
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature-responsive and biocompatible nanocarriers based on clay nanotubes for controlled anti-cancer drug release†
Hamoon
Hemmatpour
 ab,
Vahid
Haddadi-Asl
b,
Thomas C. Q.
Burgers
a,
Feng
Yan
a,
Marc C. A.
Stuart
c,
Catharina
Reker-Smit
d,
Rifka
Vlijm
a,
Anna
Salvati
d and
Petra
Rudolf
*a
ab,
Vahid
Haddadi-Asl
b,
Thomas C. Q.
Burgers
a,
Feng
Yan
a,
Marc C. A.
Stuart
c,
Catharina
Reker-Smit
d,
Rifka
Vlijm
a,
Anna
Salvati
d and
Petra
Rudolf
*a
aZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands. E-mail: p.rudolf@rug.nl
bDepartment of Polymer Engineering and Color Technology, Amirkabir University of Technology, P.O. Box 1587-4413, Tehran, Iran
cElectron Microscopy, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747AG Groningen, The Netherlands
dDepartment of Nanomedicine & Drug Targeting, Groningen Research Institute of Pharmacy, University of Groningen, A. Deusinglaan 1, Groningen, 9713AV, The Netherlands
First published on 12th January 2023
Abstract
Administration of temperature-responsive drug carriers that release anticancer drugs at high temperatures can benefit hyperthermia therapies because of the synergistic effect of anticancer drug molecules and high temperature on killing the cancer cells. In this study, we design and characterize a new temperature-responsive nanocarrier based on a naturally occurring and biocompatible clay mineral, halloysite nanotubes. Poly(N-isopropylacrylamide) brushes were grown on the surface of halloysite nanotubes using a combination of mussel-inspired dopamine polymerization and surface-initiated atom transfer radical polymerization. The chemical structure of the hybrid materials was investigated using X-ray photoelectron spectroscopy, thermogravimetric analysis and energy-dispersive X-ray spectroscopy. The hybrid material was shown to have a phase transition temperature of about 32 °C, corresponding to a 40 nm thick polymer layer surrounding the nanotubes. Cell studies suggested that grafting of poly(N-isopropylacrylamide) brushes on the polydopamine-modified halloysite nanotubes suppresses the cytotoxicity caused by the polydopamine interlayer and drug release studies on nanotubes loaded with doxorubicin showed that thanks to the poly(N-isopropylacrylamide) brushes a temperature-dependent drug release is observed. Finally, a fluorescent dye molecule was covalently attached to the polymer-grafted nanotubes and stimulated emission depletion nanoscopy was used to confirm the internalization of the nanotubes in HeLa cells.
Introduction
The ability of nanometer-sized objects to enter cells, combined with the possibility to tailor the properties of nanoparticles, has led to a growing interest in their use as drug carriers, especially in cancer therapy.1 The success of the nanomedicines currently approved for cancer therapy largely relies on passively targeting the enhanced permeability of blood vessels at tumor sites.2 By exploiting this unique feature, nanomaterials can access tumor sites much more quickly than healthy cells to deliver both hydrophilic and hydrophobic drug molecules. This targeted delivery prolongs the circulation time, and amplifies the therapeutic effect with moderate side effects.3 So far, several nanomaterials have been successfully brought to clinical application,4 with the recent vaccines against COVID-19 showcasing the potential of this technology. However, delivering drugs to their target in many cases still remains challenging.5 The focus in recent years has therefore been on developing new design strategies for smart nanocarriers, which preserve cargo during transport to the destined sites and release it there in response to a specific stimulus.6Among the stimuli that can be exploited to trigger the release of drug molecules, temperature deserves special attention because clinical studies have demonstrated that low hyperthermia therapies where the temperature locally increases to 39–41 °C for times up to 72 h, do not cause harmful side effects.7 In addition, it has been shown that cancer cells are more vulnerable to high temperatures (39–45 °C) than normal cells.8 Hence, temperature-responsive drug delivery systems are especially attractive in cancer treatment because high temperature not only accelerates the release of the cargo but also can induce apoptosis in cancer cells.9 Among temperature-responsive materials, poly(N-isopropylacrylamide) (PNIPAM) is one of the biocompatible polymers that has been widely applied in drug delivery and tissue engineering.10 The temperature responsivity of this polymer originates from its lower critical solution temperature (LCST) of around 32 °C.11 By changing the polymer composition through the addition of hydrophilic monomers such as acrylamide, the LCST can be finely tuned and increased above body temperature in order to obtain a temperature responsive behavior that can be used for triggering drug release.12,13 Thus, at temperatures below the LCST, the polymer is in a swollen state stabilized by hydrogen bonds between water molecules and amide functional groups. In contrast, at temperatures above LCST, dissociation of the hydrogen bonds and hydrophobic interactions cause the polymer chains to shrink. This phase transition can be combined with the large surface area provided by the porous nanomaterials to develop the new temperature-responsive nanocarriers, which are able to load a large dose of the cargo and release it at the desired site in a controllable manner.
One of the strategies for developing such smart nanocarriers involves using a porous nanomaterial as a core and grafting thermo-responsive polymer brushes as a shell that controls the release. In this context, many studies have exploited mesoporous silica/PNIPAM hybrid materials,14 where release accelerates at temperatures above the LCST when the PNIPAM brushes adopt the shrunken dehydrated state. Despite the great success achieved with mesoporous silica particles as drug carriers, the difficulty in developing a reproducible synthesis process on an industrial scale and the limited availability and high cost of organic templates used in the synthesis procedure lead to the limited use of these materials in practical applications.15–17 Consequently, biocompatible mesoporous nanomaterials from natural and cheap sources present an interesting alternative.
In the past decade, silica-based inorganic nanotubes have been studied to prepare new technological materials for innovative applications.18 Among the silica-based nanotubes, halloysite nanotubes (HNTs) have emerged as a promising platform for drug delivery applications due to their excellent biocompatibility, high surface area and large volume of the inner cavities.19 The unique hollow tubular structure of HNTs originates from the rolling of aluminosilicate sheets (15–20 layers) under particular geological conditions.20 The layers are rolled such that the silicon oxide layer is located on the outer surface, while the aluminum oxide layer forms the inner surface of the nanotube. Due to the different reactivity of the aluminum and silicon oxide, at pH values in the range of 2.5–8.5, the outer surface of the HNTs bares negative charges while the inner surface is positively charged.21 By exploiting this remarkable feature, different kinds of drug molecules can be loaded into HNTs by the so-called ‘vacuum assisted loading’.22 Lazzara et al. demonstrated that by applying vacuum on the aqueous suspension of HNTs/drug mixture, water confined inside the HNTs’ cavity evaporates faster than the bulk solution due to its larger vapor pressure; consequently, drug solution can be dragged inside the nanotubes.23 They indicated that utilizing such a vacuum procedure significantly enhances the amount of salicylic acid loaded into the HNTs. Regarding biocompatibility, recent studies have clearly demonstrated that pristine HNTs can easily penetrate the cell membrane and accumulate in the perinuclear areas without causing significant toxic effects.24–27
All these appealing features of HNTs encouraged us to explore the potential of these nanotubes for designing a new smart nanocarrier with temperature-mediated release behavior. For this purpose, PNIPAM brushes were grown on the surface of HNTs using a surface-initiated activator regenerated by electron transfer in atom transfer radical polymerization (ARGET-ATRP). In order to be able to use this method, the surface of the nanotubes was first functionalized with a thin layer of polydopamine (PDA), an attractive polymer with broad applications in biomedical science.28 The abundant catechol and amine functional groups in the PDA structure serve to attach the ATRP-initiator on the nanotubes surface. PNIPAM brushes were grown from the initiator-modified HNT surfaces with two different targeted polymerization degrees. The chemical characterization of the PNIPAM-grafted HNTs was performed using Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA) and electron microscopy combined with energy-dispersive X-ray spectroscopy (EDS). Cytocompatibility of the synthesized hybrid materials against HeLa cells was evaluated by 3,(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) assay, while the thermo-responsivity was verified by dynamic light scattering (DLS) analysis. Once we had established that the carrier had been realized as designed, doxorubicin (DOX) was selected as a common anticancer drug molecule to investigate the loading capacity and release feature of PNIPAM-grafted HNTs. As the final part of this study, a fluorescent dye molecule was covalently attached to the polymer-grafted nanotubes, and the localization of the nanotubes in the HeLa cells was studied by Stimulated Emission Depletion (STED) nanoscopy.
Experimental
Materials
All the chemicals used in this study were purchased from MilliporeSigma and used as received unless otherwise mentioned. For the functionalization of the HNTs surface with PDA the following materials were used: halloysite nanotubes (HNTs, Al2Si2O5(OH)4·2H2O, surface area: 64 m2 g−1), dopamine hydrochloride (DA, C8H11O2N·HCl, purity ≥ 98%), tris-(hydroxymethyl) aminomethane (Tris, C4H11O3N, purity ≥ 99.9%). 2-Bromoisobutyryl bromide (BIBB, C4H6OBr2, purity 98%), trimethylamine (TEA, C6H15N, purity 99%), 4-dimethylaminopyridine (DMAP, C7H10N2, purity 99%) and anhydrous tetrahydrofuran (THF, C4H8O, purity ≥ 99.9%) were employed for anchoring the ATRP-initiator on the HNT surface. The chemicals used to perform ARGET-ATRP reaction were tris[2-(dimethyl-amino)ethyl]amine (Me6TREN, C12H30N4, purity 97%), ethyl α-bromoisobutyrate (EBIB, C6H11O2Br, purity 98%), copper(II) chloride dihydrate (CuCl2·2H2O, purity 99%), L-ascorbic acid (AAc, C6H8O2, purity ≥ 99%), N-isopropylacrylamide (NIPAM, C6H11NO, purity 97%) and propargyl acrylate (PPA, C6H6O2, purity 98%). PPA was passed through a basic alumina column before use in order to remove the inhibitor. For drug loading and release studies, a doxorubicin hydrochloride (DOX) solution (2 mg mL−1, 25 mL vial) was purchased from ACTAVIS. Phosphate buffered saline prepared from tablets (PBS, pH 7.4) was used as the medium for the drug release experiments. Abberior STAR 635 (λexcitation: 635 nm and λemission: 655 nm) with an azide functionality acquired from Abberior was used as the fluorescent marker. Labeling of the nanotubes was performed through click reaction by using CuBr (purity 99%) as catalyst, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, C9H23N3, purity 99%) as ligand and anhydrous dimethylformamide (DMF, C3H7NO, purity > 99.9%) and dimethyl sulfoxide (DSMO, C2H6OS, purity > 99.9%) as solvents.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) were placed in a 50 mL round-bottom flask. The mixture was bubbled with argon to remove the dissolved oxygen and then sonicated for 30 min in an ice bath. Stock solutions of CuCl2/Me6TREN (72.2 mM of CuCl2 and 0.35 M of Me6TREN) and AsAc (73.1 mM) in the mixture of water/methanol (volume ratio 1:4) were prepared separately. Copper solution, EBiB and NIPAM were added to the reaction mixture to establish the molar ratios of 1500:1:1:5 and 500:1:1:5 with respect to [NIPAM]:[EBiB]:[CuCl2]:[Me6TREN], which gave us two samples with high and low molecular weights of PNIPAM brushes attached to the nanotubes. Then mixture was deoxygenized once more by argon bubbling for 15 min. After degassing and refilling the flask with argon for 3 times, 1 mL of the AsAc stock solution was added to the reaction to start the polymerization. The reaction was performed for 24 h at room temperature. After this time, polymerization was stopped by exposing the reaction mixture to air. The PNIPAM-grafted HNTs were separated using centrifugation and washed several times with methanol to remove un-attached PNIPAM polymer chains. The final products were dried under vacuum for 12 h and will be referred to as HNIP HM for the high polymerization degree of PNIPAM brushes and HNIP LM for the low polymerization degree of PNIPAM brushes. A similar procedure with that of the HNIP HM sample was performed to attach the copolymer brushes of P(NIPAM-co-PPA) to the HNTs surface by adding PPA in a molar ratio of 1:10 with respect to NIPAM. The obtained product will be referred as HNIP-PPA in the following.
