
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFerulic acid-loaded polymeric nanoparticles prepared from nano-emulsion templates facilitate internalisation across the blood–brain barrier in model membranes†
Luna
Garcia
a,
Sujey
Palma-Florez
 be,
Victor
Espinosa
a,
Fatemeh
Soleimani Rokni
a,
Anna
Lagunas
be,
Mònica
Mir
bce,
María José
García-Celma
d,
Josep
Samitier
be,
Carlos
Rodríguez-Abreu
*ae and
Santiago
Grijalvo
*e
be,
Victor
Espinosa
a,
Fatemeh
Soleimani Rokni
a,
Anna
Lagunas
be,
Mònica
Mir
bce,
María José
García-Celma
d,
Josep
Samitier
be,
Carlos
Rodríguez-Abreu
*ae and
Santiago
Grijalvo
*e
aIQAC, CSIC, Jordi Girona 18-26, E-08034-Barcelona, Spain. E-mail: carlos.rodriguez@iqac.csic.es
bInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology (BIST), E-08028-Barcelona, Spain
cDepartment of Electronics and Biomedical engineering, University of Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain
dDepartment of Pharmacy, Pharmaceutical Technology, and Physical-chemistry, IN2UB, R+D Associated Unit to CSIC, Pharmaceutical Nanotechnology, University of Barcelona, Joan XXIII 27-31, E-08028-Barcelona, Spain
eCIBER-BBN, ISCIII, Spain. E-mail: sgrgma@cid.csic.es
First published on 10th April 2023
Abstract
A hydroxycinnamic acid derivative, namely ferulic acid (FA) has been successfully encapsulated in polymeric nanoparticles (NPs) based on poly(lactic-co-glycolic acid) (PLGA). FA-loaded polymeric NPs were prepared from O/W nano-emulsion templates using the phase inversion composition (PIC) low-energy emulsification method. The obtained PLGA NPs exhibited high colloidal stability, good drug-loading capacity, and particle hydrodynamic diameters in the range of 74 to 117 nm, depending on the FA concentration used. In vitro drug release studies confirmed a diffusion-controlled mechanism through which the amount of released FA reached a plateau at 60% after 6 hours-incubation. Five kinetic models were used to fit the FA release data as a function of time. The Weibull distribution and Korsmeyer–Peppas equation models provided the best fit to our experimental data and suggested quasi-Fickian diffusion behaviour. Moderate dose–response antioxidant and radical scavenging activities of FA-loaded PLGA NPs were demonstrated using the DPPH˙ assay achieving inhibition activities close to 60 and 40%, respectively. Cell culture studies confirmed that FA-loaded NPs were not toxic according to the MTT colorimetric assay, were able to internalise efficiently SH-SY5Y neuronal cells and supressed the intracellular ROS-level induced by H2O2 leading to 52% and 24.7% of cellular viability at 0.082 and 0.041 mg mL−1, respectively. The permeability of the NPs through the blood brain barrier was tested with an in vitro organ-on-a-chip model to evaluate the ability of the FA-loaded PLGA and non-loaded PLGA NPs to penetrate to the brain. NPs were able to penetrate the barrier, but permeability decreased when FA was loaded. These results are promising for the use of loaded PLGA NPs for the management of neurological diseases.
Introduction
The development of nanotechnology in medicine has facilitated the design and preparation of all types of nanostructured materials for use in numerous biomedical applications, especially diagnostics and drug delivery systems.1–3 In contrast to traditional therapies, which have demonstrated limited efficacy and cause serious side effects, nanomedicine provides potential platforms for modulating and addressing localised therapeutic effects with improved biocompatibility.4 In this context, nanomedicine has been recognised as a promising strategy for promoting personalised medicine.5It is interesting to note that the number of clinically approved nanoparticle-based therapeutics has steadily increased over the last two decades.6,7 This success has fostered active interdisciplinary collaborations, resulting in the development of a substantial number of nanotherapeutics undergoing preclinical evaluation and clinical trials.7 Presently, liposomal and polymeric platforms are dominating the field of advanced clinical trials in nanomedicine.8
Numerous review articles have covered various protocols for preparing polymeric NPs.9,10 Nano-emulsions (NEs),11 which are formulated heterophase systems comprising kinetically stable nanodroplets dispersed in a continuous phase, have been shown to function as nanoreactors or templates12,13 for the formation of NPs from either in situ polymerisation reactions14,15 or preformed polymers.10,16 However, due to the presence of reactive initiators and the generation of by-products, the preformed polymer strategy is generally more advantageous than polymerisation reactions.10
NEs exhibit droplet sizes ranging from 20 to 200 nm and can be prepared using high-energy or low-energy methods.17 High-energy methods involve applying mechanical energy to the system, while low-energy methods use the internal energy of the system to produce uniform and smaller droplet sizes, which can be controlled by selecting the appropriate system composition.18,19 Various low-energy methods have been employed to prepare NEs, including the phase-inversion temperature (PIT) method,20 the phase inversion composition (PIC) method,21,22 emulsion inversion point23 and the bubble bursting method.24
In this article, NEs made up of a preformed FDA approved polymer, namely poly(lactic-co-glycolic acid) (PLGA) dissolved in a volatile organic solvent, were used to prepare polymeric NPs upon solvent evaporation. The selection of PLGA as a polymer is based on its biocompatibility and low cytotoxicity properties. Moreover, PLGA can be degraded into non-toxic monomers in vivo and removed from the body through specific metabolic pathways.25,26 The preparation of polymeric NPs using O/W NE templates have been widely used in our group with the aim to encapsulate and deliver both small molecule drugs and nucleic acids.27–30
Natural products have attracted attention due to their beneficial effects and potent biological activities against various diseases31 ranging from neurological disorders and cancer to inflammatory diseases.32–35 Ferulic acid (FA) and derivatives36 have demonstrated effectiveness as antioxidant scavenging radical agents,37 and neuronal protective compounds by reducing the levels of Aβ-amyloid peptide aggregation and amyloid-induced cytotoxicity in cells.38,39 Additionally, FA has demonstrated anti-inflammatory properties in animal models when exposed to mild stress.40 However, a vast number of phytochemicals including FA are phenolic compounds which tend to exhibit low water solubility, reduced levels of oral bioavailability and membrane permeability. In addition, phytochemicals have shown rapid metabolism combined with urine excretion when circulating across the bloodstream. As a consequence, these limitations have remarkably reduced the biological activity restricting the use of these small molecules in clinical applications.41 The presence of the blood–brain barrier (BBB) adds additional complexity in designing effective drugs for Central Nervous System (CNS) disorders because it prevents the free entry of small active molecules and therapeutics from the blood and thus limits their accumulation in the brain.42 To overcome these bottlenecks, the development of formulation and related pharmaceutical technologies have been widely applied as a suitable strategy to entrap not only hydrophobic drugs and improve their pharmacokinetic properties like solubility and bioavailability but also the development of strategies aimed to surpass the BBB.42,43
Several engineering drug delivery systems (DDS) have successfully designed to entrap FA to be used in multiple applications such as anticancer, antioxidant, wound healing, or respiratory disorders, among others.44 In this sense, nanostructured lipid carriers, polymers, or hydrogels have been developed to minimise the major limitations regarding FA stability in plasma, low oral bioavailability, and drug solubility.44,45
NEs are an emerging platform that can address major limitations regarding stability in plasma and drug solubility.45 Polymeric NPs prepared from NE templates have demonstrated high drug loading capacity, good biocompatibility, long circulation properties, sustained drug release, and the ability to accumulate at a target site.28 These superior features have prompted us to select and encapsulate FA within a PLGA matrix to produce the anticipated FA-loaded NPs.
Bioengineering offers advanced tools to estimate the behaviour of drug loaded NPs using advanced in vitro models.46 Currently, in vitro models are being used to mimic both blood–brain membrane barriers47 including: (i) parallel artificial membrane permeability assays (PAMPA);48 (ii) organoids;49,50 (iii) cell-based Transwell assays;51 and (iv) microfluidic devices for organ-on-chip (OoC) systems.52,53 These OoC models combine 3D cells co-culture with microfluidics to introduce the same fluidic condition in the vessel and shear stress in the cells. OoC models have superior predictive abilities than 2D cell cultures and often animal models due to the use of human cells. They are also cost-effective, less time-consuming, and allow for reduced animal testing.
In this study, we report the preparation of FA-loaded polymeric NPs from NEs using the PIC low-energy emulsification method. Various characterisation methods including dynamic light scattering (DLS), zeta (ζ)-potential, transmission electron microscopy (TEM) and dark-field microscopy, were used to characterize the materials. The release of FA from PLGA NPs was studied in vitro and fitted to various release models. The antioxidant properties, cytotoxicity, and cellular uptake of FA-loaded NPs were evaluated. Additionally, the permeation of the synthesized NPs (bare and FA-loaded NPs) through an in vitro BBB-on-a-chip (BBB-oC) model54 was studied to get insight into the potential of such drug delivery systems (DDS) to overcome the BBB (Fig. 1).
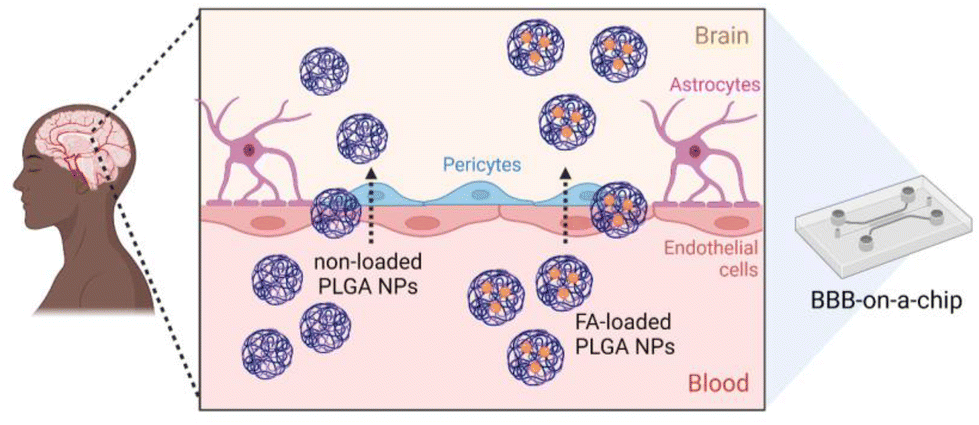 | ||
| Fig. 1 Scheme of the in vitro model that mimics the BBB54 to analyze the permeability of polymeric NPs prepared from nano-emulsions using the PIC method to the brain. | ||
Materials and methods
All reagents and solvents were used as received. Poly(lactic-co-glycolic acid; PLGA) with an average molecular weight (MW) of ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 was purchased from Boehringer Ingelheim (Ingelheim am Rhein, Germany). Surfactant Polysorbate 80 (Tween 80), fluorescein isothiocyanate isomer I, poly(ethylene glycol)bis(amine) (PEGdiamine) (MW ∼2000 g mol−1), thiazolyl blue tetrazolium bromide, hydrogen peroxide solution (30 wt% in H2O), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxy succinimide (NHS), fluorescein isothiocyanate (FITC), and FA were purchased from Merck Sigma-Aldrich (Saint Louis, MO, USA). 2,2-Diphenyl-1-picrylhydrazyl (DPPH) was purchased from TCI Europe (Zwijndrecht, Belgium). Ethanol and ethyl acetate were purchased from Panreac (Darmstadt, Germany) and were used as received. Water was Milli-Q filtered (Millipore) (Massachusetts, MA, USA). A phosphate-buffered saline (1× PBS) solution was obtained from sodium chloride (NaCl), potassium chloride (KCl), disodium monohydrogenphosphate dihydrate (Na2HPO4·2H2O) and potassium dihydrogen phosphate (KH2PO4). Salts were purchased from Merck Sigma-Aldrich (Saint Louis, MO, USA). PBS tablets were purchased from Merck Sigma-Aldrich (Saint Louis, MO, USA) and were dissolved in 1 L of RNAse-free water (DEPC-treated) to get 0.01 M phosphate buffer solution for cell culture studies. RNase-free water (1 L) was autoclaved after being incubated with diethylpyrocarbonate (DEPC) (0.1 L) (Merck Sigma-Aldrich) (Saint Louis, MO, USA) overnight to ensure sterility. Dubelcco's Modified Eagle Medium (DMEM) was supplemented with 10% Fetal Bovine Serum (FBS). SU8-2100 as photoresist was obtained from MicroChem (Ulm, Alemania), polydimethylsiloxane (PDMS; Sylgard 184) from Dow Corning (MI, USA) and coverslips from Menzel-Glaser (Germany). Pericyte growth media (pericyte medium (PM), astrocyte medium (AM), poly-L-lysine and trypsin/EDTA 0.05% were purchased from Sciencell. Collagen Type I from rat tail and Fibroblast Growth Factor-Basic (bFGF) and trypsin/EDTA 0.25% were supplied by Sigma-Aldrich (Saint Louis, MO, USA) and endoGRO™ medium was obtained from Merck Life Science S.L.U. (Madrid, Spain). CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) were provided by Promega (Madrid, Spain).
