E. Napolitano, Bioorg. Med. Chem. Lett., 1997, 7, 2919–2924 CrossRef CAS.
- R. N. Hanson, M. Ghoshal, F. G. Murphy, C. Rosenthal, R. E. Gibson, N. Ferriera, V. Sood and J. Ruch, Nucl. Med. Biol., 1993, 20, 351–358 CrossRef CAS PubMed.
- X. Huang, J. Biomol. Screening, 2003, 8, 34–38 CrossRef CAS PubMed.
- K. W. Vogel, B. D. Marks, K. R. Kupcho, K. L. Vedvik and T. M. Hallis, Lett. Drug Des. Discovery, 2008, 5, 416–422 CrossRef CAS.
- E. Napolitano, R. Fiaschi and R. N. Hanson, J. Med. Chem., 1991, 34, 2754–2759 CrossRef CAS PubMed.
- M. E. Tirri, R. J. Huttunen, J. Toivonen, P. L. Härkönen, J. T. Soini and P. E. Hänninen, J. Biomol. Screening, 2005, 10, 314–319 CrossRef CAS PubMed.
- K.-H. Lee and E.-M. Choi, Phytother. Res., 2006, 20, 952–960 CrossRef PubMed.
- X. Gao, L. Yu, L. Castro, A. B. Moore, T. Hermon, C. Bortner, M. Sifre and D. Dixon, Toxicol. Lett., 2010, 196, 133–141 CrossRef CAS PubMed.
- J. Teng, Z.-Y. Wang, D. F. Jarrard and D. E. Bjorling, Endocr.-Relat. Cancer, 2008, 15, 351–364 CrossRef CAS PubMed.
- A. Wallacides, A. Chesnel, H. Ajj, M. Chillet, S. Flament and H. Dumond, Mol. Cell. Endocrinol., 2012, 350, 61–71 CrossRef CAS PubMed.
- S. Panza, M. Santoro, F. De Amicis, C. Morelli, V. Passarelli, P. D’Aquila, F. Giordano, E. Cione, G. Passarino, D. Bellizzi and S. Aquila, Tumor Biol., 2017, 39(5) DOI:10.1177/1010428317701642.
- A. van Bokhoven, M. Varella-Garcia, C. Korch, W. U. Johannes, E. E. Smith, H. L. Miller, S. K. Nordeen, G. J. Miller and M. S. Lucia, Prostate, 2003, 57, 205–225 CrossRef CAS PubMed.
- Q. Hu, B. Zhang, R. Chen, C. Fu, J. A. X. Fu, J. Li, L. Fu, Z. Zhang and J.-T. Dong, Oncogenesis, 2019, 8, 28 CrossRef PubMed.
- H. Bonkhoff, T. Fixemer, I. Hunsicker and K. Remberger, Am. J. Pathol., 1999, 155, 641–647 CrossRef CAS PubMed.
- Q. Wu, Y. Zhou, L. Chen, J. Shi, C.-Y. Wang, L. Miao, H. Klocker, I. Park, C. Lee and J. Zhang, J. Endocrinol., 2007, 195, 89–94 CAS.
- L. Xiao, Y. Luo, R. Tai and N. Zhang, Mol. Med. Rep., 2019, 19, 3555–3563 CAS.
- C. P. Cheung, S. Yu, K. B. Wong, L. W. Chan, F. M. M. Lai, X. Wang, M. Suetsugi, S. Chen and F. L. Chan, J. Clin. Endocrinol. Metab., 2005, 90, 1830–1844 CrossRef CAS PubMed.
- H. Ide and H. Miyamoto, Cells, 2021, 10, 1169 CrossRef CAS PubMed.
- A. Ramírez-de-Arellano, A. L. Pereira-Suárez, C. Rico-Fuentes, E. I. López-Pulido, J. C. Villegas-Pineda and E. Sierra-Diaz, Front. Endocrinol., 2022, 12, 811578, DOI:10.3389/fendo.2021.
- R. W. Howell, Int. J. Radiat. Biol., 2008, 84, 959–975 CrossRef CAS PubMed.
- S. Ma, B. Kong, B. Liu and X. Liu, Int. J. Radiat. Biol., 2013, 89, 326–333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00114h |
| This journal is © The Royal Society of Chemistry 2023 |