DOI:
10.1039/D3NR02972G
(Paper)
Nanoscale, 2023,
15, 14439-14447
Multi-layer core–shell metal oxide/nitride/carbon and its high-rate electroreduction of nitrate to ammonia†
Received
21st June 2023
, Accepted 11th August 2023
First published on 12th August 2023
Abstract
The electroreduction of nitrate to ammonia is both an alternative strategy to industrial Haber–Bosch ammonia synthesis and a prospective idea for changing waste (nitrate pollution of groundwater around the world) into valuable chemicals, but still hindered by its in-process strongly competitive hydrogen evolution reaction (HER), low ammonia conversion efficiency, and the absence of stability and sustainability. Considering the unique electronic structure of anti-perovskite structured Fe4N, a tandem disproportionation reaction and nitridation–carbonation route for building a multi-layer core–shell oxide/nitride/C catalyst, such as MoO2/Fe4N/C, is designed and executed, in which abundant Fe–N active sites and rich phase interfaces are in situ formed for both suppressing HER and fast transport of electrons and reaction intermediates. As a result, the sample's NO3RR conversion displays a very high NH3 yield rate of up to 11.10 molNH3 gcat.−1 h−1 (1.67 mmol cm−2 h−1) with a superior 99.3% faradaic efficiency and the highest half-cell energy efficiency of 30%, surpassing that of most previous reports. In addition, it is proved that the NO3RR assisted by the MoO2/Fe4N/C electrocatalyst can be carried out in 0.50–1.00 M KNO3 electrolyte at a pH value of 6–14 for a long time. These results guide the rational design of highly active, selective, and durable electrocatalysts based on anti-perovskite Fe4N for the NO3RR.
 Wei Wang | Wei Wang is a lecturer at the College of Materials, Chemistry and Chemical Engineering, Hangzhou Normal University. He received his PhD degree from East China University of Science and Technology in 2020. His research interests are mainly focused on nanoscale materials and their application in photoelectrochemical energy conversion. |
Introduction
Ammonia (NH3) is an irreplaceable chemical raw material in human's manufacture and life, as the source material for nitrogen fertilizers, plastics, pharmaceuticals, explosives, synthetic fibers, resins and printing and dyeing.1,2 According to statistics, the annual growth rate of ammonia production is expected to reach 40% by 2050 (>200 million tons). At present, the main technique adopted for ammonia production is the Haber–Bosch process (350–550 °C, 15–25 MPa), which has been recognized as an energy-intensive synthetic approach (consuming about 5.50 EJ per year) with a high carbon emission (450 million tons per year of CO2).3–5 Therefore, a new economical and environment-friendly strategy for ammonia production needs to be explored. In this backdrop, the production of ammonia from nitrate has been put on the table, which is based on the idea of changing waste (nitrate) into treasures (ammonia). It is known that nitrate pollution in groundwater has become a worldwide environmental problem and endangers people's health in a direct or indirect way, such as causing infants to suffer from methemoglobinemia. Recently, the theoretical feasibility of the production of ammonia from nitrate has been realized through an electrocatalysis reaction.6,7 This direct 8-electron pathway (N5+ in nitrate to N3− in ammonia) has a low equilibrium potential of 0.69 V versus the reversible hydrogen electrode (RHE) due to the weak N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond energy (204 kJ mol−1) and effectively avoids the formation and re-cleavage of inert N
O bond energy (204 kJ mol−1) and effectively avoids the formation and re-cleavage of inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds (dissociation energy: 945 kJ mol−1), which is obviously better than the ambient electrocatalytic N2 reduction reaction (NRR).8,9 In addition, N2 has extremely low water solubility, resulting in a lower faradaic efficiency (FE) and ammonia yield in the NRR process. Comparatively speaking, the advantages in kinetics make the electrochemical nitrate reduction reaction (NO3RR) a highly economical ammonia synthesis route.10–12 Simultaneously, it should be stressed in particular that the 8-electron NO3−-to-NH3 process involves multiple intermediates, such as NO2−, NO−, NO, etc., implying that there are still challenges in optimizing the adsorption of multiple substances and accelerating the reaction.13–15
N bonds (dissociation energy: 945 kJ mol−1), which is obviously better than the ambient electrocatalytic N2 reduction reaction (NRR).8,9 In addition, N2 has extremely low water solubility, resulting in a lower faradaic efficiency (FE) and ammonia yield in the NRR process. Comparatively speaking, the advantages in kinetics make the electrochemical nitrate reduction reaction (NO3RR) a highly economical ammonia synthesis route.10–12 Simultaneously, it should be stressed in particular that the 8-electron NO3−-to-NH3 process involves multiple intermediates, such as NO2−, NO−, NO, etc., implying that there are still challenges in optimizing the adsorption of multiple substances and accelerating the reaction.13–15
To compete with the Haber–Bosch process, further improvement in the yield of NH3 (>1 mmol cm−2 h−1) and a decrease of energy consumption are highly desirable. Recently, transition metal compounds have been intensively investigated in the NO3− reduction reaction (NO3RR), including transition metal oxides (TMO), nitrides (TMN), carbides (TMC), and sulfides (TMS), because the metal centers in them with partial positive charge can effectively adsorb nitrate and nitrite anions while the partial negative charge centers, i.e., nitrogen, are the proton-acceptor centers.16–19 Iron-based nitrides with active Fe–N components on their surfaces exhibit an abnormally high conductivity (0.1–1.0 × 105 S cm−1) and show enhanced electrocatalytic performance and durability for the NO3RR. At the same time, it is also found that the Fe–N structure has a great influence on the catalytic activity of the electroreduction of nitric acid to ammonia.20,21 In particular, anti-perovskite structured Fe4N contains both Fe–N and Fe–Fe chemical bonds, resulting in its excellent metal conductivity and high spin polarizability.22 However, the electrocatalyst with only a single active component is limited by the proportion relationship; the adsorption energy of the intermediate is linearly related and can't be optimized due to the high step-by-step catalytic selectivity.23 Recently, it has been clearly demonstrated that the reaction rate and selectivity of a heterostructure catalyst system could be optimized by precisely tuning the proximity, hierarchy and content ratio of multiple active phases. Furthermore, it is also confirmed that the rationally designed heterostructures could modulate the catalyst's electronic structure to enhance the catalytic activity.24,25 The ultimate aim of these tentative efforts is to regulate the proton and/or electron transfer, as well as the binding strengths of partially reduced intermediates adsorbed on transition metal centers. Another important problem in NO3RR is the competitive side hydrogen evolution reaction (HER) under a low negative potential, resulting in a low NO3RR faradaic efficiency. So far, it has been very challenging to improve the NO3RR selectivity of the catalysts by restraining H2 generation while maintaining a high NH3 yield rate. In contrast to metal oxides, MoO2 exhibits poor HER activity due to the inappropriate hydrogen adsorption, which is beneficial for the improvement of the faradaic efficiency.26 In addition, it is also found that this HER can also be suppressed through compositing with other components, such as Mo6+-rich MoO2, which occasionally forms a passivation layer to prevent protons from transferring to the surface too quickly.27,28
Experimental
Synthesis of MFN nanospheres
0.11 g of MoCl5 and an appropriate amount of FeCl2·4H2O were added to 10 mL of deionized water with different molar ratios (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 4:1, 4:2, 4:3, 4:4). Then 0.3 g of NTA and 20 mL of isopropanol (IPA) were added and stirred until they were completely dissolved. After that the greenish solution was transferred to a 50 mL PTFE-lined stainless-steel autoclave and kept at 180 °C for 6 hours in an oven. After the reaction, the sample was adequately washed with distilled water and dried at 60 °C for 12 h. Finally, brown samples were collected and named MFN-x (x = 1, 2, 3, 4). The sample with a molar ratio of 4:0 was named MN.