000 g mol−1 was purchased from Boehringer Ingelheim (Ingelheim am Rhein, Germany). Surfactant Polysorbate 80 (Tween 80), fluorescein isothiocyanate isomer I, poly(ethylene glycol)bis(amine) (PEGdiamine) (MW ∼2000 g mol−1), thiazolyl blue tetrazolium bromide, hydrogen peroxide solution (30 wt% in H2O), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxy succinimide (NHS), fluorescein isothiocyanate (FITC), and FA were purchased from Merck Sigma-Aldrich (Saint Louis, MO, USA). 2,2-Diphenyl-1-picrylhydrazyl (DPPH) was purchased from TCI Europe (Zwijndrecht, Belgium). Ethanol and ethyl acetate were purchased from Panreac (Darmstadt, Germany) and were used as received. Water was Milli-Q filtered (Millipore) (Massachusetts, MA, USA). A phosphate-buffered saline (1× PBS) solution was obtained from sodium chloride (NaCl), potassium chloride (KCl), disodium monohydrogenphosphate dihydrate (Na2HPO4·2H2O) and potassium dihydrogen phosphate (KH2PO4). Salts were purchased from Merck Sigma-Aldrich (Saint Louis, MO, USA). PBS tablets were purchased from Merck Sigma-Aldrich (Saint Louis, MO, USA) and were dissolved in 1 L of RNAse-free water (DEPC-treated) to get 0.01 M phosphate buffer solution for cell culture studies. RNase-free water (1 L) was autoclaved after being incubated with diethylpyrocarbonate (DEPC) (0.1 L) (Merck Sigma-Aldrich) (Saint Louis, MO, USA) overnight to ensure sterility. Dubelcco's Modified Eagle Medium (DMEM) was supplemented with 10% Fetal Bovine Serum (FBS). SU8-2100 as photoresist was obtained from MicroChem (Ulm, Alemania), polydimethylsiloxane (PDMS; Sylgard 184) from Dow Corning (MI, USA) and coverslips from Menzel-Glaser (Germany). Pericyte growth media (pericyte medium (PM), astrocyte medium (AM), poly-L-lysine and trypsin/EDTA 0.05% were purchased from Sciencell. Collagen Type I from rat tail and Fibroblast Growth Factor-Basic (bFGF) and trypsin/EDTA 0.25% were supplied by Sigma-Aldrich (Saint Louis, MO, USA) and endoGRO™ medium was obtained from Merck Life Science S.L.U. (Madrid, Spain). CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) were provided by Promega (Madrid, Spain).
Preparation of polymeric NEs
Non-loaded and loaded polymeric NEs were prepared using the PIC low-energy emulsification method. The NEs contained of 90 wt% phosphate-buffered saline solution (PBS), 7 wt% oil phase (PLGA + ethyl acetate) and 3 wt% Tween 80 as described in previous works.55 For the preparation of a 4.0 mL dispersion of NPs, 120 μL of Tween 80 were firstly introduced in a test tube. Simultaneously 11.2 mg of PLGA measured in an analytical balance were dissolved in 280 μL of ethyl acetate. This solution (the oil phase) was then added to the surfactant and stirred until a homogeneous and transparent mixture was obtained. Under steady stirring at room temperature, 3.6 mL of PBS solution were added dropwise with the aid of a syringe into the oil phase previously prepared. As PBS was incorporated, the sample took a whitish turbid appearance. Towards the end of the PBS addition, the sample turned almost transparent with a slight bluish glow. FA-loaded NEs were prepared using the same procedure described above, but FA was added to the oil phase at appropriate concentrations (0.1, 0.3 and 0.4 mg mL−1) using a FA stock solution of 5 mg mL−1 in ethyl acetate.Preparation of polymeric NPs from NEs
NPs (non-loaded and FA-loaded) were obtained from their NE counterparts by solvent evaporation under reduced pressure at 25 °C and 150 rpm rotation using a Büchi R-215 V Rotavapor for 1 hour. Finally, the volume of the resultant polymeric NPs dispersion was adjusted with Milli Q water with the aim to maintain the appropriate sample osmolality at 300 mOsm kg−1.Preparation of fluorescently labelled polymeric NPs
Once PLGA and FA-loaded polymeric NPs (4.0 mL) were prepared in 1× PBS, the colloidal dispersions were acidified to pH 3 using a 1 M HCl solution. EDC (25 eq.) was added and stirred for 5 minutes at room temperature. NHS (25 eq.) was added and stirred for 2 hours. The aqueous solution was basified to pH 8 using a 1 M NaOH solution and PEGdiamine (5.0 eq.) was added. The resulting colloidal dispersions were stirred overnight at room temperature. FITC (5.0 eq.) was added, and the mixture was stirred for 5 hours at room temperature. Finally, the excess of EDC, NHS, PEGdiamine, and FITC was removed by dialysis. To calculate the PLGA-to-PEG molar ratio and estimate the %conjugation efficiency, one batch of PEGylated NPs were purified by dialysis, frozen at −80 °C, lyophilised, and finally dissolved in deuterated methanol (0.8 mL) for 1H-NMR analysis. NMR spectra was run on a Bruker Ascend 400 MHz (Bruker, Berlin, Germany) present in the core facility of the Institute for Advanced Chemistry of Catalonia (IQAC). Finally, the %conjugation efficiency was obtained according to Betancourt et al.56Biophysical characterization of polymeric NPs
For DLS measurements, a LS instrument (Freiburg, Switzerland) equipped with a He–Ne laser (633 nm) was employed. Average size (hydrodynamic diameter) and polydispersity index (PDI) of NPs in aqueous dispersion (unloaded and FA-loaded) were estimated by the method of cumulants. Samples (200 μl) were evaluated using the following conditions: 3D-Cross correlation, scattering angle of 90° and 25 °C. The surface charge (ζ-potential) of the NPs was evaluated by electrophoretic mobility measurements at 25 °C. The Smoluchkowski approximation of Henry's equation was used (eqn (1)) where εr = relative permittivity; ε0 = permittivity of vacuum; ζ = zeta potential; η = medium viscosity at experimental temperature and μe = electrophoretic mobility. ζ-Potential measurements were carried out by diluting 50 μL of the NPs dispersion with 950 μL of MilliQ water.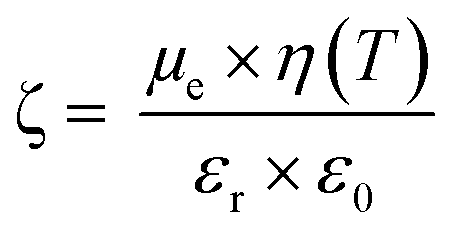 | (1) |
Data of hydrodynamic diameter, PDI, and ζ-potential correspond to the mean values of triplicate measurements for each sample.
Encapsulation efficiency (EE%)
EE% of FA within polymeric NPs was evaluated by centrifugation. Centrifugal filter units with a molecular weight cut-off (MWCO) of 3 kDa were used. FA-loaded NPs (1.0 mL) were centrifuged for 75 min at 4 °C and 6000 rpm. A calibration curve was built by consecutive HPLC measurements of FA samples with concentrations ranging from 0.04 to 8.0 × 10−6 FA mg mL−1 prepared from a 5 mg mL−1 FA stock solution in PBS:EtOH (15%) (ESI†). EE% values were determined by interpolation using the above-mentioned calibration curve and eqn (2).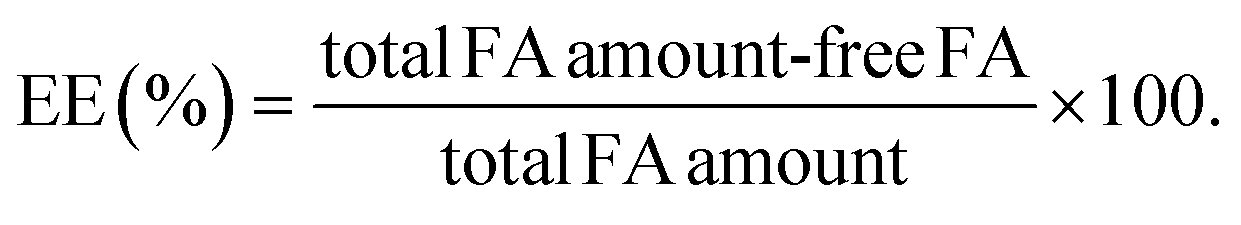 | (2) |
FA quantification essays
For FA quantification a Breeze™2 HPLC equipped with a Symmetry® C18 5 μm 4′6 × 75 mm analytical column, UV/Visible detector Waters 2489 set at 307 nm (the maximum absorption of FA), Waters 2707 autosampler and Waters 1525 binary HPLC pump was used. A gradient mode reversed-phase high-performance liquid chromatography (RP-HPLC) (Table S1†) was employed. Mobile phases used were a 0.001 wt% acetic acid solution in Milli-Q water (solvent A) and acetonitrile (solvent B) at 25 °C. Measurements were conducted at a flow rate of 1 mL min−1, injecting a volume of 50 μL, and 20 min chromatogram plotting.Transmission electron microscopy (TEM)
The size and morphology of FA-loaded PLGA NPs (FA concentration = 0.1 mg mL−1) were determined using a JEOL JEM-1400 Flash (JEOL, Japan), operating at 100 kV. Samples were diluted and deposited on glow discharged carbon coated grids (400 mesh). An adsorption on 15 μL of the sample for 2 min was carried out. A 2% uranyl acetate solution (15 μL) was used to stain TEM grids for 1 min. Finally, micrographs were taken with a One View digital camera (Gatan, USA) at various magnifications.Dark-field hyperspectral microscopy
Optical observation and spectral mapping of FA-loaded polymeric NPs were carried out using an Olympus BX-43 optical microscope coupled to a dark-field based illuminator condenser,57 which operates in the Visible-Near Infrared Range (VNIR) (400–1000 nm) with spectral imaging (Cytoviva Inc., Auburn, AL, USA). The brightness intensity was controlled using a halogen source (150 W) with a lamp voltage of 11 V. FA and PLGA solutions were separately scanned to obtain the corresponding hyperspectral reference images. FA-loaded NPs (5 μL) were then visualised and scanned obtaining their hyperspectral images which were compared with the reference library obtained above. Hyperspectral images were finally acquired with the Exponent 7 software and visualised using HyperVisual ENVI software.In vitro drug release assay
Release of FA was studied using the dialysis bag method. Experiments were performed at 37 °C using a cellulose membrane with a MWCO of 3000 kDa. Firstly, the membrane was pre-wetted with the receptor solution (1× PBS or 1× PBS:15% EtOH) for 20 minutes and the cellulose membrane was filled with 1.0 mL of the sample (aqueous FA or FA-loaded PLGA NPs). Dialysis bags were immersed in the receptor solution (40 mL) and maintained at constant temperature with steady stirring. Aliquots of 1 mL of the receptor solution were withdrawn at controlled intervals of time and FA concentration was determined by HPLC as described above. After the removal step, volume was each time adjusted with 1.0 mL of the receptor solution to maintain the osmolality of the sample. For release experiments using Tween 80-treated membranes, the dialysis membrane was soaked in a 1× PBS solution containing 3% Tween 80. The dialysis membrane was washed to eliminate the excess of Tween 80. Release experiments and FA quantification were carried out as described above with both free FA solutions and FA-loaded NPs in triplicate maintaining sink conditions over time.Mathematical models
We have used four mathematical models58 to fit the cumulative FA release data as a function of time: (i) Zero-order59 (eqn (3)); (ii) First order (eqn (4)); (iii) Higuchi60 (eqn (5)); (iv) Korsmeyer–Peppas61 (eqn (6)) and Weibull model62 (eqn (7)). Herein, M∞ and Mt are the maximum and cumulative amounts of active (FA) released at time t, respectively. K, K0, KH, and KK–P, and KWB are constants that provide information about structural and geometric properties of the dosage form. Qo is the initial amount of FA, and n is a diffusional exponent. A Fickian mechanism is considered (Case I) to be dominant when n is around 0.5. A non-Fickian anomalous diffusion is ascribed for 0.5 < n < 1 (Case II). In respect of Weibull equation, α is a constant and β describes different diffusion mechanism including (i) Fickian if β ≤ 0.75 and (ii) complex processes combined with diffusion mechanisms if 0.75 < β < 1. The DDSolver63 add-in program was used to fit the FA release data to the models described above.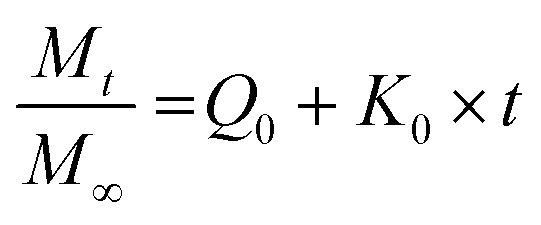 | (3) |
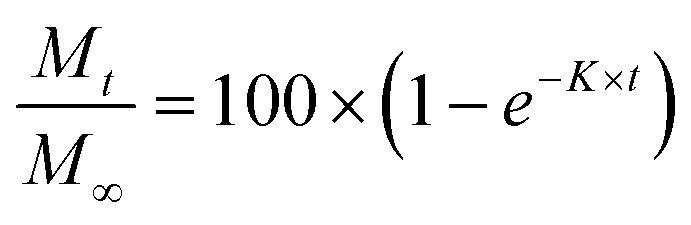 | (4) |
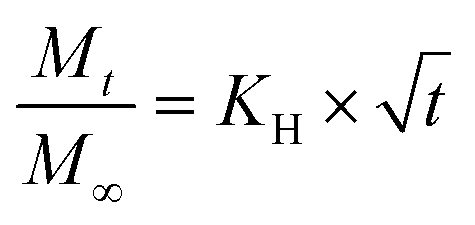 | (5) |
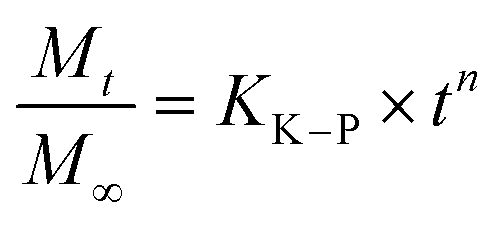 | (6) |
