
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoOx nanoparticles loaded on carbon spheres with synergistic effects for effective inhibition of shuttle effect in Li–S batteries†
Ning
Chai‡
ab,
Yujie
Qi‡
bc,
Qinhua
Gu
bc,
Junnan
Chen
bc,
Ming
Lu
bd,
Xia
Zhang
 *a and
Bingsen
Zhang
*bc
*a and
Bingsen
Zhang
*bc
aDepartment of Chemistry, College of Science, Northeastern University, Shenyang 110819, China. E-mail: xzhang@mail.neu.edu.cn
bShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, China. E-mail: bszhang@imr.ac.cn
cSchool of Materials Science and Engineering, University of Science and Technology of China, Shenyang 110016, China
dThe Joint Laboratory of MXene Materials, Key Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Key Laboratory of Preparation and Application of Environmental Friendly Materials of the Ministry of Education, Jilin Normal University, Changchun 130103, China
First published on 13th February 2023
Abstract
Lithium–sulfur (Li–S) batteries, as one of the new energy storage batteries, show immense potential due to their high theoretical specific capacity and theoretical energy density. However, there are still some problems to be solved, among which the shuttle effect of lithium polysulfides is one extremely serious issue with respect to the industrial application of Li–S batteries. Rational design of electrode materials with effective catalytic conversion ability is an effective route to accelerate the conversion of lithium polysulfides (LiPSs). Herein, considering the adsorption and catalysis of LiPSs, CoOx nanoparticles (NPs) loaded on carbon sphere composites (CoOx/CS) were designed and constructed as cathode materials. The CoOx NPs obtained, with ultralow weight ratio and uniform distribution, consist of CoO, Co3O4, and metallic Co. The polar CoO and Co3O4 enable chemical adsorption towards LiPSs through Co–S coordination, and the conductive metallic Co can improve electronic conductivity and reduce impedance, which is beneficial for ion diffusion at the cathode. Based on these synergistic effects, the CoOx/CS electrode exhibits accelerated redox kinetics and enhanced catalytic activity for conversion of LiPSs. Consequently, the CoOx/CS cathode delivers improved cycling performance, with an initial capacity of 980.8 mA h g−1 at 0.1C and a reversible specific capacity of 408.4 mA h g−1 after 200 cycles, along with enhanced rate performance. This work provides a facile route to construct cobalt-based catalytic electrodes for Li–S batteries, and promotes understanding of the LiPSs conversion mechanism.
Introduction
Rapid consumption of fossil fuels has brought about a serious energy crisis, and developing renewable energy is of fundamental importance for the sustainable development of human society. The energy demands of electric vehicles and portable devices with high energy density and low cost are growing even more rapidly.1–3,4–6 Consequently, the design and development of promising energy storage systems have received extensive attention. Most notably, lithium–sulfur (Li–S) batteries have been regarded as a new generation of rechargeable battery system due to their ultrahigh theoretical specific capacity (1675 mA h g−1), high energy density (2600 W h kg−1), low cost, and environmental friendliness.7–10So far, however, use of Li–S batteries faces tough challenges, including the shuttle effect of lithium polysulfides (LiPSs), the sluggish redox kinetics of the insulating lithium sulfide and sulfur, the dendrite effect of the lithium anode, and the volume expansion of the cathode.8,11 These issues bring about low coulombic efficiency and specific capacity, poor cycling stability, and performance degradation of the electrode materials, which hampers the industrialization process for Li–S batteries.9,12 Specifically, the shuttle effect has been considered to be one of the main obstacles, and leads to rapid capacity decay and low sulfur utilization. Therefore, rational design and synthesis of catalytic electrode materials are reasonable and effective ways to accelerate polysulfide conversion for active inhibition of the shuttle effect.13–20 So far, strenuous research efforts have been made to improve the adsorption and conversion efficiency of LiPSs, such as through the construction of self-supporting conductive substrates,21–23 heteroatom doping,23–25 introduction of quantum dots (QD),26,27 design of heterostructures,28 construction of catalytic host materials,29–36 and so on. Generally, these feasible routes can be combined into valid strategies to accelerate LiPSs conversion and restrain the shuttle effect effectively. Notably, cobalt-based materials, including Co-based single-atom catalysts (SACs),32,35 metallic Co,37,38 oxides,39,40 sulfides,41–43 nitrides,44,45 phosphides,46–48 and organic hybrid compounds,49–51 have attracted much interest in the field of Li–S battery research due to their low cost, superior electrocatalytic performance, and large anchoring capacity for LiPSs. Despite these advantages, obstacles to developing cobalt-based cathode materials still exist. For instance, poor conductivity of oxides and sulfides is unfavorable for the redox reaction;39–43 difficulty in regulating morphology and structure of sulfides;41 easy accumulation of nitrides;44,45 production of toxic gas during the synthesis process and harsh synthetic conditions for phosphides;46–48 complex reaction processes, excessive by-products and difficult extraction for organic hybrid compounds.49–51
Recently, the strategy of integrated cobalt oxides and metallic Co as electrocatalysts, such as Co@CoO@N–C/rGO membranes and CC/Co@CoO1−x composites,52,53 has been shown to be appealing and valid, by integrating the strong affinity of oxides to LiPSs and the high catalytic activity of metallic Co. Inspired by this, here we have designed and synthesized CoOx nanoparticles (NPs) loaded on carbon sphere (CoOx/CS) composites as the cathode for Li–S batteries based on the synergistic effects of Co-based materials. The size and chemical composition of CoOx NPs can be regulated by sintering conditions. The obtained CoOx NPs with ultralow Co weight ratio (0.043%) and uniform distribution consist of CoO, Co3O4, and Co phases. As polarized oxide particles exhibit intrinsic sulfiphilic properties, CoO and Co3O4 show chemical adsorption towards LiPSs. Metallic Co improves the electronic conductivity and enhances ion diffusion in the electrode simultaneously. The synergistic effects of CoO, Co3O4, and Co accelerate the redox kinetics of LiPSs conversion. In addition, the porous CS matrix provides a fast diffusion path for electrons and ions, facilitating inhibition of the shuttle effect. Compared with the CS/S electrode, the CoOx/CS/S electrode exhibits notably improved cycle performance, with an initial capacity of 980.8 mA h g−1 at 0.1C and a reversible specific capacity of 408.4 mA h g−1 after 200 cycles, along with an enhanced rate performance of 1037.6 mA h g−1 at 0.1C.