:0, 4:1, 4:2, 4:3, 4:4). Then 0.3 g of NTA and 20 mL of isopropanol (IPA) were added and stirred until they were completely dissolved. After that the greenish solution was transferred to a 50 mL PTFE-lined stainless-steel autoclave and kept at 180 °C for 6 hours in an oven. After the reaction, the sample was adequately washed with distilled water and dried at 60 °C for 12 h. Finally, brown samples were collected and named MFN-x (x = 1, 2, 3, 4). The sample with a molar ratio of 4:0 was named MN.
Synthesis of MFC nanospheres
The as-prepared MFN-x and MN precursors were calcined in a quartz tube furnace at 550 °C under an Ar atmosphere for 2 hours. Finally, black powders were collected and named MFC-x (x = 1, 2, 3, 4) and MC.
Synthesis of FC (Fe/C) porous nanorods
The steps were consistent with those of MFC-2 only without MoCl5 and the as-prepared sample was named FC.
Electrochemical nitrate reduction test
3 mg of catalyst was ultrasonically dispersed in a mixture of DI water (700 μL), isopropanol (250 μL) and Nafion (50 μL, 5 wt%) to form a catalyst ink. Then, the catalyst slurry (50 μL) was dropped onto a 1 × 1 cm2 carbon paper (catalyst loading amount: 0.15 mg cm−2). Then, it was dried naturally and used as a working electrode combined with an Ag/AgCl reference electrode and a Pt counter electrode, respectively. All nitrate reduction tests were performed using a three-electrode system in a H-type cell, in which the cathode chamber and anode chamber were separated by a commercial F117 membrane. The KOH and KNO3 electrolyte was first bubbled with a continuous high purity Ar flow for 30 min. Then the nitrate reduction reaction was triggered by a fixed potential from −0.2 to −1.0 V vs. RHE for 1 h. The as-generated NH3 in the cathode chamber was quantified by the colorimetric method and 1H-NMR spectroscopy. The by-products of NO2−, H2 and N2 were determined and quantified by a colorimetric method and gas chromatography, respectively.
Results and discussion
Synthesis and structural characterization
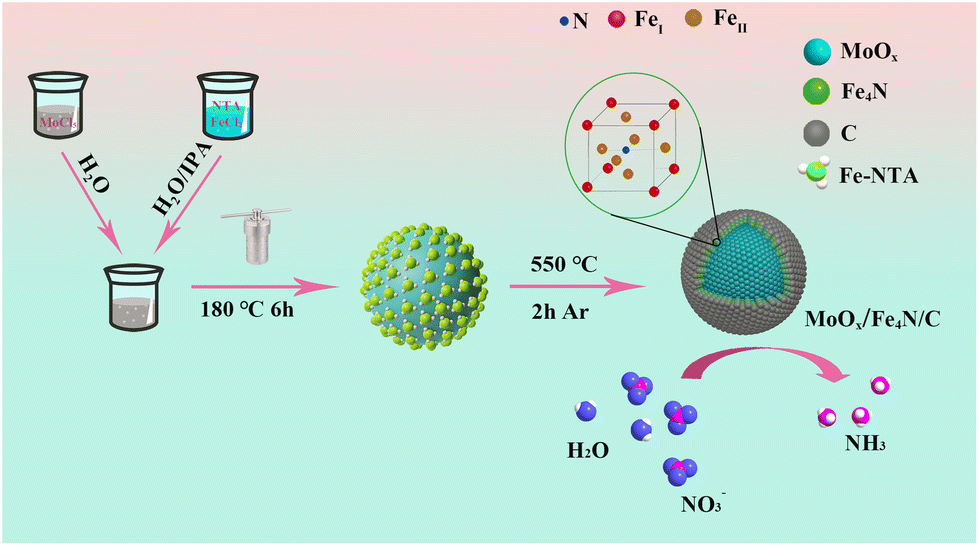
The synthesis of the multilayer core shell MFC catalyst is shown in Scheme 1. Firstly, the micro-scaled core shell of MFN was obtained by a simple hydrothermal reaction, in which a disproportionation reaction occurred: 2Mo5+ → Mo4+ + Mo6+. As a result, Mo6+-rich MoO2 spherical particles were formed and uniformly coated with Fe-NTA. The MFN sample was first identified by XRD measurement (Fig. S1†), only the peaks at 26°, 36.8° and 53.5° corresponding to the (−111), (−211) and (211) crystal planes of MoO2 (JCPDS: 32-0671) were identified, without any Fe-based compound. The composition was also confirmed by FT-IR examination. As shown in Fig. S2,† MoO, Fe–N and other typical chemical bands of NTA are depicted clearly. After comparison according to the field emission scanning electron microscopy (FE-SEM) measurement results (Fig. S3†), the MFN-2 sample with a molar ratio of MoCl5 and FeCl2·4H2O of 4:2 was selected for the following synchronous nitriding carbonization treatment due to its homogeneous structure.
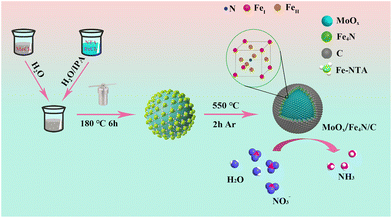 |
| | Scheme 1 A tandem disproportionation reaction (Mo5+ → Mo4+ + Mo6+) and nitridation–carbonation route (Fe4N/C) for building a multi-layer core–shell MFC catalyst. | |
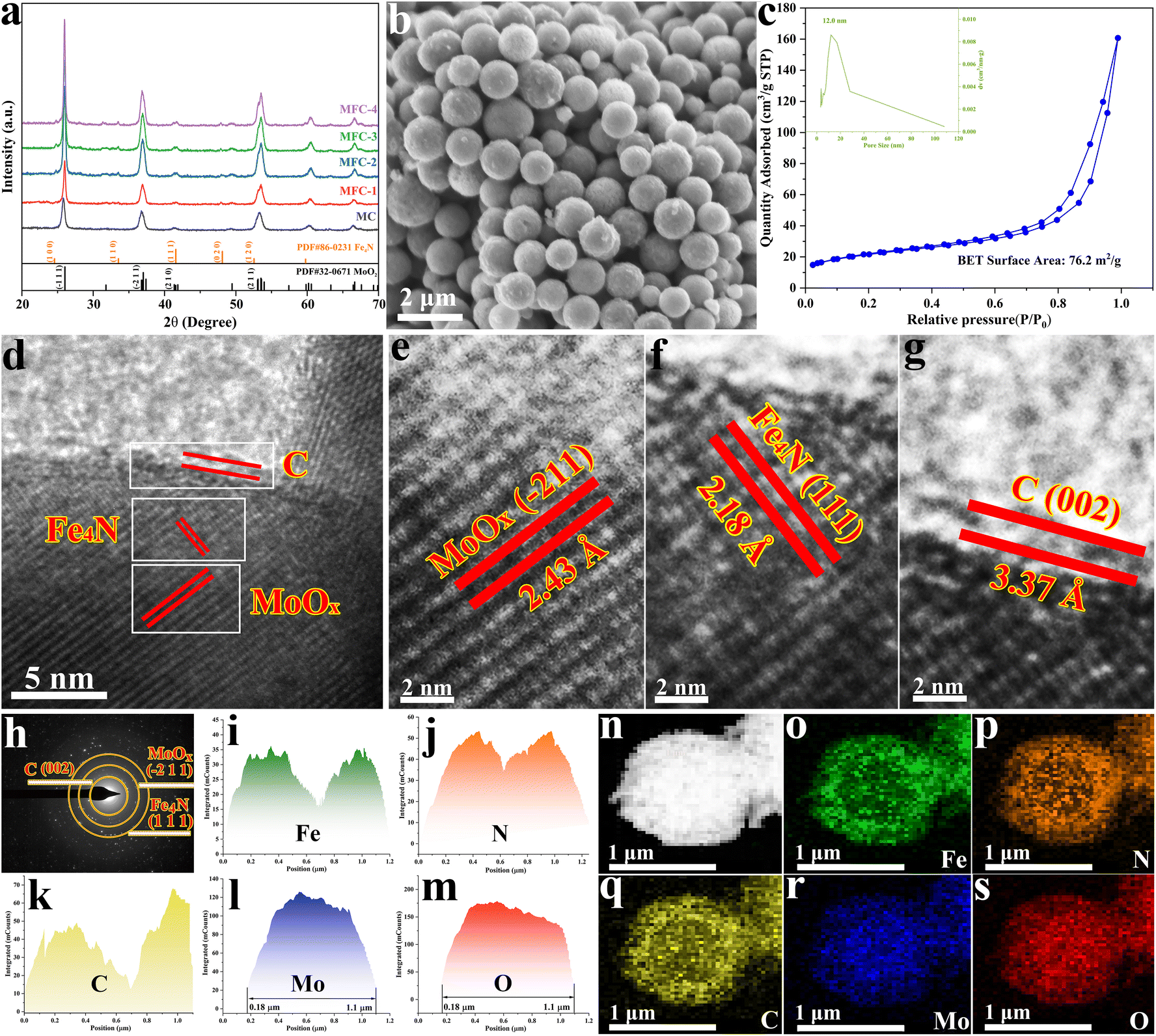
