
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic-layered V2C MXene containing bismuth elements: 2D/0D and 2D/2D nanoarchitectonics for hydrogen evolution and nitrogen reduction reaction†
Sana
Akir
 *,
Jalal
Azadmanjiri
,
Nikolas
Antonatos
,
Lukáš
Děkanovský
,
Pradip Kumar
Roy
,
Vlastimil
Mazánek
,
Roussin
Lontio Fomekong
,
Jakub
Regner
and
Zdeněk
Sofer
*
*,
Jalal
Azadmanjiri
,
Nikolas
Antonatos
,
Lukáš
Děkanovský
,
Pradip Kumar
Roy
,
Vlastimil
Mazánek
,
Roussin
Lontio Fomekong
,
Jakub
Regner
and
Zdeněk
Sofer
*
Department of Inorganic Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: akirs@vscht.cz; zdenek.sofer@vscht.cz
First published on 4th July 2023
Abstract
The exploitation of two-dimensional (2D) vanadium carbide (V2CTx, denoted as V2C) in electrocatalytic hydrogen evolution reaction (HER) and nitrogen reduction reaction (NRR) is still in the stage of theoretical study with limited experimental exploration. Here, we present the experimental studies of V2C MXene-based materials containing two different bismuth compounds to confirm the possibility of using V2C as a potential electrocatalyst for HER and NRR. In this context, for the first time, we employed two different methods to synthesize 2D/0D and 2D/2D nanostructures. The 2D/2D V2C/BVO consisted of BiVO4 (denoted BVO) nanosheets wrapped in layers of V2C which were synthesized by a facile hydrothermal method, whereas the 2D/0D V2C/Bi consisted of spherical particles of Bi (Bi NPs) anchored on V2C MXenes using the solid-state annealing method. The resultant V2C/BVO catalyst was proven to be beneficial for HER in 0.5 M H2SO4 compared to pristine V2C. We demonstrated that the 2D/2D V2C/BVO structure can favor the higher specific surface area, exposure of more accessible catalytic active sites, and promote electron transfer which can be responsible for optimizing the HER activity. Moreover, V2C/BVO has superior stability in an acidic environment. Whilst we observed that the 2D/0D V2C/Bi could be highly efficient for electrocatalytic NRR purposes. Our results show that the ammonia (NH3) production and faradaic efficiency (FE) of V2C/Bi can reach 88.6 μg h−1 cm−2 and 8% at −0.5 V vs. RHE, respectively. Also V2C/Bi exhibited excellent long-term stability. These achievements present a high performance in terms of the highest generated NH3 compared to recent investigations of MXenes-based electrocatalysts. Such excellent NRR of V2C/Bi activity can be attributed to the effective suppression of HER which is the main competitive reaction of the NRR.
1. Introduction
Currently, fossil fuels supply about 80% of the world's energy consumption.1 However, fossil fuel stocks are limited, and their intensive use will aggravate the problems of greenhouse gas emissions. Therefore, there is an urgent need to increase the utilization of renewable energy resources to meet our ever-increasing energy demands. Hydrogen gas (H2), and ammonia (NH3) are among the most produced and widely used chemicals in the world.2,3 Therefore, the production of NH3 and H2 through an economic and sustainable process is highly sought after. The electrochemical hydrogen evolution reaction (HER), and nitrogen reduction reaction (NRR) have attracted widespread attention owing to fossil-fuel-free routes for H2 and NH3 synthesis, respectively. To date, precious metals are the most outstanding electrocatalysts for electrochemical H2 and NH3 production, especially platinum-based nanomaterials for HER,4 and palladium-based nanomaterials for NRR.5 However, the high cost of those precious metals is restricting their practical applications. Therefore, it is urgent to find economical and effective NRR and HER electrocatalysts. Two-dimensional (2D) transition-metal carbides, nitrides, and carbonitrides (MXenes) have received great interest since the discovery of the first MXene (Ti3C2Tx).6 MXenes usually have a hexagonal close-packed structure with a chemical formula of Mn+1Xn (M = transition metal element, X = C, N, n = 1–3). Commonly, MXenes are obtained by selectively etching the “A” elements from their corresponding bulk MAX phases using chemicals, such as hydrofluoric acid, ammonium bifluoride, or an acidic solution of fluoride salts. Thus, their surfaces are inevitably terminated by a series of functional groups (–OH, –F, –O).6 Owing to the existence of surface terminations, most MXenes exhibit both metallic conductivity and hydrophilicity,7 which makes them promising candidates for various applications, such as energy storage and conversion, especially in electrocatalysis.8,9 Unfortunately, the NRR and HER performances with the most developed MXenes electrocatalysts are still far from satisfactory and not comparable to those of precious metals. Most of the pristine MXenes suffer from large overpotential and low stability in aqueous electrolytes which seriously hinder their large-scale practical applications.10,11 Additionally, MXenes often exhibit low ammonia yield and unsatisfactory faradaic efficiency (FE),12,13 because they are usually terminated with inactive functional groups (–F, –OH), which hinder the active metal sites for N2 binding.14,15 Therefore, many attempts have been made to improve the HER and NRR activities of MXenes. Firstly, by introducing or modifying chemical functional groups on its surface.16–18 It was proved that the electrocatalytic performances of MXenes are significantly influenced by the functional groups on the basal plane (–O, –OH, –F). For instance, MXenes terminated with O species presented relatively low hydrogen adsorption free energy (ΔGH*) and would be promising candidates for HER.11 Secondly, constructing composites of MXenes with other active components (e.g., nanoparticles, oxides, single atoms, and other 2D materials) is another efficient strategy to boost the NRR and HER performance of such 2D layered structures.19–22 In fact, the MXene-based composites could enhance the catalytic activity by increasing specific surface area (SSA). Among MXene-based composites, 2D/2D heterostructures have been identified as the best choice to ensure the largest contact area and promote electron transfer for improving electrocatalysis reactions.23Recently, Bi-based catalysts have been considered potential NRR candidates owing to their intrinsic catalytic activity, earth-abundance, low cost, and environmentally benign semi-metal.24 The density functional theory (DFT) calculation also has proved that in Bi-based catalysts, the density state near the Fermi level is mainly localized around the Bi atoms, indicating the ability of Bi to serve as active sites to donate p-electrons and thus activate N2.25 Currently, promising advances have been achieved for the NRR on Bi-based electrocatalysts including Bi nanosheets,24 2D mosaic Bi nanosheets,25 PdBi2 nanoflakes,26 and bismuthene nanostructures.27 However, the NRR performance of the reported Bi-based materials is still limited due to their poor electrical conductivity.25 Thus, combining Bi with conductive nanomaterials is considered a promising strategy to solve this issue. Very recently, Liu et al.,28 successfully anchored metallic Bi on the 2D Ti3C2Tx MXene via a simple liquid phase reduction method. The as-prepared Bi@Ti3C2Tx composite electrocatalyst achieved an excellent NH3 yield of 28.3 μg h−1 cm−2 and a large FE of 27.2% at 0.4 V vs. RHE. Their report shows a significant performance for NRR which is higher than most of the reported Bi-based catalysts. On the other hand, Bi-based catalysts possess poor activity in HER due to the unfavorable free energy of hydrogen adsorption (ΔGH*).29 Thus, a lot of studies have been applied to explore the (photo) catalytic activity of H2 production for Bi-based materials. Recently, by adopting different strategies, BiOX, (X = Cl, Br, and I),30 Bi2O3,31 and BiVO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 showed extraordinary HER performance. Among Bi compounds, BiVO4 has emerged as the most promising candidate for photocatalytic hydrogen production owing to good chemical stability, nontoxicity, and low cost.32
32 showed extraordinary HER performance. Among Bi compounds, BiVO4 has emerged as the most promising candidate for photocatalytic hydrogen production owing to good chemical stability, nontoxicity, and low cost.32
In this work, for the first time, we successfully synthesized two different MXene-based nanocomposites including V2C/BVO and V2C/Bi NPs. V2C MXene was selected because of its high electrical conductivity compared to the other reported MXenes.6 Our findings demonstrate that the first nanocomposite (V2C/BVO) consists of BiVO4 nanosheets embedded in the few layers of V2C nanosheets, and the second nanocomposite (V2C/Bi) consists of metallic Bi NPs anchored on the surface of V2C. The V2C MXene was obtained through the etching of V2AlC by HCl/NaF. The as-obtained nanocomposites were directly used as multifunctional electrodes for HER and NRR in acidic and in basic electrolytes, respectively. 2D/2D V2C/BVO showed a lower overpotential (384 mV) in 0.5 M H2SO4 aqueous electrolyte compared to pristine V2C (823 mV) when applied for HER. Also, V2C/BVO proved excellent stability for HER application. Meantime, V2C/Bi has been proven to be highly efficient NRR electrocatalyst achieving a record high-yield rate of NH3 production with 88.6 μg h−1 cm−2 and FE of 8% at −0.5 V vs. RHE. These results indicate that the decoration of a suitable matrix with Bi atoms is beneficial to improve nitrogen fixation performance. To the best of our knowledge, the HER and NRR electrocatalysis performances of the V2C/Bi and V2C/BVO have not been studied before.