:4) were placed in a 50 mL round-bottom flask. The mixture was bubbled with argon to remove the dissolved oxygen and then sonicated for 30 min in an ice bath. Stock solutions of CuCl2/Me6TREN (72.2 mM of CuCl2 and 0.35 M of Me6TREN) and AsAc (73.1 mM) in the mixture of water/methanol (volume ratio 1:4) were prepared separately. Copper solution, EBiB and NIPAM were added to the reaction mixture to establish the molar ratios of 1500:1:1:5 and 500:1:1:5 with respect to [NIPAM]:[EBiB]:[CuCl2]:[Me6TREN], which gave us two samples with high and low molecular weights of PNIPAM brushes attached to the nanotubes. Then mixture was deoxygenized once more by argon bubbling for 15 min. After degassing and refilling the flask with argon for 3 times, 1 mL of the AsAc stock solution was added to the reaction to start the polymerization. The reaction was performed for 24 h at room temperature. After this time, polymerization was stopped by exposing the reaction mixture to air. The PNIPAM-grafted HNTs were separated using centrifugation and washed several times with methanol to remove un-attached PNIPAM polymer chains. The final products were dried under vacuum for 12 h and will be referred to as HNIP HM for the high polymerization degree of PNIPAM brushes and HNIP LM for the low polymerization degree of PNIPAM brushes. A similar procedure with that of the HNIP HM sample was performed to attach the copolymer brushes of P(NIPAM-co-PPA) to the HNTs surface by adding PPA in a molar ratio of 1:10 with respect to NIPAM. The obtained product will be referred as HNIP-PPA in the following.
Drug loading and release studies
The drug loading capacity of the HNIP HM and HNIP LM samples was evaluated through the following procedure: 10 mg of PNIPAM-grafted HNTs were added to a vial containing 5 mL of an aqueous solution of DOX (2 mg mL−1). After sonication for 10 min, the vial was transferred to a vacuum jar. After establishing vacuum, a slight fizzing was observed indicating the evaporation of the water confined inside the HNT's cavity and replacing with the drug solution. Once the fizzing had stopped, the vial was transferred to atmospheric pressure and kept under stirring for 10 min; the vial was then placed again in the vacuum jar and vacuum was established. This process was repeated 3 times in order to achieve the highest possible drug loading. After that, the suspension was stirred in the dark for 24 h at room temperature, DOX-loaded nanotubes were separated by centrifugation at 11000 rpm for 10 min and washed twice with Millipore water. The same DOX loading protocol was performed with pristine HNTs in order to obtain DOX-loaded HNTs as the control sample. After each step of centrifugation, the supernatant was collected and transferred to the microplates suitable for the fluorescence-based assay. The amount of DOX remaining in the supernatant was determined using the fluorescence intensity-concentration calibration curve with an excitation at 480 nm and emission at 590 nm (see Fig. S1, ESI†). The loading capacity of the samples was calculated through the following equation:| LC = (C0 − C) × V/m | (1) |
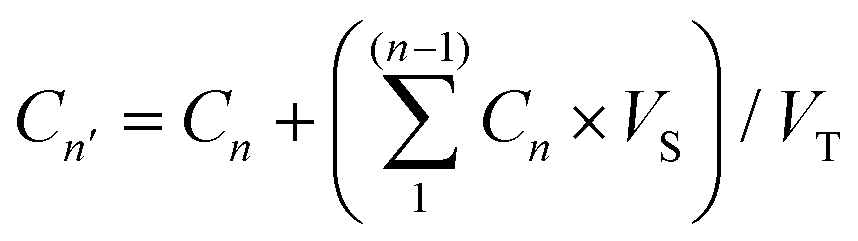 | (2) |
Cytotoxicity studies
The viability of HeLa cells after treatment with different concentrations of the pristine and modified HNTs was investigated via an MTT assay. HeLa cells were obtained from ATCC (CCL-2) and tested monthly to exclude mycoplasma contamination. In a typical procedure, HeLa cells were grown in a complete cell culture medium (cMEM) composed by MEM (Gibco™ Thermofisher Scientific, Landsmeer, Netherlands) supplemented with 10% v/v Fetal Bovine Serum (FBS, Gibco™ Thermofisher Scientific) under standard conditions (37 °C, 5% CO2). After culturing, the cells were plated in 96-well plates at a cell density of 30000 cells per well. Twenty four hours later, the medium was replaced with the suspensions of nanoparticles, which were dispersed in cMEM in different concentrations ranging from 25 to 250 μg mL−1. Then, the cells were incubated at 37 °C and 5% CO2 for different time intervals, i.e. 24, 48 and 72 h. Untreated HeLa cells seeded in the same way and incubated for the same time without adding nanoparticles were used as a control. After incubation, the medium was replaced with 200 μL of 0.5 mg mL−1 MTT solution (3,(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide acquired from MilliporeSigma) dissolved in cMEM. After incubation for another 2 h at 37 °C with 5% CO2, the precipitated formazan salt was dissolved by replacing the medium with 100 μL of DMSO followed by agitation for 15 min. Then, the absorbance of the medium was measured at 550 nm using a UV-plate reader (Molecular Devices LLC., Sunnyvale CA. USA). The cytotoxicity effects of the DOX-loaded samples on the HeLa cells viability were investigated using an analogous procedure. The results are calculated as the average and standard deviation over 3 replicate samples of the absorbance obtained in wells exposed to the nanotubes and/or doxorubicin normalized by the absorbance of untreated control cells cultured for the same time. Experiments were repeated at least 3 times to confirm reproducibility, and average results are shown.
Cellular uptake studies
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco, 31966-021, #2176345) supplemented with 10% Fetal Bovine Serum (Sigma, F7524, lotBCCC0670) and 1% Penicillin/Streptomycin (Gibco, 15140-122,#2145459). Borosilicate #1.5 18 mm coverslips (Marienfeld, 0117580, lot43862-831) were cleaned with 70% ethanol and dried inside a laminar flow hood. Next, these coverslips were placed in the wells of a 12 well plate, and HeLa cells (passage 15–30) were seeded at a density of 4.5k cells per mL in a total of 2 mL culture medium per well to achieve ∼70% cell confluency on the day of analysis. To minimize the nanotubes aggregates, F-HNIP-PPA was dissolved in PBS to a final concentration of 2.5 mg mL−1 and sonicated (VWR ultrasonic cleaner, USC-TH) for an hour before adding it to the HeLa cells. After every sonication cycle, the number of aggregates was assessed by confocal microscopy on a Abberior Expert Line microscope (solution of 0.2 μL F-HNIP-PPA mixed with 40 μL homemade mowiol was made, and 8 μL of this solution was put on a coverslip on a glass slide). F-HNIP-PPA aggregates were further reduced by going twice through the process of placing the solution in the cryogen for 10 min and letting the solution heat up to room temperature. After an additional hour of sonication, the number of aggregates was significantly reduced, and the solution became transparent (a good indication for a low number of F-HNIP-PPA aggregates). The F-HNIP-PPA were added 24 h after seeding the cells, at a final concentration of 62.5 μg mL−1 (2.5 μL of 2.5 mg mL−1 F-HNIP-PPA stock added to 1 mL of culture medium). After 48 h of incubation with the F-HNIP-PPA nanotubes, the cells were labeled, placed into live-cell imaging chambers, and analyzed. As a control to F-HNIP-PPA uptake by cells, the cells were seeded with uncoupled Abberior STAR635 dye instead of the F-HNIP-PPA nanotubes with a final concentration of 2.5 μM of the Abberior STAR 635 dye for up to 48 h. On the day of the analysis of F-HNIP-PPA nanotube uptake, the seeded and F-HNIP-PPA incubated cells were labeled inside the 12 well plate by adding 500 μL pre-warmed 37 °C PBS, with a mixture of 100 nM LysoTracker (Red DND-99, L-7528, Molecular Probes, Lot 30D1-2) and 100 nM Memglow™ 488 (MEMBRIGHT™, Cat #MG01, lot 019) during 30 min, prior to the imaging. Afterwards, the coverslips with labeled cells were transferred to live-cell imaging chambers (Live Cell Instrument, CM-B18-1, magnetic imaging chamber for 18 mm coverslips). For optimal imaging 500 μL of phenol-red free FluoBrite™ DMEM (Gibco, A18967-01, #2120559) supplemented with 10% Fetal Bovine Serum (Sigma, F7524, lotBCCC0670) and 1% Penicillin/Streptomycin (Gibco, 15140-122, #2145459) was added to the imaging chamber.STED nanoscopy