Results and discussion
The synthetic process for the CoOx/CS/S electrode material is shown schematically in Fig. 1a. After carbonization, porous carbon spheres (CS) possess abundant nanoscale pores and functional groups (e.g., carboxyl group),54 which can provide enough space and active sites for embedding cobalt ions and growing CoOx NPs. In order to improve catalytic activity, regulation of particle size is vitally important. In this work, the reaction time was optimized initially, to obtain supported Co-based NPs catalysts. As shown in Fig. S1 (ESI†), Co-based NPs with uniform size and distribution could be synthesized with a heating time of 20 min at 600 °C.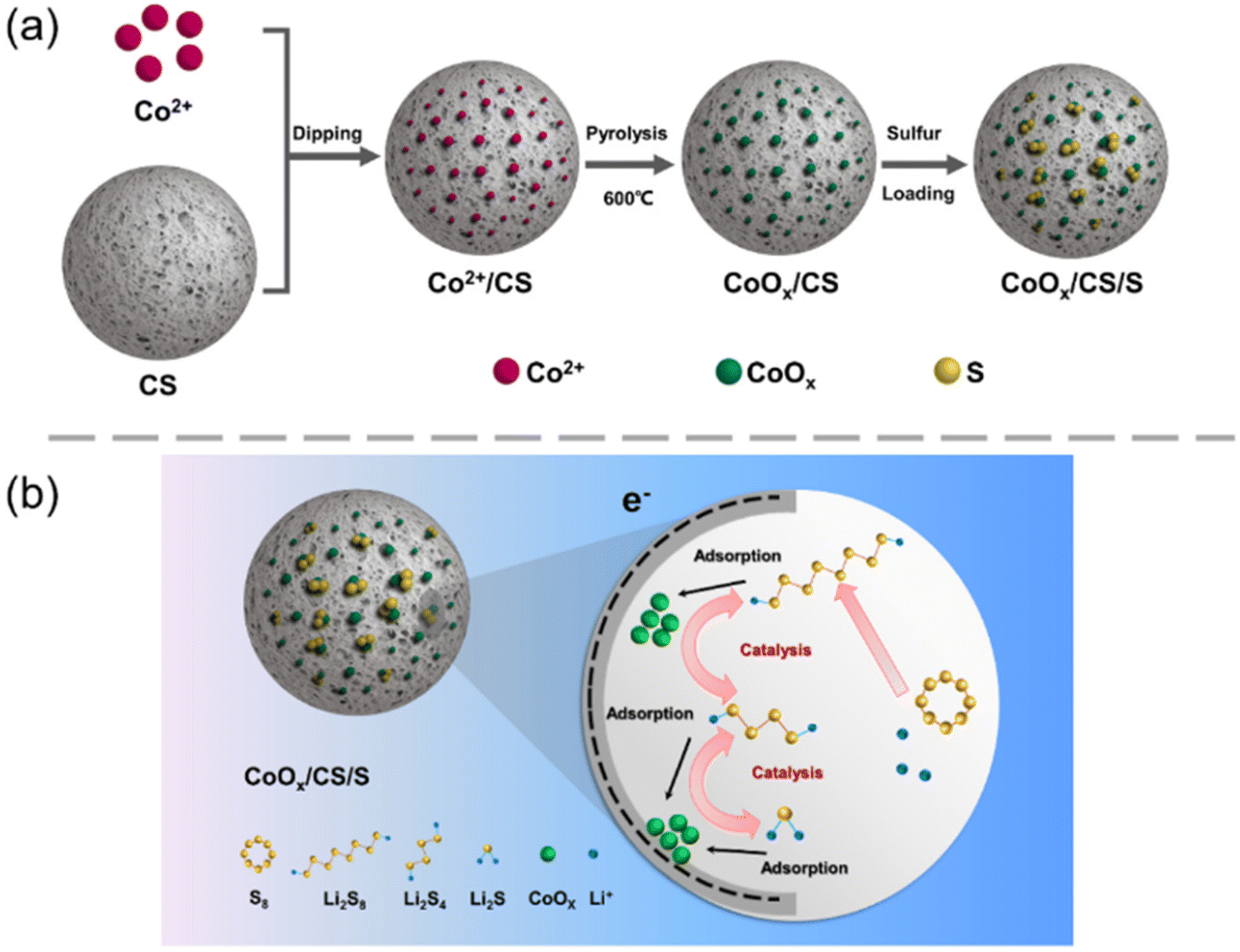 | ||
| Fig. 1 (a) Schematic diagram of synthesis process of CoOx/CS/S composites. (b) Schematic illustration of electrochemical reactions of the CoOx/CS cathode in the Li–S battery. | ||
In order to obtain microstructure information of the as-prepared composites heated at 600 °C for 20 min, transmission electron microscopy (TEM) characterization was performed. Clearly, the NPs are uniformly distributed on the surface of CS with a particle size of 5–10 nm (Fig. 2d and e, S2, and S3†), which is consistent with the high-angle annular dark-field imaging (HAADF) and energy-dispersive X-ray spectroscopy (EDX) results showing the homogeneous distribution of C, O, and Co elements (Fig. 2f). Typical high-resolution TEM (HRTEM) images of NPs with basal spacings of 2.05 Å and 1.77 Å, 2.13 Å and 1.91 Å, 2.56 Å and 1.96 Å can be seen in Fig. 2a–c, which can be assigned to (111) and (002) structures of metallic Co, (020) and (210) structures of CoO, and (310) and (041) structures of Co3O4, respectively. The NPs loaded on the surface of the CS exhibit a mixed phase structure involving CoO, Co3O4, and metallic Co. The as-prepared composites are abbreviated as CoOx/CS.
 | ||
| Fig. 2 HRTEM images of (a) Co, (b) CoO, and (c) Co3O4 NPs loaded on CoOx/CS composites heated at 600 °C for 20 min. TEM images of CoOx/CS composites (d and e), HAADF-STEM image of CoOx/CS composites and corresponding EDX elemental maps for C, O, and Co elements (f). | ||
X-ray diffraction (XRD) patterns of CS and CoOx/CS composites heated at 600 °C for 20 min and 2 h are presented in Fig. S4 and S5.† In general, the broad diffraction peak at 21.7° can be attributed to the coexistence of nanocrystalline graphite and amorphous carbon, and the diffraction peak at 43.7° corresponds to the graphite (101) structure.55 For the CoOx/CS composites heated at 600 °C for 20 min (Fig. S4†), the diffraction peak located at 36.8° corresponds to the (310) structure of Co3O4 (JCPDS 43-1003). There exist no obvious peaks for the Co and CoO phases, which can be ascribed to the low content of Co and CoO. This coincides with actual Co loading amount of 0.043 wt%, measured by inductivity coupled plasma optical emission spectrometry (ICP-OES). Simultaneously, the diffraction peaks of CoO, Co3O4, and Co can be observed in the XRD pattern of CoOx/CS composites prepared with longer heating times (Fig. S5†), indicating the stable state of the mixed phase of CoO, Co3O4, and Co. The above experimental results demonstrate that CoOx NPs with low weight ratio were successfully loaded onto CS with uniform distribution.
According to the Brunauer–Emmett–Teller (BET) results (Fig. 3a and b and Table S1†), CoOx/CS composites exhibit higher specific area than CS, which is attributed to the pyrolysis process. The scanning electron microscopy (SEM) characterization (Fig. S6†) indicates that the introduction of CoOx NPs has negligible influence on the morphology of the CS matrix. In addition, visualized adsorption tests of CoOx/CS composites and CS in Li2S6 solution have been conducted (Fig. S7†). The CS matrix exhibits weak adsorption capability towards LiPSs due to its nonpolar nature. By contrast, CoOx/CS composites show improved chemical interaction with LiPSs due to the polar CoOx NPs. As presented in Fig. S8,† the thermogravimetric analysis (TGA) curves show that the sulfur loading amount for CoOx/CS/S reaches 66.5%, which is close to the theoretical sulfur loading amount of 70% (derived from the preparation protocol). This indicates that most of the elemental sulfur penetrates into the pores of the CS matrix, and that CoOx NP blocking