2. Results and discussion
2.1. Characterization of the catalysts
The crystalline structure of V2AlC, V2C, and composite samples was examined by XRD as shown in Fig. 1a. Firstly, it should be mentioned that the as-prepared V2AlC precursor presents sharp and strong diffraction peaks at 2θ = 13.5° and 41.2°, indicating its high crystallinity and purity (PDF file 29-0101).33 After etching with HCl/NaF solution, several peaks were detected at 7.5°, 15°, 22.6°, and 30.7° corresponding to the crystal indexes (002), (004), (006), and (008) of hexagonal V2C MXene with a multilayered structure.33 The presence of small peaks observed at 13.5° and 41.2° suggests the existence of a small amount of unreacted V2AlC, indicating the incomplete etching of V2C. This observation is consistent with previous studies.34,35 The same peaks were also detected in V2C/BVO, composite confirming the unaltered hexagonal crystalline phase of V2C in the V2C/BVO. Additional diffraction peaks at 2θ = 18.6°, 28.6°, 30.7°, 34.3°, 39.9°, 42.5°, 47.2 correspond to the (011), (121), (040), (200), (211), (051) and (042), and (310), respectively, confirming the formation of monoclinic BiVO4 (JCPDS no. 14-0688).36 As well, the XRD pattern of V2C/Bi shows that all the diffraction peaks can be indexed to Bi rhombohedral crystal structure NPs (code: 01-085-1330).37 At the same time, the peaks originating from V2AlC and V2C MXene completely vanished. This can be attributed to the presence of high-density Bi nanoparticles that are fully occupied on the surface of the V2C. As a result, it becomes difficult to detect the V2AlC and V2C peaks. This phenomenon has been observed in previous reports of MXene-based nanocomposites, where the (002) peak of MXenes disappeared after being combined with other components.38–40 The loss of the (002) peak of V2C was attributed to the relatively poor crystallinity of V2CTx and the low signal-to-noise ratio.39 Low-intensity peaks of Bi2O3 can also be observed corresponding to surface oxidation.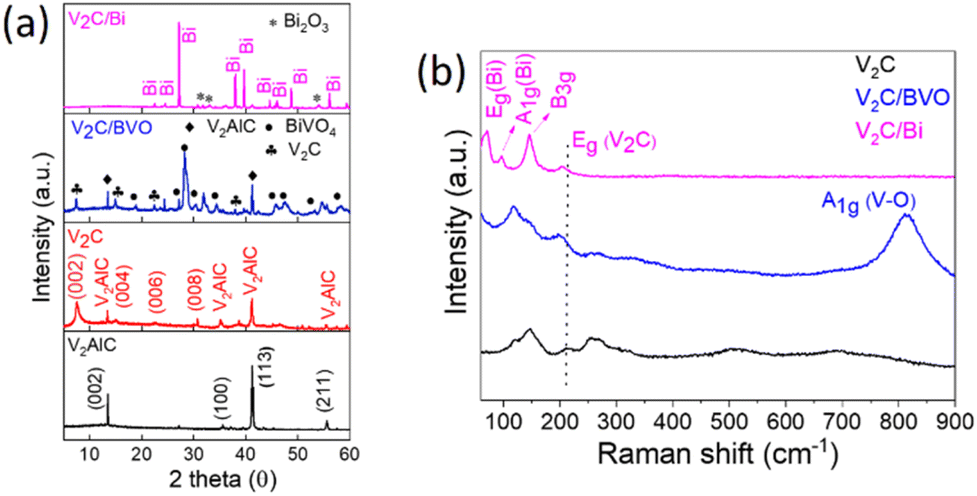 | ||
| Fig. 1 (a) XRD patterns of V2AlC, V2C, V2C/BVO, and V2C/Bi. (b) Raman spectra of V2C, V2C/BVO, and V2C/Bi. | ||
The as-prepared samples were further studied by Raman spectroscopy (Fig. 1b). For V2C, several bands appeared at approximately 147, 211, 262, 513, and 691 cm−1. According to the theoretical study of Champagne et al.,41 the peak at 211 cm−1 corresponds to the Eg mode of V2C(OH)2, the peak at 262 cm−1 corresponds to the Eg mode of V2C, and two weaker peaks at 513 and 691 cm−1 can be assigned the A1g mode of V2CF2 and V2CO(OH), respectively. The Eg mode is due to the in-plane vibration of the V atoms while the A1g mode is due to the out-of-plane vibration of the V atoms. As well, for V2C/BVO the typical Raman bands of V2C are observed with slight blue-shifting, which confirms the interface interaction between BiVO4 and V2C. Other peaks at around 814, 326, and 118 cm−1 were also detected. The most intense Raman peak at about 814 cm−1 is assigned to the symmetric V–O stretching vibrations (A1g), the sharp bond at 118 cm−1 due to external modes rotation/translation, whereas the weak band is attributed to asymmetric bending vibration (Bg) of the VO43− group in BiVO4.42,43 The above results further prove the successful synthesis of the V2C/BVO composite. For the second V2C/Bi composite, three obvious peaks at 71, 97, and 146 cm−1 are observed. The modes located at 71 and 97 cm−1 can be assigned to the first-order Eg and A1g stretching modes, respectively, of the Bi–Bi vibrational modes of rhombohedral Bi NPs.27,44 The peak at 206 cm−1 is assigned to Eg of V2C(OH)2 with slight blue-shift from that of pure V2C reveals the interaction between Bi NPs and V2C, and the predominant band at 145 cm−1 is attributed to the skeleton bent vibrational mode (B3g), characteristic of the orthorhombic phase of V2O5,45 implying the partial oxidation of the V2C MXene. The obtained XRD and Raman results confirm the synthesis of a new composite V2C/Bi through the reduction of Bi5+ in NaBiO3 to metallic Bi NPs. MXene was used as a reducing agent to donate electrons and convert NaBiO3 to Bi NPs. Previous research has shown that the surface termination groups of MXenes, such as –O, –OH, and –F, provide excellent adsorption and reduction properties.46 The reduction of Bi5+ to Bi0 during the synthesis of V2C/Bi composites occurred through a reduction process involving oxidation states of +5, +3, and 0. The presence of Bi2O3 trace in the XRD spectrum of the V2C/Bi composite confirmed the reduction of Bi5+ to Bi3+. Consequently, Bi NPs are homogeneously distributed on the surface of V2C nanosheets, which are partially oxidized during the synthesis reaction.
The investigation of the V2C and V2C/Bi elemental composition by EDS is displayed in Fig. S1.† The presence of V and C confirms the successful synthesis of MXene, and the existence of O, F, Cl, and Na further proves the presence of active groups on the surface of MXene. In addition, the peak corresponding to Bi in composites demonstrates the successful introduction of Bi into V2C and the formation of a hybrid. More information about the morphology and crystal structure of the material structure was evaluated by SEM, TEM, HRTEM, and selected-area electron diffraction (SAED).
As shown in Fig. 2a, the SEM of V2C shows the typical accordion-like multilayer nanostructure. The high-resolution TEM (HRTEM) image in Fig. S2a† reveals the crystalline lattice of V2C MXene with an interplanar distance of 0.93 nm, corresponding to its (002) planes, indicating the well-crystallized feature of the as-synthesized V2C MXene. The SAED pattern (inset of Fig. S2a†) reveals a hexagonal structure for V2C which is in good agreement with the structure determined from the XRD result. As well, for V2C/BVO, it can be observed that BiVO4 flakes were uniformly wrapped on the sheet of V2C (Fig. 2b). The TEM image in Fig. 2d further suggests that the V2C/BVO is constituted of 2D ultrathin nanosheets. A closer observation (HRTEM in Fig. 2e) revealed the presence of intimate contact and a distinguished contrast between BiVO4 and V2C. Additionally, the interplanar distance of 0.31 and 0.93 nm can be assigned to the (121) BiVO4,47 and (002) planes of V2C, respectively. This observation further corroborates that the as-prepared V2C/BVO is a 2D/2D composite with intimate coupling. This junction can increase the contact area between the two materials which can enhance the electron transfer, and as a result, improve the catalytic activity.32 In addition, the elemental mapping of V2C/BVO (Fig. 2f) reveals the existence of homogeneous distribution of C, V, O, and Bi elements.
 | ||
| Fig. 2 SEM of (a) V2C, (b) V2C/BVO and (c) V2C/Bi, (d) TEM of V2C/BVO, (e) HRTEM of V2C/BVO, (f) the main elemental components mapping of V, C, O and Bi of V2C/BVO, (g) TEM of V2C/Bi and (h) HRTEM of V2C/Bi. | ||
The selected area diffraction (SAED) image in Fig. S2b† confirms the preservation of the original hexagonal structure of MXene. For V2C/Bi, SEM, and TEM/HRTEM images (Fig. 2c, g, and h) reveal the layered structure of V2C was well maintained and Bi nanoparticles were loaded on the surface of the V2C MXene. The elemental mapping results in Fig. S2c† further present the uniform distribution of V, C, Bi, and O elements, suggesting that Bi NPs are well dispersed on V2C.