In order to realize the formation of a multi-layer core–shell oxide/nitride/C catalyst, the above prepared MFN-2 was first calcined at different temperatures from 400 to 650 °C with a 50 °C interval. As shown in Fig. S4,† after the reaction temperature was elevated up to 500 °C, three diffraction peaks of 24.7°, 33.5° and 42.8° corresponded to the (100), (110) and (111) crystal planes of anti-perovskite Fe4N (JCPDS 86-0231) were identified, respectively, meaning that high temperature was conducive to the crystallization of Fe4N. The following NO3RR for NH3 was first examined, as shown in Fig. S5,† in which the sample calcined at 550 °C displayed the most yield rate (11.10 molNH3 gcat.−1 h−1). Therefore, all MFN precursors with different Fe salt additions (MoCl5:FeCl2 = 4:0, 4:1, 4:2, 4:3, 4:4) were calcined at 550 °C, and the collected products were named MC, MFC-1, MFC-2, MFC-3 and MFC-4. Their morphologies and structures were characterized first by FE-SEM examinations. As shown in Fig. 1b and Fig. S6,† different from the spherical structure of MFC particles, FC particles display a rod-like structure composed of tiny grains (Fig. S7†). In addition, its XRD result (Fig. S8†) proves the metal Fe nature, not Fe4N or MoO2 presented in the MFC samples (Fig. 1a). This indicates that the suitable oxidizing environment around the Fe atom induced by MoO2 is beneficial for lowering the energy barrier of Fe–N bond formation. Through comparing the FE-SEM images of the MFC samples shown in Fig. S6,† it is found that the spherical particles’ surface becomes rougher with increased Fe salt addition, implying a grain growth. The exposed surface area of a catalyst is a critical factor to its catalytic performance. So the specific surface areas and pore diameters of the MFC samples were then investigated. As shown in Fig. S9 and Table S2,† the MFC-2 sample displays the largest area (76.2 m2 g−1) compared to others and a narrowly distributed pore size centered at 12 nm. The following catalytic measurements also confirm that MFC-2 has a best performance in NO3RR, which inspires us to conduct a comprehensive and detailed investigation about it. In order to fully reveal its microstructure, MFC-2 was analyzed by high-resolution transmission electron microscopy (HRTEM). As shown in Fig. 1(d–g), the (−211) crystal face of MoO2 (d = 2.43 Å), the (002) crystal face of carbon (d = 3.37 Å) and the (111) crystal face of anti-perovskite Fe4N (d = 2.18 Å) were characterized from inside out, respectively. At the same time, a typical particle's selected area electron diffraction pattern (SAED) shown in Fig. 1h proves the existence of the crystalline MoO2, C layer and Fe4N again. The multi-layered core/shell structure was further identified by energy dispersive spectroscopy (EDS), as shown in Fig. 1i–s. It is seen that the Mo and O elements are mainly distributed inside, and the elements Fe, N and C are dispersed outside, which proves that the particles are composed of MoO2, anti-perovskite Fe4N and the carbon shell from inside out.
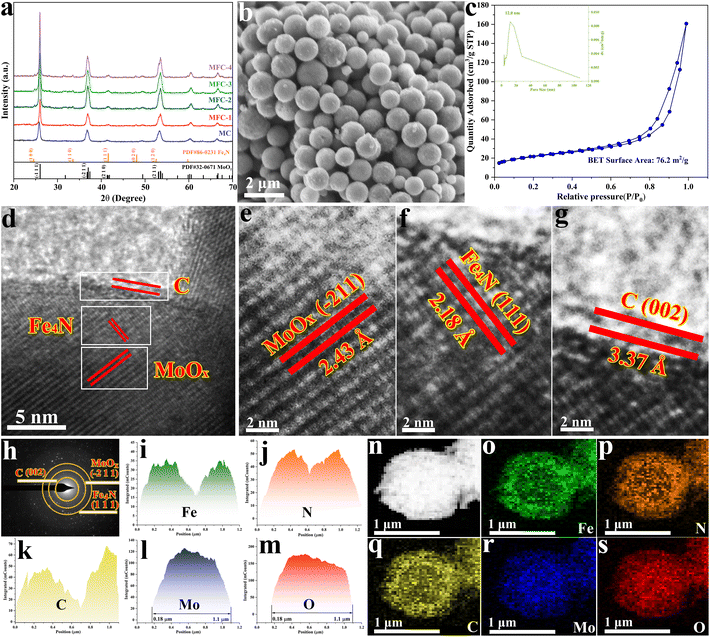 |
| | Fig. 1 Microstructure and composition characterization of the samples prepared with different Fe contents. MFC-2 is selected for detailed analysis because of its most excellent electrocatalytic activity. (a) XRD patterns of all the samples prepared with different Fe additions; (b) FE-SEM image of MFC-2; (c) N2 adsorption–desorption isotherm and pore size distribution curve of MFC-2; (d–g) HRTEM images of MFC-2; (h) SAED image of MFC-2; (i–s) EDS element distribution diagrams of Fe, N, C, Mo and O in MFC-2. | |