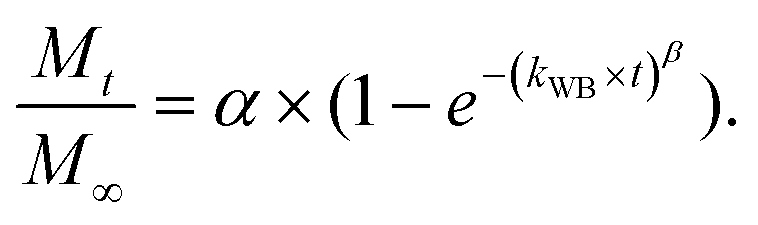 | (7) |
DPPH assay
Radical scavenging activities of free FA and FA-loaded NPs were evaluated using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay.64 FA antioxidant activity was tested following a modification of a method found in the literature.65 Assays were carried out in triplicate. Dispersions containing FA-loaded NPs (0.233 PLGA mg mL−1) with a FA concentration of 0.2 and 0.1 mg mL−1 were prepared. 200 μL of a 0.2 mM DPPH solution in ethanol (50% v/v) were mixed with 20 μl of the dispersions to be tested and introduced in a 96-well plate. Reaction mixture was incubated for 30 minutes in the darkness at room temperature. Additionally, the antioxidant activity of FA-loaded PLGA NPs was measured at the following incubation times (5, 10, 15, and 20 minutes) (Fig. S7†). Activity was determined by absorbance measurements of the samples at 517 nm using a Biotek Synergy H1 Hybrid Multi-Mode Reader (Agilent Technologies, Santa Clara, CA, USA). A control sample was prepared with 20 μL ethanol and 200 μL of the 0.2 mM DPPH solution.Fabrication of the BBB-oC microfluidic device
The microfluidic chip consists of three chambers, where the central chamber contains human pericytes, and astrocyte culture embedded in a hydrogel and human endothelial cells are injected into the side chambers to grow the barrier simulating BBB. The microfluidic device was designed by CAD software (AutoCAD 2019). The design is composed of three parallel microchambers: a central main chamber (1300 μm wide, 8800 μm long, 150 μm high) and two lateral channels (300 μm wide, 150 μm high) which are interconnected by trapezoidal pillars (300 μm base and separated 100 μm). Master moulds were fabricated in a cleanroom environment using standard photolithography techniques with 4-inch silicon wafers as substrates and the SU8-2100 as photoresist.The 3D microfluidic systems were made of PDMS mixing the elastomer base and the curing agent (10:1 w/w), which was degassed and poured onto the designed master mould and cured for 2 h at 65 °C. The PDMS was peeled off from the mould and the inlet and outlet holes were created with a biopsy punch (1 mm for the central chamber inlets and 4 mm for the media reservoirs). Then, the devices were cleaned and bonded to coverslips (0.17 mm thickness) by treating them in an air plasma chamber (Harrick Plasma PCD-002-CE) for 30 s at 10.5 W. Finally, all the chips were thermally treated overnight at 85 °C to stabilize the bonding and hydrophobize the surface (Fig. 7).
Cell culture in the BBB-oC
Human brain vascular pericytes (HBVP; at passages <7) and human astrocytes-hippocampal (HA-h; at passages <5) were cultured in T-75 cell culture flasks coated with 2 μg cm2 poly-L-lysine in their respective growth media. Pericyte medium (PM) or astrocyte medium (AM) are supplemented with 1% v/v pericyte or astrocyte growth supplement, respectively, 10% v/v fetal bovine serum (FBS) and 1% v/v penicillin/streptomycin solution. The immortalized human brain endothelial cell line hCMEC/D3 was cultured in T-75 cell culture flasks coated with a 150 μg ml−1 solution of collagen Type I from rat tail in phosphate buffered saline (PBS) in the growth medium EndoGRO™-MV Complete Media Kit supplemented with 1 ng mL−1 Fibroblast Growth Factor-Basic (bFGF). Cells were maintained in a humidified incubator (37 °C, 5% CO2) and media was replaced every 2 days.Cell seeding in the microfluidic device: BBB-oC
The bonded chip devices were sterilized by UV light prior to cell seeding. HBVP and HA-h cells were detached with 0.05% of trypsin/EDTA and centrifuged at 1000g for 5 minutes. Preparation of the cell seeded hydrogel before injection was conducted at 4 °C. HBVP and HA-h cells (4 × 104 cells of each cell type) were gently resuspended with 50 μL of a 3 mg mL−1 fibrinogen and 1 μL of thrombin solution (100 U mL1) in Dulbecco's Phosphate-Buffered Saline (DPBS). Then, this mixture was injected into the main chamber of the chip. The chip was kept inside the humidified incubator (37 °C, 5% CO2) for 15 min to allow the complete hydrogel polymerization. Then, a 50% v/v mixture of astrocyte and endothelial growth medium was perfused through the lateral fluidic channels of the chip, as supplier for the central chamber. The chip was maintained in a humidified incubator for two days. Afterwards, endothelial cell seeding was conducted. Firstly, one of the two fluidic channels flanking the central chamber was coated with a 150 μg ml−1 solution of Collagen Type I from rat tail in PBS. Then, hCMEC/D3 cells were detached with 0.25% trypsin/EDTA and 30 μL of hCMEC/D3 cells (105 cells) in 50% v/v mixture of astrocyte and endothelial growth medium were injected in the coated channel.The chip was left in vertical position (to allow the endothelial cells to contact with the hydrogel by gravity) for 1.5 hours in the humidified incubator. Then, the 50% v/v mixture of astrocyte and endothelial growth medium were perfused through the fluidic channels and the reservoirs were fulfilled. The chip was kept for 5 days in the humidified incubator (37 °C, 5% CO2) before conducting the permeability assays. Medium was replaced every day.
Cytotoxicity
The cytotoxicity of FA-loaded NPs was evaluated through cell viability assays conducted on different cell lines. SH-SY5Y cells viability was assessed via the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. Cells were seeded (about 5 × 103 cells per well) on a 96-well plate in Gibco Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% foetal bovine serum (FBS) and cultured for 24 h at 37 °C under 5% CO2 atmosphere. Culture medium was next replaced, and FA-loaded NPs were added at concentrations of 0.082, 0.041, and 0.112 PLGA mg mL−1 in 200 μL of DMEM. NPs were incubated for 24 h, and culture medium was discharged and replaced by fresh DMEM (200 μL). Further overnight incubation of the mixture was followed by 15 μL addition of [MTT] = 5 mg mL−1 solution onto the 96-well plate. After 3 h of incubation, culture medium was withdrawn carefully and 200 μL of DMSO were added to dissolve formazan crystals.The 96-well plate was shaken for 15 min at room temperature and absorbance was measured at λ = 570 nm using a Biotek Synergy H1 Hybrid Multi-Mode Reader (Agilent Technologies, Santa Clara, CA, USA). Measures were carried out in triplicate using unloaded PLGA NPs as a control. Cellular viability was calculated taking the ratio between untreated and treated cells into account.
HA-h, HBVP, and hCMEC/D3 cells viability was tested using the tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS). HA-h (12000 cells per well), HBVP and hCMEC/D3 (10000 cells per well) were seeded on 96-well plates coated with poly-L-lysine for HA-h and HBVP, and with Collagen, Type I from rat tail for hCMEC/D3 cells, in their respective growth media (total volume: 100 μL per well) as described. They were cultured for 24 h in a humidified incubator at 37 °C and 5% CO2. Cell media was then replaced, and fluorescent FITC-labelled PLGA NPs containing FA (FA-loaded PLGA_FITC NPs) and non-loaded (PLGA_FITC NPs), sterilized by filtration, were added at concentrations of 0.056, 0.090, 0.112 and 0.280 mg·mL−1 in growth medium (100 μL per well). NPs were incubated for 24 h at 37 °C and 5% CO2. Then, the cell medium was replaced and 20 μL per well of MTS reagent were added. Cells were kept in the humidified incubator at 37 °C and 5% CO2 for 1.5 h and absorbance was measured at 490 nm in a microplate reader (Infinite M200 PRO from Tecan, Männedorf, Switzerland). Measures were carried out in triplicate and cells with growth medium, cells treated with PBS and cells treated with 20% sodium dodecyl sulphate were used as live (CL), vehicle (Cvh) and dead (CD) control, respectively.
Hydrogen peroxide toxicity assay in SH-SY5Y cells
The experiment was carried out following the protocol described by Chojnacki et al. but with modifications.66 SH-SY5Y cells were seeded on a 96-well plate (35 × 103 cells per well) in DMEM supplemented with 10% FBS and cultured for 24 h at 37 °C under 5% CO2 atmosphere. DMEM was removed and bare PLGA NPs, free FA, and FA-loaded NPs were added at concentrations of 0.082 and 0.041 PLGA mg mL−1 in 200 μL of DMEM. NPs were incubated for 2 h, and hydrogen peroxide (75 μM) was added and incubated for an additional hour. Culture medium was discharged and replaced by fresh DMEM (200 μL). Cells were incubated overnight followed by the addition of a MTT solution (5 mg mL−1). After 3 h of incubation, culture medium was withdrawn and 100 μL of DMSO were added to dissolve formazan crystals. Measures were carried out in triplicate.Permeability assays in the BBB-oC
FA-loaded NPs permeability through the BBB was tested in the BBB-OoC model of 7 days of culture. Permeability assays were conducted in a Nikon Ti2 epi-fluorescence microscope optimized for long-live imaging (Nikon, NY, USA). Before injection, BBB-oC was washed with PBS to remove cell debris and supplied with cell medium. T0 images were captured every 30 s for 1 minute. Then, 70 μL of the FA-loaded PLGA_FITC NPs, sterilized by filtration, were injected into the endothelial fluidic channel of the chip at a concentration of 0.05 mg mL−1 in a 50% v/v mixture of astrocyte and endothelial growth medium. 70 μL of the mixed medium were perfused into the other fluidic channel at the same time. The liquid was dragged through the channels and the reservoirs filled. Time-lapse images were captured for 60 minutes every 3 minutes using both the bright field (BF) and fluorescent settings. Measures were carried out in triplicate using non-loaded PLGA_FITC NPs as a control. Permeability coefficients were calculated using the same methodology reported by Campisi et al.67Cellular uptake
SH-SY5Y cells (105 cells per well) were seeded on a 24-well cell plate in DMEM supplemented with 10% FBS. Cells were incubated at 37 °C and 5% CO2 until reaching ca. 70% confluency. Fluorescent FITC-labelled PLGA NPs containing FA (FA-loaded PLGA_FITC NP; 0.087 mg mL−1) were incubated for 16 hours in the presence of neuronal cells (total volume 300 μL). DMEM was discharged and cells were washed with PBS (1 × 300 μL). Trypsin-EDTA (300 μL) was added and incubated for 3 min at 37 °C. DMEM (800 μL) was added in each well and cells were centrifuged (14233 rcf, 8 min). Finally, cell pellets were resuspended in PBS (500 μL) and analysed by flow cytometry. We selected 15000 events that were collected in a R1 region. This control region corresponds to the SH-SY5Y cell population. A second region (R2) was selected to measure the amount of fluorescently labelled cells obtained. Analyses were carried out in triplicate in a Guava easyCyte 8HT instrument (Millipore, Billerica, MA, USA). The number of positive cells were quantified using Flowing Software 2.5. (University of Turku, Finland).