To examine the surface chemical state, the XPS of the samples was measured. Individual bonding states can exhibit slight variability in their position due to an inhomogeneous surface charging. Fig. S3a† shows the XPS survey spectra which confirmed the presence of O, V, F, and C in all three samples. Compared to the original V2C, the decrease of the F 1s peak and enhancement of the O 1s peak of composites may be due to the substitution of –F groups by –OH groups. The detection of the Bi 4f peak confirms the construction of V2C/BVO and V2C/Bi heterostructures. The XPS spectrum of C 1s in V2C (Fig. 3a) shows the presence of the main peak of C–V (282 eV) belonging to V2C.48 The peaks of adventitious carbon (284.4 eV), C–O (285.9 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.6 eV), and O–CO (289 eV) originate from adsorbed species on the surface of the material and from functional groups on the surface of MXene.38,49–51 For V2C/BVO, the peak from carbide V–C (282.6 eV) was still detected (Fig. 3b), indicating the presence of V2C on the surface of the material. However, in the case of V2C/Bi, the C–V bond peak disappeared (Fig. 3c), implying that the surface of MXene was highly covered with metallic bismuth. The V 2p spectrum (Fig. 3d) shows the characteristic peaks of V2+ species in V2C at 513.5 (2p3/2) and 521.4 (2p1/2) eV, as well as V4+ species in V2C at 516.5 (2p3/2) and 524.2 (2p1/2) eV.48 The obvious peak (V2+) confirms the strong metal carbide interaction (V–C) in V2C, while peaks of V4+ confirm the existence of vanadium oxide (V–O) on the surface of V2C. Simultaneously, the XPS spectrum of V 2p in V2C/BVO and V2C/Bi composites is shown in Fig. 3e and f. For both composites, the peak related to V2+ species can still be observed but is greatly decreased. Meanwhile, the peak intensity of V–O species increased, which proved the partial oxidation of the surface V atoms during the composite's preparations. Bi 4f peaks (Fig. S3b†) are deconvoluted into two pairs of peaks at 159 eV (Bi 4f7/2) and 164 eV (Bi 4f5/2) corresponding to the binding energy of Bi3+ in V2C/BVO.32 Similarly for V2C/Bi composite, the Bi 4f peaks correspond to oxidized surface (Bi3+) of the nanoparticles.48 The fitting results of the respective elements are listed in Table S1.†
O (287.6 eV), and O–CO (289 eV) originate from adsorbed species on the surface of the material and from functional groups on the surface of MXene.38,49–51 For V2C/BVO, the peak from carbide V–C (282.6 eV) was still detected (Fig. 3b), indicating the presence of V2C on the surface of the material. However, in the case of V2C/Bi, the C–V bond peak disappeared (Fig. 3c), implying that the surface of MXene was highly covered with metallic bismuth. The V 2p spectrum (Fig. 3d) shows the characteristic peaks of V2+ species in V2C at 513.5 (2p3/2) and 521.4 (2p1/2) eV, as well as V4+ species in V2C at 516.5 (2p3/2) and 524.2 (2p1/2) eV.48 The obvious peak (V2+) confirms the strong metal carbide interaction (V–C) in V2C, while peaks of V4+ confirm the existence of vanadium oxide (V–O) on the surface of V2C. Simultaneously, the XPS spectrum of V 2p in V2C/BVO and V2C/Bi composites is shown in Fig. 3e and f. For both composites, the peak related to V2+ species can still be observed but is greatly decreased. Meanwhile, the peak intensity of V–O species increased, which proved the partial oxidation of the surface V atoms during the composite's preparations. Bi 4f peaks (Fig. S3b†) are deconvoluted into two pairs of peaks at 159 eV (Bi 4f7/2) and 164 eV (Bi 4f5/2) corresponding to the binding energy of Bi3+ in V2C/BVO.32 Similarly for V2C/Bi composite, the Bi 4f peaks correspond to oxidized surface (Bi3+) of the nanoparticles.48 The fitting results of the respective elements are listed in Table S1.†
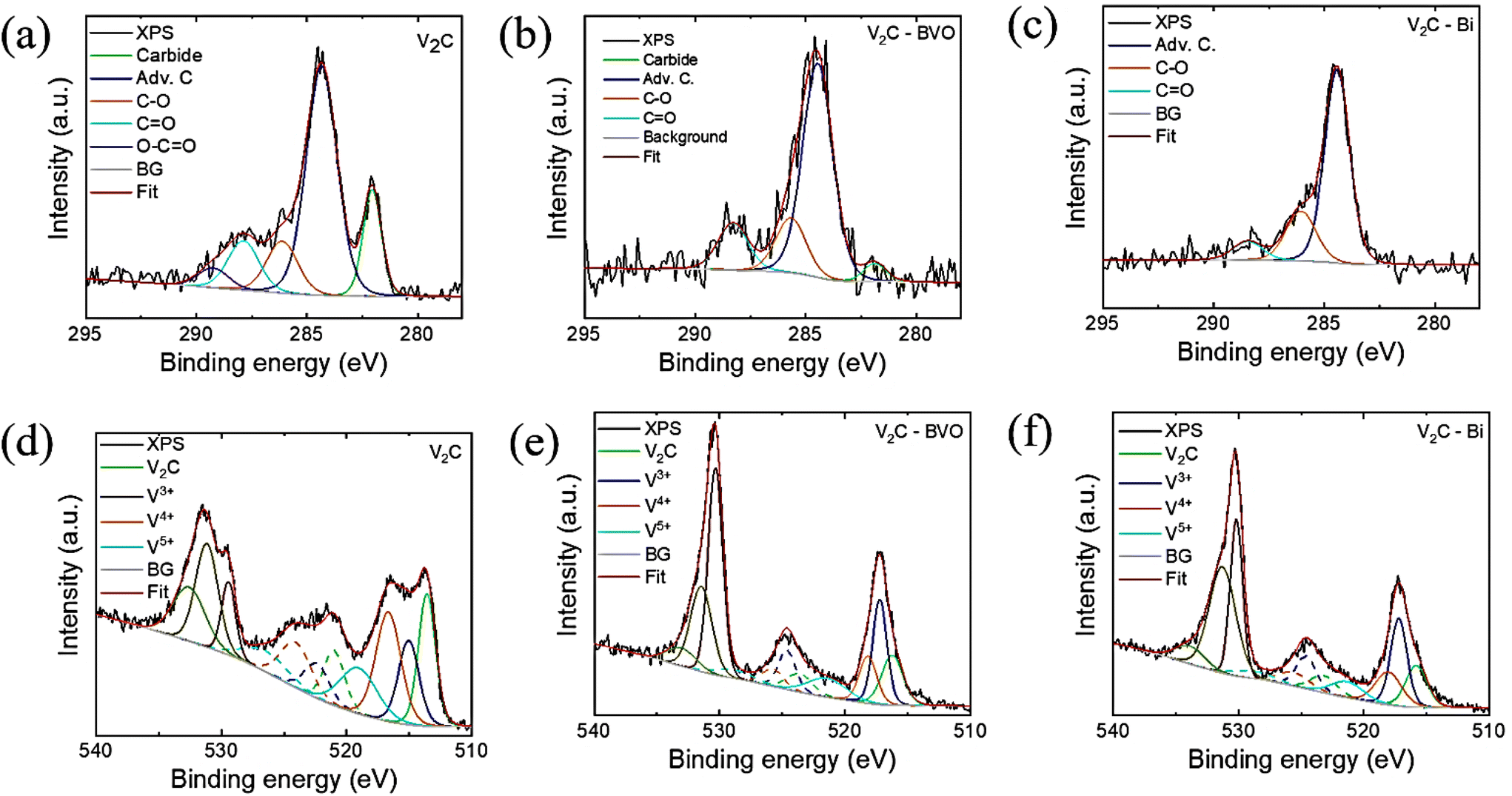 | ||
| Fig. 3 High-resolution XPS spectra of C 1s in (a) V2C, (b) V2C/BVO, and (c) V2C/Bi; high-resolution XPS spectra of V 2p in (d) V2C, (e) V2C/BVO, and (f) V2C/Bi. | ||
Since the SSA of the catalyst plays a significant role in catalytic activity, we next measured the Brunauer–Emmett–Teller (BET) SSA of the samples by nitrogen absorption and desorption test. As depicted in Fig. S4,† the composite V2C/BVO possesses a larger SSA (8.56 m2 g−1) and pore volume value (0.121 cm3 g−1) compared to V2C/Bi and pristine V2C. The larger pore volume and SSA confirmed that the 2D/2D composite with 2D/2D structure possesses an increased number of active sites for redox reactions.
