DOI:
10.1039/D3NR03375A
(Paper)
Nanoscale, 2023,
15, 19268-19281
Water-soluble ionic carbon nitride as unconventional stabilizer for highly catalytically active ultrafine gold nanoparticles†
Received
11th July 2023
, Accepted 13th November 2023
First published on 13th November 2023
Abstract
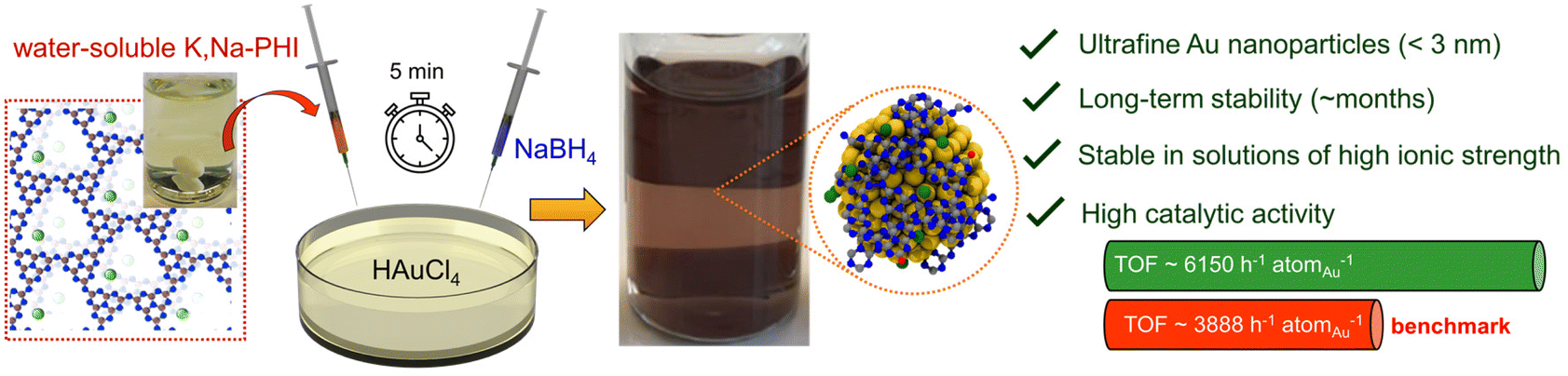
Ultrafine metal nanoparticles (NPs) hold promise for applications in many fields, including catalysis. However, ultrasmall NPs are typically prone to aggregation, which often leads to performance losses, such as severe deactivation in catalysis. Conventional stabilization strategies (e.g., immobilization, embedding, or surface modification by capping agents) are typically only partly effective and often lead to loss of catalytic activity. Herein, a novel type of stabilizers based on water-soluble ionic (K+ and Na+ containing) polymeric carbon nitride (i.e., K,Na-poly(heptazine imide) = K,Na-PHI) is reported that enables effective stabilization of highly catalytically active ultrafine (size of ∼2–3 nm) gold NPs. Experimental and theoretical comparative studies using different structural units of K,Na-PHI (i.e., cyanurate, melonate, cyamelurate) indicate that the presence of functionalized heptazine moieties is crucial for the synthesis and stabilization of small Au NPs. The K,Na-PHI-stabilized Au NPs exhibit remarkable dispersibility and outstanding stability even in solutions of high ionic strength, which is ascribed to more effective charge delocalization in the large heptazine units, resulting in more effective electrostatic stabilization of Au NPs. The outstanding catalytic performance of Au NPs stabilized by K,Na-PHI is demonstrated using the selective reduction of 4-nitrophenol to 4-aminophenol by NaBH4 as a model reaction, in which they outperform even the benchmark “naked” Au NPs electrostatically stabilized by excess NaBH4. This work thus establishes ionic carbon nitrides (PHI) as alternative capping agents enabling effective stabilization without compromising surface catalysis, and opens up a route for further developments in utilizing PHI-based stabilizers for the synthesis of high-performance nanocatalysts.
Introduction
Ultrafine and highly dispersed metal nanoparticles (NPs) exhibit unique optical, electronic and surface properties, and hold promise for application in various fields, including nanotechnology, optoelectronics, biomedicine, sensing, and catalysis.1–12 In the field of heterogeneous catalysis, ultrafine (i.e., particles with an average size of ca. ≤3 nm) metal NPs often show enhanced catalytic properties that are distinct from their larger counterparts due to the effects of electron energy quantization and surface geometry, i.e., specific surface atom arrangement and coordination.13,14 For example, ultrafine gold NPs, owing to their large surface area, exposing various surface facets, and a high number of undercoordinated atoms at edge and corner sites, are known to exhibit extraordinary performance in various catalytic conversions, such as CO oxidation, hydrogenation, oxidation of alcohols, coupling reactions, and reduction reactions.15–19 However, ultrafine metal NPs are thermodynamically and kinetically unstable and tend to aggregate in order to reduce the surface free energy, often resulting in a drastic loss of catalytic performance over time, and thus limiting their practical application.20,21 Typical strategies to stabilize NPs consist in immobilization on high surface area supports, embedding into molecular cage-like materials (e.g., metal–organic frameworks, MOFs), or surface protection by molecules or ions that can act as effective capping agents.2,22–32 However, all these strategies have also drawbacks. The supported catalysts often exhibit decreased activity as compared to colloidal NPs,33 and suffer from nanoparticle migration and aggregation, irregular distribution and regrowth, and weak affinity of metal NPs to supports, resulting in increased leaching.2 The applicability of MOF-embedded metal NPs in catalysis is often limited due to MOF stability issues under catalytic conditions.34 Finally, the surface capping agents can not only inhibit aggregation but also block the active sites on the NP surface, leading to reduced catalytic activities.22,23,35 A typical example would be the strongly inhibiting effect of low-weight thiolated polyethylene glycol (HS-PEG) as a stabilizing ligand on the catalytic activity of Au NPs towards the reduction of 4-nitrophenol.22 Considering the aforementioned aspects, the development of new stabilizers that can effectively stabilize metal NPs without blocking the active sites and the diffusion of the reactants and products to and from the metal surface is of paramount importance in catalysis.
Recently, we reported the first synthesis of fully water-soluble ionic polymeric carbon nitride, i.e., potassium and sodium ion-containing poly(heptazine imide) (K,Na-PHI),36 an ionic member of the polymeric carbon nitride family.37–43 In aqueous solutions, the K,Na-PHI material can be fully dispersed in form of very small K,Na-PHI colloidal nanoparticles (hydrodynamic diameter ∼10 nm) containing a large number of cyanamide and cyamelurate surface moieties, resulting in highly stable quasi-homogeneous solutions of K,Na-PHI with no gelation or precipitation over several months.36 Given the unique features of K,Na-PHI (i.e., water solubility, chemical stability, and ionic nature), we hypothesized that water-soluble K,Na-PHI might open a completely new and attractive possibility to stabilize catalytically active metal NPs. While various promising properties of ionic carbon nitrides, such as their photocatalytic activity and charge storage properties, are well established,36,37,39,40,44–47 the use of ionic carbon nitrides as stabilizers has not been reported so far. In this contribution, we demonstrate, for the first time, the ability of K,Na-PHI to effectively stabilize ultrafine Au NPs (<3 nm). The stabilized Au NPs exhibit very high catalytic activity in the selective reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) using NaBH4 as a reducing agent (Fig. 1), a widely used model reaction for evaluating the catalytic activity of nanocatalysts in aqueous media.22,23,48,49 Moreover, they show robust stability against agglomeration and excellent dispersibility for at least 2 months, as well as excellent accessibility in solution, as demonstrated by turnover frequencies superior to “naked” Au nanoparticles stabilized solely by excess BH4− anions. The stabilization effect of K,Na-PHI is elucidated using a comparative analysis employing different structural units of K,Na-PHI (cyanurate, melonate, and cyamelurate) and ab initio theoretical calculations, and the dominant role of functionalized heptazine moieties for the stabilization of Au NPs is discussed.
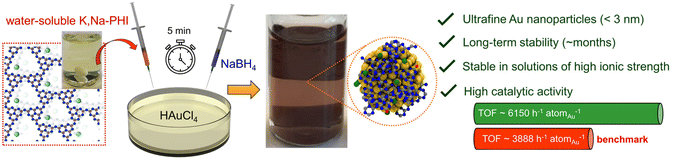 |
| | Fig. 1 Concept of effective stabilization of ultrafine Au nanoparticles using water-soluble ionic carbon nitride. The Au NPs are readily obtained by the addition of water-soluble K,Na-PHI into an aqueous solution of HAuCl4 and subsequent reduction by sodium borohydride. The resulting ultrafine Au NPs (size <3 nm) are stable over several months even in solutions of high ionic strength and outperform benchmark “naked” Au NPs stabilized solely by excess NaBH4 in a model catalytic reaction (selective reduction of 4-nitrophenol to 4-aminophenol using NaBH4 as a reducing agent). | |
Experimental procedures and computational details
All experimental procedures and computational details are provided in the ESI.†
Results and discussion
Effect of the proposed stabilizers on Au NPs
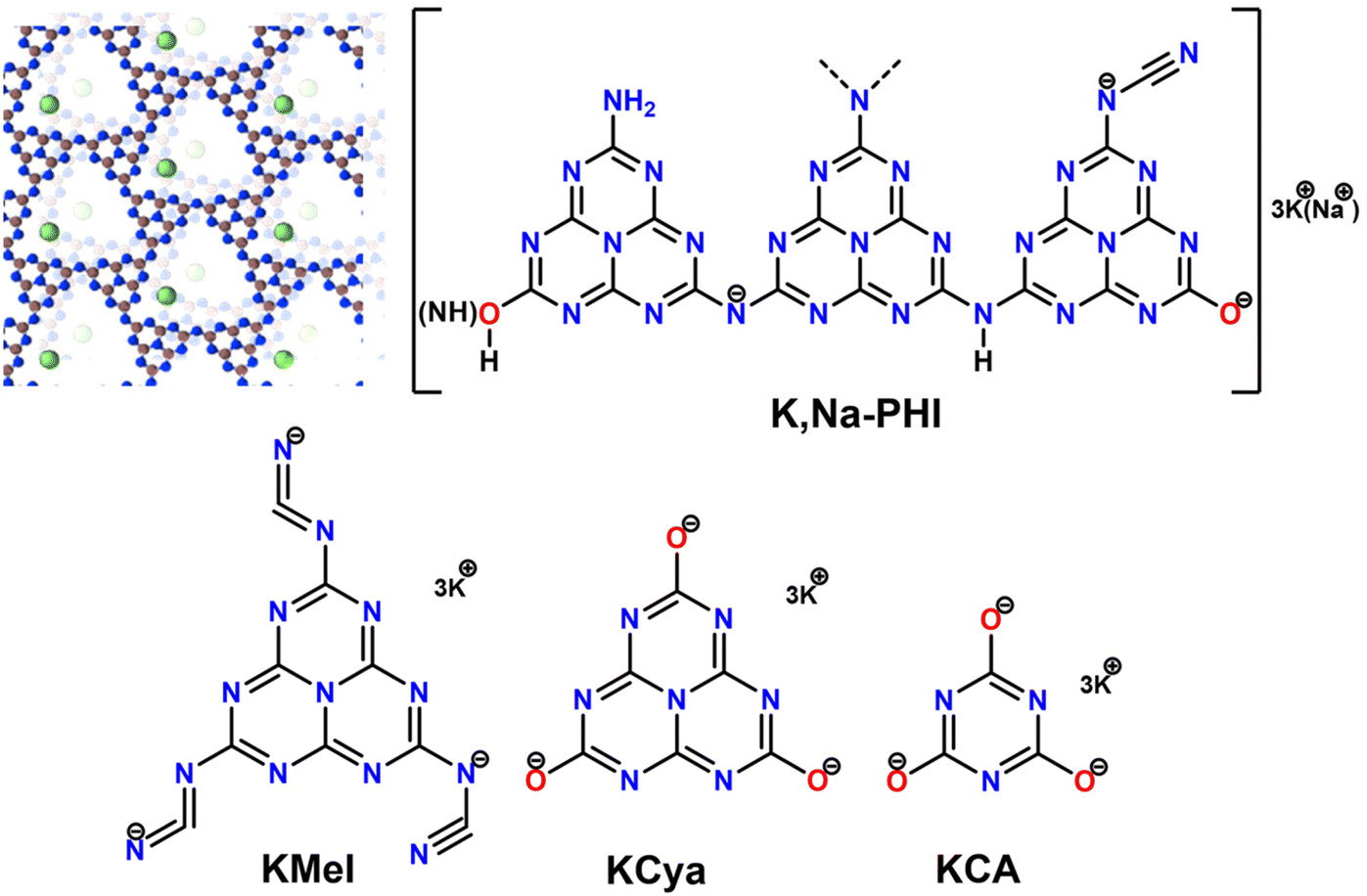
Water-soluble K,Na-PHI has various functional groups attached to the heptazine units. Our structural analyses and theoretical calculations proposed that K,Na-PHI is characterized by a decreased number of terminal NHx-groups, as compared to conventional, water-insoluble PCN, that are exchanged with cyamelurate and melonate moieties.36 Furthermore, an incomplete condensation of the PCN precursor might result in the formation of triazine species within the polymer. Therefore, to obtain a deeper insight into the possible interactions between the Au NPs and K,Na-PHI, we also studied the stabilization effect of potassium melonate (KMel), potassium cyamelurate (KCya), and potassium cyanurate (KCA) at different concentrations (Fig. 2). For all the proposed stabilizers, NaBH4 was used as a reducing agent for the synthesis of AuNPs. Notably, it is well established that the excess NaBH4 can electrostatically stabilize even very small (∼5 nm) colloidal Au NPs without any additional stabilizing agent.50 Therefore, the “naked” (i.e., ligand-free) Au NPs stabilized by excess NaBH4 represent an ideal benchmark for comparative investigations of the effect of the proposed stabilizers on the size, long-term stability, and catalytic activity of Au NPs. This choice is further substantiated by the fact that though also other weakly binding ligands (e.g., citrate) do allow for good accessibility of active sites on the Au nanoparticle surface, they are typically not able to stabilize Au NPs with sizes below 10 nm when using NaBH4 as a reducing agent,51–53 prohibiting thus comparative studies in the ultrafine size regime.