Statistical analysis
Data are expressed as the mean ± SD. A two-side Student's test was used to analyse the differences between two groups. Differences were statistically significant when ρ value was less than 0.05 (***ρ < 0.001).Results and discussion
Preparation of PLGA NPs
NP formulations based on PLGA were obtained from NE templates using the PIC method. The first step in this protocol is to dissolve the surfactant (S) in an oil (O) phase made up of a solution of PLGA in ethyl acetate, a volatile solvent. Upon addition of water to the O + S mixture, there is a phase transition from W/O microemulsions to O/W nano-emulsion at room temperature, that triggers the formation of nanosized droplets. The determination of the nano-emulsion formation region containing the appropriate (O) and (S) ratio compositions was previously optimised by our group from a ternary phase diagram. We also reported that O/S ratios of 70/30 and 90 wt% of an electrolyte solution (1× PBS) were optimal to form colloidaly stable NEs with small droplet sizes.27–29,68Once non-loaded and FA-loaded PLGA NEs containing a FA concentration of 0.1, 0.3 and 0.4 mg mL−1 were prepared, ethyl acetate was removed from the NE droplets by evaporation to obtain the expected PLGA NPs with a slight bluish shine. Interestingly, we observed that the turbidity of the samples increased with increasing FA concentration (Fig. S1†). All colloidal formulations were subjected to particle size analysis, ζ-potential measurements, and were observed under transmission electronic microscopy (TEM) (Fig. 2), and hyperspectral microscopy69 (Fig. 3).
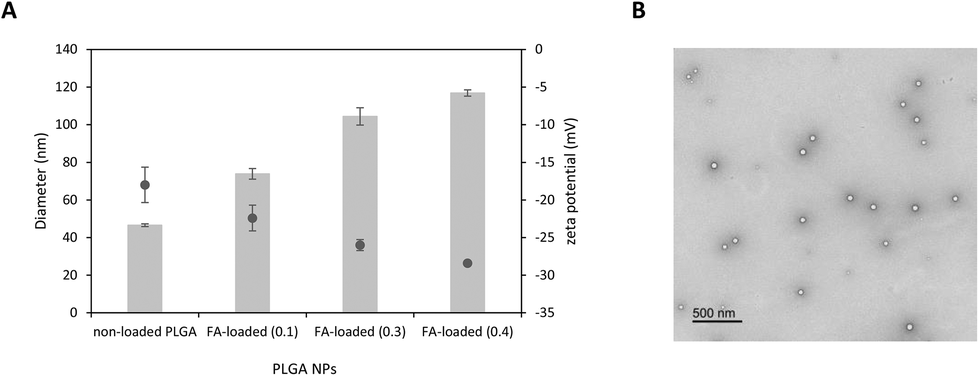 | ||
| Fig. 2 Characterisation of polymeric NPs. (A) Hydrodynamic diameter (grey bars) and zeta potential (grey dots) of non-loaded and FA-loaded PLGA NPs. The final FA concentration encapsulated within NPs was 0.1, 0.3, and 0.4 mg mL−1. (B). TEM image of FA-loaded PLGA NPs (FA concentration = 0.1 mg mL−1). Scale bar = 500 nm. | ||
 | ||
| Fig. 3 Hyperspectral characterization of polymeric NPs. (A) Dark-field image of FA-loaded PLGA NPs (40×) (scale bar = 1.67 μm); (B) mapped images of PLGA NPs; (C) mapped images of FA; (D) mapped images of FA-loaded PLGA NPs. The insets in figures B, C, and D are magnified ×10 with respect to the main image. | ||
As shown in Table 1, the particle size of all formulations measured by DLS was in the range of 40–120 nm. A significant increase in the hydrodynamic diameter (***ρ < 0.001) was noticed when FA was encapsulated within polymeric NPs if compared with their non-loaded counterparts. While the average diameter of non-loaded PLGA NPs was 46.6 ± 0.74 nm, FA-loaded NPs containing 0.1, 0.3, and 0.4 mg mL−1 of FA was 73.9 ± 2.86, 104.4 ± 4.60, and 116.9 ± 1.69 nm, respectively with PDI values in the range of 0.33 to 0.36.
| PLGA NPs | [FA] (mg mL−1) | DLS (nm) | PDI | ζ-Potential (mV) | EE% |
|---|---|---|---|---|---|
| a The total volume NPs dispersions was 4.0 mL. PLGA concentration in all cases was 2.8 mg mL−1. n.d. not determined. | |||||
| PLGA | — | 46.6 ± 0.74 | 0.28 ± 0.009 | −18.0 ± 2.36 | — |
| FA-loaded | 0.1 | 73.9 ± 2.86 | 0.36 ± 0.06 | −22.4 ± 1.71 | 90.0 ± 7.03 |
| FA-loaded | 0.3 | 104.4 ± 4.60 | 0.33 ± 0.15 | −26 ± 0.73 | 79.7 ± 2.04 |
| FA-loaded | 0.4 | 116.9 ± 1.69 | 0.35 ± 0.007 | −28.4 ± 0.27 | n.d. |
This trend observed for size distribution has been previously reported.70 From a physicochemical point of view, it is expected that the number of FA-loaded nanoparticles obtained after solvent evaporation may depend on mechanisms that govern the transition from an emulsion droplet state to a nanoparticle state.71 In this sense, one can hypothesized that emulsion droplets, which tend to be stabilized by surfactant molecules in the formulation, may display of certain colloidal stability during solvent evaporation. Therefore, it is expected that the formation of a FA-loaded nanoparticle may be produced from a single volume shrinkage of the emulsion droplet.
As described above, a PIC low-emulsification method has been used as an approach to generate the anticipated polymeric NPs.18
The incorporation of FA above certain concentrations to the oil phase may cause an increase in viscosity that hampers droplet breakdown. As a consequence, the droplet and nanoparticle sizes increase. FA may also have some interfacial activity leading to coadsorption with the surfactant at the oil/water interface, which affects emulsion stability.
As expected, all colloidal dispersions exhibited consistent negative ζ-potential values in the range −18.0 to −28.4 mV which produce electrostatic repulsion preventing NP aggregation and consequently increasing colloidal stability.72 Interestingly, we also found that the ζ-potential becomes more negative as the amount of FA loaded in NPs increases. This trend suggests that most FA is located close to the PLGA surface.73
TEM images of FA-loaded NPs containing 0.1 mg mL−1 of the entrapped drug displayed evenly distributed spherical PLGA NPs. Interestingly, TEM analysis also showed a dominant population of NPs (∼63 nm) together with a few smaller nanosized particles (∼46 and 23 nm) (Fig. S2†). The presence of such small size populations may explain the relatively high PDI values obtained in DLS measurements. The average diameter of dried FA-loaded NPs measured by TEM was found to be ∼15% smaller than the hydrodynamic diameter measured by DLS. This difference in size was likely attributed to a deflation process upon drying.74 Finally, hyperspectral microscopy was used as an analytical tool for the visualisation of FA-loaded polymeric NPs. We first obtained the scattering spectral library plots of both unloaded PLGA and FA alone (Fig. S3†). The microscopic image of the formulation was visualised, mapped, and the hyperspectral image of FA-loaded NPs was obtained. Interestingly, the location of FA coincided with the position of PLGA NPs previously analysed when FA and PLGA NPs were co-mapped (Fig. 3). This may corroborate that FA was efficiently encapsulated within polymeric NPs.
Encapsulation efficiency (EE%)
Analytical methods including reverse-phase chromatography (RP-HPLC) have played a key role in analysing the effectiveness of drug delivery systems to incorporate active molecules.75 In this sense, FA-loaded colloidal formulations were firstly centrifuged to collect the supernatant prior to HPLC analysis. Once analysed, the EE% was calculated by the percentage of FA which was incorporated into NPs with respect to the initial FA amount added. A calibration curve with different FA solutions ranging from 40 to 0.008 μg mL−1 was fitted to the linear equation Y = 198110x + 47277 (correlation coefficient, r2 = 0.998) (Fig. S4†).
To determine the EE%, two FA concentrations were initially encapsulated within PLGA NPs (0.1 and 0.3 mg mL−1). High EE% were obtained in both cases but a 0.1 mg mL−1 of FA produced higher EE% when compared with 0.3 mg mL−1 (90.0% ± 7.03 versus 79.7% ± 2.04, respectively) (Table 1).
In vitro drug release
Prior to studying the release of FA-loaded PLGA NPs, two receptor solutions: (i) PBS (pH 7.4) and (ii) a mixture of 1× PBS:EtOH (15%) were selected to monitor the diffusion of FA from a solution (0.05 mg mL−1) over time.
This was studied using a dialysis bag immersed in the receptor solution at 37 °C under sink conditions. The amount of FA released at various time intervals was determined by HPLC at 307 nm using a calibration curve (Fig. S4†). Diffusion experiments using exclusively 1× PBS as a receptor phase were unsuccessful. As a matter of fact, the major issue found was the inability to maintain sink conditions in the system due to the relatively low solubility of our FA stock in a PBS solution as well as the difficulty in monitoring its diffusion through the dialysis membrane. Some authors have used a sodium carboxymethyl cellulose solution (0.25%, w/v) or 0.01 M PBS with a FA concentration of 0.2 mg mL−1 for both release diffusion experiments and the measurement of FA stability over time, respectively.76,77
The second receptor phase combining a 1× PBS solution with EtOH has allowed us to analyse the release of hydrophobic substances from polymeric NPs.28 In this sense, we selected a mixture of 1× PBS:EtOH (15%) as an appropriate medium to study the release behaviour of both FA-loaded NPs and free FA solution. Despite these conditions favoured sink conditions allowing FA to be completely dissolved within the dialysis membrane, FA release was incomplete, reaching a plateau at only 66% release (Fig. 4A). Curiously, other reported diffusion experiments involving 20% of a free FA solution reached a plateau after 48 hours-incubation.76 To achieve the complete diffusion of FA, we decided to enlarge the incubation time,28 but high standard deviation values in the quantification measurements were obtained owing to undesirable losses of the ethanolic solvent through evaporation in the receptor phase. Limited solubility properties of the drug may also significantly impact the diffusion profile.78
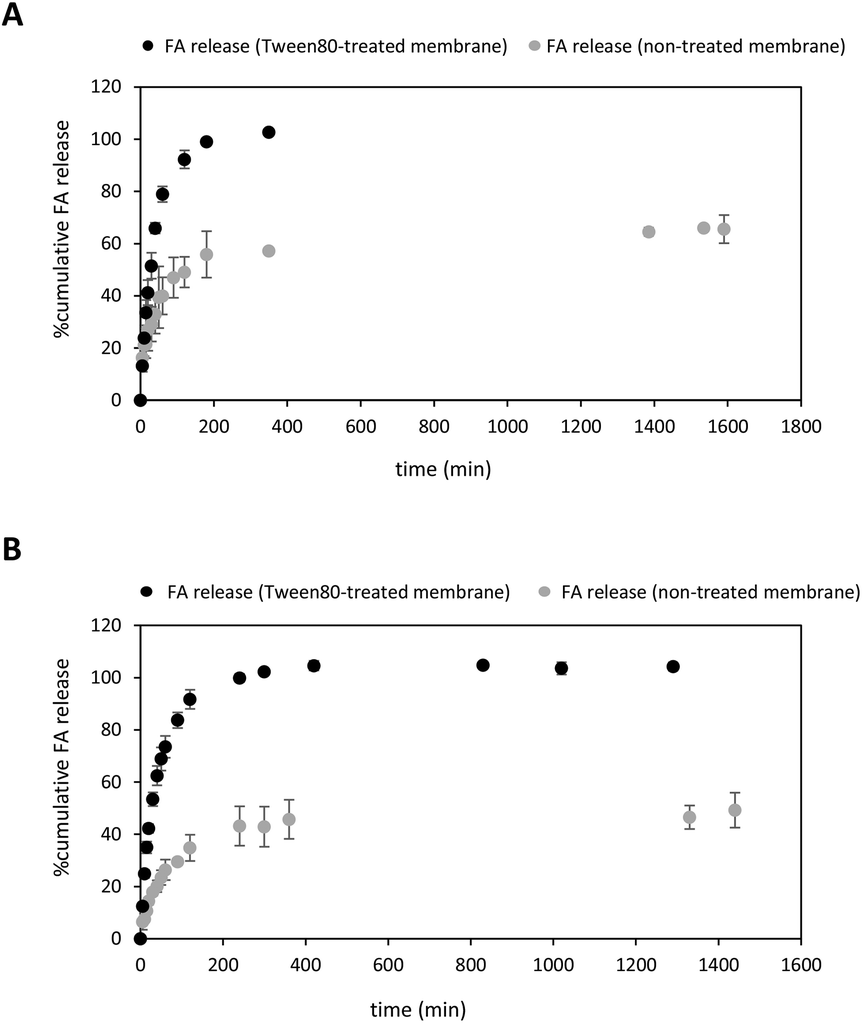 | ||
| Fig. 4
In vitro cumulative release of FA at 37 °C monitored over time. (A) FA diffusion from a FA solution through a non-treated (grey dots) and Tween-80-treated (black dots) dialysis membrane. (B) The release of FA from NPs through a non-treated (grey dots) and Tween-80-treated (black dots) dialysis membrane. The receptor phase and FA solution contained a mixture of PBS:EtOH (15%) (40 mL). Data correspond to the mean of three independent experiments (SD = 3). | ||
In our case, diffusion experiments are carried out in closed systems where potential interactions between FA and cellulose membrane might arise and affect the diffusion process to a significant extent. Similarly, other processes such as dissolution and re-precipitation might also play a key role in the diffusion, especially in closed systems.78
Thus, once an appropriate amount of FA is dissolved in a PBS:EtOH (15%) mixture, FA tends to diffuse into the receptor phase at early time points, thereby increasing the FA concentration in the ethanolic solution. However, as the incubation time is prolonged, some of the volatile solvent may evaporate thereby affecting the FA concentration. This might facilitate FA re-precipitation and lead to a dynamic equilibrium that involves the co-existence of dissolved and non-dissolved FA during the diffusion process thus affecting the sink conditions that were initially established in the system.78
To facilitate the complete release of the drug, we decided to soak first the dialysis membrane with 3% Tween 80 in a 1× PBS solution (Fig. 4A) rather than using a receptor phase containing the surfactant dissolved in a PBS solution.79 To the best of our knowledge, this pre-treatment protocol has not been reported yet in this sort of diffusion experiments. Much to our delight, the diffusion of free FA to the same ethanolic receptor phase under sink conditions was complete after 180 min-incubation at 37 °C (Fig. 4A, black dots). This was primarily due to the change not only in the surface properties of the cellulose membrane but also the presence of hydrophobic binding interactions between FA and Tween micelles.79–82 As a consequence, this process might favour the solubilization of the drug and promote its total diffusion to the receptor phase.