Images were acquired using an Abberior Expert Line microscope equipped with a large incubator chamber with temperature, humidity and CO2 control (OKO lab, Bold line) to image the cells under physiological conditions (37 °C, 5% CO2 with a 0.04 mL min−1 flow, high humidity). For high-resolution live-cell imaging, a 100× oil immersion objective (Olympus Objective UPlanSApo 100×/1.40 Oil) and 37 °C refractive index matching oil (Cargille laboratories, type37LDF) were used. The overlay positions of the confocal lasers and the STED doughnut were aligned prior to imaging using a 0.1 μm TetraSpeck™ (Invitrogen, T7279) bead sample. To minimize phototoxicity to the living cells during STED nanoscopy, the adaptive illumination strategies RESCue (Reduction of State transition Cycles) and DyMIN (Dynamic Intensity MINimum) were applied.31,32 In RESCue-mode, the number of excitation and de-excitation cycles was significantly limited by setting intensity thresholds. When these thresholds were not reached within a certain percentage of the dwell time, the lasers were automatically switched off for the remainder of the dwell time as there was no structure present at this location. In DyMIN-mode, a confocal scan was used as a probe to determine where to apply STED. When the confocal scan locations had intensities above a set threshold, first low-intensity STED was applied and only when there was still a signal from the structure, high-intensity STED was used to achieve the best attainable resolution. All STED images were measured with a pixel size of 20 nm and a pinhole size of 0.7 AU. For excitation and depletion, we used 40 MHz pulsed lasers, with a gated detection window between 12 ns and 14 ns. These lasers had powers of 200 μW (560 nm), 1 mW (640 nm), and > 2750 mW (775 nm STED) out of the laser heads. The applied excitation and depletion intensities varied between the confocal, low- and high-STED imaging: 0.5% of the 640 nm laser intensity with a dwell time of 20 μs (confocal probe); 1.2% 640 nm laser intensity and 9.6% STED775 laser intensity with a dwell time of 90 μs (STED low-intensity scan); 1.5% 640 nm laser intensity and 100% STED 775 nm laser intensity with a dwell time of 90 μs (STED high-intensity scan); 0.3% 640 nm laser intensity with a dwell time of 10 μs (confocal overview); 12% 561 nm laser intensity with a dwell time of 10 μs (lysosome); and 15% 488 nm laser intensity with a dwell time of 10 μs (membrane). The Avalanche Photodetector ranged from 650–763 nm for 640 nm laser excitation, 575–630 nm for 561 nm laser excitation and 495–550 nm for the 488 nm laser excitation.Sample characterizations
Fourier transform infrared spectroscopy (FTIR) was conducted on a Bruker IF 66/v spectrometer. Spectra were collected with a resolution of 2 cm−1 in the range from 400 to 4000 cm−1; each spectrum was the sum of 32 scans. Samples were prepared in the form of KBr pellets with 2 wt% of HNTs. X-ray photoelectron spectroscopy (XPS) was performed on a Surface Science SSX-100 ESCA instrument with a monochromatic Al Kα X-ray source (hν = 1486.6 eV). The pressure in the analysis chamber was maintained below 5 × 10−9 mbar. Samples were prepared by pressing dry powders onto a silver substrate (previously prepared by flattening Ag pearls (Goodfellow, silver lump AG006100, purity 99.999%, size: 3 mm) in a press (RHC, 30 ton pillar press)). To compensate for sample charging during data acquisition very low kinetic energy electrons from a flood gun were directed onto the sample. Spectra were collected from a spot of 1000 μm diameter; the electron take-off angle with respect to the surface normal was 37°. The energy resolution was set to 1.3 eV for both the survey spectra and the detailed spectra of the Al 2p, Si 2p, C 1s, N 1s and O 1s core levels. Binding energies are referenced to the Si 2p photoemission peak (Si–O–Si) centered at a binding energy (BE) of 102.7 eV and are accurate to ± 0.1 eV.33 The XPS data analysis, performed with the least-squares curve-fitting program Winspec (LISE laboratory, University of Namur, Belgium), included a Shirley baseline subtraction and fitting with a minimum number of peaks with profiles taken as a convolution of Gaussian and Lorentzian functions. The peak intensities for all core levels are reported with an uncertainty of 2%. All measurements were carried out in three different spots of each sample to check for homogeneity. A TGA 5500 TA instrument was used to perform the thermogravimetric analysis (TGA); samples of approximately 5 mg were heated in nitrogen atmosphere from 30 to 700 °C, at a rate of 5 °C min−1. A Micrometrics ASAP 2420 V2.05 instrument was used to acquire the N2 adsorption/desorption isotherms at −196 °C. The Brunauer, Emmett and Teller (BET) theory was employed to calculate the specific surface area of the nanotubes, while the pore size distribution was obtained using the Barrett, Joyner and Halenda (BJH) model on N2 desorption data. The nanostructure of the pristine and polymer-grafted nanotubes was investigated with a FEI Tecnai T20 transmission electron microscope (TEM), operating at 200 keV. Erythrocruorin, a giant hemoglobin from earthworm blood (sourced from an animal retrieved in the garden outside of the lab), was used to facilitate the estimation of the thickness of the polymeric shell surrounding the nanotubes because the polymeric brush was otherwise invisible by phase-contrast cryo-TEM. The samples were dispersed in water with the help of sonication and freeze-thawing. After a good dispersion was obtained, earthworm blood was added to the suspension of the nanotubes with the volume ratio of 1:25. Then the suspension was placed on a holy carbon coated grid (Quantifoil 3.5/1) and vitrified with liquid ethane (Vitrobot, FEI) prior to being examined using a Gatan cryo-stage. Images were recorded under low dose conditions on a slow scan CCD camera. Elemental analysis was done on the same microscope in STEM mode using a SDD detector (Xmax 80T, Oxford instruments). Dynamic light scattering (DLS) analysis was performed using a Malvern Zetasizer Nano ZS (Malvern Instruments Ltd, USA) instrument. Samples were suspended in Millipore water at a final concentration of 200 μg mL−1 and transferred to the polystyrene cuvettes. Measurements were conducted in back scattering mode in the range of 25 to 40 °C with steps of 2.5 °C. To ensure thermal equilibration, samples were kept at each temperature for 2 min prior the measurement. The particle sizes are reported as the average of three measurements. Drug loading and release studies were carried out using a florescent micro-plate reader (Molecular Devices, LLC, USA). The concentration of the samples was calculated based on a calibration curve obtained from 1, 5 and 25 ppm of DOX in aqueous solution reported in the ESI (Fig. S1).† After labeling HNTs, the fluorescence emission spectra were acquired using a the Abberior Expert Line microscope by varying the detection window between 400 and 700 nm, with a step and window size of 10 nm from 400 to 580 nm and a 3 nm step and 5 nm window size between 580 and 700 nm. The fluorescence emission spectrum was obtained by plotting the wavelength against the fluorescence intensity. As a control, 0.5 μL of Abberior STAR 635 dye was pipetted on a #1.5 borosilicate cover glass and air-dried while protected from light for 20 min. Afterwards, 0.5 μL of homemade mowiol was placed on a microscope slide, and the coverslip with the dried dye was placed on top. The same approach was used to create a sample with the labeled nanotubes on a glass slide. The following settings were used to acquire the spectra: 120 nm pixels, 0.7 AU, 0.2% 640 nm excitation laser with 40 μs dwell time and 3 line accumulations.
Results and discussion
HNTs were exposed to a dopamine alkaline solution buffered at pH 8.5 to modify the nanotube surface with a thin layer of PDA.29 This sample is referred to as HNT-PDA in the following. Subsequently, alkyl bromide moieties were grafted to HNT-PDA through an acylation reaction between the catechol and amine functional groups in the PDA structure and acylbromide moieties to serve as an initiator for atom transfer radical polymerization (ATRP). PNIPAM brushes were then grown from the initiator-modified HNTs (HNT-Br) surfaces by performing surface-initiated ARGET-ATRP. Fig. 1 shows the synthesis protocol used to modify the HNTs. To examine the impact of the brush length on biocompatibility and drug release behavior, samples with the two targeted polymerization degrees were synthesized by adjusting the monomer to initiator ratio to 1500 and 500 to yield respectively HNIP HM and HNIP LM. In the next step, in order to be able to attach an azide-functionalized dye molecule to the nanotubes for fluorescence imaging, alkyne functional groups were introduced through the copolymerization of NIPAM and propargyl acrylate (PPA) in the molar ratio of 100:1 via surface-initiated ARGET-ATRP reaction.
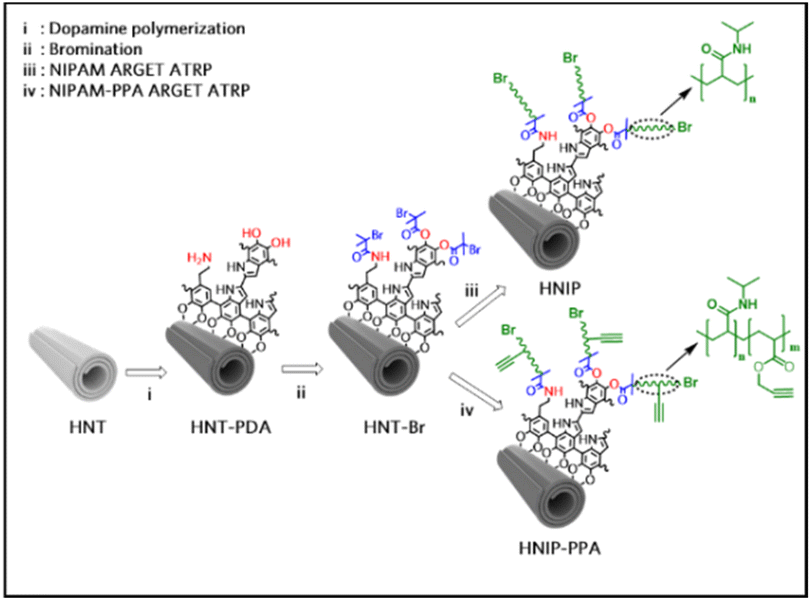 | ||
| Fig. 1 Schematic representation of the synthesis protocol for the modification of halloysite nanotubes (i) with polydopamine, followed by (ii) grafting alkyl bromide moieties and (iii) decoration with PNIPAM brushes or (iv) with poly(NIPAM-PPA) brushes. | ||
Grafting PNIPAM to HNTs surface