effect has little influence on the sulfur loading. To further confirm the composition and the chemical state of the resulting CoOx/CS composites, full-scale and corresponding high-resolution X-ray photoelectron spectroscopy (XPS) of C 1s, O 1s, and Co 2p has been carried out and the results are shown in Fig. 3c–f. The C 1s spectra can be deconvoluted into four affiliated peaks, corresponding to C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO functional groups. The O 1s spectra could be decomposed into four peaks, belonging to C–O, O–CO, CO, and Co–O bonds. The calculated contents for C and O for different chemical bonds for CS and CoOx/CS composites are summarized in Tables S2 and S3.† It was found that the relative contents of C and O change little for CS and CoOx/CS composites, demonstrating the minor influence of CS on the chemical state when introducing CoOx NPs. The Co 2p spectrum (Fig. 3f) could be divided into six individual peaks, which can be attributed to Co 2p1/2 and 2p3/2 of metallic Co (795.5 and 780.3 eV), Co 2p1/2 and 2p3/2 of Co–O band (797.1 and 782.1 eV), and satellite peaks of Co 2p1/2 and 2p3/2 (804.4 and 789.4 eV).52,53,56
O, and O–CO functional groups. The O 1s spectra could be decomposed into four peaks, belonging to C–O, O–CO, CO, and Co–O bonds. The calculated contents for C and O for different chemical bonds for CS and CoOx/CS composites are summarized in Tables S2 and S3.† It was found that the relative contents of C and O change little for CS and CoOx/CS composites, demonstrating the minor influence of CS on the chemical state when introducing CoOx NPs. The Co 2p spectrum (Fig. 3f) could be divided into six individual peaks, which can be attributed to Co 2p1/2 and 2p3/2 of metallic Co (795.5 and 780.3 eV), Co 2p1/2 and 2p3/2 of Co–O band (797.1 and 782.1 eV), and satellite peaks of Co 2p1/2 and 2p3/2 (804.4 and 789.4 eV).52,53,56
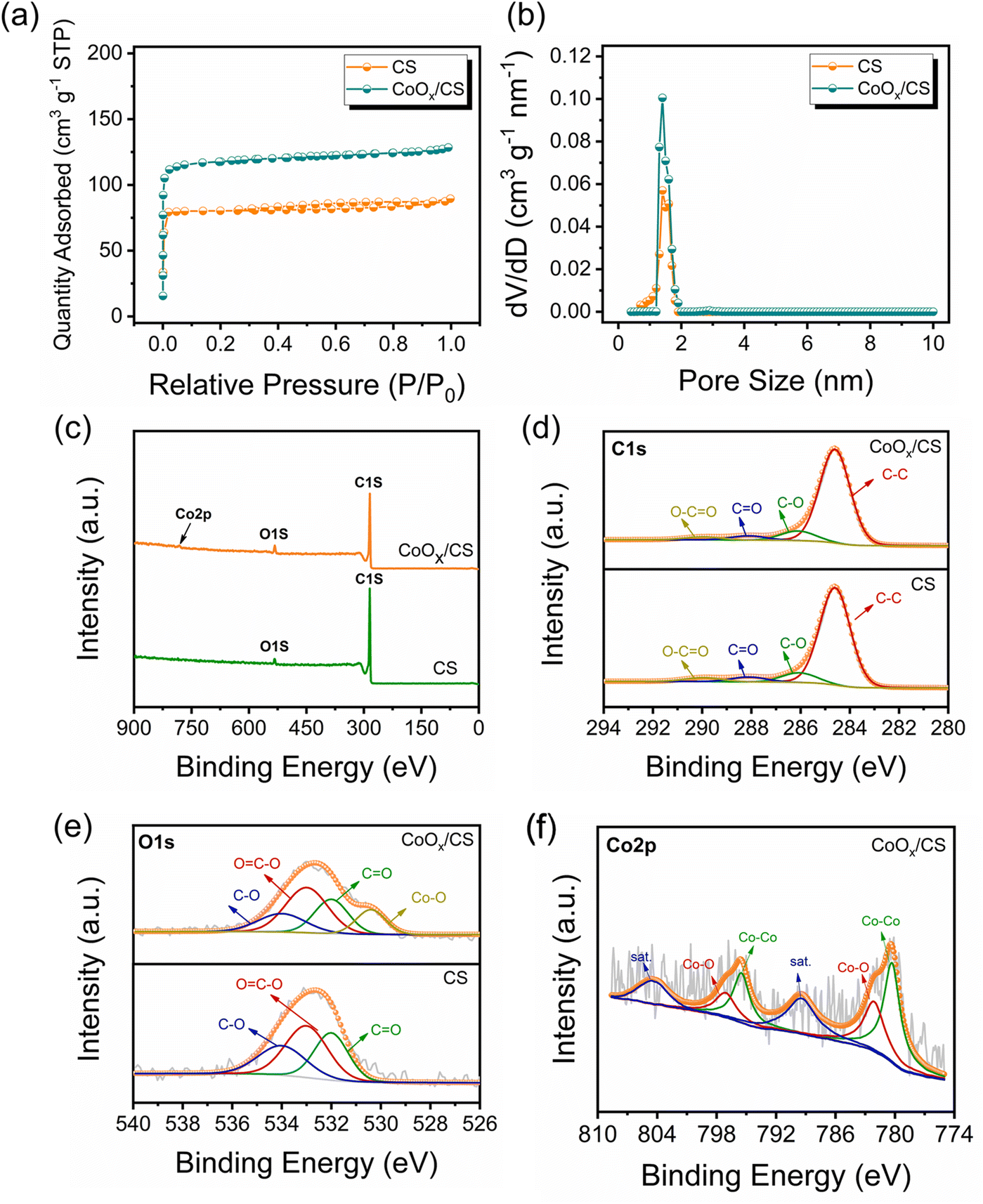 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms and (b) density functional theory pore size distributions for CS and CoOx/CS composites. XPS full spectra (c), C 1s (d), and O 1s spectra (e) of CoOx/CS composites and CS, Co 2p spectra of CoOx/CS composites (f). | ||
The diffusion coefficient of lithium ion is a key parameter to evaluate the diffusion and conversion of LiPSs. Generally, the cathodic and anodic peak currents are linear with respect to the square root of scanning rates calculated from CV curves, as shown in Fig. 4c and d. Herein, the diffusion coefficients of lithium ion for anodic reactions (peak A, Li2S → Li2Sx and peak B, Li2Sx → S8) and the cathodic reactions (peak C, S8 → Li2Sx and peak D, Li2Sx → Li2S) have been deduced using the Randles–Sevcik equation,57–61 and the calculated results are presented in Table 1. Clearly, the diffusion coefficients for the CoOx/CS/S electrode are significantly higher than those for the CS/S electrode, demonstrating that the introduction of CoOx NPs contributes to the diffusion and conversion of LiPSs. In addition, the CV curves of symmetric cells using CS and CoOx/CS as electrodes with Li2S6 electrolyte were measured, to demonstrate the catalytic performance of CoOx on redox processes of LiPSs (Fig. 4e). Compared with CS, the CoOx/CS electrode exhibits higher redox current and smaller polarization, suggesting accelerated redox kinetics and enhanced catalytic activity. Potentiostatically, Li2S precipitation tests on CS and CoOx/CS electrodes were also performed to evaluate the conversion from LiPSs to solid Li2S. Potentiostatic curves discharged at 2.05 V could be obtained with Li2S8/tetraglyme solution as catholyte, as shown in Fig. 4f, and the CoOx/CS electrode could shorten the Li2S precipitation time (≈10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s) compared with the CS electrode. In addition, the CoOx/CS electrode shows a higher Li2S nucleation capacity of 326.23 mA h g−1, while that for the CS electrode is 174.05 mA h g−1. Thus, the catalysis of CoOx NPs for the conversion of LiPSs to Li2S could be verified.
000 s) compared with the CS electrode. In addition, the CoOx/CS electrode shows a higher Li2S nucleation capacity of 326.23 mA h g−1, while that for the CS electrode is 174.05 mA h g−1. Thus, the catalysis of CoOx NPs for the conversion of LiPSs to Li2S could be verified.