After optimising the in vitro release conditions, the release of FA from PLGA NPs to the ethanolic receptor solution was studied using both non-treated and treated cellulose membranes (Fig. 4B). As expected, the release of FA was sustained over time regardless of whether the dialysis membrane was treated with the surfactant or not. Interestingly, the release rate of FA from NPs exhibited an increasing trend in the case of Tween 80-treated cellulose membrane (Fig. 4B, black dots), similar to the release rate of free FA (Fig. 4A).
Different trends were observed in the first minutes of incubation depending on the type of membrane used. When a non-treated cellulose membrane was used, an initial burst of approximately 8% of the drug was detected within the first 10 minutes of incubation. However, pre-treatment with the surfactant resulted in higher levels of uncontrolled burst of FA reaching about 25% within the same 10 min-incubation. This uncontrolled release during the first period of incubation was attributed to the FA adsorbed to the surface of the PLGA NP.83 The use of a non-treated cellulose membrane provided a prolonged release of FA from PLGA NPs which gradually reached a plateau at ca. 50% after 24 hours-incubation (Fig. 4B, grey dots). Interestingly, these experimental conditions afforded greater diffusion values when compared with free FA (ca. 66%). This clear difference in the release rates may indicate appropriate FA encapsulation within PLGA NPs, as observed in other drug delivery systems based on the same polymer.28,84 This ability of the PLGA NPs to favour controlled release FA may depend not only on PLGA degradation properties but also might be attributed to the lactic/glycolic ratio and molecular weight of the polymer.28,84,85
On the contrary, higher cumulative amount of FA released was observed using a surfactant-treated cellulose membrane. Notably, the FA release was complete after 240 minutes (Fig. 4B, black dots). Surprisingly, we did not observe significant release kinetics differences between free FA and FA-loaded NPs. This might be explained by assuming a change in the surface of the dialysis treated membrane properties which turned from hydrophobic into hydrophilic one. Indeed, lipophilic FA might adsorb the treated membrane through hydrophobic interactions and favour the amount of soluble FA to the receptor phase,86 as previously observed in Fig. 4A.
Alternatively, the EtOH content in the receptor solution was increased up to 50% with the aim to study how ethanolic solution affects drug release kinetics. As illustrated in Fig. S5,† FA release from PLGA NPs to the acceptor compartment (1× PBS:50% EtOH) was monitored for the first 90 min-incubation at 37 °C. Unfortunately, incubation times greater than 90 min afforded high deviations probably due to EtOH evaporation, as observed above. The %cumulative release of FA displayed a quasi-linear profile reaching almost the half of the initial amount of drug (ca. 48.2%) during the first incubation times (90 min).
Interestingly, FA release was remarkably higher if compared with the in vitro release previously obtained using 15% EtOH in the receptor phase (ca. 29.5%). This preliminary study might confirm the ability of EtOH to diffuse from the acceptor to the donor compartment affecting the FA release behaviour. Therefore, we hypothesized that high % of ethanol present in the receptor phase may enter the solution inside the dialysis bag, which is the donor compartment, and favour FA solubilization. As a result, a large proportion of the solubilized FA might permeate through the dialysis membrane reaching finally the receiver compartment.86
Release mechanism studies
Five mathematical models including (i) zero-order; (ii) first order; (iii) Higuchi; (iv) Korsmeyer–Peppas; and (v) Weibull distribution have been used to study the release mechanism of FA from PLGA NPs (Fig. 5 and Fig. S6†). The experimental data were fitted using the DDsolver Add-in Program63 which afforded not only the values of the constants (K0, K, KH, KK–P, and KWB), but also the correlation coefficient (r2), the Akaike's information criterion (AIC)87 and the model selection criterion (MSC)88 values. The careful examination of these values led to the identification of the best fit equation to describe the FA release from PLGA NPs. In this sense, the highest r2 value, the largest MSC value,89 and lowest AIC value90 were considered the criteria to describe the suitability of a model to fit to the experimental data. Therefore, the most accurate equation for FA-loaded NPs was the Weibull model (r2 = 0.95; AIC = 95.68, and MSC = 2.37) (Fig. 5 and Table 2).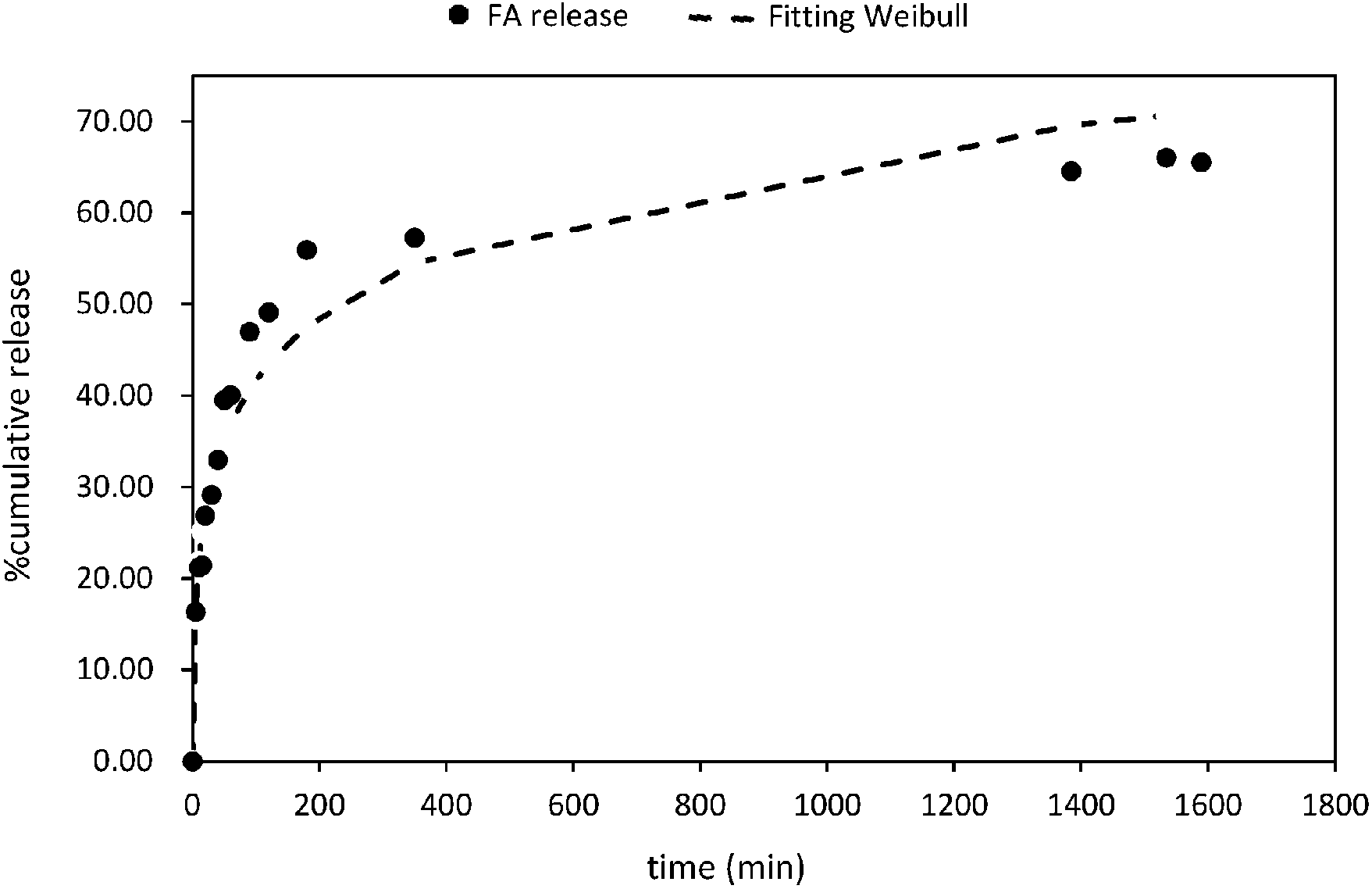 | ||
| Fig. 5 Fitting of FA release data to Weibull distribution. | ||
| Zero-order | First-order | Higuchi | Korsmeyer–Peppasa | Weibullb | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a Diffusional exponent, n = 0.169. b Constant, α = 7.24 and diffusion mechanism, β = 0.298. | ||||||||||
| Constant | K Z–O | 0.049 | K F–O | 0.001 | K H | 2.13 | K K–P | 18.089 | K WB | 3.0 |
| Correlation value (r2) | 0.73 | 0.79 | 0.85 | 0.90 | 0.95 | |||||
| AIC | 154.76 | 150.65 | 141.13 | 105.74 | 95.68 | |||||
| MSC | −1.32 | −1.06 | −0.46 | 1.74 | 2.37 | |||||
The Weibull distribution model is entirely empirical and is both related to the size and geometry of the matrix in all Euclidian spaces.62 The model can describe the transport mechanism of a drug through a polymeric matrix based on the value of the β exponent (eqn (7)). As shown in Table 2, β afforded a value of 0.298 (β < 0.75) which suggested a Fickian diffusion mechanism62 of FA through the PLGA matrix. Because this model is considered empirical, it has certain limitations with regard to the nature of the intrinsic dissolution of the active.91 Therefore, the Korsmeyer–Peppas equation was selected as a semi-empirical model to understand the release mechanisms that govern the FA diffusion to the receptor phase.
This model gave a good correlation coefficient (r2 = 0.90) and values of 105.74 and 1.74 for AIC and MSC, respectively (Fig. S5D†). Moreover, this model suggests the dependence of release on both drug concentration and incubation time.92,93
According to the Korsmeyer–Peppas kinetic model, the release exponent n describes the dominant release mechanism and affords knowledge about the diffusion and erosion produced in the matrix. In our case, n = 0.169, which is lower value than the standard value assigned to Fickian diffusion (n < 0.5). Accordingly, FA is released from the PLGA NPs with a quasi-Fickian diffusion as a dominant mechanism. This might be in line with the amount of FA encapsulated which might be located at the edge of the PLGA surface facilitating the FA diffusion.94 Other examples reported for drug-loaded PLGA NPs showing quasi-Fickian diffusion mechanisms can be found elsewhere.95–98 A linear trend in a log–log plot of FA released versus incubation time also, confirmed this diffusion mechanism (Fig. S5D,† inset). Moreover, drug release patterns and processes affecting the release of encapsulated drugs from PLGA nanoparticulate systems have been thoroughly studied.
Accordingly, parameters like nanoparticle size, geometry, polymer content, or interactions of the active with the polymer may also influence drug release behaviour.99,100 It is well known that PLGA NPs exhibiting small size contain a large surface area for the active, in this case FA, to diffuse out.101 Indeed, studies have shown that diffusion mechanisms dominate when this diffusion process takes place faster than PLGA matrix degradation.102 As time incubation time increases, PLGA NPs may undergo bulk erosion through PBS-mediated hydrolysis via autocatalytic degradation of PLGA's ester bonds at neutral pH.103,104 Hence, a combination of two mechanisms, namely diffusion and PLGA degradation, might have a contribution during the later phase of the FA release.103,105 However, additional experiments involving in vitro weight loss evaluation or NP diameter analysis during drug release might be considered to study and characterise bulk degradation over time.