The characterization of HNT-PDA and HNT-Br samples has been done in our previous study, where the PDA layer formation and the successful attachment of acyl bromide moieties on the nanotubes were confirmed by XPS and TGA analysis.29 The initiator content of about 0.4 mmol Br per g was obtained for the HNT-Br sample, which corresponds to a relatively high initiator density of 5.1 Br per nm2, as calculated from the estimated specific surface area of HNTs, 46.6 m2 g−1 (the porosimetry data are available in ESI, Fig. S2†). This value of initiator density ensures the growth of dense polymer brushes on the surface of the nanotubes in the ATRP reaction.34–37Fig. 2 shows the characterization of the samples after PNIPAM grafting; results for the two degrees of polymerization, HNIP HM and HNIP LM, are shown. As seen in Fig. 2a, the FTIR spectrum of the pristine HNTs shows the following characteristic absorption bands:38 at 534 cm−1 the band due to the bending of Al–O–Si groups; at 1032 cm−1 the one attributed to the Si–O–Si stretching vibration; at 3620 cm−1 the band related to the stretching mode of Al-OH groups on the inner surface of the nanotubes and at 3696 cm−1 that of Al-OH groups between the layers of nanotubes. The interlayer water molecules can be recognized by the bending vibration at 1640 cm−1.21 Following PNIPAM grafting, new bands appear at 2980, 1550 and 1450 cm−1, which are attributed to the stretching vibration of –CH– bonds, the in-plane bending vibration of –NH– bonds and the –CH– bonds’ bending vibration, respectively.39 In addition, the vibrational band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the amide form appears at 1650 cm−1.39 Thermogravimetric analysis was carried out to quantify the amount of polymeric brushes attached to the nanotubes in each sample (see Fig. 2b). Differently from pristine HNTs, which mainly decompose at about 475 °C due to dihydroxylation of the Al-OH groups,21 HNIP samples exhibit a multistep weight loss in the range of 200–450 °C due to PNIPAM backbone degradation.40,41 The residual weight percentage after heating to 700 °C decreased from 71.2 wt% for HNIP LM and to 65.4 wt% for HNIP HM. By subtracting these values from the residual weight percentage of the samples before grafting (79.0 wt%),29 the grafting content of the PNIPAM brushes attached to the nanotubes can be estimated as 7.8 wt% for HNIP LM and 13.6 wt% for HNIP HM. XPS was employed to investigate the surface chemical composition of the PNIPAM-grafted samples; XPS survey scan of pristine HNTs was acquired as a control sample (Fig. 2c). XPS survey scan of pristine HNTs shows signatures of the constituent elements, i.e. oxygen, silicon and aluminum; in addition, a small signal of adventitious carbon is observed. For both HNIP HM and HNIP LM samples, the intense carbon and nitrogen peaks in the survey spectra testify to the presence of PNIPAM brushes on the surface of the nanotubes. In addition, the XPS survey scans of HNIP samples show the absence the signature of Cu 2p 3/2 core level region at the BE range of 932.3 to 934.3 eV.42
O bonds in the amide form appears at 1650 cm−1.39 Thermogravimetric analysis was carried out to quantify the amount of polymeric brushes attached to the nanotubes in each sample (see Fig. 2b). Differently from pristine HNTs, which mainly decompose at about 475 °C due to dihydroxylation of the Al-OH groups,21 HNIP samples exhibit a multistep weight loss in the range of 200–450 °C due to PNIPAM backbone degradation.40,41 The residual weight percentage after heating to 700 °C decreased from 71.2 wt% for HNIP LM and to 65.4 wt% for HNIP HM. By subtracting these values from the residual weight percentage of the samples before grafting (79.0 wt%),29 the grafting content of the PNIPAM brushes attached to the nanotubes can be estimated as 7.8 wt% for HNIP LM and 13.6 wt% for HNIP HM. XPS was employed to investigate the surface chemical composition of the PNIPAM-grafted samples; XPS survey scan of pristine HNTs was acquired as a control sample (Fig. 2c). XPS survey scan of pristine HNTs shows signatures of the constituent elements, i.e. oxygen, silicon and aluminum; in addition, a small signal of adventitious carbon is observed. For both HNIP HM and HNIP LM samples, the intense carbon and nitrogen peaks in the survey spectra testify to the presence of PNIPAM brushes on the surface of the nanotubes. In addition, the XPS survey scans of HNIP samples show the absence the signature of Cu 2p 3/2 core level region at the BE range of 932.3 to 934.3 eV.42
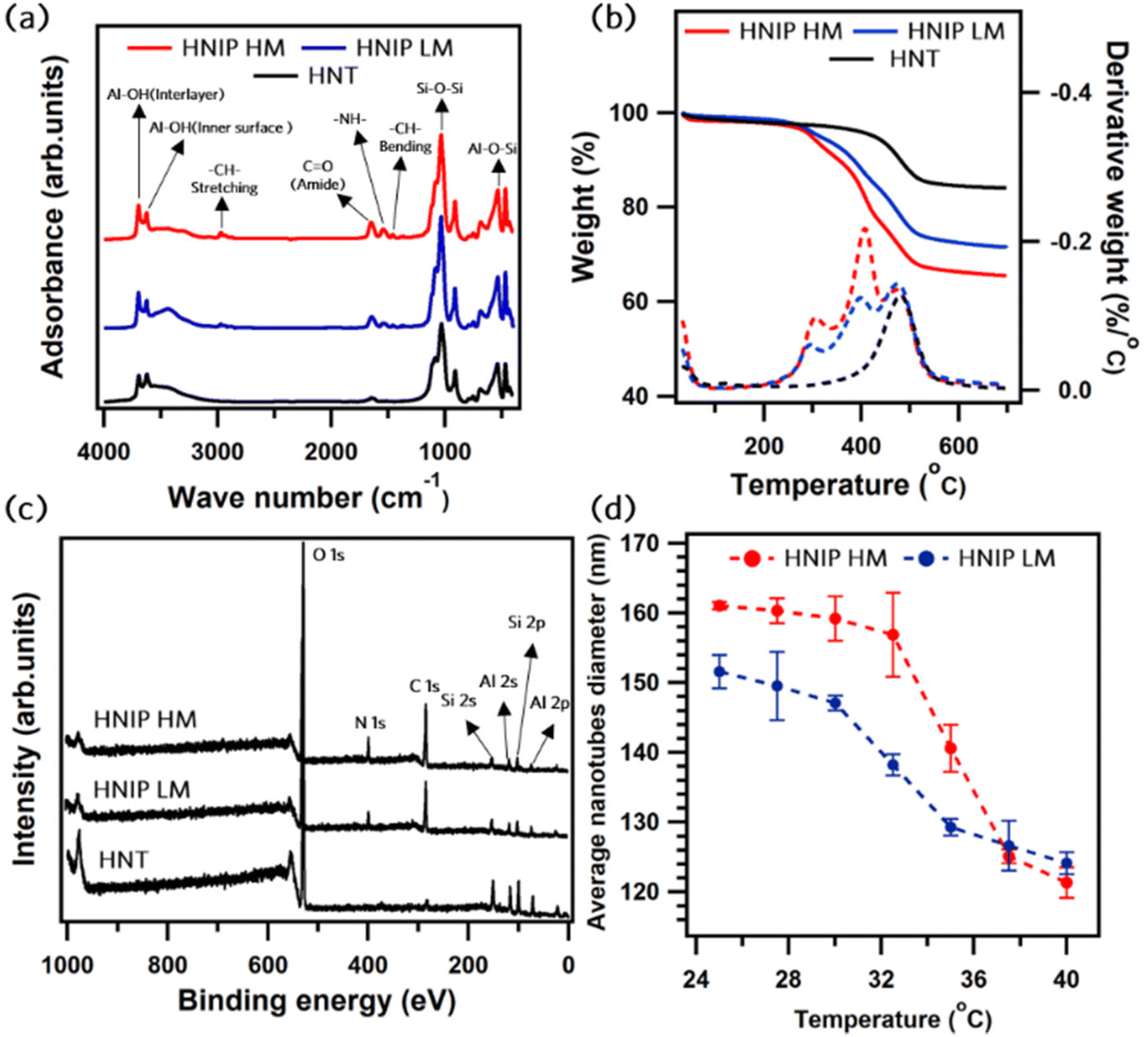 | ||
| Fig. 2 Characterization of surface-modified halloysite nanotubes after PNIPAM grafting: FTIR spectra (a), thermogravimetric analysis (b), solid lines TGA and dashed lines DTGA; XPS wide scan survey spectra (c) and temperature-dependent nanotubes diameter as determined from dynamic light scattering analysis (d). HNIP HM was prepared by SI-ARGET-ATRP with a monomer to initiator ratio of 1500, while for HNIP LM that ratio was 500 to obtain two different degrees of polymerization. | ||
This confirms the absence of copper in the polymer-grafted samples, which was indeed expected because of the ppm level of catalyst used in the ARGET-ATRP grafting reaction. The C 1s core level spectra of HNIP HM and HNIP LM require three components to obtain a good fit (Fig. S3, ESI†). The main component located at 284.8 eV (marked in red), which contributes with a relative spectral intensity of about 67% to the C 1s line, is assigned to CxHy, while the ones at 286.1 eV (relative spectral intensity of about 15%, marked in blue) and 287.8 eV (relative spectral intensity of 18%, marked in green) are attributed to the C–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –N and N–CO bonds, respectively,43 in agreement with what is expected from the stoichiometry of the NIPAM monomer. The N 1s spectra for HNIP HM and HNIP LM show a symmetrical peak at a BE of 399.8 eV, typical of N in the amide form.44Table 1 presents the surface elemental composition as calculated from the XPS data. The higher carbon and nitrogen content in HNIP HM confirms a higher degree of polymerization for PNIPAM brushes. It is worthy to note that after growing the PNIPAM brushes, Br was no longer detected in the XPS spectra. This might be due to radical termination and/or solvolysis of the terminal alkylbromide moieties upon reaction with the solvents, water and methanol, both leading to the elimination of the terminal Br.45
–N and N–CO bonds, respectively,43 in agreement with what is expected from the stoichiometry of the NIPAM monomer. The N 1s spectra for HNIP HM and HNIP LM show a symmetrical peak at a BE of 399.8 eV, typical of N in the amide form.44Table 1 presents the surface elemental composition as calculated from the XPS data. The higher carbon and nitrogen content in HNIP HM confirms a higher degree of polymerization for PNIPAM brushes. It is worthy to note that after growing the PNIPAM brushes, Br was no longer detected in the XPS spectra. This might be due to radical termination and/or solvolysis of the terminal alkylbromide moieties upon reaction with the solvents, water and methanol, both leading to the elimination of the terminal Br.45
| Sample | Chemical composition (in at%) | ||||
|---|---|---|---|---|---|
| C | Al | O | Si | N | |
| HNIP LM | 36.8 | 7.0 | 41.8 | 8.2 | 6.2 |
| HNIP HM | 43.6 | 5.5 | 36.5 | 6.6 | 7.8 |
The thermo-responsivity of the PNIPAM-grafted samples was investigated by dynamic light scattering (Fig. 2d). By monitoring the scattering from an aqueous suspension of the nanotubes with the concentration of 0.2 mg mL−1, the average diameter of the nanotubes was deduced from these data as the temperature changed from 25 to 40 °C in 2.5 °C steps (for details see Fig. S4 and S5, ESI†). For both HNIP HM and HNIP LM the average diameter was found to gradually decrease when the temperature increased from 25 to 30 °C, a behavior attributed to the dehydration of the dense inner PNIPAM brushes. When the temperature was further raised to 35 °C, a significant reduction of the diameter occurred, consistent with the collapse of the peripheral polymer brushes.46 As already mentioned in the introduction, for PNIPAM in aqueous solution, dissociation of the hydrogen bonds between PNIPAM and water at about 32 °C leads to the collapse of the extended hydrated polymeric chains into hydrophobic globules.47,48 Our data in Fig. 2d show that this phenomenon also occurs when the PNIPAM-grafted HNTs are suspended in water and the LCST, estimated in the range 30–32 °C, is very close to that of free PNIPAM chains in aqueous solutions. Moreover, HNIP HM exhibits a comparatively higher LCST value than HNIP LM. This can be explained by the higher degree of polymerization, which results in the higher molecular weight of the polymeric brushes in HNIP HM. This finding is consistent with that of Wanless et al., who demonstrated that the LCST of PNIPAM-grafted silica nanoparticles increases as a function of PNIPAM molecular weight.49 Furthermore, at temperatures below the LCTS where the PNIPAM brushes are highly swollen, the average diameter of HNIP HM is larger than that of HNIP LM, which could be attributed to the larger thickness of the polymer shell surrounding the nanotubes in HNIP HM. Comparing the average diameter at 25 °C of HNIP HM (about 161 nm) and pristine nanotubes (about 80 nm, see Fig. S5†), it can be concluded that the PNIPAM brushes attached to the nanotubes have a thickness of about 40 nm.