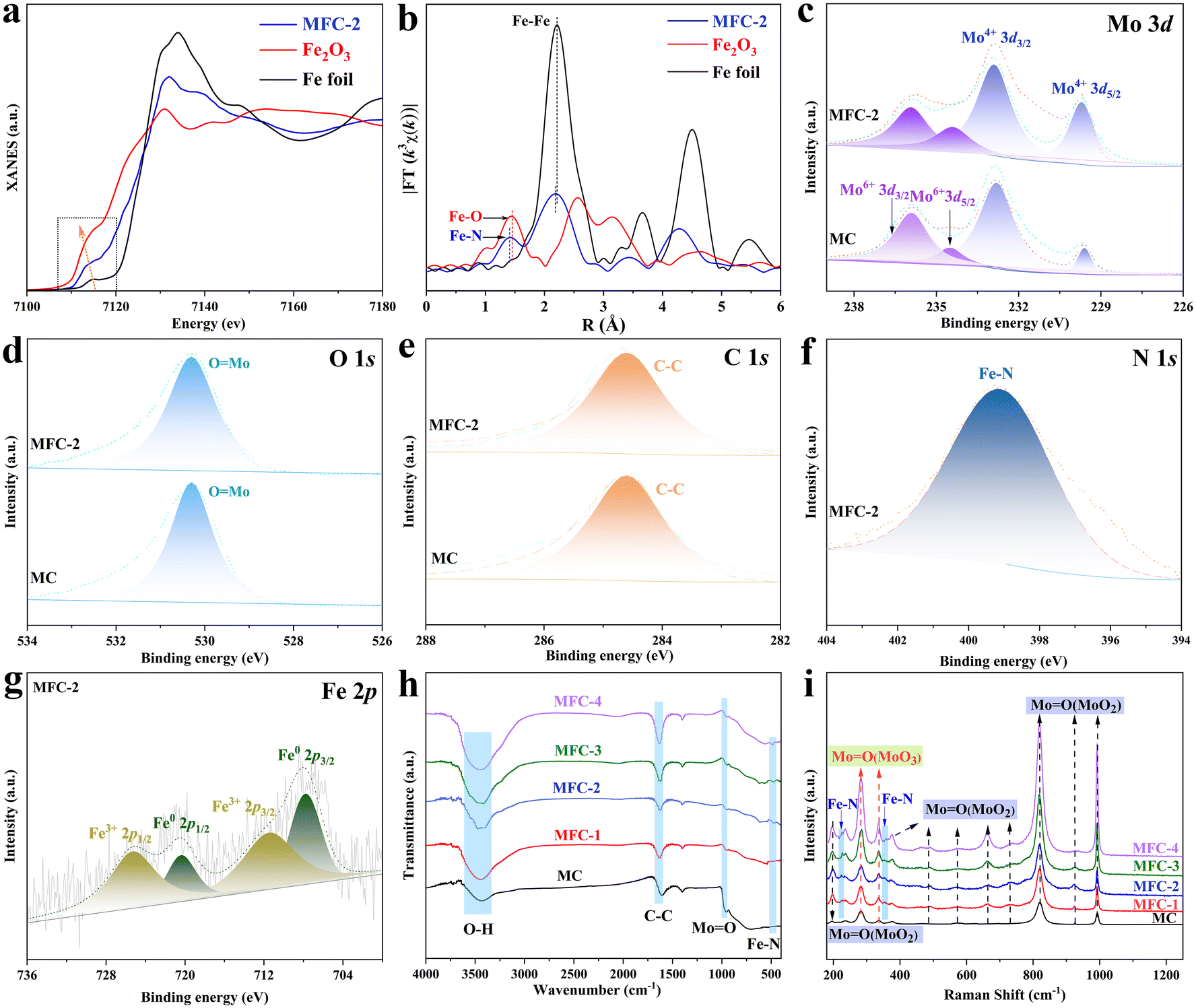
To further reveal the precise composition of MFC-2, X-ray absorption fine structure (XAFS) spectroscopy and X-ray absorption near edge structure (XANES) spectroscopy were conducted based on synchrotron radiation. As shown in Fig. 2a and b, a shoulder strap is located between 7114 eV of Fe2O3 and 7122 eV of Fe foil, which indicates that the oxidation state of Fe in Fe4N is between 0 and +3. Their Fourier transforms based on the EXAFS spectra were then depicted, in which besides the Fe–Fe in Fe4N, a specific peak at 1.41 Å was found, different from any peak in Fe2O3 or Fe foil, matched well with the reported bond length of Fe–N.29,30 Furthermore, X-ray photoelectron spectroscopy (XPS) was applied to further determine its chemical states (Fig. S11†). The Mo 3d spectra of MFC-2 and MC (Fig. 2c) show peaks at binding energies of 229.5, 232.8, 234.2 and 235.9 eV, attributed to the signals of Mo4+ 3d5/2, Mo4+ 3d3/2, Mo6+ 3d5/2 and Mo6+ 3d3/2, respectively. The peak at 530.28 eV in the O 1s spectrum belongs to MoO, which is consistent with the above conclusion (Fig. 2d).31,32 It is obvious that the disproportionation reaction (MoCl5 → MoO2 + Mo6+) expected by us has really occurred to form a Mo6+-rich MoO2 to suppress the HER. Fig. 2e shows the C 1s spectrum, in which the peak with a binding energy of 284.6 eV belongs to C–C, corresponding to the C shell formed after calcination.33 As shown in Fig. 2f, the peak at 399.2 eV in the N 1s spectrum belongs to the Fe–N in Fe4N (Fig. 2f).34 Correspondingly, the Fe 2p spectrum shows multiple valence states in MFC (Fig. 2g), in which the peaks with binding energies of 707.8 and 720.4 eV belong to Fe0, and the peaks with binding energies of 711.5 and 725.3 eV are due to Fe3+. It is worth noting that Fe0 and Fe3+ correspond to the Fe–Fe and Fe–N components in Fe4N, respectively, which is significantly different from the Fe–O bond at 709–710 eV.35,36 In order to further disclose the catalyst's composition, FT-IR spectral and Raman spectral measurements of the samples were conducted, respectively. As shown in Fig. 2h, the FT-IR spectrum results further confirm the existence of the Fe–N band appeared at 546 cm−1 for all catalysts except MC.37,38 Meanwhile, the characteristic vibration of MoO at 936 cm−1 and the vibrations of O–H and C–C with wave numbers of 3432 cm−1 and 1620 cm−1 are also observed in all the samples.39,40 In the Raman spectrum (Fig. 2i), the peaks at 195 cm−1 and 236 cm−1 are attributed to the bending vibration of MoO in MoO2, some other peaks at 657 cm−1, 819 cm−1, 924 cm−1, 991 cm−1, 489 cm−1 and 661 cm−1 come from the stretching vibration of MoO in MoO2. In addition, the bending vibration of Mo6+O was also observed at 281 cm−1 and 337 cm−1, which proved the existence of Mo6+.41,42 At the same time, it is noted that Fe–N bonds with a trend of increasing strength are detected at 228 cm−1 and 360 cm−1 in the MFC samples due to the increased Fe element addition.43 All the above results undoubtedly prove that the multi-layer core–shell MFC catalyst has been successfully prepared through a tandem disproportionation reaction and nitridation–carbonation route.