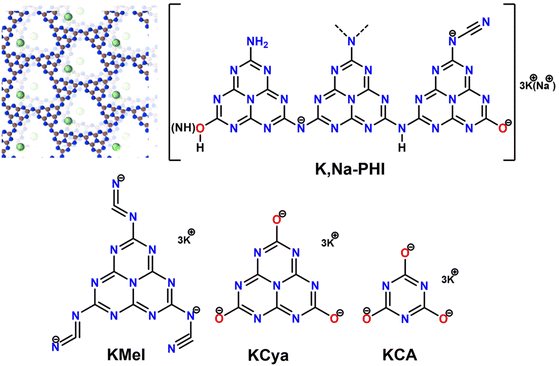 |
| | Fig. 2 Stabilizers used in the comparative analysis of the synthesis and catalytic activity of Au NPs. For K,Na-PHI both the typical 3D ionic carbon nitride structure comprising stacks of 2D heptazine-based networks and K+ and Na+ cations as well as a schematic structure highlighting the presence of charged surface moieties (e.g., cyanamide and cyamelurate) is shown. For spectroscopic characterization (FTIR and NMR) see ESI, Fig. S1.† | |
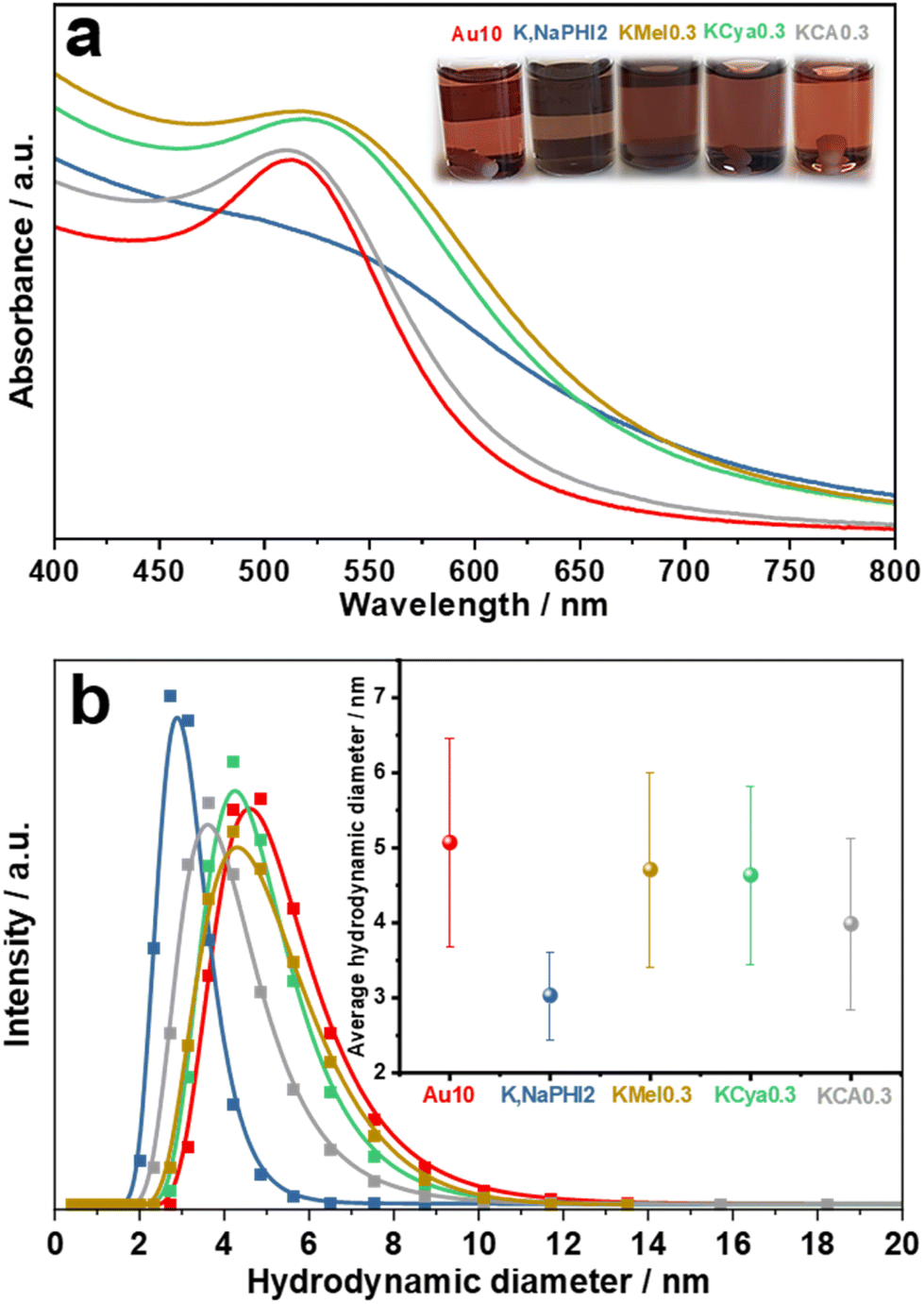
A 10-fold excess of NaBH4 with respect to HAuCl4 was selected as the optimum molar ratio of NaBH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HAuCl4 to produce the benchmark ligand-free Au NPs (denoted as Au10) without agglomeration (for details see ESI 2.1†). For a systematic and realistic comparison, the same optimum NaBH4 concentration was also used to fabricate Au NPs in the presence of K,Na-PHI and its monomeric constituents (KMel, KCya, and KCA). In general, the AuNPs stabilized by different concentrations of NaBH4, K,Na-PHI, KMel, KCya, or KCA exhibit different colours and different electronic absorption properties, which is related to their different sizes and the differently pronounced surface plasmon resonance (SPR) effects (see ESI, Fig. S2 and S3,† and the detailed discussion of the optimization studies in ESI 2.1†). For a representative overview, Fig. 3 compares the UV-vis electronic absorption spectra and optical images as well as the average hydrodynamic diameters of Au10, K,NaPHI2, KMel0.3, KCya0.3, KCA0.3 (the sample designations reflect the molar stabilizer:HAuCl4 ratios; for details see ESI, Table S1†) that were obtained at the optimum concentrations for each stabilizer with respect to the smallest achievable size of stable NPs. As presented in Fig. 3a, the SPR peak of Au NPs stabilized by monomeric heptazine-based stabilizers (K,NaPHI2, KMel0.3, and KCya0.3) is less intense and broader than in the case of triazine-based stabilizers (KCA0.3) and Au10. Moreover, K,NaPHI2 exhibits a considerably broader band with no discernible plasmonic feature at all, suggesting that most Au NPs are below ∼5 nm in size.54 These trends are consistent with the measurements of the hydrodynamic diameter using dynamic light scattering (DLS, Fig. 3b). In particular, the Au NP samples characterized by a relatively sharp and intense SPR peak show larger hydrodynamic diameters, indicating a larger particle size. More importantly, the inset of Fig. 3b demonstrates that K,NaPHI2 possesses a very narrow hydrodynamic diameter distribution (3.0 ± 0.6 nm) as compared to other samples. These observations suggest that, although all the investigated triazine- and heptazine-based compounds can stabilize Au NPs, the size as well as the size distribution of the particles strongly depend on the polymerization degree of the stabilizer.
:HAuCl4 to produce the benchmark ligand-free Au NPs (denoted as Au10) without agglomeration (for details see ESI 2.1†). For a systematic and realistic comparison, the same optimum NaBH4 concentration was also used to fabricate Au NPs in the presence of K,Na-PHI and its monomeric constituents (KMel, KCya, and KCA). In general, the AuNPs stabilized by different concentrations of NaBH4, K,Na-PHI, KMel, KCya, or KCA exhibit different colours and different electronic absorption properties, which is related to their different sizes and the differently pronounced surface plasmon resonance (SPR) effects (see ESI, Fig. S2 and S3,† and the detailed discussion of the optimization studies in ESI 2.1†). For a representative overview, Fig. 3 compares the UV-vis electronic absorption spectra and optical images as well as the average hydrodynamic diameters of Au10, K,NaPHI2, KMel0.3, KCya0.3, KCA0.3 (the sample designations reflect the molar stabilizer:HAuCl4 ratios; for details see ESI, Table S1†) that were obtained at the optimum concentrations for each stabilizer with respect to the smallest achievable size of stable NPs. As presented in Fig. 3a, the SPR peak of Au NPs stabilized by monomeric heptazine-based stabilizers (K,NaPHI2, KMel0.3, and KCya0.3) is less intense and broader than in the case of triazine-based stabilizers (KCA0.3) and Au10. Moreover, K,NaPHI2 exhibits a considerably broader band with no discernible plasmonic feature at all, suggesting that most Au NPs are below ∼5 nm in size.54 These trends are consistent with the measurements of the hydrodynamic diameter using dynamic light scattering (DLS, Fig. 3b). In particular, the Au NP samples characterized by a relatively sharp and intense SPR peak show larger hydrodynamic diameters, indicating a larger particle size. More importantly, the inset of Fig. 3b demonstrates that K,NaPHI2 possesses a very narrow hydrodynamic diameter distribution (3.0 ± 0.6 nm) as compared to other samples. These observations suggest that, although all the investigated triazine- and heptazine-based compounds can stabilize Au NPs, the size as well as the size distribution of the particles strongly depend on the polymerization degree of the stabilizer.