Cryo-TEM was used to investigate the nanostructure of the pristine HNTs and HNIP HM (Fig. 3a–d). The aqueous suspension of the samples (10 mg mL−1) was maintained at 18 °C before vitrification. In order to determine the thickness of the polymeric shell surrounding the nanotubes, a new method was employed in which the aqueous suspensions of the samples are mixed with earthworm blood in the volume ratio of 25 to 1. Earthworm blood contains giant hemoglobin particles with a globular shape and a diameter of 28.5 nm.50 As shown in Fig. 3a and b, pristine HNTs shows a hollow cylindrical morphology; their length varies in the range of 0.2 to 1.5 μm, while their inner and outer diameter can be estimated as 5–20 nm and 30–200 nm, respectively (for the distribution histograms see Fig. S5†). The relatively large diameter of the inner cavity allows to load the therapeutic molecules inside the HNTs.51 The giant hemoglobin can be clearly seen as monodispersed nano-donuts with an average diameter of 28.5 nm that are distributed homogenously between the pristine HNTs. For the HNIP HM sample a different arrangement of the hemoglobin particles can be observed (Fig. 3c and d): the polymeric shell around the nanotubes sterically hinders the hemoglobin particles from approaching the nanotube surfaces. Consequently, from the distance between the hemoglobin particles and the nanotube surface, the thickness of the polymeric shell can be estimated to be about 40 nm, which is in a good agreement with the results of the DLS analysis. EDS elemental mapping was employed as a complementary technique to investigate the distribution of the polymeric brushes around individual nanotubes. Fig. 3e displays the STEM-dark field image of the HNIP LM sample where EDS analysis was performed in the area marked by red dashed lines. For comparison, EDS analysis was carried out on HNIP HM as well, and the results are shown in Fig. 3f. As can be seen, the main constituents of HNTs, i.e. Si, Al and O, are detected homogeneously distributed over an individual nanotube. The success of polymer growth on the nanotube surface is evident from the significant amount of carbon whose signal overlaps with that of the main elements of the HNT. In addition, as expected, the elemental analysis of the scanned areas for HNIP HM and HNIP LM show a higher percentage of carbon for HNIP HM (see Table S1†).
 | ||
| Fig. 3 Cryo-TEM images of pristine halloysite nanotubes ((a) and (b)) and halloysite nanotubes decorated with poly(N-isopropylacrylamide) (HNIP HM) ((c) and (d)) with hemoglobin particles; STEM dark-field images of HNIP LM (e) and HNIP HM (f) and elemental mapping of the marked area by Energy Dispersive X-Ray Spectroscopy. | ||
Doxorubicin loading and release
Doxorubicin hydrochloride (DOX), a common anticancer drug, was used to investigate the effect of polymer grafting on the loading capacity and release profile of HNTs. DOX was loaded into the HNTs using the vacuum assisted procedure described in the introduction above.23,52 The loading capacities were calculated based on the DOX concentration remaining in the solution after loading and reported as the amount of the loaded DOX (mg) per one gram of HNTs in Table S2 (ESI).† We found that pristine HNTs have a DOX loading capacity of 77 ± 12 mg g−1, which is in a close agreement with the value reported by Li et al.53 who proposed that the electrostatic interactions between the negatively charged surface of the HNT and DOX·H+ (the soluble form of DOX) play a leading role in the loading mechanism. From the data in Table S2,† it is apparent that the loading capacity is significantly enhanced by grafting PNIPAM onto the HNTs. Since the drug loading experiments were performed at room temperature (below LCST), the PNIPAM brushes were in the hydrated and permeable state. Hence, PNIPAM-grafted HNTs load more DOX due to the formation of hydrogen bonds between the polymer brushes and DOX molecules.54 Furthermore, our findings show that the loading capacity increases with increasing degree of polymerization of the PNIPAM brushes (160 mg g−1 for HNIP LM vs. 250 mg g−1 or HNIP HM), supporting the dominant role of hydrogen bonding in the loading mechanism. Next we investigated the drug release behavior of the PNIPAM-grafted HNTs to evaluate their potential as temperature-responsive drug carrier. DOX release experiments were performed at 20 °C and 40 °C, i.e. at temperatures that are respectively below and above the LCST of PNIPAM, and the results are shown in Fig. 4a. For comparison, the release profile of the pristine HNTs at 20 °C was collected as well. Pristine HNTs show a steep initial rise in which approximately 40% of the loaded DOX molecules are released in the first hour of the experiment. This burst release behavior suggests that the majority of the drug molecules released in this period are those adsorbed on the outer surface of the HNTs via electrostatic interactions. The release profile significantly changed for the samples with PNIPAM brushes grafted on the surface. For both HNIP HM and HNIP LM, the DOX release gradually increased with time, without a noticeable burst release at the onset. Such a sustained and controlled drug release can be explained by the barrier represented by the polymer brushes: the drug molecules need more time to diffuse out from the thick shell surrounding the nanotubes. These results mirror those of previous studies, where the coating of the HNTs surface with a polymer was found to effectively reduce the burst effect and significantly delay drug release.29,55 Moreover, it can be clearly seen that the temperature influences the release behavior of PNIPAM-grafted HNTs; in fact, the accumulative release amount of the drug from HNIP HM reached 25% during the first 8 h at 40 °C, while at 20 °C, only 15% of the loaded drug was released in the same period. The enhanced release rate at higher temperature indicates the leading role of the PNIPAM brushes in controlling DOX release, as illustrated in Fig. 4b. When the temperature is below the LCST, i.e. at 20 °C, the hydrogen bonds with water molecules favor the stretching of the PNIAPM brushes to a considerable extent. In this state, the DOX molecules are stabilized by hydrogen bonds between the polar functional groups of DOX and PNIPAM, and only a small amount is released into the medium. In contrast, at temperatures above LCST, i.e. at 40 °C, the PNIPAM brushes begin to shrink because the hydrogen bonds break, leading to leakage of drug molecules in larger amounts. In addition, HNIP HM exhibits a slightly lower release rate than HNIP LM. This can be attributed to the better barrier property of the thicker PNIPAM shell present in HNIP HM. These results indicate that drug release from PNIPAM-grafted HNTs can be controlled by changing the temperature and/or the thickness of the polymeric shell. Such a behavior is highly demanded in chemotherapy where a controllable release of the anticancer drugs is crucial to decrease the side effects of the treatment.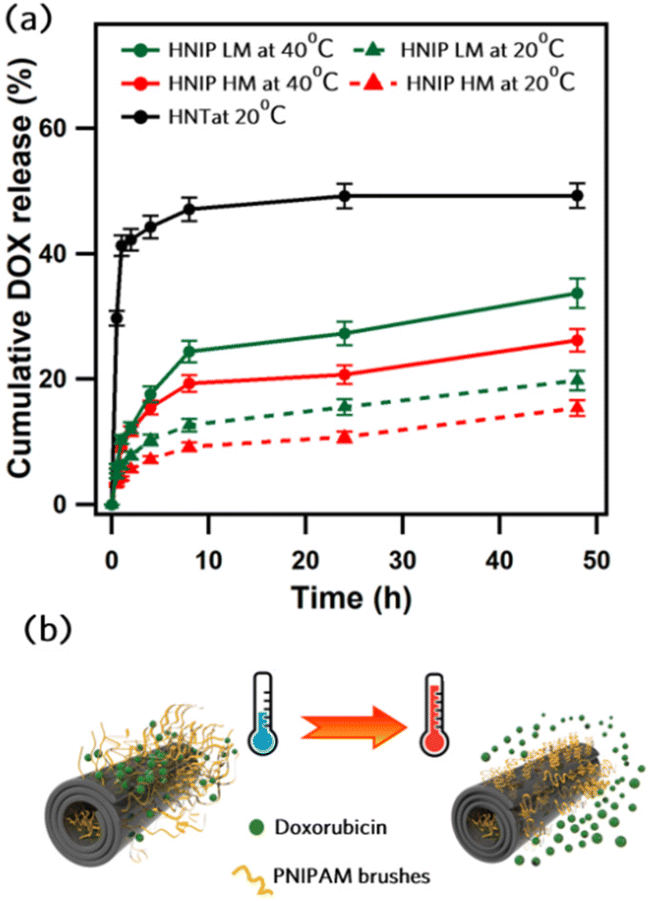 | ||
| Fig. 4 Cumulative DOX release profiles for pristine and PNIPAM-grafted HNTs at different temperatures (a) and schematic representation of temperature-responsive release of DOX (b). | ||
Cytocompatibility studies