DPPH assay
Radical scavenging activities of both FA-loaded PLGA NPs and a free FA solution as a control were investigated in vitro using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) colorimetric assay. DPPH exhibits a purple colour and has a free hydrogen radical. It also shows a characteristic peak at 517 nm.When small molecules with antioxidant properties donate free hydrogen to DPPH, it gets converted to 2,2-diphenyl-2-picrylhydrazine leading to a change colour from purple to yellow. The antioxidant activity mechanism of FA against DPPH radical has been studied before. It is believed that the stability of the phenoxy radical is increased owing to the disposal of the FA's substituents including the ortho-substituted methoxy group and the hydroxyl group which tend to favour the electron-density delocalisation through FA benzene ring.106
The antioxidant activities of drug-loaded polymeric NPs with a PLGA concentration of 0.233 mg mL−1 containing [FA] = 0.2 and 0.1 mg mL−1 were measured according to established protocols reported in the literature.65 As displayed in Fig. S7A,† the antioxidant activity of FA-loaded PLGA NPs was well-preserved exhibiting a dose–response effect with scavenging activities of 43 and 22% for [FA] = 0.2 and 0.1 mg mL−1, respectively. A free FA solution was also studied as a control, affording greater antioxidant activities than their drug-loaded counterparts (60 and 41.3% for [FA] = 0.2 and 0.1 mg mL−1, respectively).
Recently, Pham et al. observed that scavenging activities of an encapsulated active gradually increased over time, suggesting that the antioxidant effectiveness may be directly related to the release of the active from the NPs.107 Based on our drug release studies shown above, it is expected that FA would also exhibit a gradual interaction with DPPH due to its sustained release from the polymeric NPs. However, we were unable to confirm this behaviour after incubating FA-loaded PLGA NPs in the presence of an ethanolic DPPH solution (50% v/v) at shorter incubation times (5, 10, and 15 min). Indeed, the reported scavenging activity at these time intervals was not statistically different from the 30 minutes-incubation period as observed in Fig. S7A and S7B.† This was probably due to the ability of EtOH to facilitate the solubility giving rise to a rapid release of FA from the polymeric network, as illustrated in Fig. S5.†
Cytotoxicity
The cytotoxicity of FA-loaded NPs was evaluated through tetrazolium salt-based colorimetric assays.108 Three concentrations of FA-loaded NPs (0.082, 0.041, and 0.020 mg mL−1), free FA and PLGA alone as control experiments were tested in the human neuroblastoma derived SH-SY5Y cell line. As expected, PLGA NPs did not have any a negative effect on cellular proliferation or cell morphology of neuronal cells after 24 hours-incubation and cellular viabilities over 90% were obtained.The effects of FA-loaded PLGA_FITC NPs on cellular viability were tested on all the cell types included in the BBB-oC model (HA-h, HBVP, and hCMEC/D3 cells). The NPs were tested at concentrations of 0.056, 0.090, 0.112 and 0.280 mg mL−1, including non-loaded PLGA_FITC NPs as control. NPs show no significant cytotoxic effects at concentrations ≤0.056 mg mL−1, with viability values over 90% for HA-h and hCMEC/D3 cells, and around the 75% for HBVP. These results clearly support the biocompatible properties of PLGA polymeric NPs (Fig. S8†).
Cellular uptake
Internalisation studies of FA-loaded polymeric NPs were carried out in the presence of SH-SY5Y neuronal cells. The ratio between non-treated and treated cells was analysed by flow cytometry. Prior to this analysis, fluorescently labelled NPs were prepared using the same emulsification protocol as described before. In previous research, the encapsulation of Coumarin-6 allowed us to quantify the effectiveness of PLGA NPs to impart cellular uptake.84 Herein, we selected fluorescein (Flu) as an appropriate dye to be decorated in the NP surface.To conjugate both PLGA NPs (bare PLGA and FA-loaded NPs) with Flu, a poly(ethylene glycol)diamine (PEGdiamine) was selected as an appropriate linker to facilitate the conjugation reaction between PLGA NPs and fluorescein isothiocyanate (FITC). Several methodologies have been described to functionalise NPs with PEG ligands. Thus, strategies based on physical adsorption, covalent coupling, or self-assembly processes of PEG block copolymers have been reviewed.109
Carbodiimide chemistry was selected as a synthetic strategy to link the PLGA NPs’ carboxylic acid to the first amine pendent group of the PEG linker, according to well-established conjugation protocols.28 Thus, the conjugation reaction involved the activation of NPs’ carboxylic acids with (EDC/NHS) in acid conditions, whilst the PEGylation reaction with 5 eq. of PEGdiamine was carried out under basic pH (pH 8) overnight (Fig. S9†). Prior to adding FITC, the % PEGylation conjugation efficiency was calculated by determining the molar ratio between PLGA and PEG from NMR spectra (Fig. S9†).56 NMR analysis of dried NPs in MeOD showed that the PEG content decorating NPs was low. In this regard, the resulting PLGA and PEG content was 71% and 29%, respectively which resulted in a PLGA-to-PEG molar ratio of 2.45 (ca. 41% conjugation efficiency). Finally, the use of FITC favoured the final conjugation reaction through the second amine group of the linker. Finally, a dialysis process facilitated the removal of reagents used in excess and the resultant fluorescent NPs (PLGA_Flu and FA-loaded PLGA_Flu) were characterised by DLS (Fig. S10†). As expected, the covalent incorporation of additional molecules (PEG diamine linker and Flu) on the polymeric NPs’ surface did not affect the colloidal stability obtaining average hydro diameter values of 48.0 ± 4.0 (PDI = 0.26) and 65.3 ± 4.71 (PDI = 0.29) for PLGA_Flu and FA-loaded PLGA_Flu, respectively (Table S2†).
The ability of FA-loaded PLGA_Flu NPs (0.087 mg mL−1) to promote cellular uptake in SH-SY5Y neuronal cells was studied. The average ratio between cell population control and positive cells were analysed by flow cytometry. As shown in Fig. 6, we firstly selected a R1 region in a dot plot chart for our neuronal cell population (Fig. 6A). A R2 region was then selected which was used to quantify the number of positive cell populations that were transfected with FA-loaded NPs (Fig. 6B). As observed in Fig. 6C, the internalisation caused almost the totality of neuronal cells to be transfected with the fluorescent NPs. This was confirmed by displaying both non-treated and positive cell populations in an overlay histogram, that clearly showed the displacement of fluorescently labelled population in relation to non-fluorescent neuronal cells (Fig. 6C). Interestingly, this NPs efficiency was also observed when other drug-loaded polymeric NPs, prepared through nano-emulsion templates, were used.84
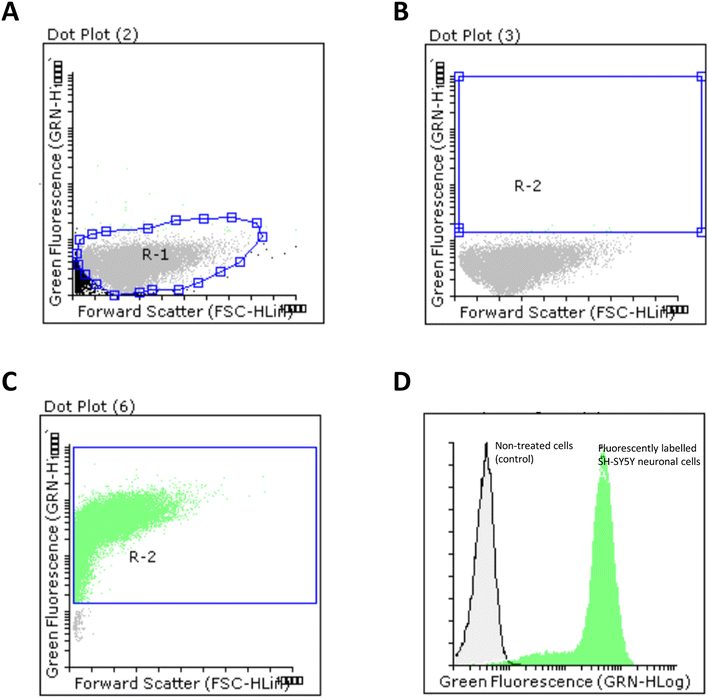 | ||
| Fig. 6 Cellular internalisation of FA-loaded PLGA NPs within SH-SY5Y neuronal cells. The ratio between non-treated fluorescent cells was analysed by flow cytometry. A. SH-SY5Y cell population. (A) R1 region was selected; (B) selection of a R2 region to analyse the number of positive cells transfected; (C) fluorescent cell population after transfection with FA-loaded PLGA_Flu; (D) combined histogram. | ||
Hydrogen peroxide toxicity assay in SH-SY5Y cells
The antioxidant effect of FA-loaded PLGA NPs was determined upon incubating hydrogen peroxide (75 μM) in SH-SY5Y cells. For that purpose, cells were treated with FA alone, bare PLGA, and FA-loaded PLGA NPs at two concentrations (0.082 and 0.041 mg mL−1). NPs and controls were firstly incubated for 2 hours at 37 °C and subjected to H2O2-treatment for one additional hour. Cellular viabilities were evaluated using the MTT assay.66 Notably, the two concentrations of FA-loaded PLGA NPs moderately suppressed the intracellular ROS-level induced by H2O2 in a dose–response manner, obtaining cellular viabilities that ranged from ca. 52% and 24.7% for 0.082 and 0.041 mg mL−1, respectively (Fig. S11†). These preliminary data may suggest not only the efficient cellular internalisation of the NPs and subsequent release of the phytochemical but also the retention of antioxidant activity by FA-loaded PLGA NPs as obtained in the DPPH assay.One of the major bottlenecks in designing NP formulations lies largely in inducing endosomal escape while retaining activity and cellular viability. Therefore, characterising and understanding the mechanisms that govern the interactions between cells and NP composition, especially polymeric NPs, is a key issue.110 For this reason, further efforts are oriented to improve endosomal escape by modulating polymer architecture, polymer disassembly or hydrophobicity in order to engineer more effective nanostructured delivery systems.
Blood–brain barrier membrane model (BBB-oC)
The capability of FA-loaded NPs towards crossing the BBB was tested in an in vitro microphysiological model (BBB-oC). This model includes three major cell types conforming the brain/blood interface: the brain cells; HA-h and HBVP embedded in a 3D scaffold of fibrin gel allocated in the central chamber and hCMEC/D3 cells in one of the two fluidic channels flanking the central chamber in directly contact with the brain cells. Permeability assays were conducted by introducing solutions of FA-loaded PLGA_Flu NPs, and non-loaded PLGA_Flu NPs as control, at a concentration of 0.05 mg mL−1 into the endothelial fluidic channel of the chip and capturing epi-fluorescence images at 3 min intervals for 60 minutes (Fig. 7). Both nanoparticles were effectively internalized towards the brain part of the model, crossing the in vitro BBB with permeability values for FA-loaded PLGA_Flu NPs, and non-loaded PLGA_Flu NPs of 3.7 × 10−6 cm s−1 and 5.1 × 10−6 cm s−1, respectively. These values indicate that FA-loading cause a significant decrease in permeability.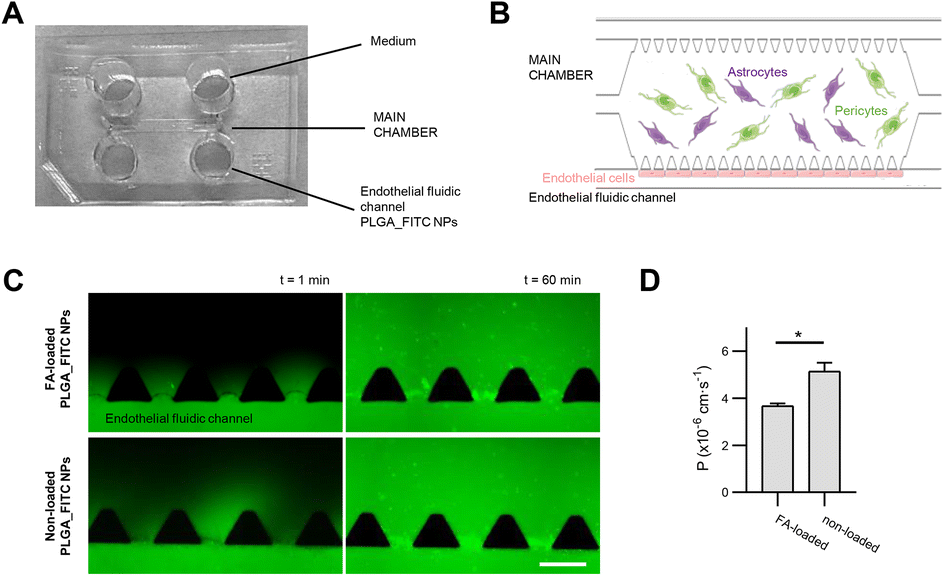 | ||
| Fig. 7 Permeability assay. (A) BBB-OoC fabricated in PDMS. The main chamber is in the central part of the chip, and it is flanked at both sides by two microfluidic channels, which are interconnected. Perfusion of the PLGA_Flu NPs is conducted in the endothelial fluidic channel. (B) Schematics of cell culture inside the chip. Human astrocytes (HA-h) and pericytes (HBVP) are embedded in a fibrin hydrogel in the main chamber, and human brain endothelial cells (hCMEC/D3) are cultured in one of the fluidic channels. Endothelial cells are in direct contact with the brain cells through the spaces between the trapezoid pillars that separate the main chamber from the fluidic channels. (C) Representative epi-fluorescence images taken at t = 1 min and t = 60 min after the injection of the FA-loaded and non-loaded PLGA_Flu NPs, respectively, showing the increase in the fluorescence intensity in the main chamber with time. Scale bar = 300 um. (D) Plot of the permeability values calculated for FA-loaded and non-loaded PLGA_Flu NPs in the BBB-OoC. *p < 0.05. | ||
PLGA_Flu NPs not targeting any specific receptor in the endothelial cells’ membrane, are assumed to cross the BBB through passive internalisation,111 which for polymeric NPs depends on several parameters, such as composition, size, surface charge and functionalization among others.112,113 Considering the size of NPs, previous studies have shown that particles with sizes between 20–50 nm have a more efficient permeation than those around 70 nm.114,115 According to that, non-loaded PLGA_Flu NPs with a size of 48 nm, compared to the 65 nm of FA-loaded PLGA_Flu NPs should be easily internalized in agreement with results from the permeability assays.