3.2. Catalytic HER activity
The HER activity of all the catalysts was measured by linear sweep voltammetry (LSV) in 0.5 M H2SO4 using three-electrode cells. The HER activity was compared to commercial Pt/C (20 wt%) and glassy carbon electrode (GC). As shown in Fig. 4a, the GC electrode displayed negligible HER activity which indicates that the observed activity is mainly due to the catalysts. As expected, the Pt/C (20 wt%) shows the highest catalytic activity with an overpotential of 68 mV at 10 mA cm−2 current density (Fig. 4b). In comparison, the V2C/BVO (with an overpotential of 384 mV at 10 mA cm−2) exhibited a much better HER performance than V2C/Bi (663 mV) and V2C (823 mV), indicating that the formed 2D/2D V2C/BVO hybrid provided the active species for the HER. The overpotential (η10) of the V2C/BVO electrocatalyst corresponding to a current density of 10 mA cm−2 is further compared to these non-noble metal electrocatalysts in Table S2.† It should be noted that the V2C/BVO catalyst needs lower overpotential compared to previously reported catalysts. Following this, we evaluated the Tafel slope which is often used to characterize the rate-limiting step of electrocatalytic reaction. The HER mechanism in acidic media is based on two possible reaction pathways namely: (a) Volmer–Heyrovsky and (b) Volmer–Tafel. HER starts with the Volmer reaction, where the hydronium protons acquire electrons on the surface of the electrode and generate an adsorbed hydrogen atom Hads as an intermediate state (H3O+ + e− → Hads + H2O). Then, H2 molecules are dominantly generated through one of two different reaction processes depending on the surface coverage of Hads. If the coverage of Hads is high, two Hads combine to form H2, so the Tafel reaction becomes the rate-determining step for the H2 production (Hads + Hads → H2). In reverse, if the coverage of Hads is low the Heyrovsky reaction becomes the rate-determining step, where a single Hads atom attracts both a proton and an electron (e−) to produce an H2 molecule (Hads + e− + H3O+ → H2 + H2O). When the Volmer step is the rate-limiting step, the Tafel slope reaches 120 mV dec−1. Whereas the Tafel slope is reduced to 40 or 30 mV dec−1 when the Heyrovsky step or Tafel step is the rate-limiting step.52,53Fig. 4c presents the Tafel plots of all samples, where V2C/BVO shows a smaller Tafel slope of 160 mV dec−1 than those of V2C/Bi (173 mV dec−1) and V2C (204 mV dec−1), revealing that the HER process of V2C/BVO is controlled by the Volmer–Heyrovsky mechanism and the Volmer step is the rate-determining step.54,55 As previously reported, under acidic conditions, the reaction between –O functional groups on the surface of MXenes and H+ ions in H2SO4 aqueous solution promotes the current before the HER process, which explains the main reason for the high Tafel slope in the current study.56,57 To further probe the origin of the enhanced HER performance of prepared electrocatalysts, the EIS measurement is a crucial method to investigate the interfacial properties and the charge-transfer resistance between the solution and the electrode. Meanwhile, the arc radius in the EIS Nyquist plots could be a credible method to estimate the charge transfer rate.58 It was commonly believed that a smaller arc in the EIS Nyquist plot implied less charge migration resistance.58–60 As displayed in Fig. 4d, V2C/BVO composite displayed a smaller arc radius, indicating faster charge transfer at the electrode/electrolyte interface, leading to enhanced HER performance. The exchange current density (j0) is another important parameter in the evaluation of HER activity, which is defined by the cathodic current density of the Tafel equation at the zero overpotential. The j0 of all the catalysts was evaluated from the Tafel plot by extending to X-axis (Fig. S5†). The j0 of the V2C/BVO hybrid is estimated to be 16 μA cm−2 which is significantly higher than the V2C/Bi (10 μA cm−2) and V2C (9 μA cm−2). It is reflected that the interfacial electronic coupling could boost the electron transfer capability of V2C/BVO hybrids for HER catalysis.61 In addition, to better understand the intrinsic activity of the catalyst, we further evaluated the double-layer capacitance (Cdl) by a series of cyclic voltammetry (CV) measurements at different scan rates from 10 to 100 mV s−1 in the capacitive range of 0 to 0.1 V (vs. RHE). As expected, the V2C/BVO electrode exhibits a higher current than that of V2C and V2C/Bi as shown in Fig. S6.† It indicates more accessible active sites are created during hydrothermal synthesis of V2C/BVO which is favourable for the enhanced HER activity. To calculate the double layer capacitance (Cdl), we first determined the capacitive density current average (Δj) using the formula Δj = |jc − ja|/2, where jc represents the cathodic density current and ja represents the anodic density current. At a potential of 0.05 V, we plotted Δj against various scan rates and determined the linear slope value, which corresponded to Cdl.62 As shown in Fig. 4e, the Cdl of V2C/BVO (6.66 mF cm−2) was much larger than that of V2C/Bi (3.23 mF cm−2) and V2C (1.56 mF cm−2) which was attributed to the high SSA of the 2D/2D V2C/BVO composite and is beneficial to the exposure of more accessible active sites for the HER and diffusion of electrocatalytic active species.63,64 We can conclude that the BiVO4 which is in situ formed and uniformly coated on the V2C layers provides additional active sites for HER reaction. Therefore, the actual active site responsible for the HER reaction in the V2C/BVO composite is the interface between the V2C and BiVO4 layers, where synergistic effects may occur.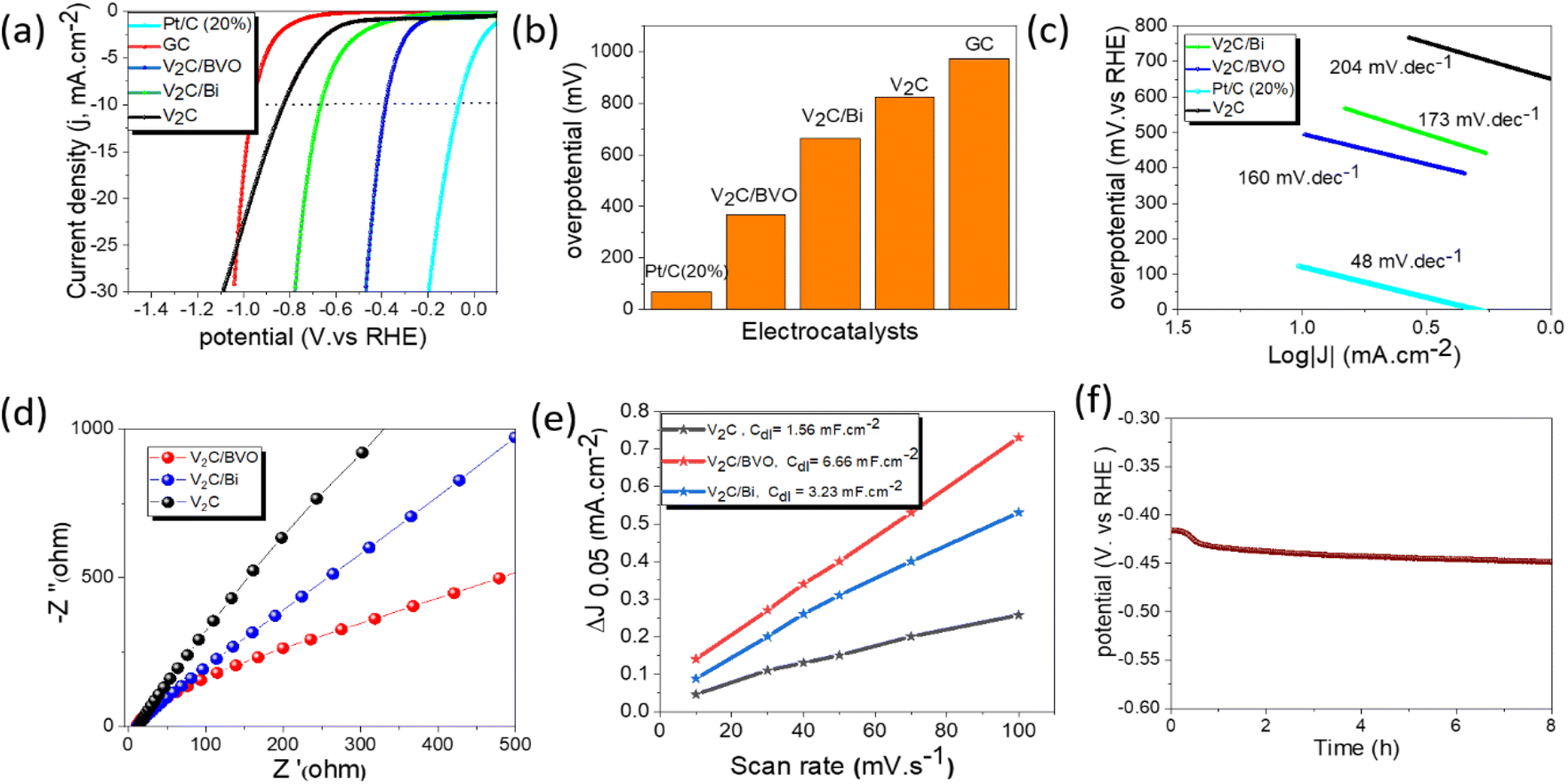 | ||
| Fig. 4 Electrochemical measurements for hydrogen evolution in 0.5 M H2SO4 acidic medium: (a) LSV curves, (b) onset overpotential at j = 10 mA cm−2 of different electrocatalysts, (c) the corresponding Tafel plots, (d) EIS Nyquist plot of different catalysts, (e) capacitive Δj as a function of scan rates for different samples, and (f) the chronoamperometric measurement for V2C/BVO at a constant current density of −10 mA cm−2. | ||
All the electrochemical measurements for HER reactions show that V2C/BVO composite exhibits impressive electrocatalytic ability in acidic media compared to pure V2C and V2C/Bi composite. The outstanding electrocatalytic performance of the V2C/BVO composite is supposed to be originating from; (i) a well-constructed 2D/2D (V2C/BVO) structure which is beneficial to increase the SSA, which allows the exposure of numerous catalytic active sites and full penetration of electrolyte, (ii) the synergistic effect of V2C and BiVO4, and their intrinsic electrocatalytic response, and (iii) the high metallic conductivity of V2C MXene sheets acts as a conductive material which facilitates electron transport. All these features confirm that the newly prepared composite (V2C/BVO) in a facile hydrothermal way could be considered as a promising non-noble catalyst for HER application. HER efficiencies of as-prepared V2C/BVO composite were also evaluated as electrocatalyst for HER in alkaline (1 M KOH, pH = 14) and neutral solution (1 M Na2SO4, pH = 7). Fig. S7a† shows that V2C/BVO composite exhibits a high overpotential of 584 and 679 mV at a current density of 10 mA cm−2 in basic and neutral media, respectively. The results suggest that the HER reactions of V2C/BVO under alkaline conditions are harder than in acidic solutions. This indicates the ability of the V2C/BVO catalyst to adsorb hydrogen rather than accelerated water dissociation.