The cytotoxicity of pristine HNTs, HNTs modified with polydopamine, as well as HNTs modified with polydopamine and further decorated with PNIPAM brushes on HeLa cells was investigated by MTT assay for three different exposure times (24, 48 and 72 h), and for different concentrations of nanoparticles (25 to 250 μg mL−1) (Fig. 5a–c). After 24 h, no noticeable decrease in cell viability was observed for the cells treated with pristine HNTs; above 85% of cells survived after 24 h of exposure to HNTs at all concentrations. For longer incubation times cell viability was found to slightly decrease, an effect that is stronger at higher nanoparticle concentrations. Only about 62% of the cells treated with HNTs at a concentration of 250 μg mL−1 are still alive after 72 h. These results corroborate those of Lvov et al.,56 who demonstrated that HNTs do not exhibit a significant impact on HeLa cells viability up to a concentration of 75 μg mL−1. After modifying the HNTs’ surface with PDA, a significant reduction in cell viability to ≈40% was observed for the lowest concentration of the nanoparticles. The observed high toxicity of PDA-coated HNTs against HeLa cells is in contrast with previous studies,57–60 in which high cytocompatibility of PDA-coated nanoparticles was demonstrated. Our finding can be explained by the fact that the negative charge of the HNTs results in a significant adsorption of the dopamine H+ cations on the surface of the nanotubes during the polymerization. In fact, we suggest that the toxicity of HNT-PDA is probably related to the release of dopamine molecules into the medium, which can trigger a variety of biological signals that cause cellular toxicity.61 Further research should be undertaken to verify this hypothesis. Instead, as can be clearly seen in Fig. 5a–c, cells show a higher survival rate when exposed to the HNIP samples, indicating that grafting the PNIPAM brushes to the nanotubes surfaces enhances the cytocompatibility of the samples because of the high biocompatibility of PNIPAM.62 What is interesting about the cell viability results is that the cytocompatibility of the samples increases with the degree of polymerization, confirming the important role of the PNIPAM brushes in reducing the cytotoxicity of HNT-PDA. Still, also for the nanotubes grafted with PNIPAM a mild decrease in cell viability was observed after 48 and 72 h (up to maximum ∼30% decrease after 72 h for cells treated with HNIP HM at the higher doses tested). Next, we performed MTT assays on pristine HNTs and HNIP samples loaded with DOX and evaluated their efficiency in killing HeLa cells after three time intervals. For comparison, the survival rate of the cells treated with the same amount of free DOX was examined as well (Fig. 5d–f). All samples exhibit a concentration-dependent toxicity on HeLa cells. However, after 24 h of the incubation a larger effect on cell viability is observed for cells exposed to the free DOX than for those in contact with the DOX-loaded halloysite nanotubes. Moreover, for the cells treated with free DOX, an increase in the incubation period has a minor effect on the survival rate of the cells; 76% of the cells were dead after treating with DOX at the concentration of 5 μg mL−1 for 72 h, which is slightly higher than that after 24 h (68%). This may be explained by the fact that free DOX molecules are readily available for uptake, as opposed to DOX loaded on the nanotubes, which first needs to release from the drug carrier.63 However, the DOX-loaded pristine HNTs showed similar cytotoxicity as free DOX for the same drug concentration. This suggests that the release of DOX from the pristine HNTs is fast, thus the cells are exposed to high drug concentrations at an early time in the experiment. In contrast, for HNIP-DOX samples, a clear difference was observed at increasing the incubation times; in fact, at the DOX concentration of 5 μg mL−1, 58% of cells survived after being treated with HNIP HM-DOX for 24 h, and this decreased to 24% after 72 h. This result is consistent with the drug release profile of the halloysite nanotubes covered by PNIAPM brushes in that the gradual release of DOX into the medium leads to stronger cytotoxicity at increasing incubation times. We stress that given that these experiments were performed at 37 °C, which is above the LCST, the PNIPAM brushes are already shrunken and a faster drug release is obtained in these conditions (as shown in Fig. 4b). In order to design nanotubes that respond to higher temperatures, and trigger drug release at T > 37 °C, the composition of the polymer brushes can be easily adjusted in order to increase the LCST, for instance by adding acrylamide.64–67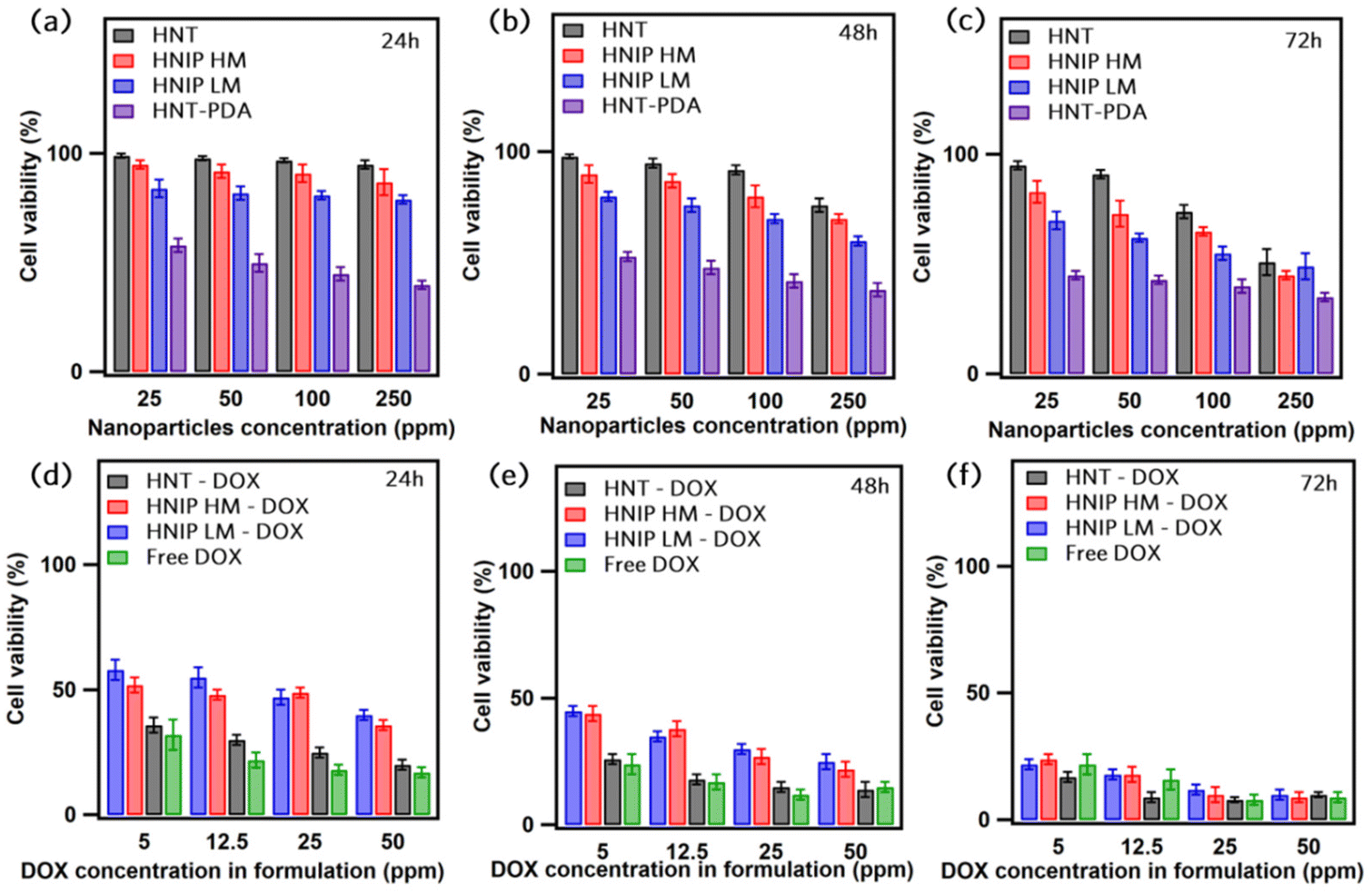 | ||
| Fig. 5 In vitro viability of HeLa cells incubated with different concentrations of pristine HNTs, HNT-PDA, HNIP LM and HNIP HM for 24 h (a), 48 h (b) and 72 h (c). In vitro viability of HeLa cells incubated for 24 h (d), 48 h (e) and 72 h (f), with different concentrations of free DOX, and of the same amount of drug loaded in pristine HNTs (HNT-DOX) and in HNTs onto which PNIPAM brushes were grafted (HNIP LM-DOX and HNIP HM-DOX). | ||
Fluorescent labeling of the nanotubes
To study the cellular uptake of the HNTs decorated with PNIPAM brushes, an azide-functionalized fluorescent molecule, Abberior STAR 635, was employed to confer fluorescent properties to the nanotubes. To attach the dye molecule to the nanotubes via click reaction, alkyne functional groups were introduced to the nanotubes by growing the copolymer brushes of P(NIPAM-co-PPA) on the nanotubes through surface-initiated ARGET-ATRP. Fig. 6a presents the schematic of the synthesis protocol employed to attach the dye molecule via click reaction. The FTIR spectrum of HNIP-PPA showed all characteristic peaks of HNTs and PNIPAM accompanied with the appearance of two new bands at 1740 and 2120 cm−1, which are attributed to the stretching mode of O–CO and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, respectively (Fig. 6b).68,69 This result confirms that the copolymer was successfully formed on the surface of the nanotubes. To confirm that the F-HNIP-PPAs were functionalized with the fluorescence dye molecule, the fluorescence emission spectra of F-HNIP-PPA and Abberior STAR 635 dye were acquired and compared. The emission spectrum of the nanotubes after binding the dye molecule corresponds to the emission spectrum of the unbound Abberior STAR 635 dye, which confirms the successful attachment of the fluorescent dye molecule to the nanotubes (Fig. S6†). The fluorescently labeled nanotubes were imaged by STED nanoscopy and the results are shown in Fig. 6c. The similar size of the nanotubes imaged by STED compared to those imaged by Cryo-TEM and STEM suggests that the STED images show individual nanotubes (Fig. 6c and Fig. S7†).
C bonds, respectively (Fig. 6b).68,69 This result confirms that the copolymer was successfully formed on the surface of the nanotubes. To confirm that the F-HNIP-PPAs were functionalized with the fluorescence dye molecule, the fluorescence emission spectra of F-HNIP-PPA and Abberior STAR 635 dye were acquired and compared. The emission spectrum of the nanotubes after binding the dye molecule corresponds to the emission spectrum of the unbound Abberior STAR 635 dye, which confirms the successful attachment of the fluorescent dye molecule to the nanotubes (Fig. S6†). The fluorescently labeled nanotubes were imaged by STED nanoscopy and the results are shown in Fig. 6c. The similar size of the nanotubes imaged by STED compared to those imaged by Cryo-TEM and STEM suggests that the STED images show individual nanotubes (Fig. 6c and Fig. S7†).
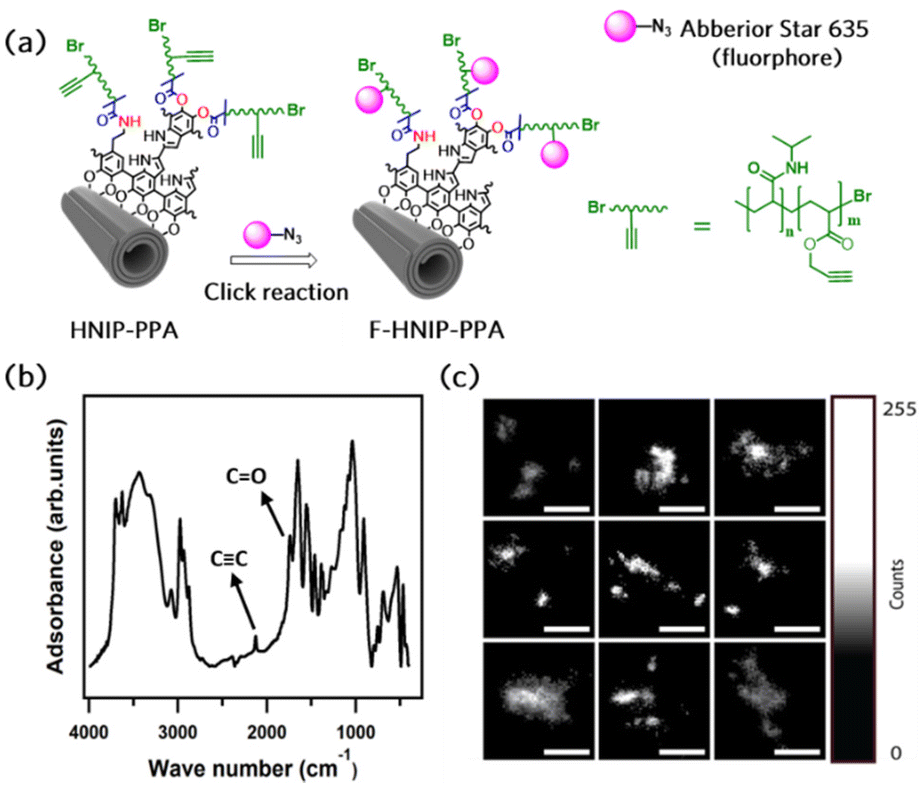 | ||
| Fig. 6 Schematic representation of the fluorophore labeling reaction (a), FTIR spectrum of halloysite nanotubes decorated with poly(NIPAM-co-PPA) brushes (b), and STED nanoscopy images of the labeled nanotubes (c); individual images in the panel are 1 × 1 μm. | ||
This provides further support for the successful labeling of the nanotubes.