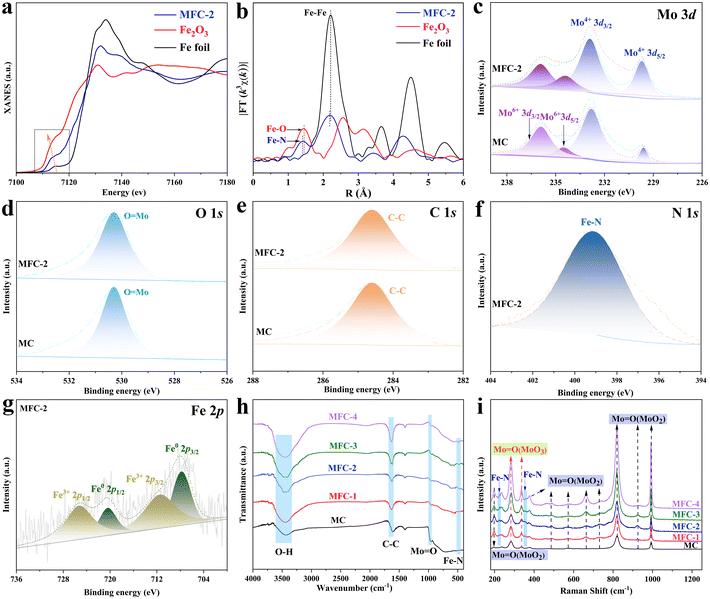 |
| | Fig. 2 Characterization of the compositions and chemical states of the as-obtained samples. (a) Normalized Fe K-edge XANES spectra of MFC-2 in reference to Fe foil and Fe2O3; (b) k3-weighted Fourier-transform Fe K-edge EXAFS spectra; (c–g) XPS spectra of MC and MFC-2; (h) FT-IR spectra; (i) Raman spectra. | |
Electrochemical NO3RR performance
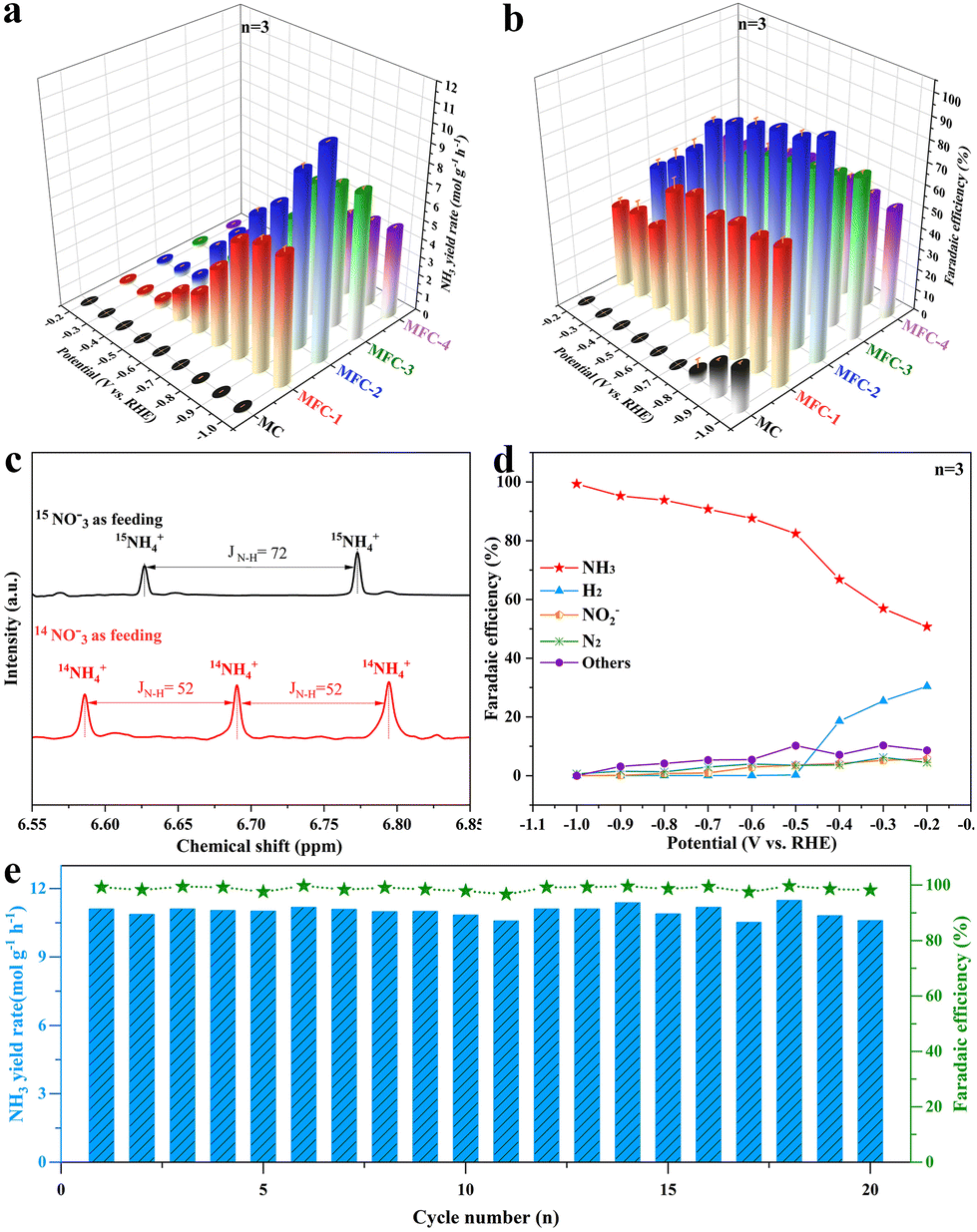
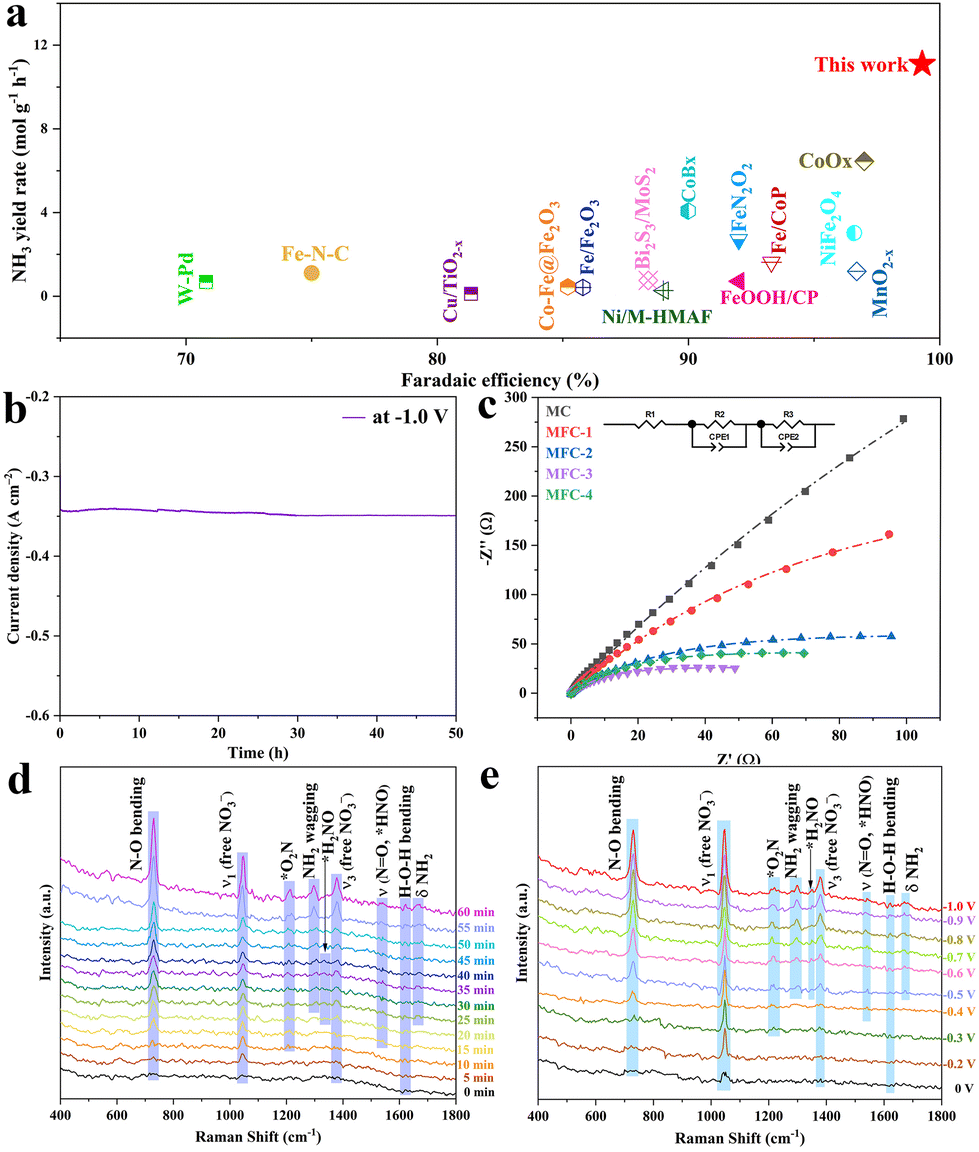
The electrochemical NO3RR performances of the above MFC and MC catalysts were investigated using H-type batteries with an electrolyte of 1 M KNO3 and 1 M KOH, and all potentials were corrected versus the reversible hydrogen electrode (RHE), as shown in Fig. S12.† Their linear sweep voltammetry (LSV) curves are shown in Fig. S13,† which are quantified by Nessler reagent colorimetry (Fig. S14†). It is seen that once anti-perovskite Fe4N is formed in the samples, all the current densities of the electrocatalytic NO3RR are significantly increased at every same voltage, proving the Fe4N promoting effect. The recorded I–t curves also reflect this information (Fig. S15†), and the maximum current density at 1 V is 360 mA cm−2. In order to confirm the fact that Fe in MFC is the critical electrocatalytic active site, LSV measurements of MFC-2 and MC in a mixture electrolyte of 1 M KNO3, 1 M KOH and 1 M KSCN were conducted, as shown in Fig. S16.† It is obvious that after introducing SCN− into the electrolyte to block the Fe3+ site, the LSV curve almost overlaps with the LSV curve of the MC sample, indicating that the Fe3+ site (not Fe0) in anti-perovskite Fe4N is indeed the critical active site for the NO3RR. Fig. 3a shows the NH3 yields and corresponding faradaic efficiencies of MFC and MC (Fig. S14†). It is found that the NH3 yield of MFC-2 increases gradually as the voltage moves to a more negative region. At −1 V, the maximum NH3 yield reaches 11.10 molNH3 gcat.−1 h−1 (1.67 mmol cm−2 h−1). In sharp contrast, at −1.0 V, the highest NH3 yields of MC, MFC-1 and MFC-3, MFC-4 samples are only 0.10, 7.04, 7.66, and 4.74 molNH3 gcat.−1 h−1, respectively, which means that the NH3 yield after Fe4N formation is mostly elevated up to 115 times. Additionally, in the 0.1 M KOH and 0.1 M KNO3 electrolyte, the ammonia yield reached 3.86 molNH3 gcat.−1 h−1 (0.58 mmol cm−2 h−1), and the faradaic efficiency reached 74.4% at −1.0 V, greatly overpassing the previous literature. Importantly, it is worth noting that besides its very high ammonia yield, our sample also has a most excellent FE value compared with the previous literature to date (Fig. 3b and Table S3†), which indicates that the competitive HER is effectively inhibited at MFC, especially at a more negative potential (−0.5 to −1 V). The substantial increase in the NH3 yield of MFC-2 proves that the Fe–N catalytic site in Fe4N is really an ideal one for NO3RR, especially after rational composition