Long-term stability testing on V2C/BVO was also carried out by a chronopotentiometry test. Fig. 4f shows low voltage increasing over 8 h at a constant current density (10 mA cm−2) operation. Fig. S7(b and g)† shows the SEM, EDS, and XPS of the catalyst after the stability test. SEM and EDS elemental mapping images show that V2C/BVO still has a multilayer structure without being damaged and the existence of all chemical composition as before electrolysis. As well, post-stability XPS measurements indicate the high structural stability of V2C/BVO toward the HER process. Furthermore, we conducted the EIS analysis after stability as shown in Fig. S7h.† The Nyquist plots displayed that the arc radius of the V2C/BVO composite after and before stability is mostly identical, revealing the same charge transfer capacity and, therefore, no considerable alteration of the electrical properties confirming the high stability of V2C/BVO.
3.3. Evaluation of NRR performance
The Electrochemical NRR measurements were performed in a two-compartment cell (H-cell) as shown in Fig. S8.† The LSV curves of catalysts were compared under an N2-saturated 0.1 M KOH electrolyte (Fig. 5a). It can be found that the current value of V2C/Bi is higher than that of V2C and V2C/BVO at the same potential, suggesting its superior catalytic activity. The result further proves that the addition of the metal Bi effectively improved the catalytic performance of the V2C. Linear sweep voltammetry (LSV) measurements of the V2C/Bi composite were then performed in Ar- and N2-saturated 0.1 M KOH aqueous under room temperature and atmospheric pressure (Fig. 5b). Obviously, the reduction of current density in the potential range of −0.4 V to −0.7 V of V2C/Bi in N2 is slightly lower than that of the Ar atmosphere, implying that NRR was more favourable over HER in the measured potential window. Next, a CV test was performed between −1 V and 1.8 V vs. RHE in N2-saturated KOH. Fig. S9a† shows 3 peaks at 0.45, 0, and −0.56 V vs. RHE. The anodic and cathodic signals at 0.45 and 0 V vs. RHE can be assigned to the surface reversible oxidation and subsequent reduction of the electrocatalyst. Specifically, these signals indicate the interconversion of species between Bi (0) and Bi (OH)3 (or BiOOH) during the electrochemical reaction.65 The broad cathodic peak that appears at −0.56 V is attributed to the reduction of Bi3+ to metallic Bi.66,67 This observation is consistent with the XRD results, which showed the presence of a small amount of Bi2O3 in the V2C/Bi composite. The complete reduction of Bi3+ to metallic Bi occurs during the first scan and subsequent scans may not show this cathodic peak due to the saturation of the surface with metallic Bi. After 3 consecutive cycles, the CV curves are almost identic, confirming the stability of the electrode before the NRR test. The electrochemical NRR performances of catalysts were further evaluated by chronoamperometry tests under potentials ranging from −0.3 to −0.7 V vs. RHE for 2 h in N2-saturated KOH aqueous solution as shown in Fig. S9b–d.† It is clearly depicted that the current density improved with the increase of potential and remains almost stable during the two hours at different potentials, implying that both V2C/Bi and V2C/BVO composites exhibit good electrochemical stability during the NRR test. While for V2C, at −0.5 V vs. RHE, the current density increased from the initial −0.13 mA cm−2 value to about −1.84 mA cm−2 after 2 h, indicating an in situ transformation during the electrocatalytic tests. It is possible that the decrease in current density observed during the electrocatalytic experiment was due to the oxidation of V2C and the formation of VOx species. These oxide layers can act as passivating layers that hinder electron transfer between the V2C and the electrolyte solution, leading to a decrease in current density over time. After electrolysis at a given potential, the generated ammonia was determined spectrophotometrically by Nessler's method. The NH3 production and corresponding FE under different potentials are displayed in Fig. 5c and d. As clearly shown, the V2C/Bi composite exhibits the highest NH3 yield of 88.6 μg h−1 cm−2 and the highest faradaic efficiency (FE) of 8% at −0.5 V vs. RHE which are both higher than the values obtained for V2C/BVO (NH3 yield: 40.5 μg h−1 cm−2, FE: 5.45%). For confirmation, the indophenol method (Fig. S10d–f†) was also used to measure the concentration of ammonia for V2C/Bi at −0.5 V (V vs. RHE), and the result was 92 μg h−1 cm−2, which is close to the value calculated from the Nessler's reagent method. This result confirms that both methods produce consistent values.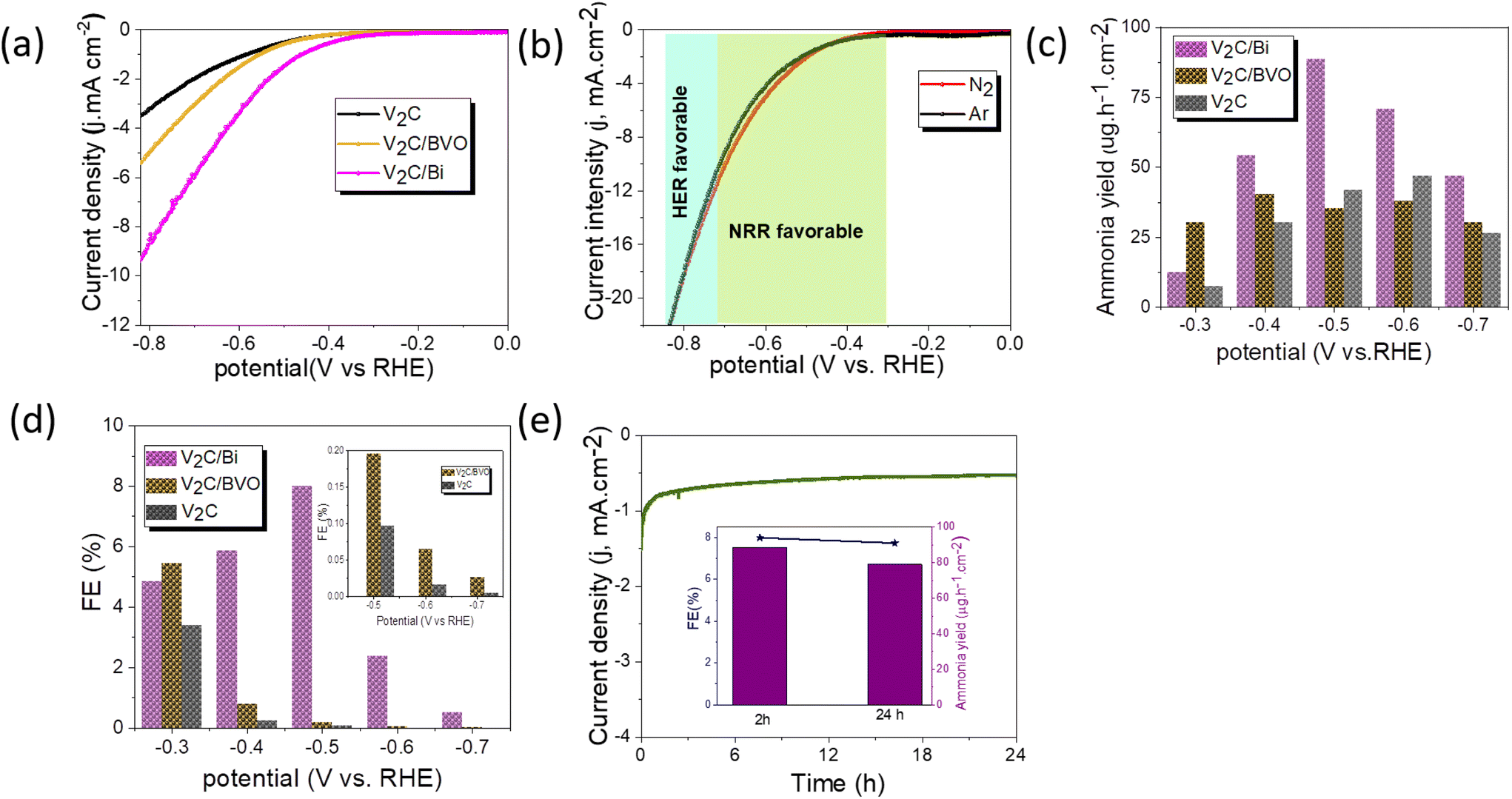 | ||
| Fig. 5 (a) Comparison of LSV curves of GC electrode, V2C, V2C/BVO, and V2C/Bi in N2-saturated 0.1 M KOH with a scan rate of 10 mV s−1, (b) LSV curves of V2C/Bi in Ar/N2-saturated KOH electrolyte with a scan rate of 10 mV s−1, (c) corresponding NH3 yields (d) corresponding FE, (e) chronoamperometric test of V2C/Bi at −0.5 V for 24 h (insert: corresponding NH3 yields and FE after stability test). | ||
We compared the ammonia yield rates and the corresponding faradaic efficiencies of V2C/Bi with those of the recently reported MXenes-based NRR catalysts. As listed in Table S3,† the ammonia yield produced over V2C/Bi is significantly higher than that of all the listed catalysts. Furthermore, we evaluated the NRR performance of the pristine V2C. Compared to V2C/Bi, V2C exhibits significantly lower NRR activity (NH3 yield: 46.8 μg h−1 cm−2, FE: 3.4%), implying that the Bi supported on V2C should be the main active sites for NRR for V2C/Bi. Obviously, the NH3 yield, and FE decreased when the applied potentials shifted to more negative values, mainly due to the competitive adsorption between N2 and H2 on the electrode surface. Furthermore, no N2H4 could be detected after the electrocatalytic NRR at different potentials. This further confirms the outstanding selectivity of the V2C/Bi catalyst. To confirm the electrochemical reduction of N2 to NH3 on V2C/Bi, three controlled experiments were conducted: (i) the working electrode in electrolyte with continual Ar flow at −0.5 V for 2 h electrolysis; (ii) the working electrode with continual N2 flow at open circuit potential (OCP) for 2 h electrolysis; and (iii) bare glassy carbon electrode in electrolyte with continual N2 flow at −0.5 V for 2 h electrolysis. As shown in Fig. S10,† very low current density and very low NH3 and corresponding FE were detected for all those control experiments, suggesting that the produced NH3 is derived from the electrochemical N2 reduction catalyzed by the V2C/Bi catalyst.