Regarding charge effects, brain microvascular endothelial cells have a net negative surface charge, thus repelling negatively charged compounds.116 DLS results indicate that large size NPs have more negative charges on the surface (Table 1). Therefore, large size FA-loaded PLGA_Flu NPs having more negative charges, are expected to experience an added difficulty in crossing the BBB. However, PLGA NPs functionalised with a PEG-linked fluorophore suffer a decrease in the ζ-potential due to charge neutralization upon derivatization (e.g., from −22.4 ± 1.71 mV to −13.6 ± 0.10 mV in FA-loaded PLGA_Flu NPs) (Table S2†). This will decrease the electrostatic repulsion, partially masking charge effects in the BBB-oC model and making permeability more sensitive to the particle size.
Conclusions
Polymeric NPs made up of PLGA and prepared from NE templates were selected as suitable carriers to encapsulate the antioxidant phytochemical FA. FA-loaded NPs had hydrodynamic diameter sizes that increased with FA concentration. Nanometric size might help FA-loaded NPs cross tissue barriers like the BBB.117 TEM analysis confirmed the spherical morphology of FA-loaded NPs. Dark-field microscopy with hyperspectral analysis also showed the presence of FA inside PLGA NPs. High EE% was obtained when a FA concentration of 0.1 mg mL−1 was used, but this efficiency dropped when the concentration increased to 0.3 mg mL−1. In vitro release experiments were carried out with the aim to characterise the release rate of FA. Unexpectedly, we were unable to monitor the FA release from NPs under physiological conditions because of the inability of FA to be dissolved in PBS. Alternative conditions that combined PBS with EtOH (15%) as a final receptor phase and coated the hydrophobic cellular membrane with Tween 80 were selected.These conditions allowed us to monitor and compare FA diffusion rates from a solution and NPs, respectively. As a result, we found that FA can be released from PLGA NPs in a sustained manner over time compared to a FA solution. In vitro FA release data were fitted to the Weibull and the Korsmeyer–Peppas kinetic models. These models suggested that Fickian diffusion governed the release of FA from the PLGA matrix. Conversely, the treatment of a dialysis cellulose membrane with a non-ionic surfactant had a profound impact on both FA diffusion from a solution and its release from NPs. We hypothesised that the adsorbed surfactant promoted the transport of FA across the membrane and also favoured its complete solubilisation in the receptor phase. Additionally, a dose–response antioxidant activity of FA-loaded NPs when the FA concentration ranged from 0.2 to 0.1 mg mL−1 was obtained using the DPPH˙ assay. FA-loaded NPs did not affect the cellular proliferation and helped cellular uptake in SH-SH5Y neuronal cells. The efficient cellular internalisation and antioxidant activity of FA-loaded PLGA NPs were confirmed subjecting a neuronal cell model to hydrogen peroxide. Interestingly, preliminary results suggested that polymeric NPs containing FA moderately suppressed the intracellular ROS-level in a dose–response manner.
Regarding the brain penetration properties of the synthesized NPs under study, their permeability was analysed using an in vitro BBB-oC microphysiological model. The results show effective internalization of FA-loaded PLGA_Flu NPs and non-loaded PLGA_Flu NPs, but with a significant decrease in the permeability of FA-loaded particles (3.7 × 10−6 cm s−1 and 5.1 × 10−6 cm s−1, respectively), which is attributed to the increase in the diameter of the loaded NPs as compared to non-loaded ones (65 nm vs. 48 nm).
These results put forward important contributions in the field of polymeric NPs prepared from NEs as suitable DDS to deliver hydrophobic antioxidant actives in a controlled way. The ability of these nanocarriers to permeate through an in vitro BBB model makes them promising vehicles to be used in the management of neurological diseases.
Author contributions
Conceptualization, S.G., M.M., A.L.; methodology, L.G., S.P., V.E., F.S.R., M.M., A.L. and S.G.; investigation and formal analysis, L.G., S.P., V.E., F.S.R., M.M, A.L., and S.G.; writing-original draft preparation, S.G., M.M., S.P. and A.L.; writing-review and editing, M.M., A.L., J.S., M.J.G-C., C.R-A., and S.G.; project administration, S.G., M.M. and A.L.; funding acquisition, C.R-A., M.M. and A.L.; supervision, C.R-A., J.S., M.M., A.L. and S.G. All authors have read and agreed to the published version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Agencia Estatal de Investigación for funding (Projects CTQ2017-84998-P and PID2021122187NB-C31) and the Instituto de Salud Carlos III (ISCIII) (CB06/01/1058 and CB06/01/0055). CIBER BBN is an initiative funded by the VI National R + D + I Plan 2008–2011, Iniciativa Ingenio 2010, Consolider Program, CIBER actions and financed by ISCIII with assistance from the European Regional Development Fund. The authors would like to thank Gemma Fabriàs’ research group for assistance and access to the cytometer instrument. The authors want to thank NANBIOSIS ICTS and the Nanostructured Liquid Characterization Unit (U12) for DLS and ζ-potential measurements, hyperspectral microscopy and dark-field imaging. The authors thank the Electron Microscopy Service from the National Centre of Biotechnology (CNB-CSIC), especially Beatriz Martín for sample preparation and TEM observations. The authors also acknowledge the “Grupo de Nanotecnología Farmacéutica” from the University of Barcelona in the Faculty of Pharmacy and Food Sciences which forms a R&D Associate Unit to CSIC.References
- L. Leon, E. J. Chung and C. Rinaldi, in Nanoparticles for Biomedical Applications, ed. E. J. Chung, L. Leon and C. Rinaldi, Elsevier, 2020, pp. 1–4 Search PubMed.
- W. Zhang, M. Zhao, Y. Gao, X. Cheng, X. Liu, S. Tang, Y. Peng, N. Wang, D. Hu and H. Peng, Chem. Eng. J., 2022, 433, 133498 CrossRef CAS.
- T. Tong, Y. Qi, L. D. Bussiere, M. Wannemuehler, C. L. Miller, Q. Wang and C. Yu, Nanoscale, 2020, 12, 16339–16347 RSC.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- C. Fornaguera and M. J. García-Celma, J. Pers. Med., 2017, 7, 12 CrossRef PubMed.
- J. M. v. Makabenta, A. Nabawy, C.-H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19, 23–36 CrossRef CAS PubMed.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761–769 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2016, 1, 10–29 CrossRef PubMed.
- C. I. C. Crucho and M. T. Barros, Mater. Sci. Eng., C, 2017, 80, 771–784 CrossRef CAS PubMed.
- C. Vauthier and K. Bouchemal, Pharm. Res., 2009, 26, 1025–1058 CrossRef CAS PubMed.
- C. Solans, P. Izquierdo, J. Nolla, N. Azemar and M. J. Garcia-Celma, Curr. Opin. Colloid Interface Sci., 2005, 10, 102–110 CrossRef CAS.
- K. Landfester, Macromol. Rapid Commun., 2001, 22, 896–936 CrossRef.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- K. Landfester, Adv. Mater., 2001, 13, 765–768 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, Langmuir, 2001, 17, 908–918 CrossRef CAS.
- S. Desgouilles, C. Vauthier, D. Bazile, J. Vacus, J.-L. Grossiord, M. Veillard and P. Couvreur, Langmuir, 2003, 19, 9504–9510 CrossRef CAS.
- H. Jasmina, O. Džana, E. Alisa, V. Edina and R. Ognjenka, in CMBEBIH 2017, Springer, 2017, pp. 317–322 Search PubMed.
- C. Solans and I. Solé, Curr. Opin. Colloid Interface Sci., 2012, 17, 246–254 CrossRef CAS.
- A. Gupta, H. B. Eral, T. A. Hatton and P. S. Doyle, Soft Matter, 2016, 12, 2826–2841 RSC.
- P. Izquierdo, J. Esquena, T. F. Tadros, C. Dederen, M. J. Garcia, N. Azemar and C. Solans, Langmuir, 2002, 18, 26–30 CrossRef CAS.
- I. Solè, C. M. Pey, A. Maestro, C. González, M. Porras, C. Solans and J. M. Gutiérrez, J. Colloid Interface Sci., 2010, 344, 417–423 CrossRef PubMed.
- M. Hessien, N. Singh, C. Kim and E. Prouzet, Langmuir, 2011, 27, 2299–2307 CrossRef CAS PubMed.
- P. Fernandez, V. André, J. Rieger and A. Kühnle, Colloids Surf., A, 2004, 251, 53–58 CrossRef CAS.
- J. Feng, M. Roché, D. Vigolo, L. N. Arnaudov, S. D. Stoyanov, T. D. Gurkov, G. G. Tsutsumanova and H. A. Stone, Nat. Phys., 2014, 10, 606–612 Search PubMed.
- R. Dinarvand, N. Sepehri, S. Manoochehri, H. Rouhani and F. Atyabi, Int. J. Nanomed., 2011, 6, 877 Search PubMed.
- S. Grund, M. Bauer and D. Fischer, Adv. Eng. Mater., 2011, 13, B61–B87 CrossRef.
- C. Fornaguera, S. Grijalvo, M. Galán, E. Fuentes-Paniagua, F. J. de la Mata, R. Gómez, R. Eritja, G. Calderó and C. Solans, Int. J. Pharm., 2015, 478, 113–123 CrossRef CAS PubMed.
- C. Fornaguera, A. Dols-Perez, G. Calderó, M. J. García-Celma, J. Camarasa and C. Solans, J. Controlled Release, 2015, 211, 134–143 CrossRef CAS PubMed.
- C. Fornaguera, N. Feiner-Gracia, G. Calderó, M. J. García-Celma and C. Solans, Nanoscale, 2015, 7, 12076–12084 RSC.
- M. Monge, C. Fornaguera, C. Quero, A. Dols-Perez, G. Calderó, S. Grijalvo, M. J. García-Celma, C. Rodríguez-Abreu and C. Solans, Eur. J. Pharm. Biopharm., 2020, 156, 155–164 CrossRef CAS PubMed.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- Z. Liang, A. Currais, D. Soriano-Castell, D. Schubert and P. Maher, Pharmacol. Ther., 2021, 221, 107749 CrossRef CAS PubMed.
- M. Erkisa, M. Sariman, O. G. Geyik, C. Geyik, T. Stanojkovic and E. Ulukaya, Curr Med Chem., 2022, 29(4), 741–783 CrossRef CAS PubMed.
- X. Cao, L. Cao, W. Zhang, R. Lu, J.-S. Bian and X. Nie, Pharmacol. Ther., 2020, 216, 107687 CrossRef CAS PubMed.
- M. T. Kabir, M. H. Rahman, M. Shah, M. R. Jamiruddin, D. Basak, A. Al-Harrasi, S. Bhatia, G. M. Ashraf, A. Najda and A. F. El-Kott, Biomed. Pharmacother., 2022, 146, 112610 CrossRef CAS PubMed.
- Y. P. Singh, H. Rai, G. Singh, G. K. Singh, S. Mishra, S. Kumar, S. Srikrishna and G. Modi, Eur. J. Med. Chem., 2021, 215, 113278 CrossRef CAS PubMed.
- M. Srinivasan, A. R. Sudheer and V. P. Menon, J. Clin. Biochem. Nutr., 2007, 40, 92–100 CrossRef CAS PubMed.
- R. Sultana, Biochim. Biophys. Acta, Mol. Basis Dis., 2012, 1822, 748–752 CrossRef CAS PubMed.
- K. Ono, M. Hirohata and M. Yamada, Biochem. Biophys. Res. Commun., 2005, 336, 444–449 CrossRef CAS PubMed.
- Y.-M. Liu, J.-D. Shen, L.-P. Xu, H.-B. Li, Y.-C. Li and L.-T. Yi, Int. Immunopharmacol., 2017, 45, 128–134 CrossRef CAS PubMed.
- M. Rychlicka, A. Rot and A. Gliszczyńska, Foods, 2021, 10, 1417 CrossRef CAS PubMed.
- W. M. Pardridge, J. Cereb. Blood Flow Metab., 2012, 32, 1959–1972 CrossRef CAS PubMed.
- N. Poovaiah, Z. Davoudi, H. Peng, B. Schlichtmann, S. Mallapragada, B. Narasimhan and Q. Wang, Nanoscale, 2018, 10, 16962–16983 RSC.