design. In addition, through etching the MoO2 species of MFC-2 in a high concentration KOH (5 M) solution, pure Fe4N was obtained to confirm the effect of MoO2 on NO3RR. It is seen in the XRD pattern (Fig. S17†) that the characteristic peaks of MoO2 completely disappear. The following NO3RR measurements indicate that the electrochemical Faraday efficiency decreased from 99.3% to 82.0% with a lowered ammonia production from 11.10 to 10.02 molNH3 gcat.−1 h−1 at −1.0 V. It is worth noting that the Faraday efficiency of H2 increased significantly (13.24%) compared with that before etching (0.04%). This phenomenon further proves that MoO2 species are the main force to inhibit the side reaction of HER, and Fe4N is the critical catalytic center for NO3RR. In order to further confirm that the generated NH3 really comes from the reduction of NO3−, 15N isotope labelling experiments were carried out. As shown in Fig. 3c, the 1H-NMR spectrum recorded in 14NO3− shows a triplet mode with a coupling constant of JN–H = 52 Hz, which can be specified as standard NH4Cl. When 15NO3− is used as a N source, a dipole pattern with coupling constant JN–H = 72 Hz is detected, which is attributed to 15NH4+.44 The possible by-products (NO2−, N2 and H2) in the NO3RR are also quantified by Griess reagent colorimetry and gas chromatography (GC), as shown in Fig. S19–S21.† As shown in Fig. 3d, the H2 FE of MFC-2 is 30.4% at −0.2 V and gradually drops to near zero once over −0.5 V. Simultaneously, the FE values of N2 and NO2− are also lowered over −0.5 V, indicating that the MFC samples have good NH3 selectivity under suitable potential. To reveal the widespread applicability of MFC in NO3RR, we further evaluated the performance of MFC-2 under different NO3− concentrations and pH environments (Fig. S22 and S23†). The results showed that NO3RR had higher NH3 yield and faradaic efficiency under high nitrate concentrations (0.5–1.0 M) and pH environments (pH = 7–14). Besides the above measurements, stability tests were also carried out. Fig. 3e shows the cycling stability of durable MFC-2, in which it is seen that the MFC-2 sample can last for 20 consecutive cycles without obvious NH3 yield and faradaic efficiency degradation. After 20 cycles (20 hours) of tests, the structure of MFC-2 (Fig. S25†) remained unchanged, thanks to the protection of a carbon layer under such a strong alkaline environment.45 The XRD and XPS results of MoO2/Fe4N/C after the electrocatalytic test, as shown in Fig. S26 and S27,† reveal that there is no evidence of structure and phase transformation appeared in the sample after the durability test, which further proves that the catalyst has excellent composition and structural stability in the long-term electrochemical process. In view of the excellent NO3RR activity and stability of the MFC-2 sample, we can look forward with optimism that it has broad application prospects in future industrial catalysis.