For electrocatalytic NRR, long-term catalytic stability is critical for the possible practical application. Fig. 5e shows a very steady current density over 24 h of continuous chronoamperometric test at −0.5 V, with a 90.5% retention of its initial NH3 yield and almost unchanged FE after 24 h (inset Fig. 5e). Additionally, the SEM images and corresponding EDS elemental mapping (Fig. S11†) of the V2C/Bi after stability revealed that the morphology and the chemical composition have no obvious changes compared to the fresh one, proving the robustness of the heterostructure for stable NRR performance. To further evaluate the long-term stability of V2C/Bi catalyst, EIS analysis was recorded before and after 24 h of chronoamperometry analysis. Fig. S11e† indicates that the catalyst exhibits negligible deviation of the arc radius after stability which confirms its excellent long-term stability. Furthermore, the XPS spectra after NRR stability (Fig. S11f–h†) confirm that the V2C/Bi well retains its initial chemical bonding state for V 2p and C 1s. The appearance of two new peaks with low intensity at 157.4 and 162.4 eV in the Bi 4f spectra indicates the presence of Bi0. As discussed earlier, V2C/Bi composite exhibits reversible redution at 0 V vs. RHE (Fig. S9a†) of bismuth nanoparticles. Thus, these nanoparticles were reduced during the long term experiment to metalic Bi. Subsequently when exposed to air/oxygen, their surface was again oxidized. All the above results unambiguously demonstrate V2C/Bi to be a promising NRR catalyst for efficient NH3 electro-synthesis. Generally, the NRR reaction process involves continuous proton–electron pairs interacting with N2. As already proven HER is the main competitive reaction of the NRR. Therefore, increasing N2 coverage on the electrocatalyst and suppressing the HER process during NRR is an effective strategy to increase the selectivity to NH3. Hence, we study the HER activity of the V2C/Bi nanocomposite and compare it with that of V2C MXene. As shown in Fig. 6a, V2C/Bi exhibited lower HER performance compared to V2C. The Tafel slopes of the samples were then calculated. As demonstrated in Fig. 6b, the Tafel slopes of the V2C/Bi composite were higher than that of the pristine V2C MXene, indicating a less favourable kinetics process for HER over the V2C/Bi electrode. In addition, the EIS measurement (Fig. 6c) shows that the arc radius of V2C/Bi is significantly larger than that of V2C, indicating low electron transfer in the composite and resulting in lower HER performance. This result confirms the negative effect of the Bi nanoparticles on HER, which, on the other hand, is highly beneficial for the improvement of NRR performance for gradually elevated ammonia yields. Thus, we proved that the HER process can be significantly repressed with Bi by inhibiting the adsorption of H on active sites. The same result has already been proven by previous researchers when studying the NRR activity of the Mo-doped V2C system.68 Theoretical calculations confirmed that ΔGN2* > ΔGH+* on Mo-doped V2C, which confirms the restriction of the HER reaction after doping of V2C with Mo.
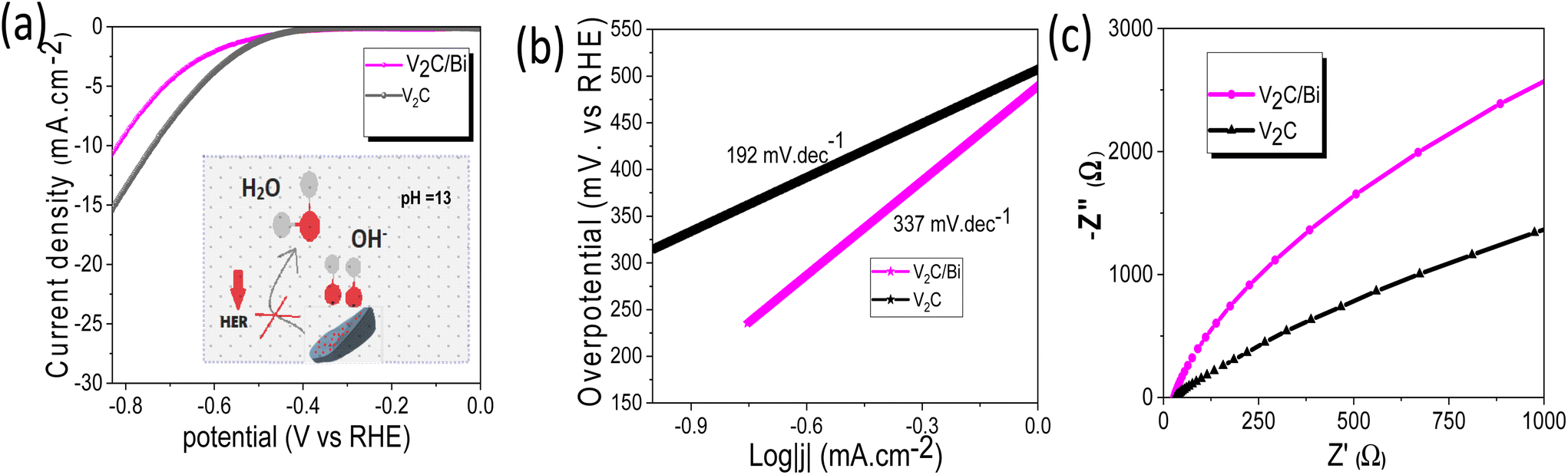 | ||
| Fig. 6 (a) LSV of V2C/Bi and V2C in Ar saturated KOH (insert: the deactivation mechanism of the HER process over V2C/Bi), (b) the corresponding Tafel plots analysis, and (c) EIS of V2C and V2C/Bi. | ||
In this study, the V2C/Bi heterojunction is produced by supporting the Bi-metal on O-terminated V2C, indicating that V2C can offer electrons that can be transported to Bi through the heterojunction interface (Fig. S12†). The electrons provided by V2C are mainly accumulated at the V2C/Bi interface. Such electron-rich interfaces facilitate the downshift of the p-band centre of interfacial Bi atoms,69 which serve as the main active sites to promote the N2 activation and hydrogenation while impeding the competing hydrogen evolution. This suggests the outstanding selectivity of V2C/Bi for N2-to-NH3 conversion.