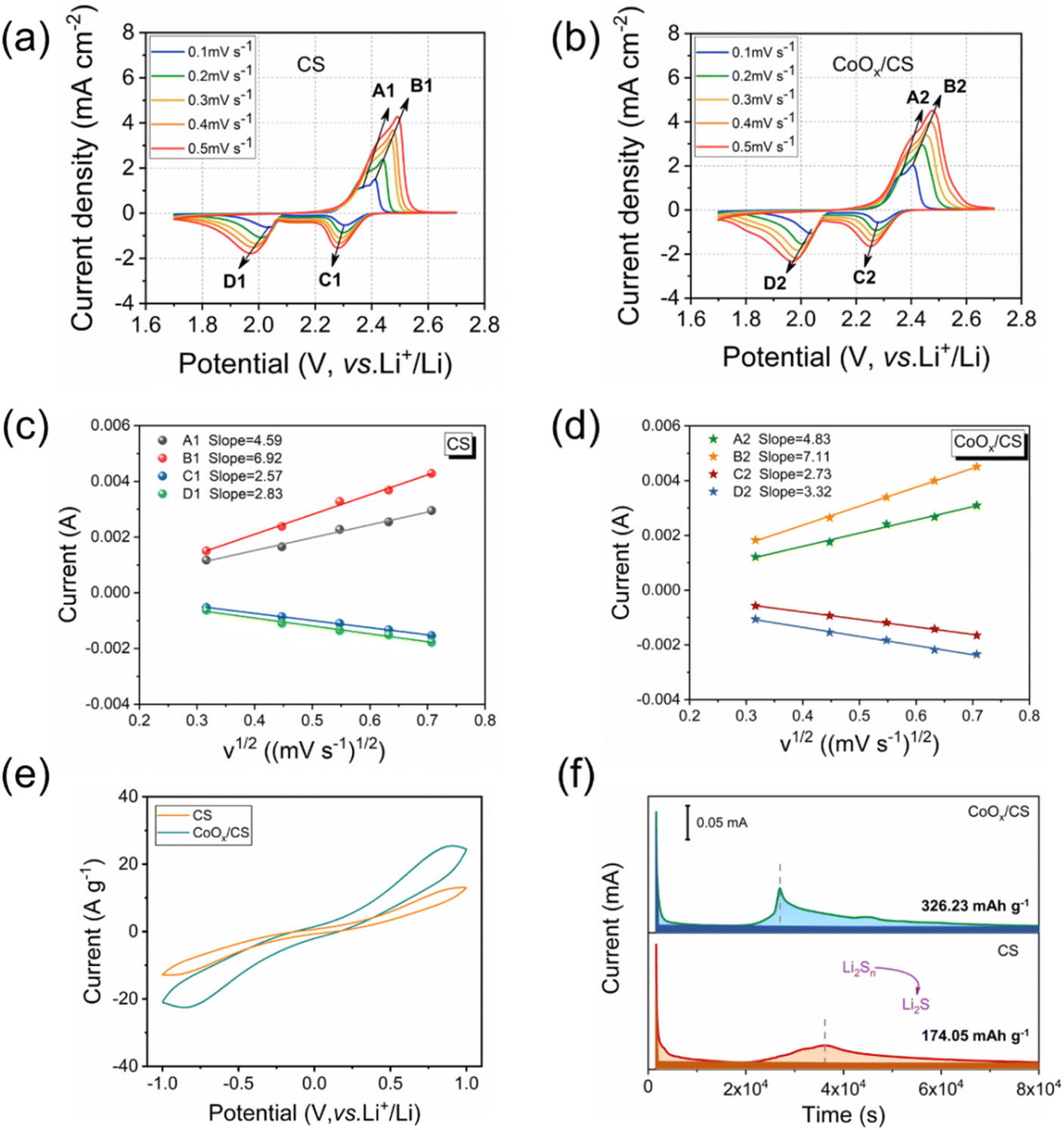 | ||
| Fig. 4 CV curves of CS (a) and CoOx/CS electrodes (b) at various scanning rates. (c and d) Corresponding plot of square root of CV peak current versus scanning rate. (e) CV curves of CS and CoOx/CS symmetrical cells. (f) Potentiostatic discharge curves of Li2S8/tetraglyme solution at 2.05 V on CS and CoOx/CS electrodes. | ||
| D Li+ (cm2 s−1) | CS | CoOx/CS |
|---|---|---|
| A (anodic peak at 2.35 V) | 1.873 × 10−10 | 1.931 × 10−10 |
| B (anodic peak at 2.4 V) | 8.434 × 10−11 | 9.822 × 10−11 |
| C (cathodic peak at 2.3 V) | 3.293 × 10−11 | 3.737 × 10−11 |
| D (cathodic peak at 2.0 V) | 1.343 × 10−10 | 2.053 × 10−10 |
In order to demonstrate the catalytic polysulfide conversion of CoOx, electrochemical measurements of CoOx/CS/S and CS/S electrodes were performed on CR2032-type coin cells. Fig. 5a presents initial CV curves of CoOx/CS/S and CS/S electrodes at 0.1 mV s−1. Clearly, the anodic peak for the CoOx/CS/S electrode displays a negative shift to 2.40 V and the cathodic peak shows a positive shift to 2.04 V, demonstrating the improved reaction kinetics and higher utilization rate of active materials. The catalytic activity of CoOx NPs was further investigated by Tafel plots (Fig. S9†), which were calculated from the reduction peak and oxidation peak in Fig. 5a. The CoOx/CS/S electrode exhibits lower Tafel slopes of 71.2 mV dec−1 and 140.9 mV dec−1 for reduction and oxidation processes, while the CS/S electrode shows higher Tafel slopes of 93.0 mV dec−1 and 183.5 mV dec−1 calculated from the reduction peak at 2.0 V and the oxidation peak at 2.4 V, indicating noticeable kinetic promotion of sulfur species during the charging and discharging processes. Electrochemical impedance spectra of cells with CoOx/CS/S and CS/S as cathodes before cycling and after 10 cycles at 0.1C are shown in Fig. 5b. Two semicircles can be seen in the high frequency region, which can be attributed to the resistance of the Li2S layer and the formation of a solid–electrolyte interface (SEI) (Rg) and the charge transfer resistance (Rct).62 The low frequency line represents the ion diffusion resistance in the cathode (W), and the intersection between the real axis and the first semicircle involves electrolyte resistance (Re).63
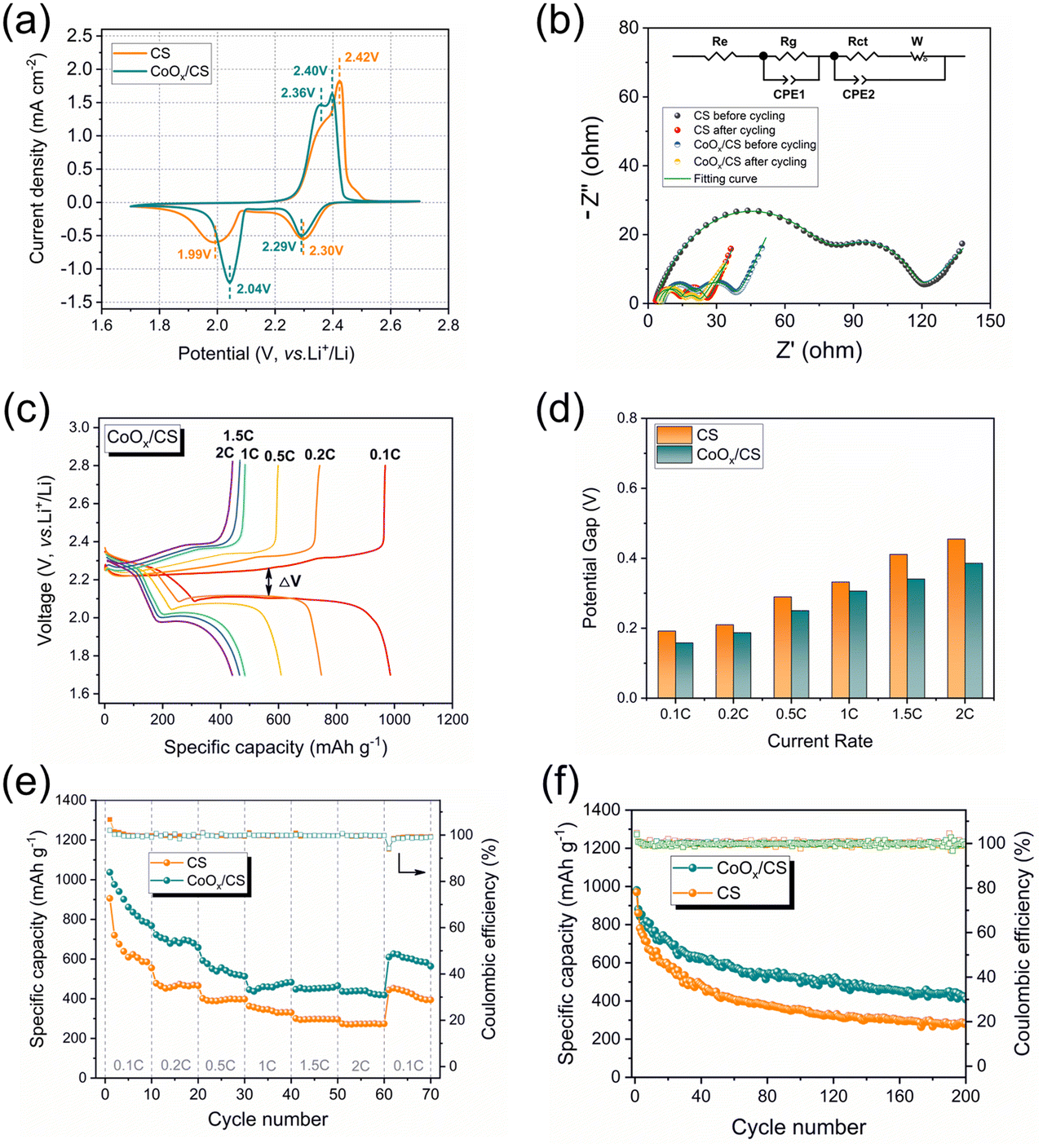 | ||
| Fig. 5 (a) Initial CV curves of CS/S and CoOx/CS/S electrodes with scanning rate of 0.1 mV s−1. (b) Nyquist plots of CS/S and CoOx/CS/S electrodes. (c) Charge and discharge profiles of CoOx/CS/S electrode at various current densities. (d) Potential differences (ΔV) between discharge and charge plateaus of CoOx/CS/S and CS/S electrodes at different current rates. Rate performances (e) and cycling performances (f) of CoOx/CS/S and CS/S electrodes. | ||
The smaller semicircles for the CoOx/CS/S electrode indicate more rapid electron transportation and faster faradaic reaction, compared with the CS/S electrode. In addition, the galvanostatic charge–discharge profiles of the CoOx/CS/S and CS/S electrodes under various current densities were measured, as shown in Fig. 5c and S10.† The charge–discharge profiles for the CoOx/CS/S electrode show smooth curves and stable plateaus, suggesting fast kinetic reaction and rapid flow of electrons and ions. The potential differences (ΔV) between the discharge and charge plateaus of the electrodes at different current rates are presented in Fig. 5d. The CoOx/CS/S electrode exhibits smaller potential differences compared with the CS/S electrode, indicating that the introduction of CoOx accelerates the redox reaction and promotes conversion of LiPSs.