Cellular uptake studies
Cytocompatibility of the fluorescent dye-labeled nanotubes was investigated by comparing the morphology of HeLa cells incubated with F-HNIP-PPA at a concentration of 62.5 μg mL−1 with the control sample without F-HNIP-PPA. As can be seen in Fig. S8,† functionalized nanotubes do not have significant impact on cell the viability, as no significant difference in the cell's morphology was observed. The percentage of cells in a round state was comparable with 9.3 ± 1.0% for the control, versus 10.3 ± 1.2% for cells incubated for 48 h with F-HNIP-PPA, confirming the findings of MTT assay. To study the cellular uptake and localization of F-HNIP-PPA in living HeLa cells, the cell membrane and lysosomes were labeled using Memglow and Lysotracker RED DND-99, respectively, and 3-D z-scans were acquired using confocal microscopy. XZ and YZ scans showed that the F-HNIP-PPAs were internalized by the HeLa cells (Fig. 7a). The F-HNIP-PPA clusters internalized by an individual cell were imaged using STED nanoscopy, and the results showed that the nanotube clusters were engulfed by intracellular organelles showing different intensities of Lysotracker signal (Fig. 7b–d and Fig. S9†). Lysotracker predominantly accumulates in the lysosomes, however, it can also stain other acidic organelles; the more acidic the organelle, the brighter the staining.70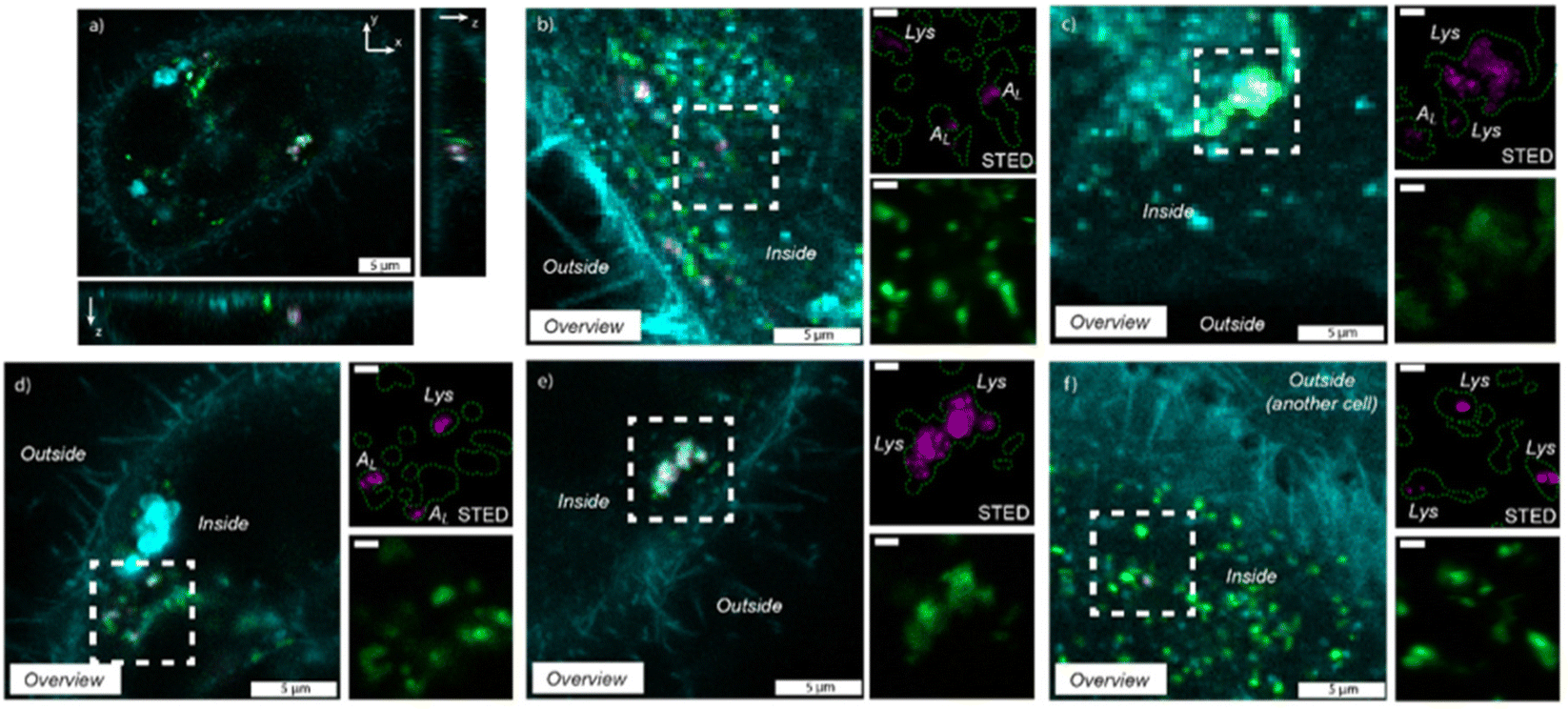 | ||
| Fig. 7 Uptake of F-HNIP-PPA (67.5 μg mL−1) after 48 h of incubation in HeLa cells. For all panels, magenta indicates the F-HNIP-PPA, green indicates the LysoTracker RED DND-99 (acidic organelles – lysosomes) staining and cyan represents the cellular membrane. XZ and YZ panels of confocal image of an individual cell (a), three examples of imaged cells where differently sized F-HNIP-PPA clusters were localized inside the cells in low-acidic organelles (in figure: AL) based on the obtained intensity upon imaging (b, c and d) and two examples of imaged cells where differently sized F-HNIP-PPA clusters were located in organelles with a high intensity of LysoTracker, indicating a high-acidic vesicle, which are the lysosomes, indicated as ‘Lys’ in figure (e and f). All zoomed-in region scale bars are 1 μm. | ||
Considering the constant decrease of pH in endocytosis compartments from 6.3 to 5.5 and 4.7 for early and late endosomes and lysosomes respectively,71–73 we can conclude that the nanotubes that colocalized with the low-intensity lysotracker region (typically < 10% of the maximum intensity of lysotracker in the cell) were engulfed by low-acidic vesicles, i.e. early endosomes and late endosomes (Fig. 7b–d), while nanotubes colocalized with a high-intensity Lysotracker region were engulfed by lysosomes (Fig. 7e and f). This suggests that F-HNIP-PPAs were internalized by active mechanisms of endocytosis into endosomes and finally trafficked to the lysosomes as indeed observed for most nanomaterials.74,75 These findings are also consistent with previous studies on the cellular uptake of PNIPAM-functionalized nanoparticles in which the accumulation of nanoparticles in lysosomes was observed in various cells.76–80 Analyzing the colocalization of the Lysotracker and F-HNIP-PPA signals by microscopy showed that large F-HNIP-PPA clusters (>1 × 1 μm, 24 measured HNTs aggregates in out of 1528 cells from 9 different samples) always reside in the lysosomes of the HeLa cells. For small F-HNIP-PPAs clusters (<1 × 1 μm), 77.2% (57 HNTs, in 1528 cells from n = 9 repeats) were engulfed by lysosomes and the remaining nanotube regions were located in the earlier vesicles with a lower acidity. A more detailed analysis of the size distribution of nanotube regions either engulfed by lysosomes or early vesicles was performed based on STED images (Fig. 8a and b). According to this analysis, nanotube regions that were engulfed by low-acidic vesicles had an average size of 494 ± 140 nm, which is smaller than the average size of the nanotube regions in the lysosomes (940 ± 497 nm). By considering an average size of 233 ± 113 nm, which was found for the nanotubes in the stock solution (Fig. S10†), it can be concluded that, large F-HNIP-PPA regions (>1 × 1 μm), which based on their size, are likely comprised of a few aggregated nanotubes, end up in the lysosome. Smaller F-HNIP-PPA regions (<1 × 1 μm), which are, based on their size, more likely to be comprised of single nanotubes end up likely in other vesicles along the endosomal pathways, after active uptake by endocytosis (Fig. 8c). This is consistent with what is usually observed after nanoparticle endocytosis, where nanoparticles transit into different endosomes while they accumulate over time in the lysosomes, thus leading to an overall higher amount of nanoparticles observed in these end-compartments.81,82
 | ||
| Fig. 8 Average size (a) and size distribution (b) of the nanotube clusters end up in the lysosomes and low acidic vesicles (e.g. early and late endosomal vesicles); schematic representation of the nanotube uptake by HeLa cells (c). | ||
Conclusions
In this study, we demonstrated how a combination of polydopamine surface modification and ARGET-ATRP could be exploited for grafting PNIPAM brushes on the HNTs surface to design a new temperature-responsive drug carrier. Successful formation of the PNIPAM brushes on the surface of HNTs was confirmed using XPS, FTIR and TGA, cryo-TEM and EDS analysis. We found that the grafting of PNIPAM brushes on the HNTs surface leads to a temperature-responsive hybrid material with a thermal phase transition temperature of about 32 °C.The polymer-grafted HNTs exhibit high loading capacity for doxorubicin and a temperature-dependent release behavior. Cytocompatibility studies reveal that PNIPAM-grafted HNTs have no impact on the viability of HeLa cells up to 100 μg mL−1 for 48 h. In the next step, PNIPAM-grafted HNTs were successfully labeled with a fluorescent dye as confirmed fluorescence spectroscopy and STED nanoscopy. Under our exposure conditions of 62.5 μg mL−1 by fluorescence labeled HNTs during at least 48 h, the nanotubes proved to be non-toxic against HeLa cells. A combination of confocal microscopy and STED nanoscopy showed that nanotubes were internalized by HeLa cells and accumulated in the lysosomes. Of the internalized nanotubes, all regions with a size above 1 μm2 were found to be in the lysosomes, whereas HNT regions below 1 μm2 could be located in lysosomes or other low acidic organelles, e.g. early or late endosomes. This is consistent with an active uptake mechanism of endocytosis and trafficking to the lysosomes, as observed for most nanomaterials. Further studies are required to elucidate the mechanisms.
Author contributions
H. H. proposed, planned and executed the synthesis, cytocompatibility and drug release experiments, analyzed the data and wrote the manuscript; V. H. A. revised the paper; T. C. Q. B. performed the cell imaging experiments, analyzed data and wrote the cellular uptake interpretation; F. Y. participated in the experiments and theoretical discussion; M. C. A. S. performed the Cryo-TEM, STEM and EDS elemental mapping experiments and revised the paper; C. R. S. performed cytocompatibility experiments with H. H.; R. V. designed and supervised the cellular uptake experiments, supervised T. C. Q. B. and revised the paper; A. S. designed and supervised the cytocompatibility and drug release experiments and revised the paper; P. R. designed this project together with H. H., supervised him in all experiments, and revised the paper. All authors critically reviewed and approved the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. H. acknowledges the Ubbo Emmius programme for supporting his PhD studies at the University of Groningen. This work benefitted from financial support by the Advanced Materials Research program of the Zernike National Research Centre under the Bonus Incentive Scheme of the Dutch Ministry for Education, Culture and Science. Additionally, we would like to thank Frank Mol for his contribution to assisting with the automated imaging of large overviews of HeLa cell samples.References
- S. Tran, P. J. DeGiovanni, B. Piel and P. Rai, Clin. Transl. Med., 2017, 6, 1–21 CrossRef PubMed.
- L. E. Gerlowski and R. K. Jain, Microvasc. Res., 1986, 31, 288–305 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nano-Enabled Med. Appl., 2020, 61–91 Search PubMed.
- D. Kalyane, N. Raval, R. Maheshwari, V. Tambe, K. Kalia and R. K. Tekade, Mater. Sci. Eng., C, 2019, 98, 1252–1276 CrossRef CAS PubMed.
- X. Zhao, J. Bai and W. Yang, Cancer Biol. Med., 2021, 18, 319 Search PubMed.
- M. Hei, J. Wang, K. Wang, W. Zhu and P. X. Ma, J. Mater. Chem. B, 2017, 5, 9497–9501 RSC.
- L. R. Hirsch, R. J. Stafford, J. Bankson, S. R. Sershen, B. Rivera, R. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS PubMed.
- B. Hildebrandt, P. Wust, O. Ahlers, A. Dieing, G. Sreenivasa, T. Kerner, R. Felix and H. Riess, Crit. Rev. Oncol. Hematol., 2002, 43, 33–56 CrossRef PubMed.
- X. Xu, Y. Liu, W. Fu, M. Yao, Z. Ding, J. Xuan, D. Li, S. Wang, Y. Xia and M. Cao, Polymers, 2020, 12, 580 CrossRef CAS PubMed.
- S. Ashraf, H. K. Park, H. Park and S. H. Lee, Macromol. Res., 2016, 24, 297–304 CrossRef CAS.
- T. E. de Oliveira, D. Mukherji, K. Kremer and P. A. Netz, J. Chem. Phys., 2017, 146, 034904 CrossRef PubMed.
- L. Hao, H. Yang and Z. Lei, Mater. Lett., 2012, 70, 83–85 CrossRef CAS.
- E. Aznar, M. Oroval, L. Pascual, J. R. Murguía, R. Martinez-Manez and F. Sancenon, Chem. Rev., 2016, 116, 561–718 CrossRef CAS PubMed.
- P. Mora-Raimundo, M. Manzano and M. Vallet-Regí, AIMS Bioeng., 2017, 4, 259–274 CAS.