4. Experimental procedures
4.1 Synthesis of layered V2C
Multilayered V2C MXene powder was obtained by etching Al from V2AlC powder according to a previous report.70 Briefly, 6 g NaF was dissolved in 120 mL of deionized water, then 140 mL HCl solution (6 mol L−1) was added to obtain the etching solution. After that, 6 g of as-received V2AlC MAX phase powder was slowly added to the etching solutions in Teflon-lined stainless-steel autoclaves (500 mL). The Teflon bottle was kept in an oil bath with magnetic stirring at 90 °C for 5 days. After the end of the etching process, the dispersion was then centrifuged at 5000 rpm for 5 min to harvest the sediment, which was further centrifuged and washed with deionized (DI) water and ethanol six times to raise the pH of the supernatant to almost neutral (pH ∼ 7). Finally, the solid product was collected and then placed in a vacuum freeze dryer at −50 °C for 24 h to remove the residual water and obtain a powder of V2C MXene.4.2. Synthesis of V2C/BVO by hydrothermal method
The composite V2C/BVO was synthesized through a facile hydrothermal method (Fig. 7). To do this, 100 mg of the prepared V2C MXene powder was put in 40 mL of deionized water and ultrasonically dispersed for 30 minutes to obtain a uniform MXene suspension. Another solution was prepared by dissolving 100 mg NaBiO3 in 25 mL of deionized water. Then, the NaBiO3 aqueous solution was slowly added to the uniform MXene suspension. The mixed suspension was vigorously stirred for 30 minutes, then transferred to a 100 mL Teflon-lined autoclave for 4 h at 200 °C. The dark green precipitate was centrifuged and thoroughly washed with deionized water and absolute ethanol several times. The V2C/BVO composite was finally obtained by drying it in a vacuum oven at 60 °C overnight.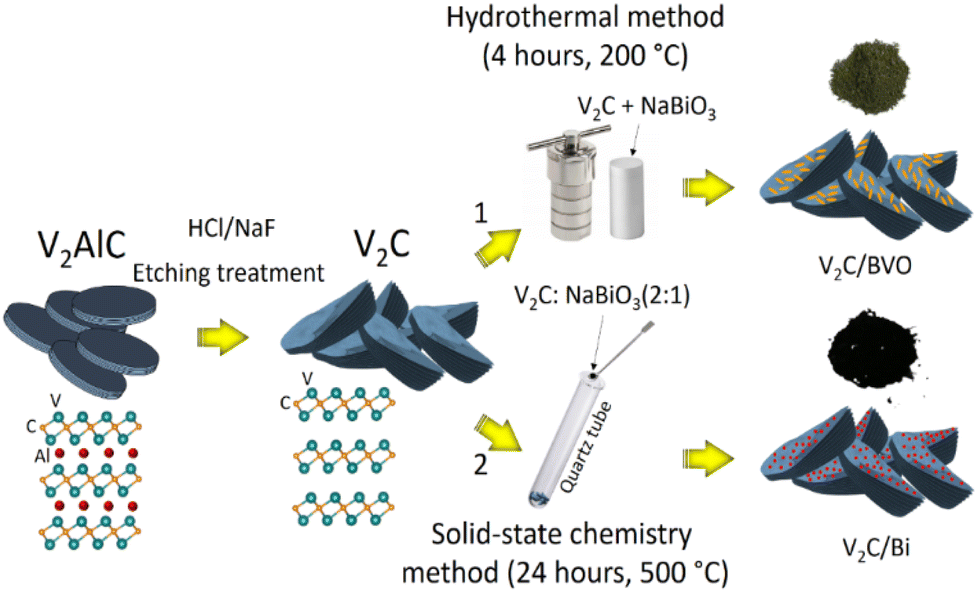 | ||
| Fig. 7 A schematic illustration of the synthesis of composites V2C/BVO and V2C/Bi. | ||
4.3. Synthesis of V2C/Bi by solid-state annealing technique
The composite V2C/Bi was synthesized through a solid-state annealing method following the steps described in the previous report (Fig. 7).71 In this process, the obtained V2C MXene and NaBiO3 were mixed and crushed for 20 minutes with an agate mortar. After uniformly mixing the two components, the mixture powder was inserted inside a quartz ampoule and then was vacuum sealed to a pressure of approximately 10−5 mbar using a vacuum pump. The vacuum sealing of the ampoule was required to prevent the mixture from moisture. Finally, the sealed ampoule including the mixture was placed in a furnace and heated at 500 °C for 24 h using a 5 °C min−1 heating and cooling rate. After annealing, the ampoule was removed from the furnace and cooled in the air. Then, it was opened, and the black precipitate was manually collected.4.4. Characterization
X-ray diffraction (XRD) patterns of the synthesized materials were acquired on a Bruker D8 diffractometer operating at 40 kV with Cu Kα radiation (λ = 1.54056 Å). The diffraction patterns were collected between 5° and 60° with a step size of 0.02°. Scanning electron microscopy (SEM) was performed by a field-emission scanning electron microscope (FESEM, Tescan Lyra dual microscope) with attached energy-dispersive X-ray spectroscopy of a 20 mm2 SDD detector (Oxford Instruments). Transmission electron microscopy (TEM, EFTEM Jeol 2200 FS microscope) equipped with energy dispersive spectrometry (EDS) at an acceleration voltage of 200 kV. Raman spectra were performed by Raman spectrometer (Renishaw, England) in backscattering geometry with a CCD detector and a DPSS laser (532 nm, 50 mW) with an applied power of 0.5 mW. The samples were prepared by drop-casting a film on a nickel substrate, directly prior to the testing. X-ray photoelectron spectroscopy (XPS) analysis was recorded using a SPECS spectrometer equipped with an Al Kα X-ray source (1486.7 eV). The Brunauer–Emmett–Teller (BET) and pore size distribution of the samples were measured by a Micro-metrics ASAP 2020 apparatus at liquid nitrogen temperature.4.5. Electrochemical measurements
| ERHE = EAg/AgCl + 0.197 + 0.059 pH |
Linear sweep voltammetry (LSV) was performed in 0.5 M H2SO4 with a sweep rate of 5 mV s−1. The Tafel slope of the catalyst can be calculated by the formula η = a + blog|j| (η, a, b, and j are standing for overpotential, Tafel constant, Tafel slope, and current density, respectively). The overpotential for HER was calculated using the following equation η = 0 − E (vs. RHE (V)). Electrochemical impedance spectroscopy (EIS) was recorded by applying an AC potential amplitude of 10 mV within the frequency range from 0.1–1000 Hz. The double-layer capacitances were obtained through cyclic voltammetry (CV) with various scan rates in the voltage range of 0–0.1 V vs. RHE.
The ammonia formation rate was calculated according to eqn (1):
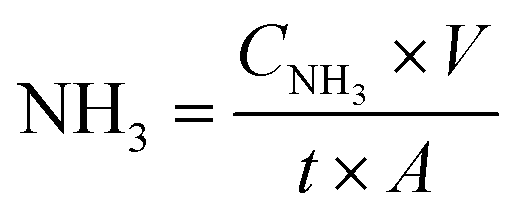 | (1) |
The faradaic efficiency was calculated based on eqn (2):
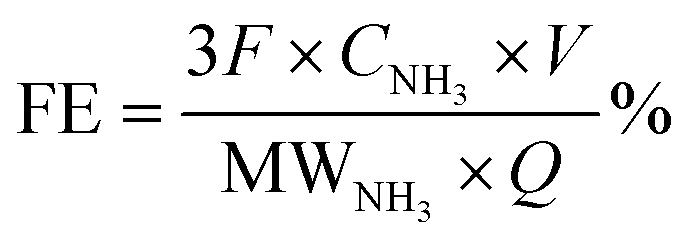 | (2) |
The amount of Hydrazine (N2H4) after the electrolysis reaction was detected by the D1385 method.26 For determination of the generated hydrazine, 10 mL of the electrolyte after the NRR test was taken out and twelve drops of hydrazine reagent were subsequently added. The absorbance was measured at a wavelength of 466 nm, after 12 minutes.
5. Conclusions
Two nanocomposites consisting of 2D/2D V2C/BVO and 2D/0D V2C/Bi NPs were successfully prepared and applied for electrocatalytic HER and NRR. The V2C/BVO composite has been synthesized via a facile hydrothermal method and applied as the catalyst electrode for HER in acidic conditions. Compared to pristine V2C, the HER activity of V2C/BVO has been greatly increased due to the strong interfacial interactions between BVO sheets and V2C MXene nanosheets. This 2D/2D structure provided sufficient active sites and ensured favorable electrochemical kinetics, improving the HER activity. Furthermore, the optimized composite showed excellent long-term stability over 8 h in acidic media at the current density of 10 mA cm−2. Also, V2C/Bi was proven to be a highly efficient NRR electrocatalyst, achieving a FE and NH3 yield of 8% and 88.6 μg cm−2 h−1, respectively. The obtained values are among the highest NRR selectivity for MXenes-based NRR catalysts. We also proved that impeding the competing HER by doping V2C with p block metal (Bi) is an interesting strategy to achieve high N2-to-NH3 conversion.This work provides inexpensive strategies for fabricating novel V2C-based catalysts and may shed some light on energy-related applications.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. A. would like to acknowledge the European Structural and Investment Funds, “CHEMFELLS IV” (CZ.02.2.69/0.0/0.0/20_079/0017899). This work was supported by ERC-CZ program (project LL2101) from Ministry of Education Youth and Sports (MEYS).References
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC.
- H. Liu, L. Syama, L. Zhang, C. Lee, C. Liu, Z. Dai and Q. Yan, SusMat, 2021, 1, 482–505 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- D. A. Kuznetsov, Z. Chen, P. V. Kumar, A. Tsoukalou, A. Kierzkowska, P. M. Abdala, O. V. Safonova, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2019, 141, 17809–17816 CrossRef CAS PubMed.
- A. Liu, X. Liang, X. Ren, W. Guan, M. Gao, Y. Yang, Q. Yang, L. Gao, Y. Li and T. Ma, Adv. Funct. Mater., 2020, 30, 2003437 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- X. Wu, Z. Wang, M. Yu, L. Xiu and J. Qiu, Adv. Mater., 2017, 29, 1607017 CrossRef PubMed.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- J. Zhao, L. Zhang, X.-Y. Xie, X. Li, Y. Ma, Q. Liu, W.-H. Fang, X. Shi, G. Cui and X. Sun, J. Mater. Chem. A, 2018, 6, 24031–24035 RSC.
- Y. Luo, G.-F. Chen, L. Ding, X. Chen, L.-X. Ding and H. Wang, Joule, 2019, 3, 279–289 CrossRef CAS.
- S. Li, P. Tuo, J. Xie, X. Zhang, J. Xu, J. Bao, B. Pan and Y. Xie, Nano Energy, 2018, 47, 512–518 CrossRef CAS.
- L. Li, X. Wang, H. Guo, G. Yao, H. Yu, Z. Tian, B. Li and L. Chen, Small Methods, 2019, 3, 1900337 CrossRef CAS.
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2018, 1, 173–180 CrossRef CAS.
- L. R. Johnson, S. Sridhar, L. Zhang, K. D. Fredrickson, A. S. Raman, J. Jang, C. Leach, A. Padmanabhan, C. C. Price, N. C. Frey, A. Raizada, V. Rajaraman, S. A. Saiprasad, X. Tang and A. Vojvodic, ACS Catal., 2020, 10, 253–264 CrossRef CAS.
- C. Ling, L. Shi, Y. Ouyang, Q. Chen and J. Wang, Adv. Sci., 2016, 3, 1600180 CrossRef PubMed.
- W. Kong, F. Gong, Q. Zhou, G. Yu, L. Ji, X. Sun, A. M. Asiri, T. Wang, Y. Luo and Y. Xu, J. Mater. Chem. A, 2019, 7, 18823–18827 RSC.
- J. Liu, Y. Liu, D. Xu, Y. Zhu, W. Peng, Y. Li, F. Zhang and X. Fan, Appl. Catal., B, 2019, 241, 89–94 CrossRef CAS.