The rate capacities and cyclabilities of CoOx/CS/S and CS/S electrodes were also evaluated on cells with an S loading of 1.2 mg cm−2, as displayed in Fig. 5e and f. The CoOx/CS/S electrode presented enhanced rate performance at various current densities, and achieved capacities of 1037.6 mA h g−1, 722.9 mA h g−1, 592.6 mA h g−1, 483.4 mA h g−1, 465.4 mA h g−1, and 440.3 mA h g−1 at 0.1C, 0.2C, 0.5C, 1C, 1.5C, and 2C, respectively. By contrast, the CS/S electrode showed lower rate capacities of 906.2 mA h g−1, 478.2 mA h g−1, 402.3 mA h g−1, 363.5 mA h g−1, 300.8 mA h g−1, and 275.3 mA h g−1 at 0.1C, 0.2C, 0.5C, 1C, 1.5C, and 2C, respectively. Meanwhile, the enhanced cycling performance of the CoOx/CS/S electrode can be seen in Fig. 5f and S11,† with the CoOx/CS/S electrode delivering a maximum capacity of 980.8 mA h g−1 at a current density of 0.1C and retaining a stable reversible capacity of 408.4 mA h g−1 after 200 cycles with almost 100% coulombic efficiency. The CS/S electrode exhibits decayed capacity from 970.5 to 281.3 mA h g−1 after 200 cycles with 28.9% retention, demonstrating the preferable cycling stability and active suppression of the shuttle effect for the CoOx/CS/S electrode. Furthermore, the cycling performance of the CoOx/CS/S electrode with a high S loading of 4.4 mg cm−2 was also measured (Fig. S12†). The CoOx/CS/S electrode exhibited a maximum capacity of 703.0 mA h g−1 and maintained a stable capacity retention over 70 cycles with a relatively low capacity decay of 0.397% per cycle, which demonstrates the good cycling stability of the CoOx/CS/S electrode at high S loading.
To explore the catalytic mechanism of CoOx NPs, XPS and SEM characterization of the CoOx/CS/S electrode before cycling and after 10 cycles at 0.2C was conducted, as shown in Fig. 6, S13, and S14.† The C 1s, O 1s, and S 2p decomposed spectra for the CoOx/CS/S electrode before and after cycling are displayed in Fig. 6a, b and c, respectively. The C 1s spectra could be deconvoluted into C–C, C–O/C–S, CO, and O–CO peaks.64–66 The O 1s spectra are composed of five peaks, corresponding to Co–O, S–O, CO, O–CO, and C–O groups.67,68 The content of the C–C, C–O/C–S, CO, and O–CO groups in the C 1s spectra remain substantially unchanged before and after cycling (Table S4†); however, increased concentration of the S–O group and attenuated content of the CO bond can be found in the O 1s spectrum after cycling (Table S5†). As for the S 2p spectra, there are five deconvoluted peaks at 162.0, 163.6, 166.6, 167.2, and 168.8 eV, corresponding to sulfide, S–S, sulfite, thiosulfate, and sulfate groups.69–73 Most notably, the relative content of metallic Co for the CoOx/CS/S electrode after cycling reduced significantly, from 25.6% to 15.4%, compared to the CoOx/CS/S electrode before cycling (Fig. 6d). Simultaneously, the content of the Co–O bond increased substantially from 10.0% to 17.2%, the content of the Co–S bond rose to 32.6% from a primary of 22.8%, and the content of the satellite peak decreased slightly to 34.8% from 41.6% (Table S7†). The declining content of metallic Co, and the increasing content of the Co–S bond and Co–S bond indicate the strong affinity between CoOx NPs and LiPSs, and the improved adsorption effect.74
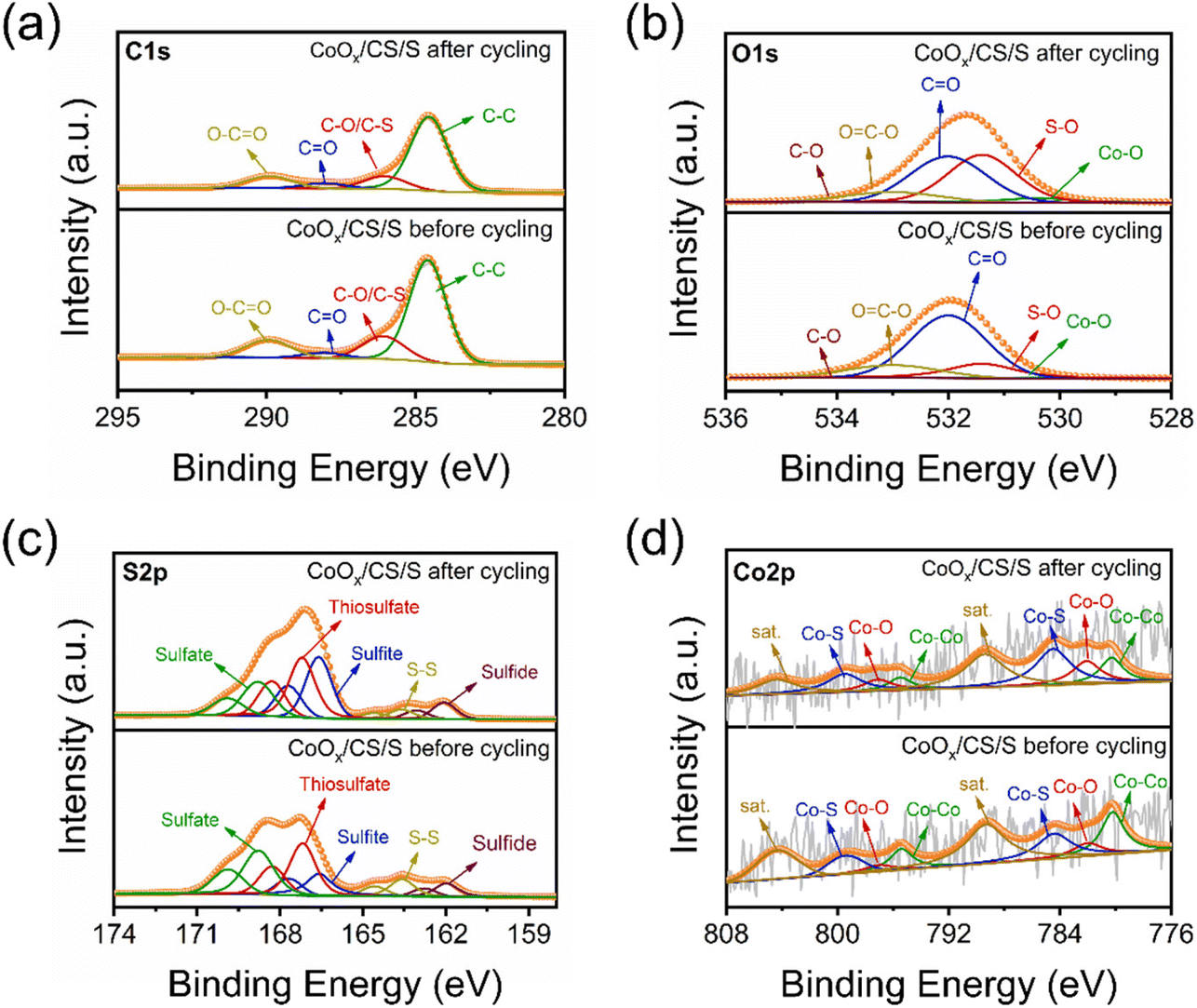 | ||
| Fig. 6 Decomposed XPS spectra of C 1s (a), O 1s (b), S 2p (c), and Co 2p (d) for CoOx/CS/S electrodes before and after cycling. | ||
In summary, the enhanced electrochemical performance of CoOx/CS composites can be illustrated for the following reasons. First, CoOx NPs loaded on CS with ultralow weight ratio and uniform distribution were obtained in this work. CoOx NPs avoid agglomeration with sizes in the range of 5–10 nm, resulting from precise regulation of the experimental parameters, which ensures efficient exposure of active sites and a low proportion of Co-based catalysts. Second, as shown by HRTEM and XPS analysis, CoOx NPs are composed of a stable mixed phase of CoO, Co3O4, and Co. The synergistic effects of the Co-based materials, including strong adsorption of LiPSs from polar CoO and Co3O4, good electrical conductivity of Co, along with charge transfer between CoOx NPs and the CS matrix, can contribute to the catalytic conversion of LiPSs during the cycling process, leading to enhanced electrochemical performance. Third, the strong physical confinement of LiPSs from the CS matrix, which has been verified by our recent work,54 could also play an important role in inhibiting the shuttle effect. Moreover, CoOx/CS composites with porous structure improve sulfur utilization and provide short diffusion routes for transportation of electrons and ions in the electrolyte, promoting redox kinetics.