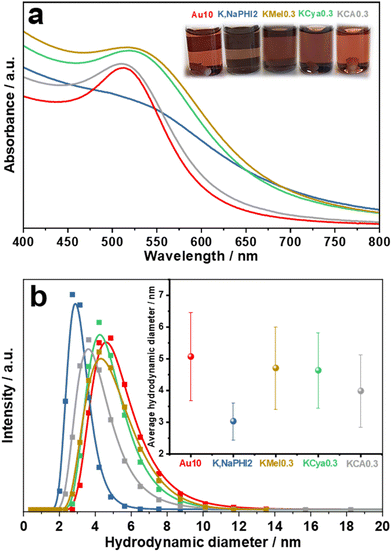 |
| | Fig. 3 Optical properties and hydrodynamic diameter of Au NPs: (a) UV-vis electronic absorption spectra and (b) hydrodynamic diameter data determined from the dynamic light scattering (DLS) of Au10, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3. The inset in (a) shows the optical images of the samples; the inset in (b) shows the average hydrodynamic diameters. The samples were measured after 1 day of their synthesis. The error bars were constructed as +/− the double of standard deviation (95% confidence interval). | |
Zeta potential (ζ-potential) is the electric potential (Galvani potential) at the shear plane (i.e., at the interface separating the mobile fluid from the fluid that remains attached to the NP surface), and represents an important indicator of the stability of colloidal suspensions by electrostatic repulsion forces. ζ-Potential is normally influenced by the nature of the NPs and the dispersion medium.55 We measured the ζ-potential of the prepared suspensions of the Au NPs at the native pH value after dialysis against water to remove the excess NaBH4 hydrolysis products. Au10 displays a negative ζ-potential value of −32 ± 1 mV (pH 6.8) because the surface of AuNPs is surrounded by a layer of BH4− or B(OH)4−. Similarly, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3 show comparable negative ζ-potential values of −32 ± 2 (pH 6.0), −30 ± 1 (pH 5.7), −31 ± 1 (pH 5.5), and −32 ± 1 mV (pH 5.6), respectively, due to the negativity charged surface moieties (e.g., cyanamide and cyamelurate) of heptazine and triazine-based stabilizers (see Fig. 2). The observed values of ζ-potential of ca. −30 mV are in line with the very good stability of NPs.56
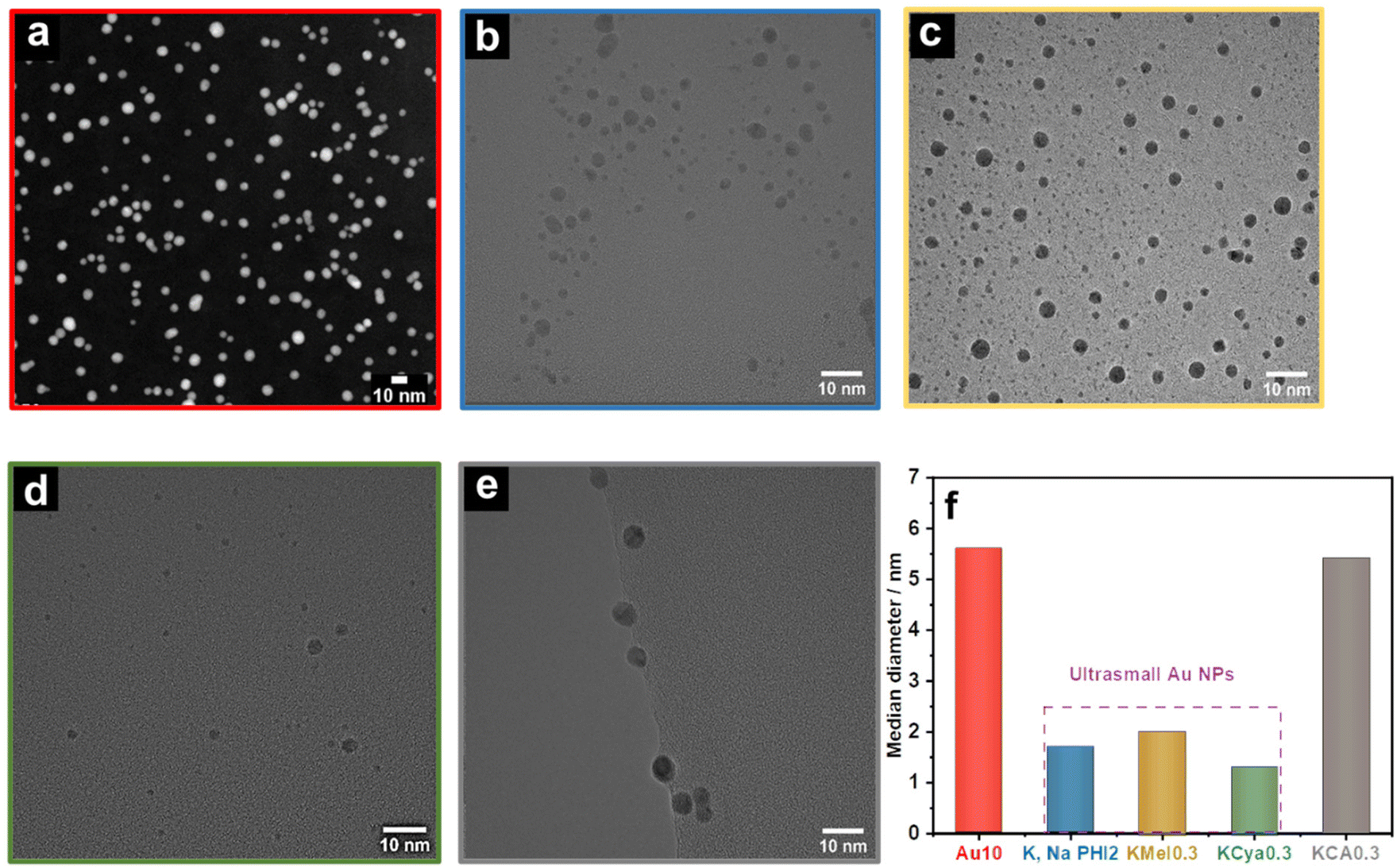
Next, we analyzed the as-prepared samples using scanning transmission electron microscopy (STEM) as well as high-resolution transmission electron microscopy (HRTEM). The (S)TEM images of Au10, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3 are shown in Fig. 4. The produced Au NPs with the proposed stabilizers have similar morphology, but the median diameter and the size distributions are different (Fig. 4f and ESI, Fig. S4†). The HAADF-STEM image of Au10 shows the formation of dispersed Au NPs with a median diameter of around 5.6 nm, which is at least 2.5 times larger than that of K,NaPHI2, KMel0.3, and KCya0.3, but comparable to KCA0.3. Furthermore, the estimated particle size distributions reveal that Au10 and KCA0.3 have a broader size distribution than K,NaPHI2, KMel0.3, and KCya0.3 (ESI, Fig. S4†). Notably, for samples stabilized by monomeric stabilizers (KMel0.3, KCya0.3, and KCA0.3) the observed trends are not fully in line with the trends of hydrodynamic diameters derived from the DLS measurements (compare Fig. 3b and Fig. 4f), which clearly indicates that the hydrodynamic diameter is strongly influenced by the nature of the stabilizer. Most importantly, in contrast to Au NPs stabilized by NaBH4 only (Au10) or by cyanurate (KCA0.3), only K,Na-PHI (K,NaPHI2) and the monomeric heptazine-based stabilizers (KMel0.3, and KCya0.3) provide Au NPs in the ultra-small regime (sub-3 nm). The median diameter of Au NPs stabilized by K,Na-PHI is ∼1.7 nm. This size is larger than the estimated cavity size of pol(heptazine imides) (∼0.78 nm;57 for a schematic representation of heptazine cages see ESI Fig. S5†), indicating that Au NPs are not confined in the heptazine cages.
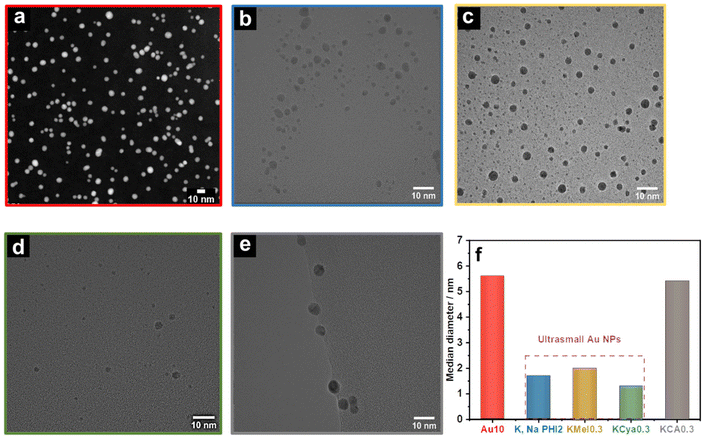 |
| | Fig. 4 Size of NPs. HAADF STEM image of (a) Au10, and HRTEM images of (b) K,NaPHI2, (c) KMel0.3, (d) KCya0.3, and (e) KCA0.3, and their corresponding median diameter (f) derived from the TEM images (for size distribution histograms see ESI, Fig. S4†). | |
X-ray photoelectron spectroscopy (XPS) analyses were performed to identify the oxidation state of gold and the chemical composition of Au10, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3. As shown in Fig. 5, the two distinct peaks centered at 83.6 and 87.3 eV correlate to the spin–orbit doublet of Au 4f7/2 and 4f5/2, respectively, demonstrating the presence of metallic Au in the samples.58 Interestingly, the stabilizers do not significantly affect the binding energy of Au 4f7/2 and Au 4f5/2 electrons (ESI, Table S2†), which implies that the stabilizer moieties interact rather weakly with the surface of the Au NPs, most likely via electrostatic attractions.