- K. Li, T. Jiao, R. Xing, G. Zou, Q. Zhao, J. Zhou, L. Zhang and Q. Peng, Green Energy Environ., 2018, 3, 147–155 CrossRef.
- P. Li, J. Zhu, A. D. Handoko, R. Zhang, H. Wang, D. Legut, X. Wen, Z. Fu, Z. W. Seh and Q. Zhang, J. Mater. Chem. A, 2018, 6, 4271–4278 RSC.
- T. A. Shifa, F. Wang, Y. Liu and J. He, Adv. Mater., 2019, 31, 1804828 CrossRef CAS PubMed.
- R. Zhang, L. Ji, W. Kong, H. Wang, R. Zhao, H. Chen, T. Li, B. Li, Y. Luo and X. Sun, Chem. Commun., 2019, 55, 5263–5266 RSC.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- N. Antonatos, J. D. Elliott, V. Mazánek, P. Marvan, P. Carbone, W. Geng, Y. Jing and Z. Sofer, J. Mater. Chem. A, 2022, 10, 11904–11916 RSC.
- S. Mourdikoudis, N. Antonatos, V. Mazánek, I. Marek and Z. Sofer, Inorg. Chem., 2022, 61, 5524–5538 CrossRef CAS PubMed.
- A. Liu, X. Liang, H. Zhu, X. Ren, L. Gao, M. Gao, Y. Yang, G. Li and T. Ma, ChemCatChem, 2022, 14, e202101683 CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- H.-x. Pan, L.-p. Feng, W. Zeng, Q.-c. Zhang, X.-d. Zhang and Z.-t. Liu, Inorg. Chem., 2019, 58, 13195–13202 CrossRef CAS PubMed.
- Z. Wu, J. Mei, Q. Liu, S. Wang, W. Li, S. Xing, J. Bai, J. Yang, W. Luo, O. Guselnikova, A. P. O'Mullane, Y. Gu, Y. Yamauchi, T. Liao and Z. Sun, J. Mater. Chem. A, 2022, 10, 808–817 RSC.
- Y. Li, Y. Liu, D. Xing, J. Wang, L. Zheng, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Appl. Catal., B, 2021, 285, 119855 CrossRef CAS.
- Y. Guan, S. Jiang, Y. Cong, J. Wang, Z. Dong, Q. Zhang, G. Yuan, Y. Li and X. Li, 2D Mater., 2020, 7, 025010 CrossRef CAS.
- S. A. Zahra, B. Anasori, M. Z. Iqbal, F. Ravaux, M. Al Tarawneh and S. Rizwan, APL Mater., 2022, 10, 060901 CrossRef CAS.
- H. Chand, M. Sharma and V. Krishnan, Sep. Purif. Technol., 2022, 292, 121032 CrossRef CAS.
- G. Zou, Z. Zhang, J. Guo, B. Liu, Q. Zhang, C. Fernandez and Q. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 22280–22286 CrossRef CAS PubMed.
- S. Dadashi, H. Delavari and R. Poursalehi, Procedia Mater. Sci., 2015, 11, 679–683 CrossRef CAS.
- W. Lv, G. Wu, X. Li, J. Li and Z. Li, Energy Storage Mater., 2022, 46, 138–146 CrossRef.
- D. Sha, C. Lu, W. He, J. Ding, H. Zhang, Z. Bao, X. Cao, J. Fan, Y. Dou, L. Pan and Z. Sun, ACS Nano, 2022, 16, 2711–2720 CrossRef CAS PubMed.
- L. Qin, S. Xu, Y. Liu, S. Zhu, L. Hou and C. Yuan, Chin. Chem. Lett., 2020, 31, 1030–1033 CrossRef CAS.
- A. Champagne, L. Shi, T. Ouisse, B. Hackens and J.-C. Charlier, Phys. Rev. B, 2018, 97, 115439 CrossRef CAS.
- M. V. Malashchonak, E. A. Streltsov, D. A. Kuliomin, A. I. Kulak and A. V. Mazanik, Mater. Chem. Phys., 2017, 201, 189–193 CrossRef CAS.
- M. Gotić, S. Musić, M. Ivanda, M. Šoufek and S. Popović, J. Mol. Struct., 2005, 744–747, 535–540 CrossRef.
- S. Onari, M. Miura and K. Matsuishi, Appl. Surf. Sci., 2002, 197–198, 615–618 CrossRef CAS.
- Q. Su, C. K. Huang, Y. Wang, Y. C. Fan, B. A. Lu, W. Lan, Y. Y. Wang and X. Q. Liu, J. Alloys Compd., 2009, 475, 518–523 CrossRef CAS.
- Y. Wang, S. Wang, N. Dong, W. Kang, K. Li and Z. Nie, Anal. Chem., 2020, 92, 4623–4629 CrossRef CAS PubMed.
- P. Subramanyam, T. Khan, G. Neeraja Sinha, D. Suryakala and C. Subrahmanyam, Int. J. Hydrogen Energy, 2020, 45, 7779–7787 CrossRef CAS.
- W. Hong, A. Wang, L. Li, T. Qiu, J. Li, Y. Jiang, G. Zou, H. Peng, H. Hou and X. Ji, Adv. Funct. Mater., 2021, 31, 2000756 CrossRef CAS.
- Y. Guo, T. Wang, Q. Yang, X. Li, H. Li, Y. Wang, T. Jiao, Z. Huang, B. Dong, W. Zhang, J. Fan and C. Zhi, ACS Nano, 2020, 14, 9089–9097 CrossRef CAS PubMed.
- H. Yuan, Y. Jin, X. Chen, J. Lan, Y. Yu and X. Yang, ACS Sustainable Chem. Eng., 2019, 7, 6033–6042 CrossRef CAS.
- T. Wu, C. Zhang, H. Hou, P. Ge, G. Zou, W. Xu, S. Li, Z. Huang, T. Guo, M. Jing and X. Ji, Adv. Funct. Mater., 2018, 28, 1705744 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. Ding, H. Yang, S. Zhang, Q. Liu, H. Cao, J. Luo and X. Liu, Small, 2022, 18, 2204524 CrossRef CAS PubMed.
- T. Wei, W. Liu, S. Zhang, Q. Liu, J. Luo and X. Liu, Chem. Commun., 2023, 59, 442–445 RSC.
- H. Chen, S. Zhang, Q. Liu, P. Yu, J. Luo, G. Hu and X. Liu, Inorg. Chem. Commun., 2022, 146, 110170 CrossRef CAS.
- Y. Tan, Z. Zhu, X. Zhang, J. Zhang, Y. Zhou, H. Li, H. Qin, Y. Bo and Z. Pan, Int. J. Hydrogen Energy, 2021, 46, 1955–1966 CrossRef CAS.
- M. Hu, Z. Li, T. Hu, S. Zhu, C. Zhang and X. Wang, ACS Nano, 2016, 10, 11344–11350 CrossRef CAS PubMed.
- B. Shao, Z. Liu, G. Zeng, Z. Wu, Y. Liu, M. Cheng, M. Chen, Y. Liu, W. Zhang and H. Feng, ACS Sustainable Chem. Eng., 2018, 6, 16424–16436 CrossRef CAS.
- B. Shao, J. Wang, Z. Liu, G. Zeng, L. Tang, Q. Liang, Q. He, T. Wu, Y. Liu and X. Yuan, J. Mater. Chem. A, 2020, 8, 5171–5185 RSC.
- Y. Bai, H. Bai, Z. Fang, X. Li, W. Fan and W. Shi, Chem. Commun., 2021, 57, 10568–10571 RSC.
- L. Li, D. Yu, P. Li, H. Huang, D. Xie, C.-C. Lin, F. Hu, H.-Y. Chen and S. Peng, Energy Environ. Sci., 2021, 14, 6419–6427 RSC.
- Y. Li and C. Zhao, ACS Catal., 2017, 7, 2535–2541 CrossRef CAS.
- G. Meng, H. Cao, T. Wei, Q. Liu, J. Fu, S. Zhang, J. Luo and X. Liu, Chem. Commun., 2022, 58, 11839–11842 RSC.
- D. Qi, F. Lv, T. Wei, M. Jin, G. Meng, S. Zhang, Q. Liu, W. Liu, D. Ma, M. S. Hamdy, J. Luo and X. Liu, Nano Res. Energy, 2022, 1, e9120022 CrossRef.
- W. Zheng, Y. Li, C. S. Tsang, P. K. So and L. Yoon Suk Lee, iScience, 2021, 24, 102342 CrossRef CAS PubMed.
- Z. Fang, P. Wu, Y. Qian and G. Yu, Angew. Chem., Int. Ed., 2021, 60, 4275–4281 CrossRef CAS PubMed.
- J. Wu, Y. Miao, X. Liang, Z. Yang, Y. Yang and R. Ouyang, Electroanalysis, 2014, 26, 856–863 CrossRef CAS.
- Y. Cao, Y. Tan, X. T. Zhu, H.-L. Li, Y.-Q. Zhao and Y. Xu, Phys. E, 2021, 134, 114875 CrossRef CAS.
- X. Li, Y. Luo, Q. Li, Y. Guo and K. Chu, J. Mater. Chem. A, 2021, 9, 15955–15962 RSC.
- M. Wu, Y. He, L. Wang, Q. Xia and A. Zhou, J. Adv. Ceram., 2020, 9, 749–758 CrossRef CAS.
- P. K. Singh, S. K. Sharma, S. K. Tripathi and D. K. Dwivedi, Results Phys., 2019, 12, 223–236 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr01144e |
| This journal is © The Royal Society of Chemistry 2023 |