Conclusions
We employed CoOx/CS composites as cathode materials to catalyze the conversion of LiPSs in Li–S batteries. The microstructure of CoOx has been investigated and confirmed, and the nature of the CoOx NPs with ultralow weight ratio and uniform distribution is a stable mixed phase of CoO, Co3O4, and Co. The synergistic effects of polar CoO, Co3O4, and metallic Co, along with efficient charge transfer between CoOx NPs and the CS matrix could contribute to promoting the redox kinetics of sulfur species. In addition, the porous structure of the CS matrix is beneficial to the strong physical confinement of LiPSs, and provides a short diffusion route for ions and electrons, which also further suppresses the shuttle effect. As a result, the CoOx/CS/S electrode presents enhanced cycling performance with specific capacity of 980.8 mA h g−1 at 0.1C, and retains a stable reversible capacity of 408.4 mA h g−1 after 200 cycles, with 0.292% capacity decay per cycle. This work highlights a facile and rational method to construct cobalt-based catalytic electrode materials to accelerate polysulfide conversion and restrain the shuttle effect, which could pave the way for the application of high-performance Li–S batteries.Experimental
Synthesis of CoOx/CS composites
CoOx/CS composites were prepared by the wet impregnation method using cobalt tetrahydrate ((CH3COO)2Co·4(H2O)) as the cobalt source and carbon spheres (CS) as the active material carrier. CSs were synthesized by hydrothermal carbonization using sucrose as carbon source.75 In general, 0.2 g porous carbon spheres and 0.03113 g cobalt acetate tetrahydrate were evenly dispersed in 50 mL ethanol solution, stirred vigorously, and treated with ultrasound for 1 h to obtain a uniform solution. Afterwards, the mixed solution was filtered and dried to obtain the precursor powder. Finally, the CoOx/CS composites were prepared by calcining precursor powder at 600 °C for 20 min in an argon atmosphere.Synthesis of CS/S and CoOx/CS/S electrode materials
CoOx/CS/S electrode was prepared by the melt diffusion method. Sublimated sulfur and CoOx/CS composite materials were mixed and ground evenly at a mass ratio of 7:3, and transferred to a 10 mL high-pressure reactor. CoOx/CS/S electrode material was obtained by heating at 155 °C for 12 h and naturally cooling to room temperature. CS/S electrode material was prepared in the same way as the control group.
Electrochemical measurements
Electrode material powders (CoOx/CS/S or CS/S composites), super-P, and polyvinylidene fluoride (PVDF) were weighed in a mortar with appropriate amounts of N-methyl-2-pyrrolidone (NMP) and mixed evenly to obtain a slurry (mass ratio 7:2:1). The slurry was evenly coated on aluminum foil, vacuum dried at 55 °C for 12 h, and pressed into a circular electrode sheet with a diameter of 10 mm using a tablet press.
CR2032 button cells with CS/S and CoOx/CS/S cathode materials were assembled in an argon-filled MBraun glovebox. Lithium foil was used as the anode, Celgard 2400 polypropylene film as the diaphragm, 1.0 M DME/DOL (V:V = 1:1), and 1 wt% LiNO3 as the electrolyte. The areal sulfur loading was 1.2 mg cm−2 or 4.4 mg cm−2, and the electrolyte/sulfur (E/S) ratio was 10:1. Cyclic voltammetry and electrochemical impedance spectroscopy tests were carried out on a VMP3 electrochemical workstation (BioLogic). The constant current discharge–charge (GCD) test was carried out in a NEWARE battery test system (CT-4800).
The CS and CoOx/CS symmetrical cells were fabricated as follows: CS or CoOx/CS powders and PVDF were mixed in 2 ml NMP with a weight ratio of 5:1, then the slurry was coated on carbon paper. The carbon paper was cut into circular discs with a diameter of 10 mm and used as working and counter electrodes. Afterwards, two identical electrodes, polypropylene films, and 40 μl electrolyte of 0.2 M Li2S6 were assembled in CR2032 cells. CV measurements of symmetrical cells were performed at a scanning rate of 50 mV s−1. All the electrochemical tests were carried out at room temperature.
Li2S nucleation measurement
The 0.2 M Li2S8 electrolyte was prepared by mixing sulfur powder and lithium sulfide according to a stoichiometric ratio of 7:1 in tetraglyme with vigorous stirring. The CS and CoOx/CS powders were dispersed in NMP and loaded on carbon papers with a diameter of 10 mm as cathodes, and lithium foil was used as the anode. During the process of cell assembly, 20 μl Li2S8 electrolyte was added as catholyte and 20 μl electrolyte without Li2S8 was added as anolyte. The assembled cells were first galvanostatically discharged to 2.06 V with a current of 0.112 mA, then discharged potentiostatically at 2.05 V for Li2S nucleation and growth.76
Li2S6 visualized adsorption experiment
The 0.3 mM Li2S6 solution was initially prepared by dissolving Li2S and sulfur powders with a molar ratio of 1:5 in DOL/DME (v:v = 1:1). In order to conduct visualized adsorption tests, 50 mg of CoOx/CS and CS powders were added into 2 ml Li2S6 solution, respectively.
Instruments and characterization
SEM images were collected on an FEI Nova NanoSEM 450. TEM images were obtained on an FEI Tecnai G2 F20 with an acceleration voltage of 200 kV. The XRD patterns were acquired using an X-ray diffractometer (Rigaku D/MAX-2400) equipped with CuKα radiation (λ = 1.5406 Å). X-ray photoelectron spectra were obtained on a Thermo ESCALAB 250 equipped with a monochromatic AlKα of 1486.6 eV. The cobalt loading amount was determined using an ICP-OES device (PerkinElmer Optima 7300 DV). BET analysis was carried out using a micropore physisorption analyzer (TriStar II 3020). TGA was performed using a NETZSCH STA 449F5 apparatus from 20 °C to 500 °C with a heating rate of 5 °C min−1.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (No. 51932005, 22072164), Liaoning Revitalization Talents Program (No. XLYC1807175), and the Research Fund of Shenyang National Laboratory for Materials Science.References
- L. P. Hou, X. Q. Zhang, B. Q. Li and Q. Zhang, Mater. Today, 2021, 45, 62–76 CrossRef CAS.