- S. Jafari, H. Derakhshankhah, L. Alaei, A. Fattahi, B. S. Varnamkhasti and A. A. Saboury, Biomed. Pharmacother., 2019, 109, 1100–1111 CrossRef CAS PubMed.
- N. Iturrioz-Rodríguez, M. A. Correa-Duarte and M. L. Fanarraga, Int. J. Nanomed., 2019, 14, 3389 CrossRef PubMed.
- F. Pierini, E. Foresti, G. Fracasso, I. G. Lesci and N. Roveri, Isr. J. Chem., 2010, 50, 484–499 CrossRef CAS.
- M. Fizir, P. Dramou, N. S. Dahiru, W. Ruya, T. Huang and H. He, Microchim. Acta, 2018, 185, 1–33 CrossRef CAS PubMed.
- B. Singh, Clays Clay Miner., 1996, 44, 191–196 CrossRef CAS.
- H. Hemmatpour, V. Haddadi-Asl and H. Roghani-Mamaqani, Polymer, 2015, 65, 143–153 CrossRef CAS.
- R. R. Price, B. P. Gaber and Y. Lvov, J. Microencapsulation, 2001, 18, 713–722 CrossRef CAS PubMed.
- L. Lisuzzo, G. Cavallaro, P. Pasbakhsh, S. Milioto and G. Lazzara, J. Colloid Interface Sci., 2019, 547, 361–369 CrossRef CAS PubMed.
- Z. Long, Y. P. Wu, H. Y. Gao, J. Zhang, X. Ou, R.-R. He and M. Liu, J. Mater. Chem. B, 2018, 6, 7204–7216 RSC.
- B. Wu, M. Jiang, X. Liu, C. Huang, Z. Gu and Y. Cao, Nanotoxicology, 2020, 14, 1017–1038 CrossRef CAS PubMed.
- D. Sawicka, L. Zapor, L. Chojnacka-Puchta and K. Miranowicz-Dzierzawska, Toxicol. Res., 2021, 37, 301–310 CrossRef CAS PubMed.
- G. Biddeci, G. Spinelli, M. Massaro, S. Riela, P. Bonaccorsi, A. Barattucci and F. Di Blasi, Int. J. Nanomed., 2021, 16, 4755 CrossRef CAS PubMed.
- R. Batul, T. Tamanna, A. Khaliq and A. Yu, Biomater. Sci., 2017, 5, 1204–1229 RSC.
- H. Hemmatpour, V. Haddadi-Asl, F. Khanipour, M. C. Stuart, L. Lu, Y. Pei, H. Roghani-Mamaqani and P. Rudolf, Eur. Polym. J., 2022, 180, 111583 CrossRef CAS.
- C. E. Tas, E. B. Sevinis Ozbulut, O. F. Ceven, B. A. Tas, S. Unal and H. Unal, ACS Omega, 2020, 5, 17962–17972 CrossRef CAS PubMed.
- T. Staudt, A. Engler, E. Rittweger, B. Harke, J. Engelhardt and S. W. Hell, Opt. Express, 2011, 19, 5644–5657 CrossRef PubMed.
- J. Heine, M. Reuss, B. Harke, E. D'Este, S. J. Sahl and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9797–9802 CrossRef CAS PubMed.
- M. Liu, B. Guo, M. Du, X. Cai and D. Jia, Nanotechnology, 2007, 18, 455703 CrossRef.
- S. Edmondson, C.-D. Vo, S. P. Armes, G.-F. Unali and M. P. Weir, Langmuir, 2008, 24, 7208–7215 CrossRef CAS PubMed.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497–7505 CrossRef CAS PubMed.
- K. Ohno, T. Morinaga, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2005, 38, 2137–2142 CrossRef CAS.
- Y. Liu, V. Klep, B. Zdyrko and I. Luzinov, Langmuir, 2004, 20, 6710–6718 CrossRef CAS PubMed.
- M. Amjadi, A. Samadi, J. L. Manzoori and N. Arsalani, Anal. Methods, 2015, 7, 5847–5853 RSC.
- M. H. Futscher, M. Philipp, P. Müller-Buschbaum and A. Schulte, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- M. Kurdtabar, G. Baghestani and G. R. Bardajee, Gold Bull., 2019, 52, 9–17 CrossRef CAS.
- X. Jiang, S. Zhai, X. Jiang, G. Lu and X. Huang, Polymer, 2014, 55, 3703–3712 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- Q. Feng, D. Tang, H. Lv, W. Zhang and W. Li, RSC Adv., 2015, 5, 62024–62032 RSC.
- Y. Duan, J. Ma, J. Liu, L. Qiang and J. Xue, Fibers Polym., 2020, 21, 717–723 CrossRef CAS.
- W. D. Castro-Godoy, J. E. Argüello, M. Martinelli and D. A. Caminos, J. Chem. Educ., 2018, 95, 1827–1831 CrossRef CAS.
- B. A. Humphreys, E. J. Wanless and G. B. Webber, J. Colloid Interface Sci., 2018, 516, 153–161 CrossRef CAS PubMed.
- Z. Chen, Z. M. Cui, C. Y. Cao, W. D. He, L. Jiang and W. G. Song, Langmuir, 2012, 28, 13452–13458 CrossRef CAS PubMed.
- S. A. Jadhav, R. Nistico, G. Magnacca and D. Scalarone, RSC Adv., 2018, 8, 1246–1254 RSC.
- B. A. Humphreys, S. W. Prescott, T. J. Murdoch, A. Nelson, E. P. Gilbert, G. B. Webber and E. J. Wanless, Soft Matter, 2019, 15, 55–64 RSC.
- S. Xu and M. Arnsdorf, J. Microsc., 1997, 187, 43–53 CrossRef CAS PubMed.
- N. Danyliuk, J. Tomaszewska and T. Tatarchuk, J. Mol. Liq., 2020, 309, 113077 CrossRef CAS.
- L. Lisuzzo, G. Cavallaro, S. Milioto and G. Lazzara, J. Nanostruct. Chem., 2021, 11, 663–673 CrossRef CAS.
- L. Li, H. Fan, L. Wang and Z. Jin, RSC Adv., 2016, 6, 54193–54201 RSC.
- N. Huang, J. Wang, X. Cheng, Y. Xu and W. Li, J. Inorg. Biochem., 2020, 211, 111216 CrossRef CAS PubMed.
- S. Levis and P. Deasy, Int. J. Pharm., 2003, 253, 145–157 CrossRef CAS PubMed.
- V. Vergaro, E. Abdullayev, Y. M. Lvov, A. Zeitoun, R. Cingolani, R. Rinaldi and S. Leporatti, Biomacromolecules, 2010, 11, 820–826 CrossRef CAS PubMed.
- J. Si and H. Yang, Mater. Chem. Phys., 2011, 128, 519–524 CrossRef CAS.
- C. Tao, F. Ma, T. Chen, X. Li, W. Guan and A. Zhang, J. Alloys Compd., 2017, 715, 154–160 CrossRef CAS.
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384–9395 CrossRef CAS PubMed.
- D. Chang, Y. Gao, L. Wang, G. Liu, Y. Chen, T. Wang, W. Tao, L. Mei, L. Huang and X. Zeng, J. Colloid Interface Sci., 2016, 463, 279–287 CrossRef CAS PubMed.
- M. V. Clement, L. H. Long, J. Ramalingam and B. Halliwell, J. Neurochem., 2002, 81, 414–421 CrossRef CAS PubMed.
- Z. Guo, S. Li, C. Wang, J. Xu, B. Kirk, J. Wu, Z. Liu and W. Xue, J. Bioact. Compat. Polym., 2017, 32, 17–31 CrossRef CAS.
- X. Wang, J. Zhang, Y. Wang, C. Wang, J. Xiao, Q. Zhang and Y. Cheng, Biomaterials, 2016, 81, 114–124 CrossRef CAS PubMed.
- Y. Feng, Y. Cheng, Y. Chang, H. Jian, R. Zheng, X. Wu, K. Xu, L. Wang, X. Ma and X. Li, Biomaterials, 2019, 217, 119327 CrossRef CAS PubMed.
- M. Ji, X. Qiu, L. Hou, S. Huang, Y. Li, Y. Liu, S. Duan and Y. Hu, Int. J. Nanomed., 2018, 13, 1773 CrossRef CAS PubMed.
- A. Li, Y. Wang, T. Chen, W. Zhao, A. Zhang, S. Feng and J. Liu, Eur. Polym. J., 2017, 92, 51–60 CrossRef CAS.
- Z. Wei, Z. Liu, X. Wang, S. Long and J. Yang, Eur. Polym. J., 2019, 114, 1–10 CrossRef CAS.
- L. Q. Xu, H. Jiang, K. G. Neoh, E. T. Kang and G. D. Fu, Polym. Chem., 2012, 3, 920–927 RSC.
- S. Khoee and N. Abedini, Polymer, 2014, 55, 5635–5647 CrossRef CAS.
- X. Chao, Y. Qi and Y. Zhang, ACS Sens., 2020, 6, 786–796 CrossRef PubMed.
- J. Llopis, J. M. McCaffery, A. Miyawaki, M. G. Farquhar and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6803–6808 CrossRef CAS PubMed.
- F.-X. Theillet, A. Binolfi, T. Frembgen-Kesner, K. Hingorani, M. Sarkar, C. Kyne, C. Li, P. B. Crowley, L. Gierasch and G. J. Pielak, Chem. Rev., 2014, 114, 6661–6714 CrossRef CAS PubMed.
- A. H. Ponsford, T. A. Ryan, A. Raimondi, E. Cocucci, S. A. Wycislo, F. Fröhlich, L. E. Swan and M. Stagi, Autophagy, 2021, 17, 1500–1518 CrossRef CAS PubMed.
- V. Francia, D. Montizaan and A. Salvati, Beilstein J. Nanotechnol., 2020, 11, 338–353 CrossRef CAS PubMed.
- A. Salvati, C. Åberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Nanomedicine, 2011, 7, 818–826 CrossRef CAS PubMed.
- C. Gerecke, A. Edlich, M. Giulbudagian, F. Schumacher, N. Zhang, A. Said, G. Yealland, S. B. Lohan, F. Neumann and M. C. Meinke, Nanotoxicology, 2017, 11, 267–277 CrossRef CAS PubMed.
- P. C. Naha, K. Bhattacharya, T. Tenuta, K. A. Dawson, I. Lynch, A. Gracia, F. M. Lyng and H. J. Byrne, Toxicol. Lett., 2010, 198, 134–143 CrossRef CAS PubMed.
- J. McMasters, S. Poh, J. B. Lin and A. Panitch, J. Controlled Release, 2017, 258, 161–170 CrossRef CAS PubMed.
- S. Poh, J. B. Lin and A. Panitch, Biomacromolecules, 2015, 16, 1191–1200 CrossRef CAS PubMed.
- W. Zhang, Z. Mao and C. Gao, J. Colloid Interface Sci., 2014, 434, 122–129 CrossRef CAS PubMed.
- H. Garcia Romeu, S. Deville and A. Salvati, Small, 2021, 17, 2100887 CrossRef CAS PubMed.
- N. Vtyurina, C. Åberg and A. Salvati, Nanoscale, 2021, 13, 10436–10446 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06801j |
| This journal is © The Royal Society of Chemistry 2023 |