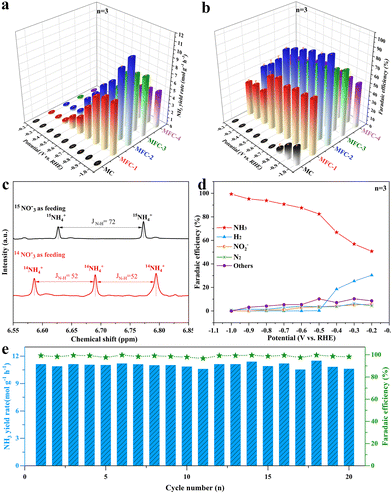 |
| | Fig. 3 (a) NH3 yield rate; (b) the corresponding NO3−-to-NH3 faradaic efficiency of MFC-2 in reference to other samples; (c) 1H-NMR spectra recorded in 14NO3− or 15NO3− solution; (d) potential dependent faradaic efficiencies of NH3, NO2−, N2 and H2 for MFC-2; (e) cycling stability test of MFC-2 at −1.0 V in 1 M KNO3 and 1 M KOH. (Data are presented as the mean values ± standard deviation derived from n = 3 independent experiments.) | |
Electrocatalytic mechanism
It is worth noting that, through investigation, this is the high ammonia output so far (Fig. 4a). In view of the excellent electro catalytic performance of MFC-2, we further evaluated its durability (Fig. 4b). After continuous testing for 50 hours, its current density has no significant change, which indicates its excellent stability. Electrochemical impedance spectroscopy (EIS) (Fig. 4c) shows that with the increasing Fe content, the samples’ charge transfer resistances are obviously decreased due to the high conductivity of Fe4N (∼1.0 × 105 S cm−1). Though MFC-4 has the smallest electrochemical impedance, this also makes its corresponding faradaic efficiency lowered. In contrast, the appropriate electrochemical impedance of MFC-2 effectively suppresses the side reactions with a high performance, and the faradaic efficiency reaches nearly 100%. According to previous research, there are five possible pathways for the NO3RR on Fe-based catalysts (see the ESI† for details).46,47 In order to further explore the inherent activity of the catalyst, the electrochemical active surface area (ECSA) of the catalyst was measured using a double-layer capacitance (Cdl) measurement method. As shown in Fig. S28,† the Cdl of MFC-2 (4.17 mF cm−2) showed no significant change compared to that of MC (3.85 mF cm−2), indicating that the greatly enhanced electrochemical performance of MFC-2 samples should be due to the higher intrinsic activity of Fe4N, rather than the ECSA. To analyze the NO3RR mechanism of MFC, we carried out in situ Raman measurements of MFC-2 and MC on rotating ring disk glassy carbon electrodes using a mixture of 1 M potassium nitrate and 1 M potassium hydroxide as the electrolyte, as shown in Fig. S29.† Firstly, in the in situ Raman spectrum of MC under a continuously regulated voltage from 0 to −1 V, it is found that no intermediates related to NO3RR are detected between 0 V and −0.7 V except the peaks related to the catalyst and nitrate, which agrees with the NH3 yield results under different potentials shown in Fig. 3a. Once the potential is scanned up to −0.8 V, the bending vibration peak of NH3 appears at 1298 cm−1 though the signal is relatively weak.48 Due to the low concentration or nonexistence of intermediates, no signal of related intermediates is detected, which further confirms that MC is not the critical active site for the NO3RR. As shown in Fig. 4d, in situ Raman spectra were recorded at −1.0 V with different reaction times to reveal this electrocatalytic dynamic mechanism. The vibration peaks at 730, 1047 and 1380 cm−1 are attributed to the bending mode of N–O (NO3−) and free NO3−, respectively. Obviously, with the reaction time extended from 0 to 60 minutes, the intensity of the vibration peak gradually increases, indicating that NO3− and intermediates are aggregated on the catalyst. In addition, the two vibration peaks at 1218 and 1548 cm−1 appeared at 10 and 15 min, respectively, corresponding to adsorbed NO2 (expressed as −*O2N) and adsorbed HNO (expressed as the NO stretching mode of *HNO species). The appearance of these two intermediate species indicates that NO3− is first transformed into *NO2, and then into *HNO. As an important intermediate species, the peaks of *H2NO appears at 1322 cm−1 until 20 minutes, meaning that *HNO was further transformed into *H2NO. The conversion of *HNO to *H2NO is a characteristic process of pathway 1 (see the ESI†).49,50 In addition, the swinging mode (1298 cm−1) and bending mode (1664 cm−1) of NH2 were observed at 25 minutes, indicating the formation of ammonia. The H–O–H bending mode at 1620 cm−1 also increases with the extended reaction time, implying that that the NO3RR is assisted by proton coupling.51 Then, in order to explore the thermodynamic mechanism, in situ Raman tests were carried out from −0.2 to −1.0 V potentials. As shown in Fig. 4e, the H–O–H bending pattern at 1620 cm−1 also increases with the negative shift of voltage, indicating that the NO3RR is assisted by proton coupling. Between 0 and −0.2 V, vibrational peaks appear at 730, 1047 and 1380 cm−1, attributed to the bending mode of N–O and the ν1 and ν3 modes of freely adsorbed NO3−, respectively. With a negative voltage shift to −0.3 V, the signal of adsorbed NO2 (denoted as −*O2N) appears at 1218 cm−1, indicating the conversion of NO3− to NO2, followed by adsorbed HNO (denoted as the NO stretching mode of *HNO species) emerged at 1548 cm−1 at −0.4 V, indicating the further conversion of −*O2N to *HNO. When the voltage is further negatively shifted to −0.5 V, the characteristic peaks of *H2NO intermediate species as well as the oscillatory and bending modes of NH2 are observed simultaneously at 1322 cm−1, 1298 cm−1 and 1664 cm−1, indicating the formation of ammonia. It is worth noting that the conversion of *HNO to *H2NO is the pathway 1 characteristic process (see the ESI†), again confirming the reaction course as follows:
| * + HNO3 + H+ + e− = *NO2 + H2O |
| *HNO2 + H+ + e− = *NO + H2O |
| *H2NOH + H+ + e− = *NH2 + H2O |
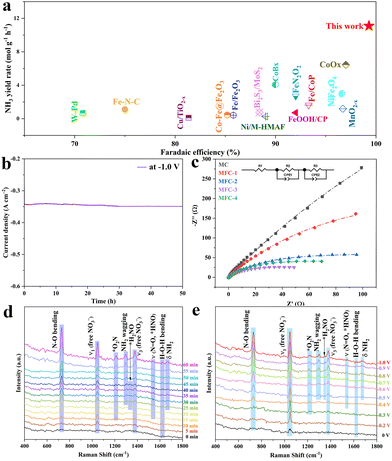 |
| | Fig. 4 (a) Comparation of ammonia yields reported in the literature and in this work; (b) current stability of MFC-2 tested at −0.1 V potential for 50 h; (c) EIS Nyquist plots of MFC-X and MC samples; (d) electrochemical in situ Raman spectra of MFC-2 in the range of 0–60 min; (e) electrochemical in situ Raman spectra of MFC-2 in the range of 0–1.0 V. | |
Conclusions
This paper reports a tandem disproportionation reaction and nitridation–carbonation route for building an anti-perovskite Fe4N-based multi-layer oxide/nitride/C catalyst, which enables the in situ formation of abundant active sites and rich phase interfaces for both fast transport of reaction intermediates and suppressing HER. After detailed examinations, it is proved that Fe–N in Fe4N is the active site for the NO3RR and the existence of MoO2 can effectively suppress the HER side reaction. As a result, the sample's NO3RR conversion displays a NH3 yield rate of 11.10 molNH3 gcat.−1 h−1 (1.67 mmol cm−2 h−1) with a 99.3% faradaic efficiency, and the highest half-cell energy efficiency of 30%. This study provides a new idea for constructing efficient and stable catalysts based on anti-perovskite Fe4N, which has multiple electrochemical and catalytic properties.
Author contributions
P. Gao, X. Y. Li and P. Deng conceived the idea and designed and performed the experiments as well as data analysis. M. Q. Xu performed the in situ Raman spectroscopy test and helped in analyzing the electrocatalytic mechanism. Z. B. Peng and Y. H. Zhou assisted with the materials synthesis, characterization, and the electrochemical performance test. P. Gao and X. Y. Li co-wrote the manuscript. P. Gao, W. Wang, W. Ye and G. Jia performed the analysis, supervised, and supported the work.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work was supported by the Innovation and Entrepreneurship Project of High-level Overseas Students in Hangzhou in 2021 (no. 4095C5021920466), the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2023KY1009), Innovative Leading Talents in Colleges and Universities of Zhejiang Province and the Open Project of Key Laboratory of Superlight Materials and Surface Technology of Ministry of Education of Harbin Engineering University.
Notes and references
- J. Liang, Z. Li, L. Zhang, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, H. Yan, B. Ying, S. Sun, Q. Liu, M. S. Hamdy, B. Tang and X. Sun, Chem, 2023, 9, 1768–1827 CAS.
- X. Xu, L. Hu, Z. Li, L. Xie, S. Sun, L. Zhang, J. Li, Y. Luo, X. Yan, M. S. Hamdy, Q. Kong, X. Sun and Q. Liu, Sustainable Energy Fuels, 2022, 6, 4130–4136 RSC.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC.
- J. Liu, W. Kong, Z. Jin, Y. Han, J. Sun, L. Ma, Y. Niu and Y. Xu, J. Mater. Chem. A, 2020, 8, 19278–19282 RSC.
- J. Liang, Q. Liu, A. A. Alshehri and X. Sun, Nano Res. Energy, 2022, 1, e9120010 CrossRef.
- I. Mukherjee and U. K. Singh, Sci. Total Environ., 2021, 787, 147657 CrossRef CAS PubMed.
- T. Zhu, Q. Chen, P. Liao, W. Duan, S. Liang, Z. Yan and C. Feng, Small, 2020, 16, e2004526 CrossRef PubMed.