- M. Lu, W. J. Han, H. J. Li, W. Shi, J. H. Wang, B. S. Zhang, Y. Zhou, H. B. Li, W. Zhang and W. T. Zheng, Energy Storage Mater., 2019, 16, 163–168 CrossRef.
- M. Lu, W. J. Han, H. B. Li, W. Zhang and B. S. Zhang, J. Energy Chem., 2020, 48, 344–363 CrossRef.
- F. F. Liu, Z. W. Zhang, S. F. Ye, Y. Yao and Y. Yu, Acta Phys.-Chim. Sin., 2021, 37, 2006021 Search PubMed.
- C. X. Bi, M. Zhao, L. P. Hou, Z. X. Chen, X. Q. Zhang, B. Q. Li, H. Yuan and J. Q. Huang, Adv. Sci., 2022, 9, 2103910 CrossRef CAS PubMed.
- J. F. Ding, R. Xu, X. X. Ma, Y. Xiao, Y. X. Yao, C. Yan and J. Q. Huang, Angew. Chem., Int. Ed., 2022, 61, e202115602 CAS.
- Y. F. Zhang, S. C. Wu and Q. H. Yang, EnergyChem, 2021, 3, 100063 CrossRef CAS.
- S. Feng, Z. H. Fu, X. Chen and Q. Zhang, InfoMat, 2022, 4, e12304 CrossRef CAS.
- C. X. Bi, M. Zhao, L. P. Hou, Z. X. Chen, X. Q. Zhang, B. Q. Li, H. Yuan and J. Q. Huang, Adv. Sci., 2022, 9, 2103910 CrossRef CAS PubMed.
- Z. X. Chen, M. Zhao, L. P. Hou, X. Q. Zhang, B. Q. Li and J. Q. Huang, Adv. Mater., 2022, 34, 2201555 CrossRef CAS PubMed.
- X. Y. Yue, C. Ma, J. Bao, S. Y. Yang, D. Chen, X. J. Wu and Y. N. Zhou, Acta Phys.-Chim. Sin., 2021, 37, 2005012 Search PubMed.
- X. G. Qiu, W. Liu, J. D. Liu, J. Z. Li, K. Zhang and F. Y. Cheng, Acta Phys.-Chim. Sin., 2021, 37, 2009012 Search PubMed.
- R. Xu, J. F. Ding, X. X. Ma, C. Yan, Y. X. Yao and J. Q. Huang, Adv. Mater., 2021, 33, 2105962 CrossRef CAS PubMed.
- M. Zhao, B. Q. Li, X. Q. Zhang, J. Q. Huang and Q. Zhang, ACS Cent. Sci., 2020, 6, 1095–1104 CrossRef CAS PubMed.
- Y. Chen, T. Y. Wang, H. J. Tian, D. W. Su, Q. Zhang and G. X. Wang, Adv. Mater., 2021, 33, 2003666 CrossRef CAS PubMed.
- R. Xiao, T. Yu, S. Yang, K. Chen, Z. Li, Z. B. Liu, T. Hu, G. Hu, J. Li, H. M. Cheng, Z. H. Sun and F. Li, Energy Storage Mater., 2022, 51, 890–899 CrossRef.
- C. Y. Zhang, C. Zhang, J. L. Pan, G. W. Sun, Z. Shi, C. Li, X. Chang, G. Z. Sun and J. Y. Zhou, eScience, 2022, 2, 405–415 CrossRef.
- H. Shi, Z. Sun, W. Lv, S. Xiao, H. Yang, Y. Shi, K. Chen, S. Wang, B. Zhang, Q. H. Yang and F. Li, J. Energy Chem., 2020, 45, 135–141 CrossRef.
- R. Xiao, K. Chen, X. Zhang, Z. Z. Yang, G. Hu, Z. Sun, H. M. Cheng and F. Li, J. Energy Chem., 2021, 54, 452–466 CrossRef CAS.
- X. Wang, X. Zhao, C. Ma, Z. Yang, G. Chen, L. Wang, H. Yue, D. Zhang and Z. Sun, J. Mater. Chem. A, 2020, 8, 1212–1220 RSC.
- Q. H. Q. Xiao, J. L. Yang, X. D. Wang, Y. R. Deng, P. Han, N. Yuan, L. Zhang, M. Feng, C. A. Wang and R. P. Liu, Carbon Energy, 2021, 3, 271–302 CrossRef CAS.
- Z. T. Wu, T. X. Shang, Y. Q. Deng, Y. Tao and Q. H. Yang, Adv. Sci., 2020, 7, 1903077 CrossRef CAS PubMed.
- M. D. Zhang, B. Chen and M. B. Wu, Acta Phys.-Chim. Sin., 2022, 38, 2101001 Search PubMed.
- Q. H. Gu, Y. J. Qi, W. X. Hua, T. X. Shang, J. N. Chen, L. Z. Jiang, L. N. Li, M. Lu, Y. X. Zhang, X. Liu, Y. Wan and B. S. Zhang, J. Energy Chem., 2022, 69, 490–496 CrossRef CAS.
- Y. Gao, Z. C. Xiao, D. B. Kong, R. Iqbal, Q. H. Yang and L. J. Zhi, Nano Energy, 2019, 64, 103879 CrossRef CAS.
- Y. Liu, Z. Y. Ma, G. Yang, Z. Wu, Y. T. Li, J. Gu, J. Gautam, X. X. Gong, A. N. Chishti, S. Q. Duan, C. Chen, M. Chen, L. B. Ni and G. W. Diao, Adv. Funct. Mater., 2022, 32, 2109462 CrossRef CAS.
- Z. B. Liu, L. Wang and W. T. Yang, Chin. Chem. Lett., 2021, 32, 2919–2922 CrossRef CAS.
- B. Guan, X. Sun, Y. Zhang, X. Wu, Y. Qiu, M. X. Wang, L. S. Fan and N. Q. Zhang, Chin. Chem. Lett., 2021, 32, 2249–2253 CrossRef CAS.
- Z. Liang, J. Shen, X. Xu, F. Li, J. Liu, B. Yuan, Y. Yu and M. Zhu, Adv. Mater., 2022, 34, 2200102 CrossRef CAS PubMed.
- J. Shen, X. Xu, J. Liu, Z. Wang, S. Zuo, Z. Liu, D. Zhang, J. Liu and M. Zhu, Adv. Mater., 2021, 11, 2100673 CAS.
- J. Shen, X. Xu, J. Liu, Z. Liu, F. Li, R. Hu, J. Liu, X. Hou, Y. Feng, Y. Yan and M. Zhu, ACS Nano, 2019, 13, 8986–8996 CrossRef CAS PubMed.
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed.
- Z. Wang, J. Shen, J. Liu, X. Xu, Z. Liu, R. Hu, L. Yang, Y. Feng, J. Liu, Z. Shi, L. Ouyang, Y. Yu and M. Zhu, Adv. Mater., 2019, 31, 1902228 CrossRef PubMed.
- Y. Zhang, J. Liu, J. Wang, Y. Zhao, D. Luo, A. Yu, X. Wang and Z. W. Chen, Angew. Chem., Int. Ed., 2021, 60, 26622–26629 CrossRef CAS PubMed.
- E. Andritsos, C. Lekakou and Q. Cai, J. Phys. Chem. C, 2021, 125, 18108–18118 CrossRef CAS.
- H. Zhao, B. Tian, C. Su and Y. Li, ACS Appl. Mater. Interfaces, 2021, 13, 7171–7177 CrossRef CAS PubMed.
- Z. S. Wang, J. D. Shen, X. J. Xu, J. J. Yuan, S. Y. Zuo, Z. B. Liu, D. C. Zhang and J. Liu, Small, 2022, 18, 2106640 CrossRef CAS PubMed.
- H. D. Shi, Y. G. Li, P. F. Lu and Z. S. Wu, Acta Phys.-Chim. Sin., 2021, 37, 2008033 Search PubMed.
- R. L. Li, D. W. Rao, J. B. Zhou, G. Wu, G. Z. Wang, Z. X. Zhu, X. Han, R. B. Sun, H. Li, C. Wang, W. S. Yan, X. S. Zheng, P. X. Cui, Y. E. Wu, G. M. Wang and X. Hong, Nat. Commun., 2021, 12, 3102 CrossRef CAS PubMed.