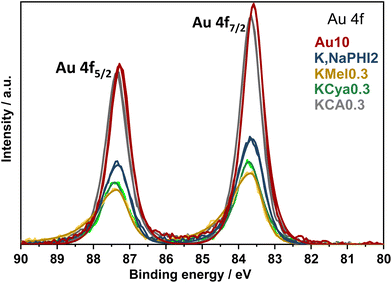 |
| | Fig. 5 XPS analysis. High-resolution Au 4f XP spectra of Au10, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3. Further details regarding the XPS analysis of all samples are provided in ESI, Fig. S6 and S7.† | |
Long-term stability of Au NPs
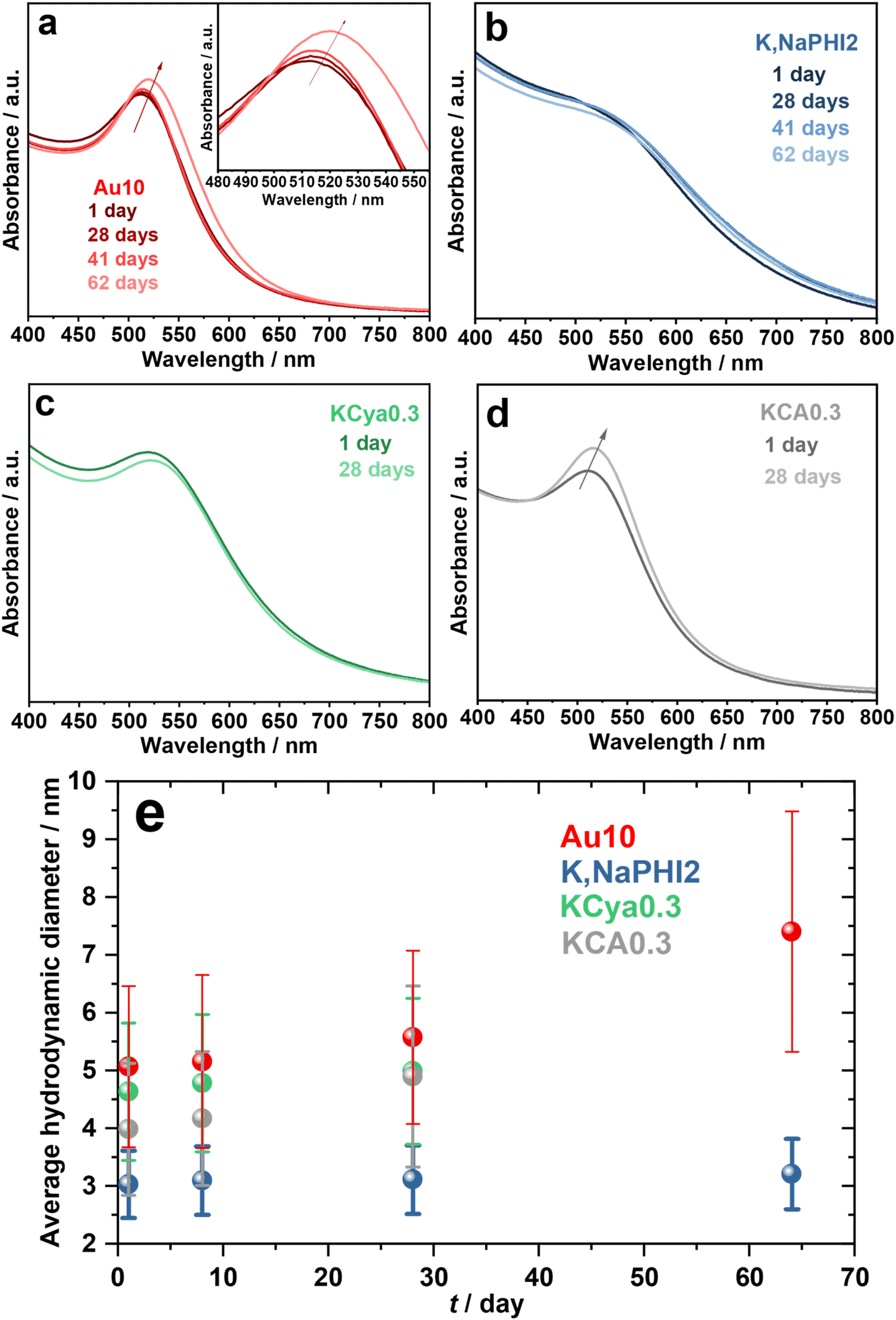
First, we have investigated the long-term stability of the produced Au NPs against agglomeration under ambient storage conditions. Fig. 6 compares the UV-vis spectra and average hydrodynamic diameters of Au10, K,NaPHI2, KCya0.3, and KCA0.3 over time. The SPR peak and hydrodynamic diameter of Au10, K,NaPHI2, and KCya0.3 did not change significantly over 28 days, indicating high stability against agglomeration (Fig. 6a–c and e). In contrast, KCA0.3 tends to agglomerate over time, as demonstrated by a sharper SPR peak and larger hydrodynamic diameter after 28 days. Notably, Au10 possesses a higher tendency to agglomeration after about two months of storage, as indicated by the red-shift of the SPR peak by about 10 nm (inset of Fig. 6a) and the larger hydrodynamic diameter (Fig. 6e). It should be noted that the K,NaPHI2 shows outstanding stability for at least two months, suggesting that K,Na-PHI is a remarkably effective stabilizer that endows the Au NPs with long-term stability.
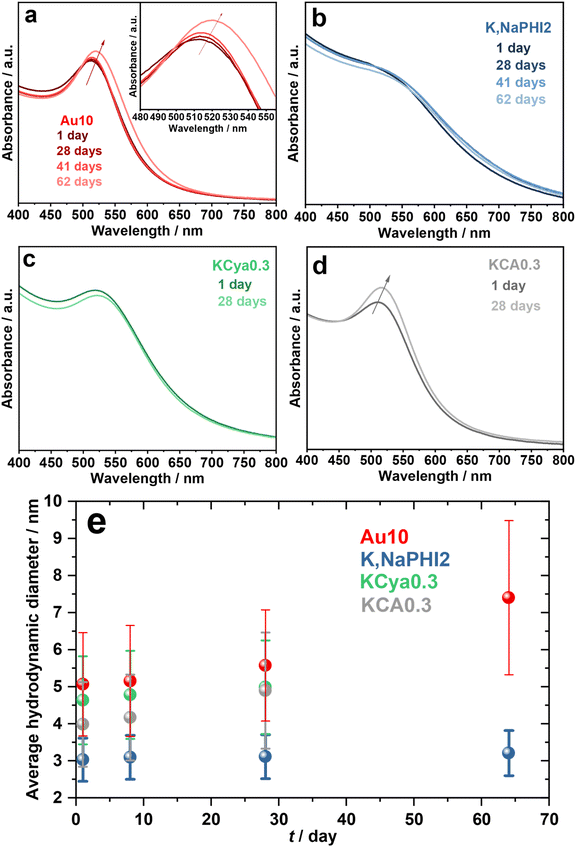 |
| | Fig. 6 Long-term stability. (a–d) UV-vis electronic absorption spectra and (e) average hydrodynamic diameter determined from the DLS results of Au10, K,NaPHI2, KCya0.3, and KCA0.3 as a function of storage time under ambient conditions. | |
Further, we investigated the influence of various concentrations of K,Na-PHI, KCya, and KCA stabilizers on the stability of Au NPs over time (see ESI, Fig. S8a–c and Fig. S9a–f†). Notably, no changes in the UV-vis spectra of the Au NPs stabilized by K,Na-PHI are observed after two months of storage for all the investigated concentrations (ESI, Fig. S8a†). The DLS measurements demonstrate that the hydrodynamic diameter of Au NPs increases as a function of K,Na-PHI concentration (ESI, Fig. S9a and b†), which suggests that the hydrodynamic diameter is strongly influenced not only by the nature of the stabilizer but also by its amount. Moreover, all the K,Na-PHI stabilized Au NPs are highly dispersed and stable for at least two months except for K,NaPHI5, where the hydrodynamic diameter is slightly increasing over time, possibly due to an excessively high amount of the stabilizer. In the case of KCya and KCA-stabilized Au NPs, the stability of the produced Au NPs decreases over time, especially when the molar ratios of the stabilizer with respect to HAuCl4 are ≥10 (see ESI, Fig. S8b, c,† and Fig. 9c–f). The increased tendency to the agglomeration of Au NPs at higher concentrations of KCya and KCA is most likely due to the shorter Debye-length of the electrical double layer as a consequence of the higher ionic strength.59,60
Stability of Au NPs at high ionic strength
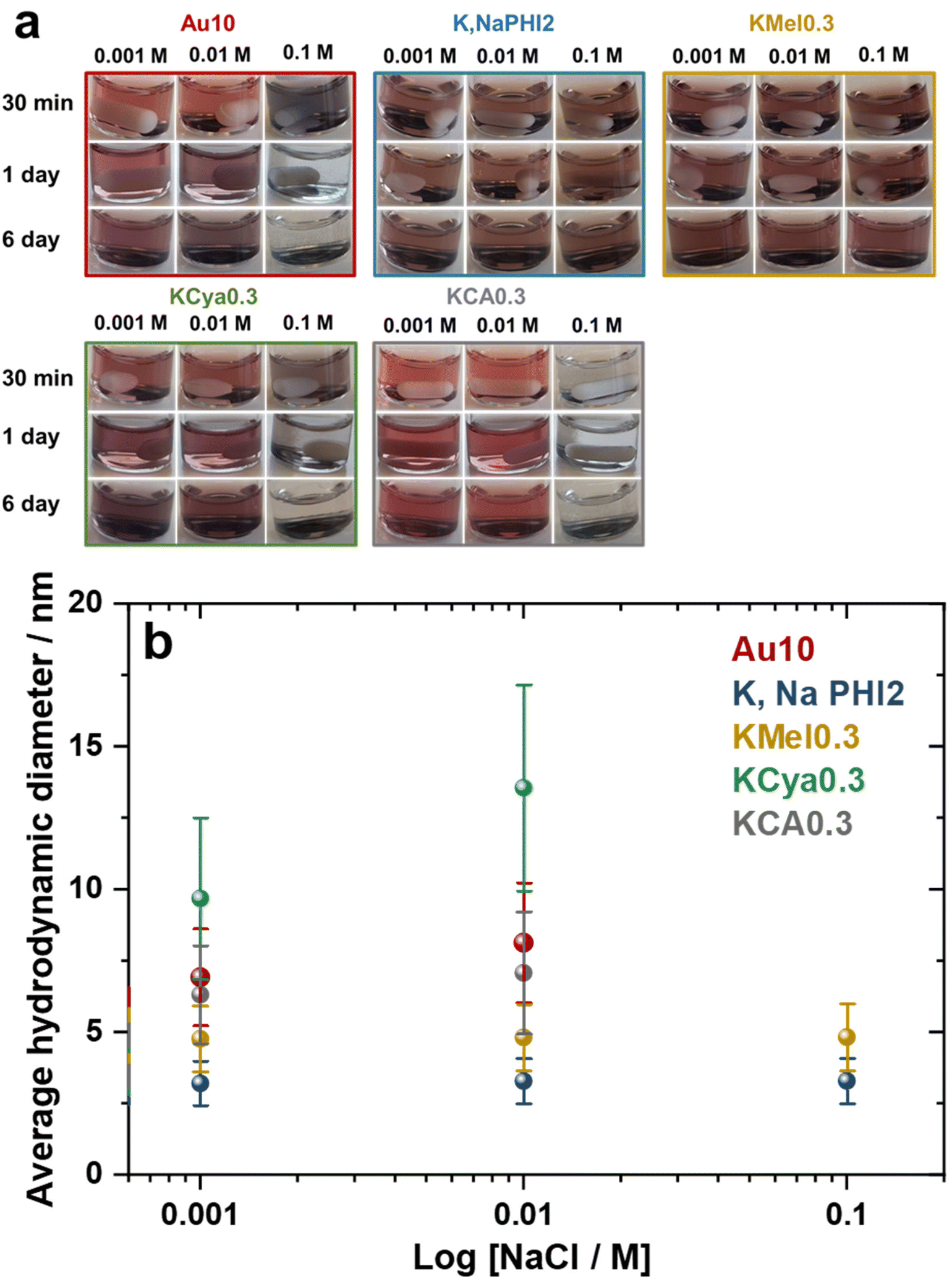
The stability of metal nanoparticles in solutions of high ionic strength is crucial for their application in catalysis and biology.61 Therefore, the effect of ionic strength on the stability of Au10, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3 was investigated by the addition of NaCl at different concentrations (0.001, 0.01, and 0.1 M). The corresponding optical images and average hydrodynamic diameter of the samples are shown in Fig. 7. Remarkably, K,NaPHI2 and KMel0.3 are stable and agglomeration-free for at least 6 days even in the presence of 0.1 M NaCl. In contrast, Au10, KCya0.3, and KCA0.3 tend to agglomerate over time in the presence of 0.001 and 0.01 M NaCl, as indicated by a larger hydrodynamic diameter and darker brown colour after 6 days of storage. Moreover, the optical images of these samples demonstrate a complete agglomeration in the presence of 0.1 M NaCl after less than 24 h (Fig. 7a). Similar behaviour is well-known for Au NPs stabilized by electrostatic repulsion, such as citrate-capped Au NPs, and can be ascribed to the increased screening of the surface charge and compression of the double layer, resulting in a more attractive particle–particle van der Waals interaction and agglomeration.62–64 In this context, the strongly enhanced stability of Au NPs stabilized by K,Na-PHI and KMel can be likely explained by the ability of these stabilizers to act as a hydrated protective layer that is, due to the more pronounced delocalization of charges (see the discussion below), less prone to the double layer compression effects and thus more effectively diminishes the attractive forces between the particles at high ionic strength.59
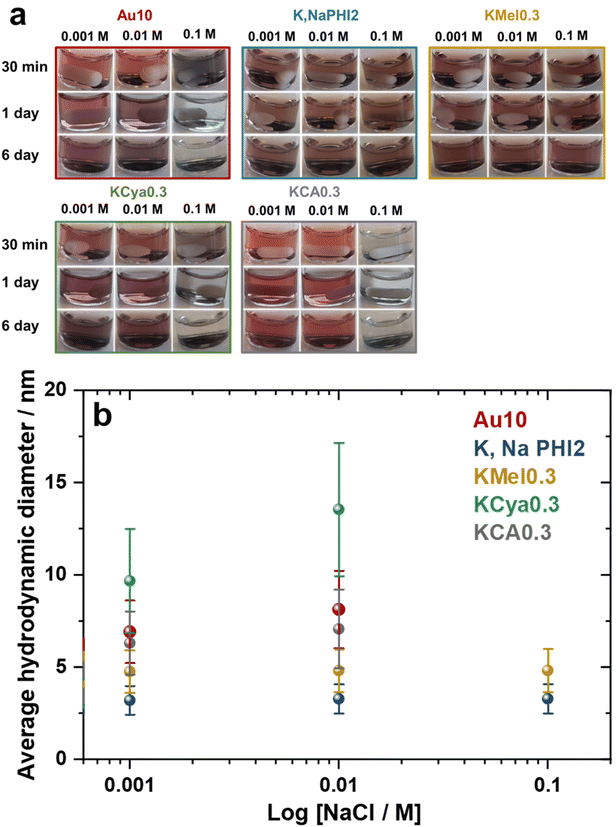 |
| | Fig. 7 Stability at high ionic strength. (a) Optical images of Au10, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3 in the presence of different concentrations of NaCl and (b) the corresponding average hydrodynamic diameter measured 6 days after the addition of NaCl. | |
Stability of Au NPs at elevated temperature
The thermal stability of colloidal nanoparticles is of paramount importance for practical applications.65,66 Therefore, Au10, K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3 were heated at 85 °C for 4 h, 8 h, 24 h, 48 h, and 72 h to determine the thermal stability. Au10 and KCA0.3 exhibit a notable broadening of the plasmon band, along with a slight red shift of the maximum, after heating at 85 °C for 4 h and 48 h, respectively (ESI, Fig. S10a and e†). This indicates that at higher temperatures, the nanoparticles experience growth and aggregation, resulting in larger particle sizes due to nanoparticle coalescence.67 In comparison, the SPR peak of K,NaPHI2, KMel0.3, and KCya0.3 did not change considerably after heating at 85 °C even after 72 h, revealing excellent dispersion stability against heating (ESI, Fig. S10b–d†). These results highlight the beneficial effects of the heptazine-based stabilizers that are assumed to form a hydrated protective layer that effectively prevents nanoparticle coalescence and further growth during heating.