- W. Song, L. Yue, X. Fan, Y. Luo, B. Ying, S. Sun, D. Zheng, Q. Liu, M. S. Hamdy and X. Sun, Inorg. Chem. Front., 2023, 10, 3489–3514 RSC.
- Y. Xu, Y. Sheng, M. Wang, T. Ren, K. Shi, Z. Wang, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 16883–16890 RSC.
- Y. Xu, Y. Wen, T. Ren, H. Yu, K. Deng, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2023, 320, 12981 Search PubMed.
- Z. Li, J. Liang, Q. Liu, L. Xie, L. Zhang, Y. Ren, L. Yue, N. Li, B. Tang, A. A. Alshehri, M. S. Hamdy, Y. Luo, Q. Kong and X. Sun, Mater. Today Phys., 2022, 23, 100619 CrossRef CAS.
- X. He, Z. Li, J. Yao, K. Dong, X. Li, L. Hu, S. Sun, Z. Cai, D. Zheng, Y. Luo, B. Ying, M. S. Hamdy, L. Xie, Q. Liu and X. Sun, iScience, 2023, 26, 107100 CrossRef CAS PubMed.
- Z. Wang, D. Richards and N. Singh, Catal. Sci. Technol., 2021, 11, 705–725 RSC.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726 RSC.
- Z. Deng, C. Ma, X. Fan, Z. Li, Y. Luo, S. Sun, D. Zheng, Q. Liu, J. Du, Q. Lu, B. Zheng and X. Sun, Mater. Today Phys., 2022, 28, 100854 CrossRef CAS.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D. H. Nam, C. S. Tan, Y. Ding, J. Wu, Y. Lum, C. T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- H. Wang, J. Li, K. Li, Y. Lin, J. Chen, L. Gao, V. Nicolosi, X. Xiao and J. M. Lee, Chem. Soc. Rev., 2021, 50, 1354–1390 RSC.
- S. Wang, L. Li, Y. Shao, L. Zhang, Y. Li, Y. Wu and X. Hao, Adv. Mater., 2019, 31, e1806088 CrossRef PubMed.
- T. Ren, Z. Yu, H. Yu, K. Deng, Z. Wang, X. Li, H. Wang, L. Wang and Y. Xu, Appl. Catal., B, 2022, 318, 121805 CrossRef CAS.
- D. Malko, A. Kucernak and T. Lopes, J. Am. Chem. Soc., 2016, 138, 16056–16068 CrossRef CAS.
- G. Ye, K. Zhao, Z. He, R. Huang, Y. Liu and S. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15624–15633 CrossRef CAS.
- H. K. Singh, I. Samathrakis, N. M. Fortunato, J. Zemen, C. Shen, O. Gutfleisch and H. Zhang, npj Comput. Mater., 2021, 7, s41524 Search PubMed.
- H. Zhu, L. Gu, D. Yu, Y. Sun, M. Wan, M. Zhang, L. Wang, L. Wang, W. Wu, J. Yao, M. Du and S. Guo, Energy Environ. Sci., 2017, 10, 321–330 RSC.
- N. R. Singstock and C. B. Musgrave, J. Am. Chem. Soc., 2022, 144, 12800–12806 CrossRef CAS PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- J. Wang, Y.-p. Liu, H. Zhang, D.-j. Huang and K. Chu, Catal. Sci. Technol., 2019, 9, 4248–4254 RSC.
- Y. Wan, Z. Wang, J. Li and R. Lv, ACS Nano, 2022, 16, 643–654 CrossRef CAS PubMed.
- A. Kumar, J. Lee, M. G. Kim, B. Debnath, X. Liu, Y. Hwang, Y. Wang, X. Shao, A. R. Jadhav, Y. Liu, H. Tuysuz and H. Lee, ACS Nano, 2022, 16, 15297–15309 CrossRef CAS.
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long, M. Chen, J. Zhu, P. Gao, L. Song, J. Jiang and Y. Xiong, Chem, 2019, 5, 2865–2878 CAS.
- X. Ao, W. Zhang, Z. Li, L. Lv, Y. Ruan, H.-H. Wu, W.-H. Chiang, C. Wang, M. Liu and X. C. Zeng, J. Mater. Chem. A, 2019, 7, 11792–11801 RSC.
- T. Hu, M. Wang, C. Guo and C. M. Li, J. Mater. Chem. A, 2022, 10, 8923–8931 RSC.
- F. Rehman, S. Kwon, C. B. Musgrave, M. Tamtaji, W. A. Goddard and Z. Luo, Nano Energy, 2022, 103, 107866 CrossRef CAS.
- X. Yang, H. Wang, D. Y. W. Yu and A. L. Rogach, Adv. Funct. Mater., 2018, 28, 1706609 CrossRef.
- W. Kiciński, S. Dyjak and W. Tokarz, J. Power Sources, 2021, 513, 230537 CrossRef.
- A. M. Demin, A. V. Mekhaev, A. A. Esin, D. K. Kuznetsov, P. S. Zelenovskiy, V. Y. Shur and V. P. Krasnov, Appl. Surf. Sci., 2018, 440, 1196–1203 CrossRef CAS.
- Y. Zhou, Z. Zhou, R. Shen, R. Ma, Q. Liu, G. Cao and J. Wang, Energy Storage Mater., 2018, 13, 189–198 CrossRef.
- S. R. Hormozi Jangi and M. Akhond, Microchem. J., 2020, 158, 105328 CrossRef CAS.
- B. Liu, H. Zhao, J. Yang, J. Zhao, L. Yan, H. Song and L. Chou, Microporous Mesoporous Mater., 2020, 293, 109820 CrossRef CAS.
- J. Chen, K. Sun, Y. Zhang, D. Wu, Z. Jin, F. Xie, X. Zhao and X. Wang, Anal. Bioanal. Chem., 2019, 411, 2781–2791 CrossRef CAS PubMed.
- T. N. Rudneva, N. S. Emel'yanova and S. V. Kurmaz, Chem. Pap., 2018, 73, 95–104 CrossRef.
- C. Gu, D. Li, S. Zeng, T. Jiang, X. Shen and H. Zhang, Nanoscale, 2021, 13, 5620–5651 RSC.
- M. A. Camacho-López, L. Escobar-Alarcón, M. Picquart, R. Arroyo, G. Córdoba and E. Haro-Poniatowski, Opt. Mater., 2011, 33, 480–484 CrossRef.
- F. Liu, N. Yan, G. Zhu, Z. Liu, S. Ma, G. Xiang, S. Wang, X. Liu and W. Wang, New J. Chem., 2021, 45, 13004–13014 RSC.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- H. Guo, N. Youliwasi, L. Zhao, Y. Chai and C. Liu, Appl. Surf. Sci., 2018, 435, 237–246 CrossRef CAS.
- T. Ren, K. Ren, M. Wang, M. Liu, Z. Wang, H. Wang, X. Li, L. Wang and Y. Xu, Chem. Eng. J., 2021, 426, 130759 CrossRef CAS.
- Z. Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. T. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- C. Byram, S. S. B. Moram, A. K. Shaik and V. R. Soma, Chem. Phys. Lett., 2017, 685, 103–107 CrossRef CAS.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- F. Lei, K. Li, M. Yang, J. Yu, M. Xu, Y. Zhang, J. Xie, P. Hao, G. Cui and B. Tang, Inorg. Chem. Front., 2022, 9, 2734–2740 RSC.
- Y. H. Wang, S. Zheng, W. M. Yang, R. Y. Zhou, Q. F. He, P. Radjenovic, J. C. Dong, S. Li, J. Zheng, Z. L. Yang, G. Attard, F. Pan, Z. Q. Tian and J. F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Peng
Gao
*a,
Peng
Gao