- D. Shukla, N. K. Nandi, B. Singh, A. Singh, B. Kumar, R. K. Narang and C. Singh, J. Drug Delivery Sci. Technol., 2022, 103621 CrossRef CAS.
- C. Lovelyn and A. A. Attama, J. Biomater. Nanobiotechnol., 2011, 2, 626 CrossRef CAS.
- S. Tang, Z. Davoudi, G. Wang, Z. Xu, T. Rehman, A. Prominski, B. Tian, K. M. Bratlie, H. Peng and Q. Wang, Chem. Soc. Rev., 2021, 50, 12679–12701 RSC.
- E. Jagtiani, M. Yeolekar, S. Naik and V. Patravale, J. Controlled Release, 2022, 343, 13–30 CrossRef CAS PubMed.
- A. Avdeef, Expert Opin. Drug Metab. Toxicol., 2005, 1, 325–342 CrossRef CAS PubMed.
- Z. Davoudi, N. Peroutka-Bigus, B. Bellaire, A. Jergens, M. Wannemuehler and Q. Wang, Mar. Drugs, 2021, 19, 282 CrossRef CAS PubMed.
- Z. Zhao, X. Chen, A. M. Dowbaj, A. Sljukic, K. Bratlie, L. Lin, E. L. S. Fong, G. M. Balachander, Z. Chen and A. Soragni, Nat. Rev. Methods Primers, 2022, 2, 94 CrossRef CAS.
- M. J. Gomes, B. Mendes, S. Martins and B. Sarmento, in Concepts and models for drug permeability studies, Elsevier, 2016, pp. 169–188 Search PubMed.
- L. M. Griep, F. Wolbers, B. de Wagenaar, P. M. ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS PubMed.
- A. Oddo, B. Peng, Z. Tong, Y. Wei, W. Y. Tong, H. Thissen and N. H. Voelcker, Trends Biotechnol., 2019, 37, 1295–1314 CrossRef CAS PubMed.
- S. Palma-Florez, A. Lopez-Canosa, F. Moralez-Zavala, O. Castaño, M. J. Kogan, J. Samitier, A. Lagunas and M. Mir, J. Nanobiotechnol., 2023, 21, 115–132 CrossRef CAS PubMed.
- T. Tadros, P. Izquierdo, J. Esquena and C. Solans, Adv. Colloid Interface Sci., 2004, 108, 303–318 CrossRef PubMed.
- T. Betancourt, J. D. Byrne, N. Sunaryo, S. W. Crowder, M. Kadapakkam, S. Patel, S. Casciato and L. Brannon-Peppas, J. Biomed. Mater. Res., Part A, 2009, 91, 263–276 CrossRef PubMed.
- S. Lawrence, Microsc. Today, 2021, 29, 50–55 CrossRef CAS.
- J. H. Lee and Y. Yeo, Chem. Eng. Sci., 2015, 125, 75–84 CrossRef CAS PubMed.
- M.-L. Laracuente, H. Y. Marina and K. J. McHugh, J. Controlled Release, 2020, 327, 834–856 CrossRef CAS PubMed.
- T. Higuchi, J. Pharm. Sci., 1963, 52, 1145–1149 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 37–42 CrossRef CAS.
- V. Papadopoulou, K. Kosmidis, M. Vlachou and P. Macheras, Int. J. Pharm., 2006, 309, 44–50 CrossRef CAS PubMed.
- Y. Zhang, M. Huo, J. Zhou, A. Zou, W. Li, C. Yao and S. Xie, AAPS J., 2010, 12, 263–271 CrossRef CAS PubMed.
- O. P. Sharma and T. K. Bhat, Food Chem., 2009, 113, 1202–1205 CrossRef CAS.
- G. Djiokeng Paka, S. Doggui, A. Zaghmi, R. Safar, L. Dao, A. Reisch, A. Klymchenko, V. G. Roullin, O. Joubert and C. Ramassamy, Mol. Pharm., 2016, 13, 391–403 CrossRef CAS PubMed.
- J. E. Chojnacki, K. Liu, X. Yan, S. Toldo, T. Selden, M. Estrada, M. I. Rodríguez-Franco, M. S. Halquist, D. Ye and S. Zhang, ACS Chem. Neurosci., 2014, 5, 690–699 CrossRef CAS PubMed.
- M. Campisi, Y. Shin, T. Osaki, C. Hajal, V. Chiono and R. D. Kamm, Biomaterials, 2018, 180, 117–129 CrossRef CAS PubMed.
- C. Fornaguera, G. Calderó, M. Mitjans, M. P. Vinardell, C. Solans and C. Vauthier, Nanoscale, 2015, 7, 6045–6058 RSC.
- G. A. Roth, S. Tahiliani, N. M. Neu-Baker and S. A. Brenner, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 565–579 CAS.
- F. Madani, S. S. Esnaashari, B. Mujokoro, F. Dorkoosh, M. Khosravani and M. Adabi, Adv. Pharm. Bull., 2018, 8, 77 CrossRef CAS PubMed.
- S. Desgouilles, C. Vauthier, D. Bazile, J. Vacus, J.-L. Grossiord, M. Veillard and P. Couvreur, Langmuir, 2003, 19, 9504–9510 CrossRef CAS.
- S. Lazzari, D. Moscatelli, F. Codari, M. Salmona, M. Morbidelli and L. Diomede, J. Nanopart. Res., 2012, 14, 1–10 CrossRef PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 265–273 Search PubMed.
- H. R. Marsden, L. Gabrielli and A. Kros, Polym. Chem., 2010, 1, 1512–1518 RSC.
- A. Gomez-Hens and J. M. Fernandez-Romero, TrAC, Trends Anal. Chem., 2006, 25, 167–178 CrossRef CAS.
- S. Saini, T. Sharma, A. Jain, H. Kaur, O. P. Katare and B. Singh, Colloids Surf., B, 2021, 205, 111838 CrossRef CAS PubMed.
- M. L. Bondi, G. Montana, E. F. Craparo, P. Picone, G. Capuano, M. D. Carlo and G. Giammona, Curr. Nanosci., 2009, 5, 26–32 CrossRef CAS.
- J. Siepmann and F. Siepmann, Int. J. Pharm., 2020, 577, 119009 CrossRef CAS PubMed.
- Z. Zhang and S.-S. Feng, Biomacromolecules, 2006, 7, 1139–1146 CrossRef CAS PubMed.
- V. Mohylyuk, T. Pauly, O. Dobrovolnyi, N. Scott, D. S. Jones and G. P. Andrews, J. Drug Delivery Sci. Technol., 2021, 63, 102416 CrossRef CAS.
- S. M. Wong, I. W. Kellaway and S. Murdan, Int. J. Pharm., 2006, 317, 61–68 CrossRef CAS PubMed.
- X. Wang and Y. Gao, Food Chem., 2018, 246, 242–248 CrossRef CAS PubMed.
- S. Lappe, D. Mulac and K. Langer, Int. J. Pharm., 2017, 517, 338–347 CrossRef CAS PubMed.
- J. García-Melero, J.-J. López-Mitjavila, M. J. García-Celma, C. Rodriguez-Abreu and S. Grijalvo, Materials, 2022, 15, 4572 CrossRef PubMed.
- Y. Morikawa, T. Tagami, A. Hoshikawa and T. Ozeki, Biol. Pharm. Bull., 2018, 41, 899–907 CrossRef CAS PubMed.
- W. Ren, G. Tian, S. Jian, Z. Gu, L. Zhou, L. Yan, S. Jin, W. Yin and Y. Zhao, RSC Adv., 2012, 2, 7037–7041 RSC.
- K. Yamaoka, T. Nakagawa and T. Uno, J. Pharmacokinet. Biopharm., 1978, 6, 165–175 CrossRef CAS PubMed.
- J. G. Wagner, Pharmacokinetics for the pharmaceutical scientist, CRC Press, 2018 Search PubMed.
- O. I. Corrigan and X. Li, Eur. J. Pharm. Sci., 2009, 37, 477–485 CrossRef CAS PubMed.
- A. M. Agrawal, S. H. Neau and P. L. Bonate, AAPS PharmSci, 2003, 5, 48–60 CrossRef PubMed.
- M. L. Bruschi, Strategies to modify the drug release from pharmaceutical systems, Woodhead Publishing, 2015 Search PubMed.
- S. Gholizadeh, J. A. A. M. Kamps, W. E. Hennink and R. J. Kok, Int. J. Pharm., 2018, 548, 747–758 CrossRef CAS PubMed.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- R. T. C. Silva, L. F. Dalmolin, J. A. Moreto, C. G. Oliveira, A. E. H. Machado, R. F. v Lopez and P. I. S. Maia, J. Nanopart. Res., 2020, 22, 1–13 CrossRef.
- V. D. Wagh and D. U. Apar, J Nanotechnol., 2014, 683153 Search PubMed.
- M. Fathi-Achachelouei, D. Keskin, E. Bat, N. E. Vrana and A. Tezcaner, J. Biomed. Mater. Res., Part B, 2020, 108, 2041–2062 CrossRef CAS PubMed.
- J. Siepmann and F. Siepmann, Int. J. Pharm., 2008, 364, 328–343 CrossRef CAS PubMed.
- U. D. Laddha and S. J. Kshirsagar, Heliyon, 2020, 6, e04589 CrossRef PubMed.
- S. Fredenberg, M. Wahlgren, M. Reslow and A. Axelsson, Int. J. Pharm., 2011, 415, 34–52 CrossRef CAS PubMed.
- V. Nandakumar, V. Geetha, S. Chittaranjan and M. Doble, Biomed. Pharmacother., 2013, 67, 431–436 CrossRef CAS PubMed.
- J. Panyam, M. M. Dali, S. K. Sahoo, W. Ma, S. S. Chakravarthi, G. L. Amidon, R. J. Levy and V. Labhasetwar, J. Controlled Release, 2003, 92, 173–187 CrossRef CAS PubMed.
- M. A. Akl, A. Kartal-Hodzic, T. Oksanen, H. R. Ismael, M. M. Afouna, M. Yliperttula, A. M. Samy and T. Viitala, J. Drug Delivery Sci. Technol., 2016, 32, 10–20 CrossRef CAS.
- J. L. Italia, D. K. Bhatt, V. Bhardwaj, K. Tikoo and M. N. V. R. Kumar, J. Controlled Release, 2007, 119, 197–206 CrossRef CAS PubMed.
- F. von Burkersroda, L. Schedl and A. Göpferich, Biomaterials, 2002, 23, 4221–4231 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- D. Goujot, M.-E. Cuvelier, P. Soto and F. Courtois, Talanta, 2019, 196, 284–292 CrossRef CAS PubMed.
- D. T. Pham, D. X. T. Nguyen, R. Lieu, Q. C. Huynh, N. Y. Nguyen, T. T. B. Quyen and V. De Tran, Drug Delivery, 2023, 30, 2168793 CrossRef PubMed.
- J. C. Stockert, R. W. Horobin, L. L. Colombo and A. Blázquez-Castro, Acta Histochem., 2018, 120, 159–167 CrossRef CAS PubMed.
- L. Shi, J. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- K. I. Cupic, J. J. Rennick, A. P. R. Johnston and G. K. Such, Nanomedicine, 2019, 14, 215–223 CrossRef CAS PubMed.
- C. S. Lee and K. W. Leong, Curr. Opin. Biotechnol, 2020, 66, 78–87 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- K. Zhi, B. Raji, A. R. Nookala, M. M. Khan, X. H. Nguyen, S. Sakshi, T. Pourmotabbed, M. M. Yallapu, H. Kochat and E. Tadrous, Pharmaceutics, 2021, 13, 500 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- A. Malugin and H. Ghandehari, J. Appl. Toxicol., 2010, 30, 212–217 Search PubMed.
- L. del Amo, A. Cano, M. Ettcheto, E. B. Souto, M. Espina, A. Camins, M. L. García and E. Sánchez-López, Appl. Sci., 2021, 11, 4305 CrossRef CAS.
- Y. Chen, G. Dalwadi and H. A. E. Benson, Curr. Drug Delivery, 2004, 1, 361–376 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Physical appearance and DLS size distributions of non-loaded and FA-loaded NPs; electron microscopy images of FA-loaded PLGA NPs; scattering spectra of FA and PLGA, FA calibration curves, gradient mode for FA quantification, in vitro release of FA using 1× PBS:50% EtOH as a receptor phase, fitting the FA release data to zero-order, Higuchi, and Korsmeyer–Peppas kinetic models, radical DPPH˙ scavenging activities, preparation of fluorescently labelled FA-loaded PLGA NPs, 1H-NMR of PEGylated PLGA NPs, DLS size distributions of unloaded and FA-loaded fluorescently labelled NPs, average hydrodynamic diameter and PDI values for PLGA_Flu and FA-loaded PLGA_Flu, ROS scavenging assay. See DOI: https://doi.org/10.1039/d2nr07256d |
| This journal is © The Royal Society of Chemistry 2023 |