Effect of the reductant-derived impurities on the Au NPs stability
It is known that the stabilization mechanism of pristine Au NPs (Au10) stabilized by the excess of NaBH4 relies on the formation of a layer (incorporating water molecules) of anions (BH4−, B(OH)4−, Cl−) and cations (Na+; in a second layer) formed during the reduction of HAuCl4 by NaBH4 on the surface of Au NPs.50 Hence, to evaluate the true stabilization ability of K,Na-PHI, the prepared suspensions of the Au NPs were dialyzed against water to remove the NaBH4 hydrolysis products. The results unambiguously show that K,Na-PHI can efficiently stabilize ultrafine AuNPs also in the absence of the NaBH4 hydrolysis products; for details see ESI, Fig. S11 and S12,† as well as Tables S3 and S4.†
Stabilization mechanism
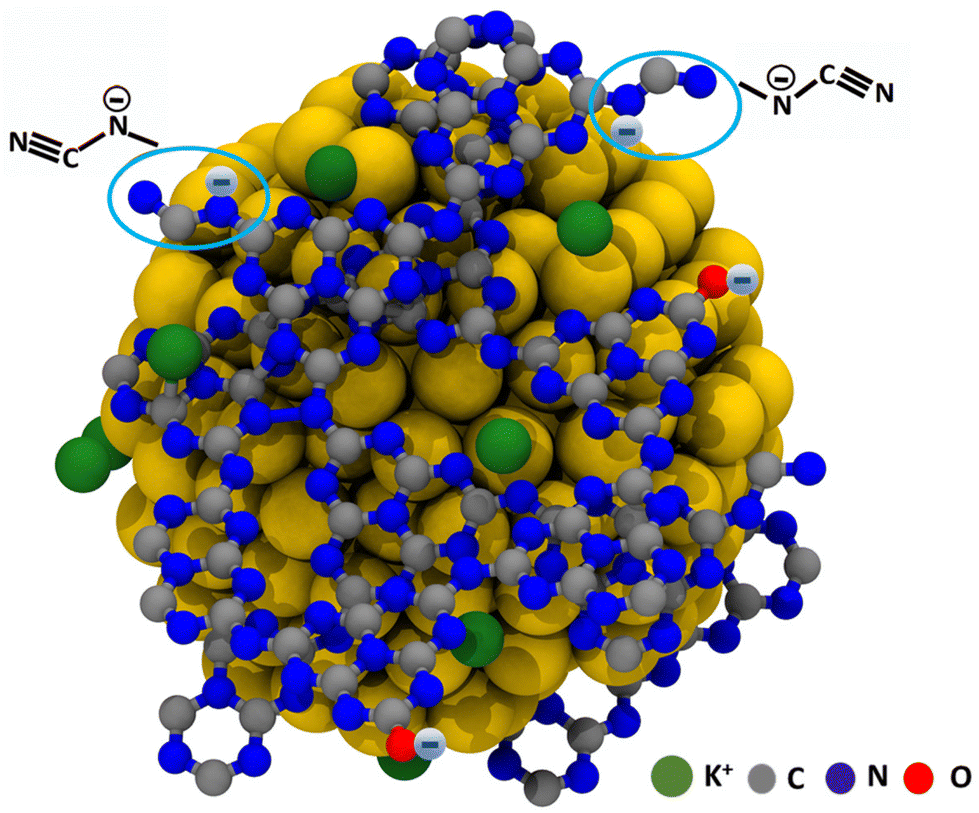
The studied stabilizers K,Na-PHI, KMel, KCya, and KCA are water-soluble ionic compounds bearing negatively charged functional groups (see Fig. 2). In particular, K,Na-PHI consists of a poly(heptazine imide) network incorporating, apart from NH and –NH2 groups or uncondensed NHx-including triazine species, also terminal cyanamide N–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N− and cyamelurate C–O− functional groups. We have reported that the dissolved K,Na-PHI has a zeta potential of −31 mV at a pH of 7.5,36 confirming the negatively charged surface under our reaction conditions. The presence of the negatively charged functional groups on the stabilizers allows for electrostatic stabilization and solubilization of Au NPs in water. The stabilization of the NPs by repulsive electrostatic interactions originates from a diffuse double layer at the surface of NPs coated with charged molecules. Presumably, K,Na-PHI, KMel, KCya, and KCA establish a hydrophilic protective layer around the growing metal nuclei that is permeable to tetrachloroaurate ions (to allow for self-limiting growth) while preventing the diffusional coupling of the particles. A representative model for the electrostatic stabilization of Au NPs by K, Na-PHI is shown in Fig. 8. These considerations are mainly based on the TEM and XPS results revealing that (i) the particle size of Au NPs stabilized by K,Na-PHI is larger than the estimated cavity size of heptazines (ESI, Fig. S5†), excluding the confinement of Au NPs into the heptazine cages, and (ii) the presence of K,Na-PHI does not affect the binding energy of Au 4f7/2 and Au 4f5/2 electrons of the Au(0) species, revealing that the stabilizer functional groups only weakly interact with the surface of the Au NPs. Although the stabilization of the Au NPs in water is conditioned by the charged functional groups, the size of the obtained Au NPs depends mostly on the employed stabilizer, i.e., on the monomeric unit and the polymerization degree. Most notably, as compared to the triazine-based stabilizer (KCA), the stabilizers containing heptazine units (K,Na-PHI, KMel, KCya) lead to smaller Au NPs which also exhibit better stability (except for cyanurate) in high ionic strength electrolytes.
N− and cyamelurate C–O− functional groups. We have reported that the dissolved K,Na-PHI has a zeta potential of −31 mV at a pH of 7.5,36 confirming the negatively charged surface under our reaction conditions. The presence of the negatively charged functional groups on the stabilizers allows for electrostatic stabilization and solubilization of Au NPs in water. The stabilization of the NPs by repulsive electrostatic interactions originates from a diffuse double layer at the surface of NPs coated with charged molecules. Presumably, K,Na-PHI, KMel, KCya, and KCA establish a hydrophilic protective layer around the growing metal nuclei that is permeable to tetrachloroaurate ions (to allow for self-limiting growth) while preventing the diffusional coupling of the particles. A representative model for the electrostatic stabilization of Au NPs by K, Na-PHI is shown in Fig. 8. These considerations are mainly based on the TEM and XPS results revealing that (i) the particle size of Au NPs stabilized by K,Na-PHI is larger than the estimated cavity size of heptazines (ESI, Fig. S5†), excluding the confinement of Au NPs into the heptazine cages, and (ii) the presence of K,Na-PHI does not affect the binding energy of Au 4f7/2 and Au 4f5/2 electrons of the Au(0) species, revealing that the stabilizer functional groups only weakly interact with the surface of the Au NPs. Although the stabilization of the Au NPs in water is conditioned by the charged functional groups, the size of the obtained Au NPs depends mostly on the employed stabilizer, i.e., on the monomeric unit and the polymerization degree. Most notably, as compared to the triazine-based stabilizer (KCA), the stabilizers containing heptazine units (K,Na-PHI, KMel, KCya) lead to smaller Au NPs which also exhibit better stability (except for cyanurate) in high ionic strength electrolytes.
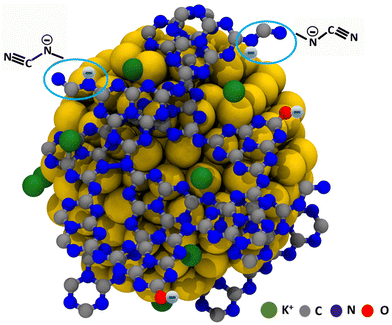 |
| | Fig. 8 Schematic representation of the stabilization of Au NPs by K,Na-PHI. | |
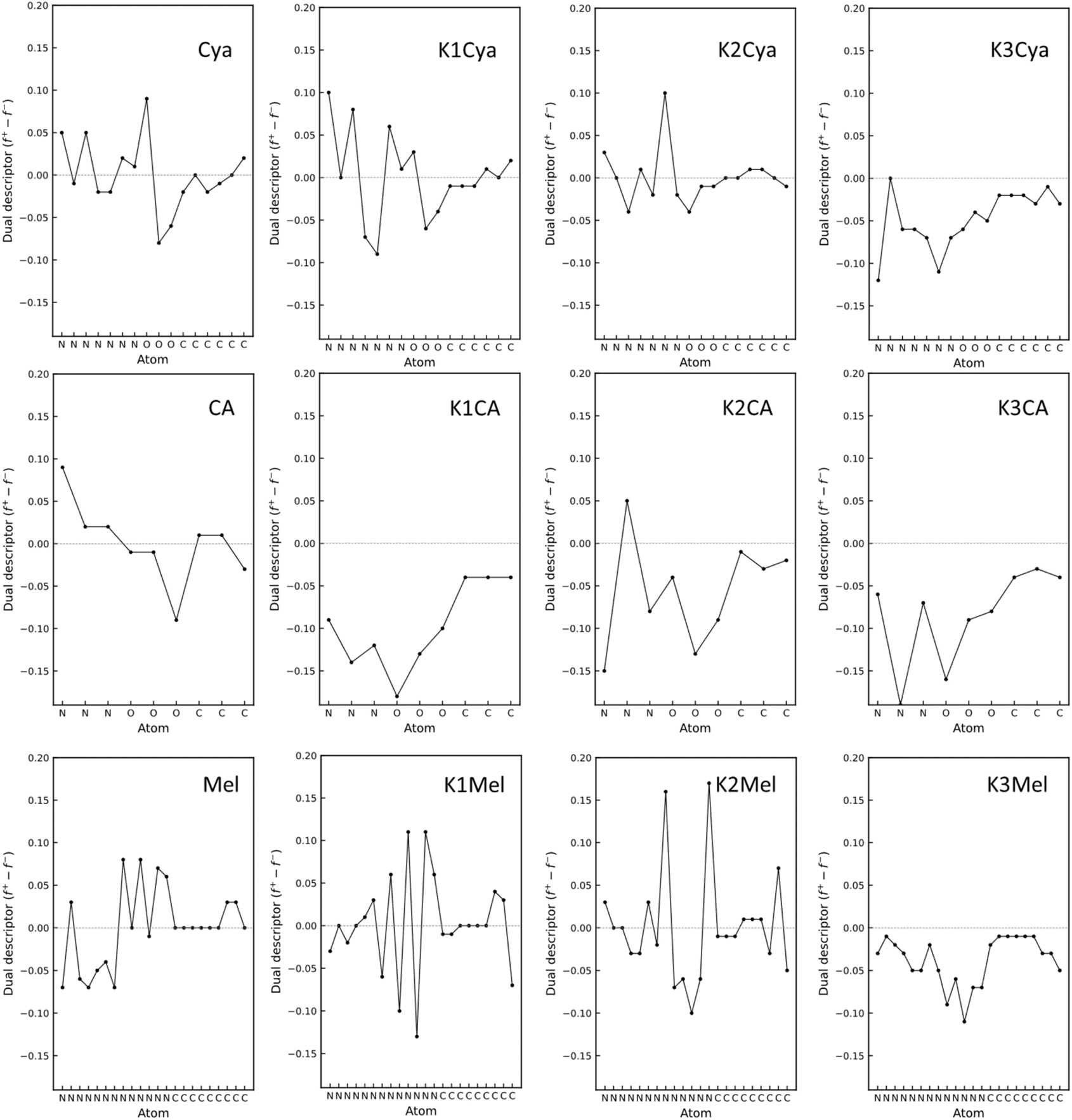
We hypothesized that these beneficial effects of larger heptazine-based stabilizers might be due to the larger charge delocalization in the aromatic system, resulting in more effective electrostatic stabilization of Au NPs which are typically positively charged.50 To obtain further insight into the stabilization mechanism, we performed density functional theory (DFT) calculations to find the optimized geometry of KCA, KCya, and KMel structures and to investigate the impact of the conjugation size, negatively charged cyamelurate or cyanamide functional groups, and the nature and number of counterions (Fig. 9). Particularly, we modelled the structures with a different number of counterions based on the assumption that the counterions initially stabilize the carbon nitride species and may detach after the stabilization of Au NPs. In addition, we performed a Fukui function analysis of the carbon nitride species to gain a deeper understanding of the stabilization mechanism of Au NPs.68–71 This analysis provides an estimate of the expected interaction between the stabilizer and Au-NP without the need to directly include the extended Au-NP (having a broad size and shape distribution) into the simulation. More specifically, the electron density of the f− Fukui function of the carbon nitride represents the electron density for the electrophilic attack from the Au NPs (Fig. 9). The optimized geometries of the carbon nitride species functionalized by the oxygen-containing cyamelurate groups (CA and Cya) reveal a structural out-of-plane deformation near the cyamelurate groups when the counterions are absent. However, in the presence of the counterions, the structures become more planar. In this context, it should be mentioned that the out-of-plane structural distortion translates into a breaking of the π-electron conjugation, which effectively reduces the size of electron delocalization. On the other hand, the cyanamide (NCN−)-functionalized species (melonate) tends to retain an in-plane structure. The presence of potassium counterions renders the structure even more planar, resulting in an extension of the electron conjugation as compared to the situation without the counterions. Therefore, the primary role of the counterions (potassium) can be understood as facilitating the flattening of the structures with functional groups. Specifically, the electron densities of f− Fukui functions reveal that the presence of counterions causing the structural flattening leads to an alignment of the directional orbitals from the previously distorted structure into perpendicular π-orbitals within the flat structure. This orbital alignment translates into an extension of the delocalized electron cloud to the functional groups, facilitating thus the stabilization of the positive surface of Au NPs. The effectiveness of the extended π orbitals in stabilizing Au NPs is supported by the experimental observation that the stabilization (in terms of size and long-term stability) of Au NPs by the structurally more distorted KCA is similar to the case of stabilization solely by NaBH4, while the flatter carbon nitride species, such as KCya, K,Na-PHI, and KMel, can effectively stabilize Au NPs smaller than 3 nm in size (see Fig. 4f and the histograms in ESI, Fig. S4†). In other words, these results suggest that the more effective stabilization of Au NPs by KCya, K,Na-PHI, and KMel as compared to KCA can be attributed to the availability of a delocalized electron cloud with a size equivalent to or larger than the heptazine moiety. In addition, from the analysis of the condensed Fukui function (for details see section 1.5. in ESI†), also known as the dual descriptor, we conclude that this phenomenon is a result of the synergistic effects of the functional groups and the counterions (Fig. 10). The condensed Fukui function describes the local reactivity per atom, and its value is negative at sites prone to the electrophilic attack from an electrophile, and positive at sites preferable for the nucleophilic attack.68–73 Notably, the condensed Fukui functions for all atoms in structures with the same number of K+ counterions as the number of functional groups yield all negative values, which corroborates their ability to effectively stabilize the electrophilic Au NPs (Fig. 10). In contrast, the pristine (non-ionic, non-functionalized) heptazine and triazine moieties show positive values of the condensed Fukui functions despite their positive net charge (Fig. S13 and S14 in ESI†), which highlights the significance of the presence of the counterion-balanced cyamelurate and cyanamide functional groups in ionic carbon nitride for the effective stabilization of Au NPs.
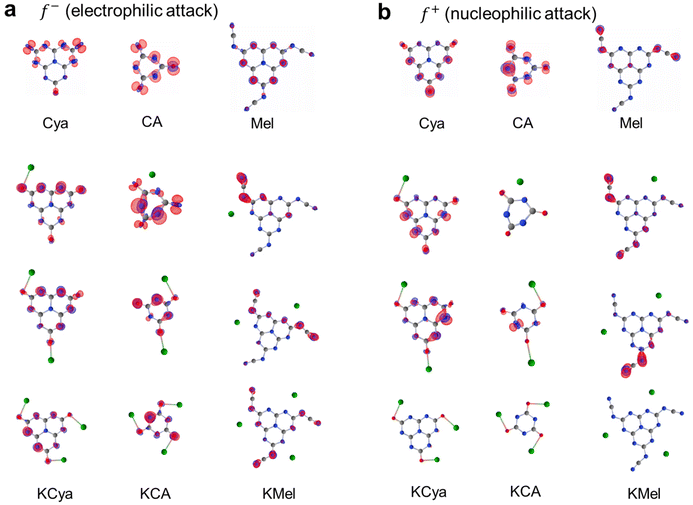 |
| | Fig. 9 Theoretical calculations. The electron density from Fukui function (a) negative f− and (b) positive f+. The electron density shows the potential frontier reactive site for the respective carbon nitride species. The isosurface value of the electron density is fixed to 0.005787 e Å−3 over the structures. The structure is illustrated without the cation (top) and with the different numbers (1–3) of potassium cations (bottom). The blue, the gray, and the green balls stand for N, C, and K atoms, respectively. | |
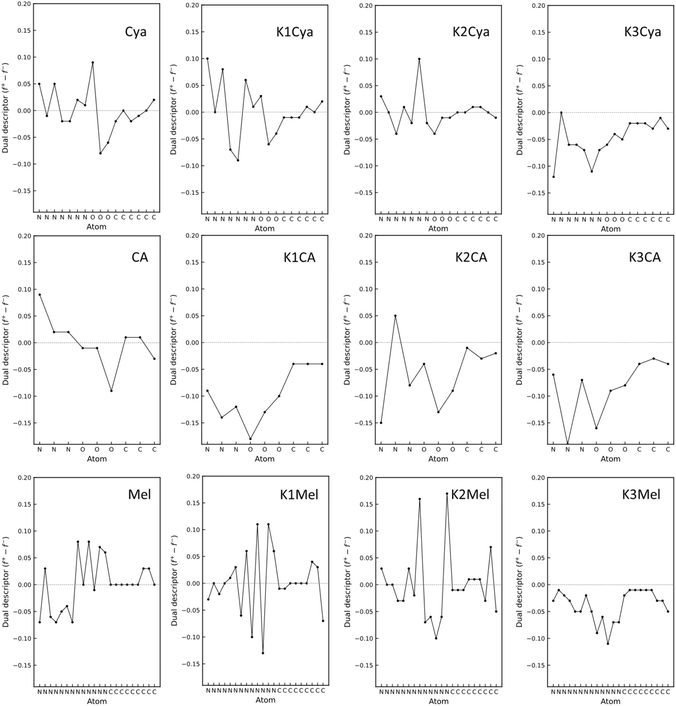 |
| | Fig. 10 Condensed Fukui functions. The dual descriptor of the carbon nitride species (Cya, CA, and Mel) in the presence of the different numbers of potassium cations. The negative value corresponds to the electrophilic attack site and the positive value corresponds to the nucleophilic attack site. The values for potassium ions have been excluded in order to emphasize the variations in other atoms. All values, including those for potassium ions, are shown in Table S5 in ESI.† | |
Catalytic performance
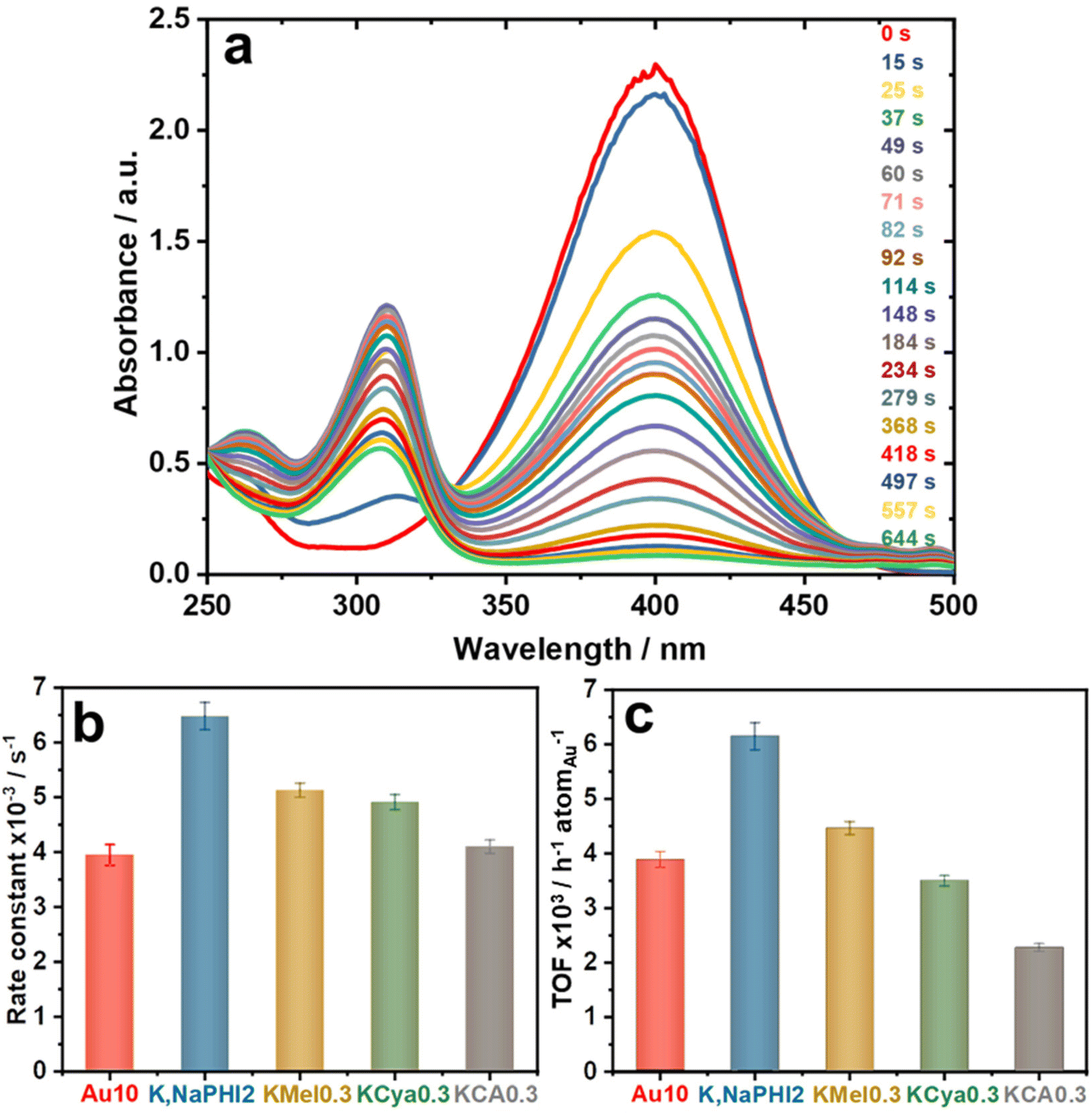
It is known that strongly interacting stabilizing agents might block the utilization and accessibility of the active sites on the surface of catalytic metal NPs, resulting in catalyst deactivation/poisoning.22,35 However, the stabilizers that electrostatically stabilize Au NPs typically do not interfere significantly with the mass transport processes towards the catalytic surface, thus allowing for high diffusion rates of the reactants and products without completely blocking the active sites.22,23 As a proof of concept, the catalytic activity of K,NaPHI2, KMel0.3, KCya0.3, and KCA0.3 was evaluated in the selective reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) by NaBH4, using the “naked” Au NPs electrostatically stabilized by excess NaBH4 (Au10) as a benchmark. The catalytic activity of all investigated samples was evaluated one day after their synthesis. The catalytic hydrogenation of nitroarenes has been extensively employed as a model reaction to evaluate the activity of Au NP catalysts, and it has the advantage of easy monitoring by UV-vis spectroscopy.16,22,23,58 As the reaction proceeds after the addition of the catalyst, the absorption peak at 400 nm decreases, while the absorption peak at 300 nm is increasing, showing the conversion of 4-NP to 4-AP (Fig. 11a). The time dependence of the relative absorbance (At/A0%) of the 4-nitrophenolate peak at 400 nm in the presence of Au10, K,NaPHI2, KMel0.3, KCya0.3, or KCA0.3 is shown in ESI, Fig. S15a,† where At and A0 represent the absorbance at the reaction time t and at time t = 0, respectively. An important feature of the reduction of 4-NP catalyzed by ligand-stabilized nanoparticles is the induction period, an initial time interval needed until the reaction takes place.23 The induction period is related to either the formation of a diffusion pathway through the ligand shell on the NP surface or the restructuring of the NP surface induced by the adsorbed reactant molecules.22,23,74,75 Interestingly, very short (∼2–20 s) induction times are observed for all investigated materials except for KCA0.3 (∼45 s; Fig. S15a†), which indicates that especially the heptazine-based stabilizing agents can both effectively stabilize the Au NPs and allow for fast diffusion of reactants and products needed for effective surface catalysis.
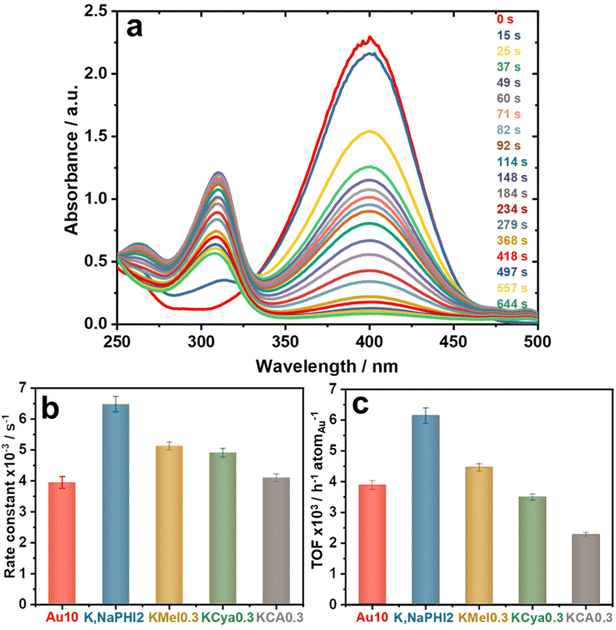 |
| | Fig. 11 (a) UV-vis electronic absorption spectra recorded during the reduction of 4-NP in the presence of K,NaPHI2 catalyst and the calculated (b) rate constant and (c) TOF values for Au10, K,Na PHI2, KMel0.3, KCya0.3, and KCA0.3. Experimental conditions: 0.1 mL NaBH4 (2.16 mol L−1), 0.3 mL 4-NP (1.2 × 10−3 mol L−1), and 2.5 mL distilled water were mixed into a cuvette at ambient conditions. Then, the UV-Vis spectrum was recorded, followed by the addition of 0.025 mL of AuNP catalyst (1.14 mg L−1) and shaking quickly the cuvette before recording the subsequent spectra. | |
The rate constants (Fig. 11b) were determined by fitting the data from Fig. S15a† to a pseudo-first-order kinetic model concerning the concentration of 4-NP (Fig. S15b†). The turnover frequencies (TOF) for investigated samples (Fig. 11c) were calculated using the linear range of substrate conversion at the beginning of the reaction, taking the end of the induction period as time 0. The TOF value is given with respect to one atom of Au, hence its unit is h−1 atomAu−1. The benchmark sample Au10 has a TOF value of 3888 h−1 atomAu−1, which is comparable to the value reported in the literature.50 Notably, the K,NaPHI2 catalyst exhibits a remarkably high TOF value of 6149 h−1 atomAu−1, which is superior to TOFs of the benchmark Au10 (3888 h−1 atomAu−1), KMel0.3 (4464 h−1 atomAu−1), KCya0.3 (3498 h−1 atomAu−1), and KCA0.3 (2276 h−1 atomAu−1), as shown in Fig. 11c. The sample KCA0.3, comprising a triazine-based stabilizer, possesses the lowest TOF value, which is likely because KCA leads to larger Au NPs. Moreover, among the proposed stabilizers, KCA has the highest density of negatively charged groups where all carbons bear one negatively charged O− group. Therefore, the AuNPs surface is likely crowded by the stabilizing molecules, partially blocking the surface catalytic activity. The outstanding catalytic performance of the Au NPs stabilized by K,Na-PHI can be, in our view, attributed to two main effects. Firstly, K,Na-PHI favours the formation of ultrasmall Au NPs, allowing for higher specific surface area and more optimal utilization of Au atoms. Secondly, the flexible and extended framework structure of the polymeric K,Na-PHI enables both effective stabilization and – at the same time – facilitates the diffusion of reactants and products to and from the surface of gold NPs. In this context, it should be noted that NaBH4 at high concentrations is known to increase significantly the ionic strength and thus to induce aggregation of Au NPs.76 Indeed, under our reaction conditions, both Au10 and K,NaPHI2 are prone to aggregation, as apparent by a blue shift of the SPR band (see ESI, Fig. S16†), which can be ascribed to a high concentration of NaBH4 in the reaction solution as NaBH4 is used in a large excess (∼600 times the concentration of 4-NP, i.e. ∼0.1 M). Higher stability against aggregation is expected to be achievable in catalytic reactions operating under optimized conditions with alternative reagents and/or at lower ionic strengths.
Conclusions
In this work, we demonstrate for the first time that water-soluble ionic (K+ and Na+ containing) polymeric carbon nitride (K,Na-PHI) can act as a facile, scalable, and highly effective stabilizer in the synthesis of highly catalytically active ultrafine (<3 nm) gold nanoparticles. In the selective reduction of 4-nitrophenol to 4-aminophenol by NaBH4 employed as a model catalytic reaction, the K,Na-PHI-stabilized Au NPs not only outperform Au NPs stabilized by monomeric structural units of K,Na-PHI (cyanurate, melonate, cyamelurate), but their activity is even superior (TOF higher by the factor of ∼1.6) to that of the benchmark “naked” (i.e., ligand-free) Au NPs electrostatically stabilized by excess NaBH4. These results suggest that K,Na-PHI can provide an effective electrostatic stabilization against the aggregation of Au NPs, without blocking the diffusion of the reactants and products to and from the active sites at the Au catalyst surface. A truly outstanding feature of our K,Na-PHI-stabilized Au NPs is their excellent stability both under ambient conditions (over at least two months), as well as under high ionic strength conditions (up to 0.1 M NaCl solutions) and at elevated temperatures (85 °C for at least 72 hours). Based on our experimental evidence and theoretical calculations, we propose that the strongly enhanced stability of Au NPs stabilized by K,Na-PHI can be explained by the ability of K,Na-PHI to act as a hydrated protective layer that is, due to the more pronounced delocalization of charges, less prone to the double layer compression effects and thus more effectively diminishes the attractive forces between the nanoparticles. Moreover, a detailed comparative study using different structural units of K,Na-PHI (cyanurate, melonate, cyamelurate) shows that in particular the presence of functionalized heptazine moieties allowing for better charge delocalization is crucial for the synthesis and more effective stabilization of small Au NPs. This is corroborated by theoretical DFT calculations of Fukui functions which reveal the beneficial effect of the counterion-balanced cyamelurate and cyanamide functional groups in the ionic carbon nitride on the orbital alignment and electron delocalization, resulting in more effective stabilization of Au NPs. Thus, this work establishes the ionic carbon nitrides (PHI) as alternative capping agents for nanocatalysts that enable effective stabilization without compromising surface catalysis. More generally, it opens up a route for further developments in utilizing PHI-based stabilizers for the synthesis of high-performance ultrafine nanoparticles, especially for applications in which both the stability and the effective transport of species between the interfacial region of nanoparticles and the environment is mandatory.
Author contributions
M.E., L.K., I.K., T.J., R.B. and D.M. conceived and supervised the project, J.L., M.E. and I.K. synthesised the materials, characterized them and performed catalytic experiments. C.N. and A.T. performed XPS investigation of the materials and analysed the results. R.L. and U.K. performed TEM-EDX analyses and processed the data. C.I. and T.J. performed the theoretical calculations. M.E. wrote the first draft of the manuscript, and M.E., J.L., C.I., I.K. and R.B. finalized the manuscript with inputs from all authors.
Data availability
All datasets related to this work are available from the repository: https://doi.org/10.5281/zenodo.8130796.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project No. 364549901 – TRR 234 CataLight (Projects B6, B7, B10, Z2) as well as JA 1972/27-1 and BE 5102/5-1 (Project No. 428764269). Computational resources were provided by the state of Baden–Württemberg through bwHPC and the German Science Foundation (DFG) under Grant No. INST 40/467-1 FUGG. C. N. and A. T. acknowledge financial support of the DFG through a research infrastructure grant INST 275/257-1 FUGG. I. K. acknowledges support by Spanish MCINN (PID2020-113558RB-C41), Gobierno del Principado de Asturias (IDI-2021-000048), and the Alexander von Humboldt Foundation through the Humboldt Research Fellowship.
References
- N. Wang, Q. Sun and J. Yu, Adv. Mater., 2019, 31, 1803966 CrossRef PubMed.
- X. Yang, J.-K. Sun, M. Kitta, H. Pang and Q. Xu, Nat. Catal., 2018, 1, 214–220 CrossRef CAS.
- Q. L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- L. M. Liz-Marzán, Langmuir, 2006, 22, 32–41 CrossRef PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- V. Karthick, L. Kumar Shrestha, V. G. Kumar, P. Pranjali, D. Kumar, A. Pal and K. Ariga, Nanoscale, 2022, 14, 10630–10647 RSC.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
- O. J. H. Chai, Z. Liu, T. Chen and J. Xie, Nanoscale, 2019, 11, 20437–20448 RSC.
- H. T. K. Duong, A. Abdibastami, L. Gloag, L. Barrera, J. J. Gooding and R. D. Tilley, Nanoscale, 2022, 14, 13890–13914 RSC.
- M. E. King, M. V. Fonseca Guzman and M. B. Ross, Nanoscale, 2022, 14, 602–611 RSC.
- C. Spedalieri and J. Kneipp, Nanoscale, 2022, 14, 5314–5328 RSC.
- C. Adler, D. Mitoraj, I. Krivtsov and R. Beranek, J. Chem. Phys., 2020, 152, 244702 CrossRef CAS PubMed.
- J. Yang, M. Yuan, D. Xu, H. Zhao, Y. Zhu, M. Fan, F. Zhang and Z. Dong, J. Mater. Chem. A, 2018, 6, 18242–18251 RSC.
- R. R. Nasaruddin, T. Chen, N. Yan and J. Xie, Coord. Chem. Rev., 2018, 368, 60–79 CrossRef CAS.
- L.-W. Guo, P.-P. Du, X.-P. Fu, C. Ma, J. Zeng, R. Si, Y.-Y. Huang, C.-J. Jia, Y.-W. Zhang and C.-H. Yan, Nat. Commun., 2016, 7, 13481 CrossRef CAS PubMed.
- X. Gou, T. Liu, Y. Wang and Y. Han, Angew. Chem., Int. Ed., 2020, 59, 16683–16689 CrossRef CAS PubMed.
- H. Yang, Z. Yu, S. Li, Q. Zhang, J. Jin and J. Ma, J. Catal., 2017, 353, 256–264 CrossRef CAS.
- Y. Jiang, X. Zhang, X. Dai, W. Zhang, Q. Sheng, H. Zhuo, Y. Xiao and H. Wang, Nano Res., 2017, 10, 876–889 CrossRef CAS.
- B. Weng, K.-Q. Lu, Z. Tang, H. M. Chen and Y.-J. Xu, Nat. Commun., 2018, 9, 1543 CrossRef PubMed.
- A. Mondal and N. R. Jana, ACS Catal., 2014, 4, 593–599 CrossRef CAS.
- Y. Wang, J. He, C. Liu, W. H. Chong and H. Chen, Angew. Chem., Int. Ed., 2015, 54, 2022–2051 CrossRef CAS PubMed.
- S. M. Ansar and C. L. Kitchens, ACS Catal., 2016, 6, 5553–5560 CrossRef CAS.
- R. D. Neal, R. A. Hughes, P. Sapkota, S. Ptasinska and S. Neretina, ACS Catal., 2020, 10, 10040–10050 CrossRef CAS.
- A. M. Alam and Y. S. Shon, ACS Appl. Nano Mater., 2021, 4, 3294–3318 CrossRef CAS PubMed.
- Q. L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- J. A. Singh, S. H. Overbury, N. J. Dudney, M. Li and G. M. Veith, ACS Catal., 2012, 2, 1138–1146 CrossRef CAS.
- G. González-Rubio, J. Mosquera, V. Kumar, A. Pedrazo-Tardajos, P. Llombart, D. M. Solís, I. Lobato, E. G. Noya, A. Guerrero-Martínez, J. M. Taboada, F. Obelleiro, L. G. MacDowell, S. Bals and L. M. Liz-Marzán, Science, 2020, 368, 1472–1477 CrossRef PubMed.
- J. Feng, C. Gao and Y. Yin, Nanoscale, 2018, 10, 20492–20504 RSC.
- L. E. González García, N. Ninan, J. Simon, R. Madathiparambil Visalakshan, R. Bright, S. K. Wahono, K. Ostrikov, V. Mailänder, K. Landfester, N. Goswami and K. Vasilev, Nanoscale, 2021, 13, 19936–19945 RSC.
- H. R. Moon, D.-W. Lim and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807–1824 RSC.
- W. Yuan and C. M. Li, Langmuir, 2009, 25, 7578–7585 CrossRef CAS PubMed.
- W. Yuan, Z. Lu and C. M. Li, J. Mater. Chem., 2011, 21, 5148 RSC.
- S. Chakraborty, S. M. Ansar, J. G. Stroud and C. L. Kitchens, J. Phys. Chem. C, 2018, 122, 7749–7758 CrossRef CAS.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262 RSC.
- I. A. Safo, C. Dosche and M. Özaslan, ChemPhysChem, 2019, 20, 3010–3023 CrossRef CAS PubMed.
- I. Krivtsov, D. Mitoraj, C. Adler, M. Ilkaeva, M. Sardo, L. Mafra, C. Neumann, A. Turchanin, C. Li, B. Dietzek, R. Leiter, J. Biskupek, U. Kaiser, C. Im, B. Kirchhoff, T. Jacob and R. Beranek, Angew. Chem., Int. Ed., 2020, 59, 487–495 CrossRef CAS PubMed.
- A. Savateev and M. Antonietti, ChemCatChem, 2019, 11, 6166–6176 CrossRef CAS.
- H. Schlomberg, J. Kröger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. v. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. v. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- V. W. Lau and B. v. Lotsch, Adv. Energy Mater., 2022, 12, 2101078 CrossRef CAS.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- C. Im, B. Kirchhoff, I. Krivtsov, D. Mitoraj, R. Beranek and T. Jacob, Chem. Mater., 2023, 35, 1547–1559 CrossRef CAS.
- D. Zheng, J. Zhou, Z. Fang, T. Heil, A. Savateev, Y. Zhang, M. Antonietti, G. Zhang and X. Wang, J. Mater. Chem. A, 2021, 9, 27370–27379 RSC.
- C. Li, E. Hofmeister, I. Krivtsov, D. Mitoraj, C. Adler, R. Beranek and B. Dietzek, ChemSusChem, 2021, 14, 1728–1736 CrossRef CAS PubMed.
- C. Adler, S. Selim, I. Krivtsov, C. Li, D. Mitoraj, B. Dietzek, J. R. Durrant and R. Beranek, Adv. Funct. Mater., 2021, 31, 2105369 CrossRef CAS.
- C. Adler, I. Krivtsov, D. Mitoraj, L. Santos-Gómez, S. García-Granda, C. Neumann, J. Kund, C. Kranz, B. Mizaikoff, A. Turchanin and R. Beranek, ChemSusChem, 2021, 14, 2170–2179 CrossRef CAS PubMed.
- I. Krivtsov, A. Vazirani, D. Mitoraj, M. M. Elnagar, C. Neumann, A. Turchanin, Y. Patiño, S. Ordóñez, R. Leiter, M. Lindén, U. Kaiser and R. Beranek, J. Mater. Chem. A, 2023, 11, 2314–2325 RSC.
- J. Strachan, C. Barnett, A. F. Masters and T. Maschmeyer, ACS Catal., 2020, 10, 5516–5521 CrossRef CAS.
- D. Patra, S. R. Nalluri, H. R. Tan, M. S. M. Saifullah, R. Ganesan and B. Gopalan, Nanoscale Adv., 2020, 2, 5384–5395 RSC.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- B. Saini, L. Khamari and T. K. Mukherjee, J. Phys. Chem. B, 2022, 126, 2130–2141 CrossRef CAS PubMed.
- C. Liang, J. Y. Cheong, G. Sitaru, S. Rosenfeldt, A. S. Schenk, S. Gekle, I. Kim and A. Greiner, Adv. Mater. Interfaces, 2022, 9, 2100867 CrossRef CAS.
- S. Karimi, A. Moshaii and M. Nikkhah, Mater. Res. Express, 2019, 6, 1150f2 CrossRef.
- I. Hussain, S. Graham, Z. Wang, B. Tan, D. C. Sherrington, S. P. Rannard, A. I. Cooper and M. Brust, J. Am. Chem. Soc., 2005, 127, 16398–16399 CrossRef CAS PubMed.
-
R. J. Hunter, Zeta Potential in Colloid Science: Principles and Applications, Academic Press, 1st edn, 1988 Search PubMed.
- A. Carone, S. Emilsson, P. Mariani, A. Désert and S. Parola, Nanoscale Adv., 2023, 5, 2017–2026 RSC.
- H. Schlomberg, J. Kröger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. v. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- X. Hang, S. Wang, H. Pang and Q. Xu, Chem. Sci., 2022, 13, 461–468 RSC.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- D. Zopes, B. Stein, S. Mathur and C. Graf, Langmuir, 2013, 29, 11217–11226 CrossRef CAS PubMed.
- Y. Wang, J. E. Q. Quinsaat, T. Ono, M. Maeki, M. Tokeshi, T. Isono, K. Tajima, T. Satoh, S. Sato, Y. Miura and T. Yamamoto, Nat. Commun., 2020, 11, 6089 CrossRef CAS PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759 RSC.
- R. Pamies, J. G. H. Cifre, V. F. Espín, M. Collado-González, F. G. D. Baños and J. G. de la Torre, J. Nanopart. Res., 2014, 16, 2376 CrossRef.
- S. Christau, T. Moeller, J. Genzer, R. Koehler and R. von Klitzing, Macromolecules, 2017, 50, 7333–7343 CrossRef CAS.
- Y. Wang, J. E. Q. Quinsaat, T. Ono, M. Maeki, M. Tokeshi, T. Isono, K. Tajima, T. Satoh, S. Sato, Y. Miura and T. Yamamoto, Nat. Commun., 2020, 11, 6089 CrossRef CAS PubMed.
- M. Ranka, P. Brown and T. A. Hatton, ACS Appl. Mater. Interfaces, 2015, 7, 19651–19658 CrossRef CAS PubMed.
- L. M. Farigliano, M. A. Villarreal, E. P. M. Leiva and S. A. Paz, J. Phys. Chem. C, 2020, 124, 24009–24016 CrossRef CAS.
-
A. D. McNaught and A. Wilkinson, Compendium of chemical terminology, Blackwell Science Oxford, 1997, vol. 1669 Search PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- R. R. Contreras, P. Fuentealba, M. Galván and P. Pérez, Chem. Phys. Lett., 1999, 304, 405–413 CrossRef CAS.
- W. Yang and R. G. Parr, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 6723–6726 CrossRef CAS PubMed.
-
P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2010, 18, 015012 CrossRef.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS.
- Z. Zhang and Y. Wu, Langmuir, 2010, 26, 9214–9223 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Johannes
Liessem‡
a,
Changbin
Im‡
a,
Dariusz
Mitoraj
a,
Johannes
Liessem‡
a,
Changbin
Im‡
a,
Dariusz
Mitoraj