- W. C. Ren, W. Ma, M. M. Umair, S. F. Zhang and B. T. Tang, ChemSusChem, 2018, 11, 2695–2702 CrossRef CAS PubMed.
- Q. Wang, H. Q. Zhao, B. Y. Li, C. H. Yang, M. Y. Li, Y. N. Li, P. Han, M. M. Wu, T. Y. Li and R. P. Liu, Chin. Chem. Lett., 2021, 32, 1157–1160 CrossRef CAS.
- J. T. Zhang, L. Yu and X. W. Lou, Nano Res., 2017, 10, 4298–4304 CrossRef CAS.
- R. J. Li, Z. Bai, W. S. Hou, J. S. Qiao, W. Sun, Y. Bai, Z. H. Wang and K. N. Sun, Chin. Chem. Lett., 2021, 32, 4063–4069 CrossRef CAS.
- H. Wu, H. Jiang, Y. Q. Yang, C. Y. Hou, H. T. Zhao, R. Xiao and H. Z. Wang, J. Mater. Chem. A, 2020, 8, 14498–14505 RSC.
- H. Zhang, D. X. Tian, Z. B. Zhao, X. G. Liu, Y. N. Hou, Y. J. Tang, J. J. Liang, Z. C. Zhang, X. Z. Wang and J. S. Qiu, Energy Storage Mater., 2019, 21, 210–218 CrossRef.
- Y. R. Zhong, L. C. Yin, P. He, W. Liu, Z. S. Wu and H. L. Wang, J. Am. Chem. Soc., 2018, 140, 1455–1459 CrossRef CAS PubMed.
- A. M. Elshahawy, C. Guan, X. Li, H. Zhang, Y. T. Hu, H. J. Wu, S. J. Pennycook and J. Wang, Nano Energy, 2017, 39, 162–171 CrossRef CAS.
- S. L. Yu, W. L. Cai, L. Chen, L. X. Song and Y. Z. Song, J. Energy Chem., 2021, 55, 533–548 CrossRef CAS.
- S. Y. Hu, M. J. Yi, X. Y. Huang, D. Wu, B. B. Lu, T. S. Wang, N. Li, Z. Y. Zhu, X. L. Liu and J. H. Zhang, J. Mater. Chem. A, 2021, 9, 2792–2805 RSC.
- C. X. Zhao, X. Y. Li, M. Zhao, Z. X. Chen, Y. W. Song, W. J. Chen, J. N. Liu, B. Wang, X. Q. Zhang, C. M. Chen, B. Q. Li, J. Q. Huang and Q. Zhang, J. Am. Chem. Soc., 2021, 143, 19865–19872 CrossRef CAS PubMed.
- A. A. Razzaq, G. W. Chen, X. H. Zhao, X. T. Yuan, J. P. Hu, Z. W. Li, Y. F. Chen, J. B. Xu, R. H. Shah, J. Zhong, Y. Peng and Z. Deng, J. Energy Chem., 2021, 61, 170–178 CrossRef.
- H. H. Li, Y. X. Wang, H. Q. Chen, B. X. Niu, W. C. Zhang and D. P. Wu, Chem. Eng. J., 2021, 406, 126802 CrossRef CAS.
- D. L. Fang, G. Z. Wang, S. Z. Huang, T. C. Li, J. Z. Yu, D. B. Xiong, D. Yan, X. L. Li, J. T. Zhang, Y. V. Lim, S. A. Yang and H. Y. Yang, Chem. Eng. J., 2021, 411, 128546 CrossRef CAS.
- Y. J. Qi, N. Chai, Q. H. Gu, J. N. Chen, M. Lu, X. Zhang and B. S. Zhang, Chem. Eng. J., 2022, 435, 135112 CrossRef CAS.
- Y. J. Qi, L. Qi, L. P. Liu, B. S. Dai, D. C. Wei, G. M. Shi and Y. Qi, Carbon, 2019, 150, 259–267 CrossRef CAS.
- C. Y. Fan, S. Y. Liu, H. H. Li, Y. H. Shi, H. C. Wang, H. F. Wang, H. Z. Sun, X. L. Wu and J. P. Zhang, J. Mater. Chem. A, 2017, 5, 11255–11262 RSC.
- M. F. Chen, Q. Lu, S. X. Jiang, C. Huang, X. Y. Wang, B. Wu, K. X. Xiang and Y. T. Wu, Chem. Eng. J., 2018, 335, 831–842 CrossRef CAS.
- X. Y. Tao, J. G. Wang, C. Liu, H. T. Wang, H. B. Yao, G. Y. Zheng, Z. W. Seh, Q. X. Cai, W. Y. Li, G. M. Zhou, C. X. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- X. D. Ren, Q. Zhao, W. D. McCulloch and Y. Y. Wu, Nano Res., 2017, 10, 1313–1321 CrossRef CAS.
- A. Ottmann, G. S. Zakharova, B. Ehrstein and R. Klingeler, Electrochim. Acta, 2015, 17, 4682–4687 Search PubMed.
- K. Tang, X. Q. Yu, J. P. Sun, H. Li and X. J. Huang, Electrochim. Acta, 2011, 56, 4869–4875 CrossRef CAS.
- H. Wang, S. A. He, Z. Cui, C. T. Xu, J. Q. Zhu, Q. Liu, G. J. He, W. Lou and R. J. Zou, Chem. Eng. J., 2021, 420, 129693 CrossRef CAS.
- H. L. Wu, Y. Li, J. Ren, D. W. Rao, Q. J. Zheng, L. Zhou and D. M. Lin, Nano Energy, 2019, 55, 82–92 CrossRef CAS.
- Y. J. Qi, D. C. Wei, G. M. Shi, M. Zhang and Y. Qi, Sci. Rep., 2019, 9, 12429 CrossRef PubMed.
- H. L. Xu, X. W. Yin, M. Zhu, M. K. Han, Z. X. Hou, X. L. Li, L. T. Zhang and L. F. Cheng, ACS Appl. Mater. Interfaces, 2017, 9, 6332–6341 CrossRef CAS PubMed.
- W. L. Song, M. S. Cao, L. Z. Fan, M. M. Lu, Y. Li, C. Y. Wang and H. F. Ju, Carbon, 2014, 77, 130–142 CrossRef CAS.
- W. Shi, B. S. Zhang, Y. M. Lin, Q. Wang, Q. Zhang and D. S. Su, ACS Catal., 2016, 6, 7844–7854 CrossRef CAS.
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and N. M. D. Brown, Carbon, 2005, 43, 153–161 CrossRef CAS.
- L. L. Peng, Z. Y. Wei, C. Z. Wan, J. Li, Z. Chen, D. Zhu, D. Baumann, H. T. Liu, C. S. Allen, X. Xu, A. I. Kirkland, I. Shakir, Z. Almutairi, S. Tolbert, B. Dunn, Y. Huang, P. Sautet and X. F. Duan, Nat. Catal., 2020, 3, 762–770 CrossRef CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- Z. L. Xu, J. K. Kim and K. Kang, Nano Today, 2018, 19, 84–107 CrossRef CAS.
- L. Zhang, L. W. Ji, P. A. Glans, Y. G. Zhang, J. F. Zhu and J. H. Guo, Phys. Chem. Chem. Phys., 2012, 14, 13670–13675 RSC.
- C. X. Zu and A. Manthiram, Adv. Energy Mater., 2013, 3, 1008–1012 CrossRef CAS.
- Z. Zhang, L. L. Kong, S. Liu, G. R. Li and X. P. Gao, Adv. Energy Mater., 2017, 7, 1602543 CrossRef.
- Y. J. Qi, M. Zhang, L. Qi and Y. Qi, RSC Adv., 2016, 6, 20814–20823 RSC.
- W. X. Hua, H. Li, C. Pei, J. Y. Xia, Y. F. Sun, C. Zhang, W. Lv, Y. Tao, Y. Jiao, B. S. Zhang, S. Z. Qiao, Y. Wan and Q. H. Yang, Adv. Mater., 2021, 33, 2101006 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr07194k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |