
Mapping the reaction zones for CdTe magic-sized clusters and their emission properties†
Saryvoudh A.
Mech
,
Fuyan
Ma
and
Chenjie
Zeng
 *
*
Department of Chemistry, University of Florida, Gainesville, FL 32611, USA. E-mail: zeng@chem.ufl.edu
First published on 12th December 2022
Abstract
CdTe magic-sized clusters (MSCs) are promising building blocks for semiconductor devices because of their single size, consistent properties, and reproducible synthesis. However, the synthetic conditions for CdTe MSCs vary significantly in different reports, which hinders the general understanding of their formation mechanisms. Here, we employed Cd(oleate)2, trioctylphosphine telluride (TOPTe), and oleylamine, which are commonly used for larger quantum dot (QD) synthesis, as standard reaction precursors, and systematically investigated the effects of solvent, phosphine amount, oleylamine amount, Cd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Te ratio, and temperature on the evolution of MSCs with time. These conditions compose the “reaction coordinates” to map out the “reaction zones” for CdTe MSCs and QDs. We found that CdTe MSCs with the first excitonic transition (E1) at 449 nm can be synthesized in high purity with excess TOPTe using toluene as the solvent at 100 °C. Whereas higher temperature, excess of Cd(oleate)2, or more viscous solvent led to the aggregation of 449 nm MSC into larger magic-sized species with E1 at 469 nm as well as QDs with E1 > 500 nm. Increasing phosphine concentration simply enhanced the rate and yield of CdTe MSCs, while a critical amount of oleylamine was required to “turn on” the MSC formation. Interestingly, the pure 449 nm MSCs were non-emissive, but colorful emissions were observed for the reaction mixtures containing both MSCs and QDs. The emissions could be attributed to a small amount of QDs formed during the reaction. The mapping of reaction zones is a crucial step towards the rational synthesis of CdTe MSCs and further understanding of their formation mechanism.
:Te ratio, and temperature on the evolution of MSCs with time. These conditions compose the “reaction coordinates” to map out the “reaction zones” for CdTe MSCs and QDs. We found that CdTe MSCs with the first excitonic transition (E1) at 449 nm can be synthesized in high purity with excess TOPTe using toluene as the solvent at 100 °C. Whereas higher temperature, excess of Cd(oleate)2, or more viscous solvent led to the aggregation of 449 nm MSC into larger magic-sized species with E1 at 469 nm as well as QDs with E1 > 500 nm. Increasing phosphine concentration simply enhanced the rate and yield of CdTe MSCs, while a critical amount of oleylamine was required to “turn on” the MSC formation. Interestingly, the pure 449 nm MSCs were non-emissive, but colorful emissions were observed for the reaction mixtures containing both MSCs and QDs. The emissions could be attributed to a small amount of QDs formed during the reaction. The mapping of reaction zones is a crucial step towards the rational synthesis of CdTe MSCs and further understanding of their formation mechanism.
 Chenjie Zeng | Dr Chenjie Zeng received her Ph.D. in Chemistry from Carnegie Mellon University in 2016 and was a NatureNet Science postdoctoral fellow at the University of Pennsylvania from 2017–2019. She joined the Department of Chemistry at the University of Florida as an assistant professor in 2019. Research in her group centers around precision nanochemistry and nanomaterials, with emphasis on developing synthetic methods for atomically precise nanomaterials, understanding the structure and property correlations of nanomaterials at the atomic level, as well as assembling the nanoparticle building blocks into thin films and bulk solids for better energy conversion and exotic physical properties. |
Introduction
Semiconductor nanocrystals (also called quantum dots, QDs), are important building blocks for solution-processible optoelectronic devices.1,2 An interesting observation during the colloidal synthesis of QDs is the formation of magic-sized clusters (MSCs).3,4 They are single-sized clusters with enhanced stability due to closed geometric structures compared with non-magic ones with random sizes and less stable structures.5–10 Because of their single sizes and stability, MSCs can be reproducibly synthesized in different reactions. This contrasts with conventional QDs, which usually show size distributions and batch-to-batch variations in synthesis. MSCs are usually identified spectroscopically by (i) sharp, intense, and consistent absorption peaks and (ii) quantized redshift of absorption peaks as MSCs grow from one size to another. Despite these intriguing features, the synthesis and formation mechanisms of semiconductor MSCs remain unexplored compared to that of QDs.Cadmium telluride (CdTe) is a promising semiconductor material for optoelectronic devices and is the second most used semiconductor after Si for commercialized solar cells.11 Solar cells based on CdTe QDs have reached an efficiency of >10%.12 In addition, the relatively smaller band gap of CdTe (1.50 eV, 829 nm) compared to the related CdSe (1.73 eV, 716 nm) and CdS (2.42 eV, 512 nm)4 provides a wider room for the ultrasmall CdTe clusters to show absorption and emission in the visible light region (400 nm to 700 nm), which makes them potential candidates for multicolor light-emitting diodes.13,14 Despite the potential applications, CdTe MSCs are less studied than CdSe and CdS MSCs15–21 due to the limited tunability in Te precursor compared to S and Se precursors.22,23
Nonetheless, several CdTe MSCs have been discovered in the past 25 years using colloidal synthetic methods with various precursors and reaction conditions. For example, CdTe MSCs were first observed by Rogach et al. in the aqueous phase synthesis employing NaHTe, Cd(ClO4)2, and thiols (as protecting ligands).24 A relatively sharp absorption peak at 460 nm corresponding to the first excitonic transition (E1) was observed after reacting at 96 °C for 2 hours. Later, Wuister et al. reported CdTe MSCs with E1 at ∼420 and ∼450 nm by reacting Cd(CH3)2 and trioctylphosphine telluride (TOPTe) in dodecylamine (DDA) at 40 °C and 100 °C, respectively.25 Dagtepe et al. replaced the organometallic Cd(CH3)2 precursor with cadmium hexylphosphonate and used amine and phosphine oxide as the coordinating solvent; CdTe MSCs with E1 at 449 nm and 491 nm were obtained at >200 °C under different reaction times.26 The following work showed that a Te-rich environment (Cd:Te of 0.2:1) was critical for the formation of CdTe MSCs, while a Cd-rich (Cd:Te of 2:1) one resulted in larger QDs.27 CdTe MSCs with E1 at 445 nm and 483 nm were also observed by using dialkylphosphinic acid.28 Based on the previous success in the sequential growth of CdSe MSCs,17 Zanella et al. extended the synthetic method to CdTe MSCs and obtained 445 nm, 488 nm, and 506 nm MSCs by reacting cadmium nonanoate in decylamine at 130 °C.29 The classical cadmium oleate precursor can also be used to obtain 447 nm and 483 nm MSCs from 50 °C to 180 °C.30 Using a Cd(acetate)2-amine precursor and TOPTe together with super-hydride, Buhro and coworker obtained 374 nm MSCs at 40 °C and a 450 nm one at 70 °C.31,32 The two clusters were assigned to (CdTe)13 and (CdTe)34, respectively, based on previous mass spectrometry anaylsis.15 Recently, Yu and coworkers discovered a two-step method to obtain CdTe MSCs with E1 at 371 nm, 417 nm, and 448 nm by firstly preparing a precursor mixture of Cd(acetate)2-oleylamine and TOPTe at various temperatures and then injecting it into the solvent mixture of toluene and amine. The size control was achieved by tuning amine concentration in the solution, in which a lower concentration led to larger MSCs.33–35
The above examples show that the CdTe MSCs can be prepared in a wide range of reaction conditions, including reaction temperatures (from room temperature33–35 to more than 200 °C26–28), Cd:Te ratios (from Cd-rich25 to Te-rich27), and reaction time (from one second34,35 to more than one day30). While the tellurium precursors used in these reactions are mostly TOPTe, the cadmium precursors vary significantly, including alkyl,25 phosphonate,26–28 carboxylate,29,30 and amine31–35 complexes of cadmium. The different reactivities of cadmium precursors result in various reaction conditions for CdTe MSC formation. Despite the variations, alkylamine has been employed in all CdTe MSCs syntheses and a large amount of phosphine was often used as the coordinating solvent. This can be distinguished from the synthesis of CdTe QDs, where only cadmium complex, tellurium precursor, and non-coordinating solvents are involved in typical synthesis.4,36,37
To better investigate the critical reaction conditions for CdTe MSCs and minimize kinetic variations from different precursor conversion chemistry,38,39 we first identified a model reaction employing Cd(oleate)2, TOPTe, and oleylamine as precursors. These reagents are commonly used in QDs synthesis, which facilitates the comparison of critical conditions between MSC and QD synthesis. The effects of reaction parameters on the evolution of MSC were systematically investigated, including (i) reaction solvent and environment such as octadecene (ODE) and toluene, (ii) the amount of additional trioctylphosphine (TOP), (iii) the amount of oleylamine (OAM), (iv) the Cd:Te ratio, and (v) the temperature (Fig. 1). These parameters serve as “reaction coordinates” to map out “reaction zones” between the formation of CdTe MSCs and QDs.
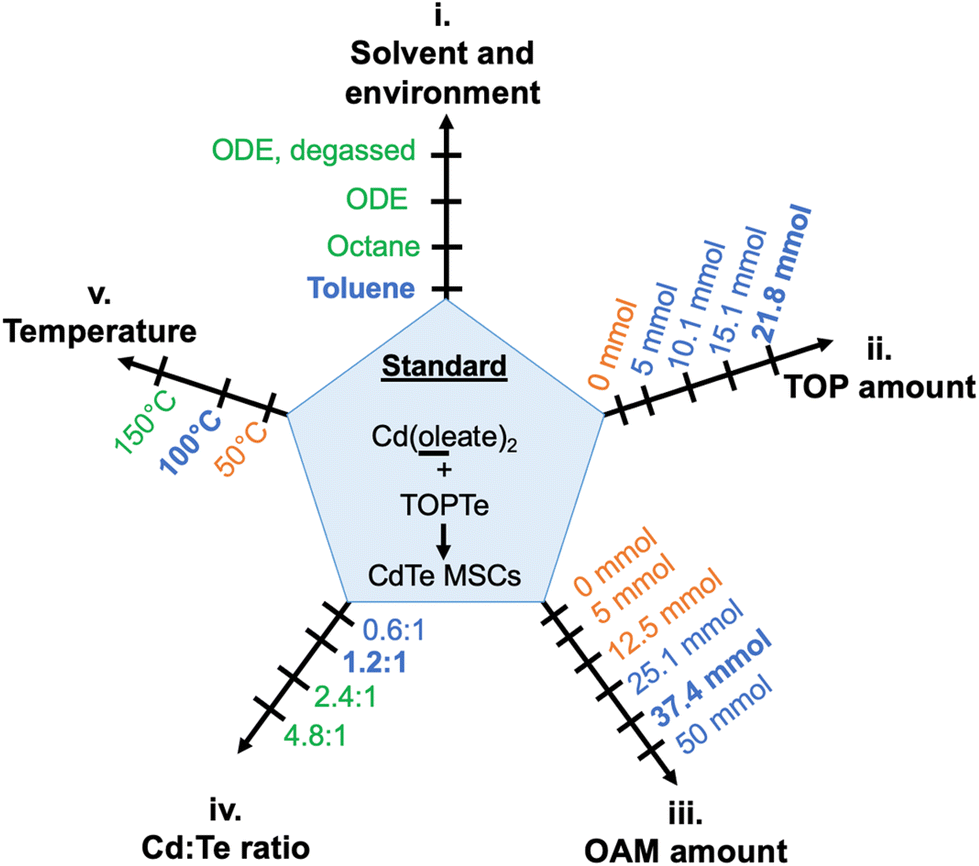 | ||
| Fig. 1 Diagram showing different reaction conditions explored in this work. Standard conditions are highlighted in bold. The conditions that produce MSCs, a mixture of MSCs and QDs, and no MSCs are labeled in blue, green, and orange, respectively. | ||
Results and discussion
To set the standard conditions, we first modified the reaction that employed Cd(oleate)2, TOPTe, and oleylamine.30 The amount of Te precursor was fixed at 1 mmol by using 1 ml of 1 M TOPTe stock solution (Fig. 2A). A slight excess of Cd(oleate)2 precursor (1.2 mmol, from 2 ml of 0.6 M of Cd(oleate)2 stock solution) was added together with TOP (10 ml, 21.2 mmol) and OAM (17.6 ml, 37.4 mmol). ODE (19.4 ml) was added as the non-coordinating solvent to balance the total reaction volume to 50 ml, resulting in a reaction concentration of 20 mM based on Te. The reaction mixture was degassed at room temperature under ∼0.5 torr vacuum for 30 minutes. Then N2 was introduced and the temperature was set to 100 °C. About 2 ml aliquots were withdrawn from the reaction mixture at 0.5, 1, 1.5, 2, 4, 8, 23, and 24 hours, respectively. The aliquot was diluted 100 times by injecting 50 μl of the reaction mixture into 5 ml of toluene for absorption and photoluminescence characterization (Fig. 2B–E).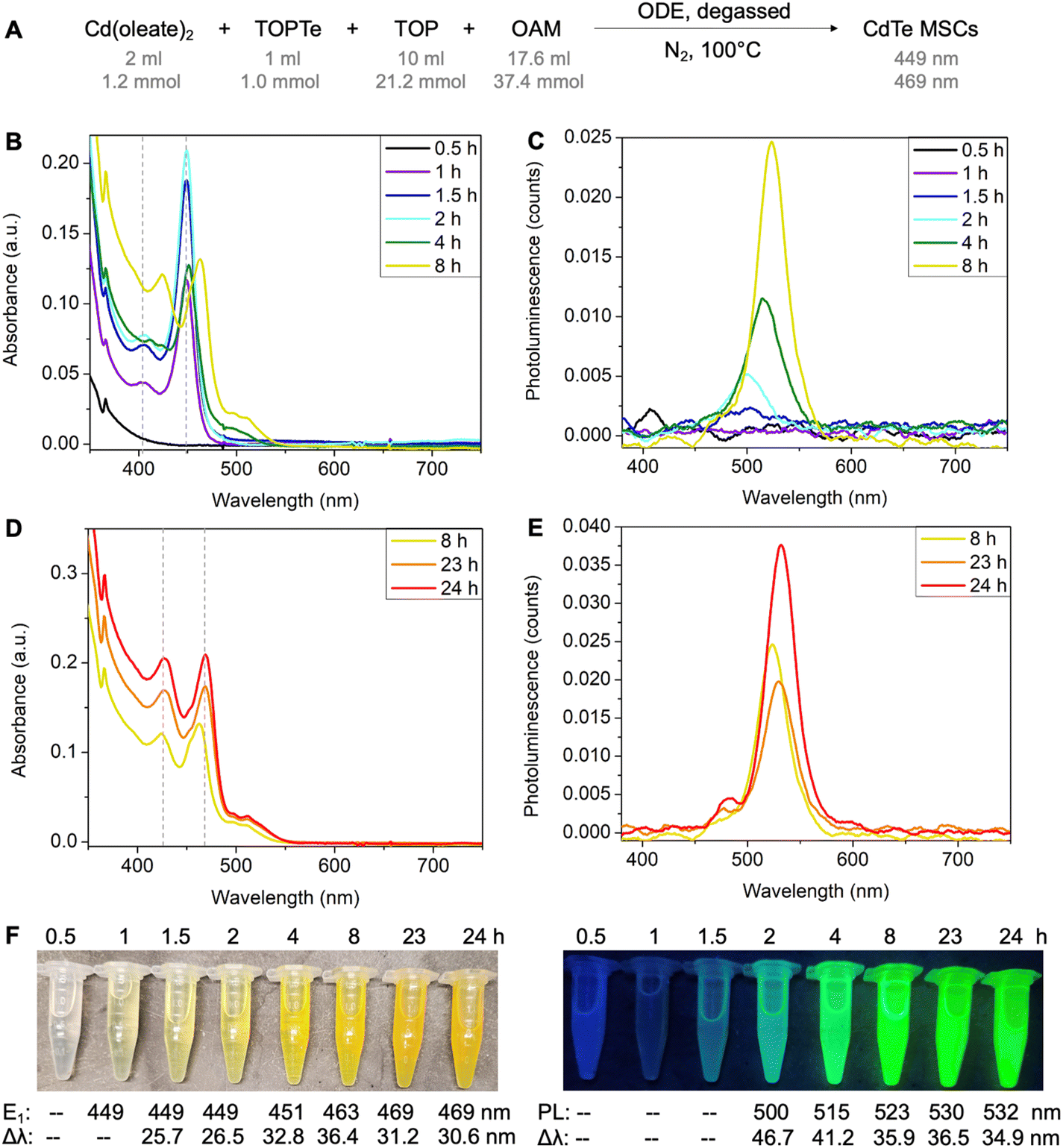 | ||
| Fig. 2 (A) Reaction scheme for CdTe MSCs synthesis in ODE. (B and D) UV-vis spectra of the reaction aliquots from 0.5 to 24 hours. Each aliquot is diluted 100 times in toluene before measurement. (C and E) Corresponding emission spectra of diluted aliquots (excitation wavelength is 405 nm). (F) Photos of aliquots under ambient light and UV light (wavelength = 366 nm). | ||
While the 0.5-hour aliquot showed no absorption (Fig. 2B, black line), a prominent peak at 449 nm and a shoulder at ∼405 nm were observed after 1 hour of reaction (purple line). The peak positions remained constant at 449 nm and 405 nm, but the absorbance of the two peaks continuously increased and reached the maximum at ∼2 hours (blue and cyan lines). The consistent peak positions and absorption profiles indicated a MSC was formed. The absorption spectra are similar to previously reported ones,25,27–29,32 and the 449 nm and 405 nm peaks are assigned to the first (E1) and second (E2) excitonic transitions of the MSC, respectively. For the 4-hour aliquot, the absorbance at 449 nm decreased with a broader peak emerging at 500 nm (green line). After 8 hours, the peaks redshifted to 463 nm and 424 nm (yellow line), and further stabilized at 469 nm and 427 nm at 24 hours, respectively (orange and red lines, Fig. 2D). We tentatively assign the two peaks as E1 and E2 of the second magic-sized species.
Interestingly, bright photoluminescence (PL) with color ranging from cyan to green was observed for the aliquots from 2 hours to 24 hours under UV light (Fig. 2F). The corresponding emission peak redshifted continuously from 500 to 532 nm with an overall increase in PL intensity and a decrease in the full width at half maximum (Fig. 2C and E). We assign the emission to the small amount of QDs formed during the reaction rather than the MSCs. This is based on (i) no PL was observed for the 1-hour aliquot, which showed a purer 449 nm MSC peak with negligible tail absorbance (Fig. S1A†); (ii) the onset of emission was accompanied by the appearance of the tail absorbance at ∼500 nm, which were assigned to larger QDs impurities (Fig. S1B–H†); and (iii) the emissions redshifted continuously with reaction time, which was characteristic of the emissions of QDs with increasing sizes. To further probe the origin of the emissions, PL excitation spectra were recorded for the aliquots (Fig. S2†). Similar to the emission peaks, the excitation peaks continuously redshifted from 474 nm to >520 nm with increasing reaction time (Fig. S2D†), which further confirmed that the bright emissions originated from QDs impurities instead of MSCs. Note that a small emission peak at 480 nm was observed in 23–24 hours aliquots (Fig. S2C†), which could possibly originate from the 469 nm species. But its intensity is much lower compared to the main emission peak.
We also performed the reaction in ODE without degassing and similar results were observed (Fig. S3A†). Considering MSC synthesis did not require high temperatures (>200 °C) and was tolerant to moisture and air, we further modified the reaction solvent from ODE to the less viscous toluene. The reaction in toluene led to purer 449 nm MSCs without forming second magic-sized species and QDs (Fig. S3B†). Also, no strong PL was observed, further confirming the non-emissive nature of the 449 nm MSCs. Performing the reaction in octane led to similar results as shown in ODE, with the formation of second magic-sized species at 469 nm in 24 hours (Fig. S3C†). These results suggest that solvents with long hydrocarbon groups can facilitate the aggregation of small clusters into larger magic-sized species and QDs. We hypothesize that the long hydrocarbon chain of the solvent can possibly form “bundles” with oleate, OAM, or TOP with similar hydrocarbon groups, therefore, increasing the interaction of CdTe clusters carried by these ligands and facilitating their aggregation into larger species. In contrast, small toluene molecules lack such interactions thus MSCs can be stabilized for a long period of time without aggregation at 100 °C.
Since the reaction in toluene gave purer 449 nm MSCs, we selected toluene as the standard solvent for the systematic investigation of reaction conditions. Compared to typical CdTe QDs synthesis,36,37 a large amount of TOP is often required as a coordinating solvent in MSCs synthesis. To investigate the effect of TOP, its amount was systematically varied from 0 ml to 10 ml (0 mmol to 21.7 mmol). The amount of OAM was kept at 50 mmol and the total volume at 50 ml (balanced with toluene). Without additional TOP, MSCs were not observed, and the reaction solution remained colorless over 24 hours (Fig. 3A and Fig. S4A†). When increasing TOP amount to 5 mmol, 449 nm MSC appeared after 4 hours of reaction (Fig. S4B†). Adding more TOP increased the rate and yield of MSC, as shown in the faster increase of 449 nm absorbance (Fig. 3A) and a higher concentration of 449 nm MSC at 4 hours (Fig. 3B). A possible explanation for the increased reaction rate is the presence of a larger amount of secondary phosphine as an impurity when more TOP is used. Secondary phosphine is known to increase the reaction rate and yield of QDs and is considered the reactive component in precursor conversion.40,41
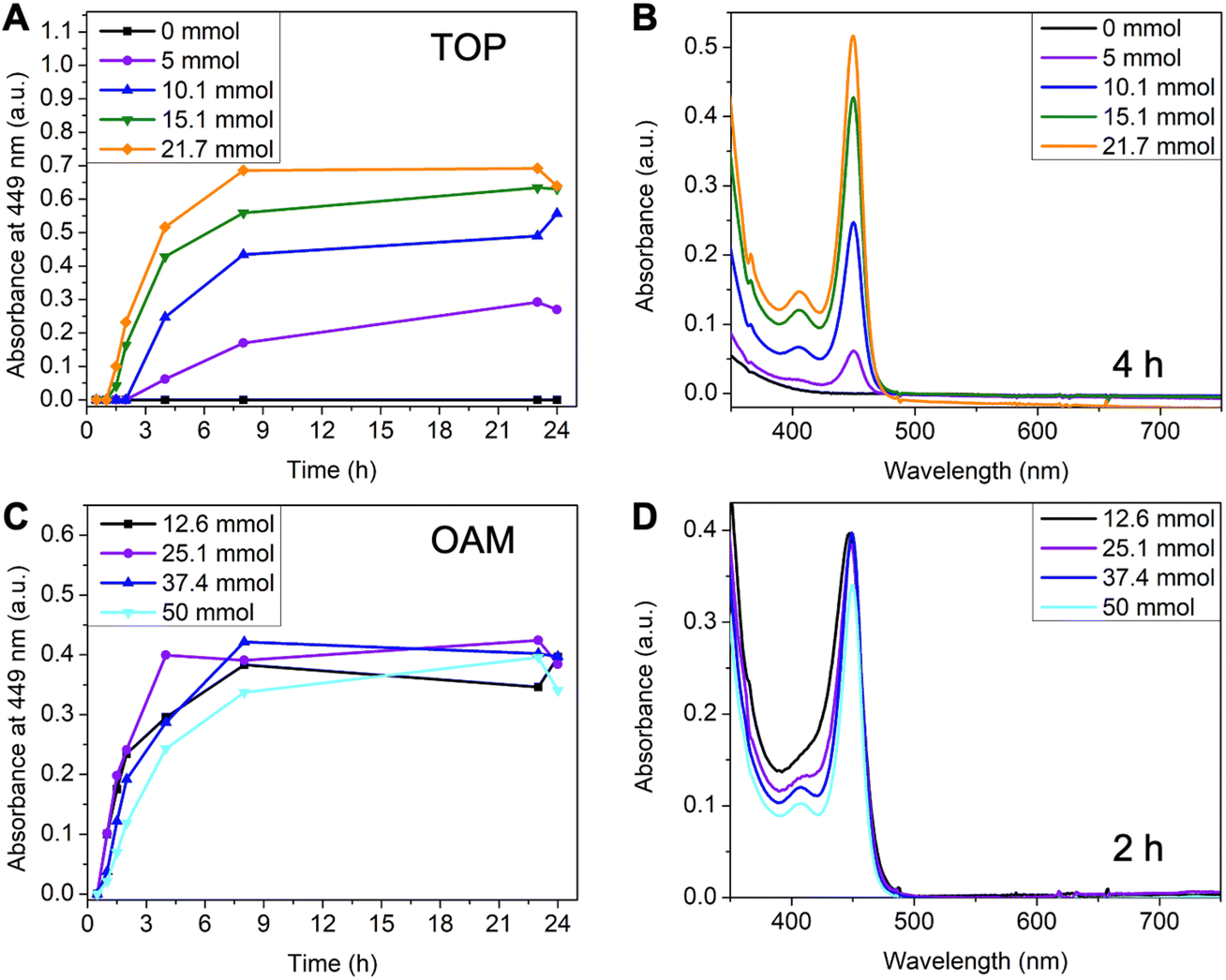 | ||
| Fig. 3 Effect of trioctylphosphine and oleylamine on the formation of CdTe MSCs. (A) The evolution of peak absorbance at 449 nm with time under different TOP amounts. (B) Corresponding absorption spectra at 4 hours. (C) The evolution of 449 nm absorbance with time under different OAM amounts. (D) Corresponding absorption spectra at 2 hours. | ||
The effect of oleylamine is more complicated. First of all, a critical amount of OAM was required.30 With 0 mmol of OAM, no CdTe MSCs were observed. Instead, a broad peak centered at ∼430 nm with its intensity continuously increased over 24 hours (Fig. S5A†). Increasing OAM to 6 mmol led to a mixture of 449 nm MSC and the ∼430 nm species (Fig. S5B†). Only when the amount of OAM exceeded 12.6 mmol, pure MSCs were observed (Fig. S6†). Secondly, further increasing the amount of OAM reduced the formation rate and yield of MSCs. This can be shown in the decrease of absorbance at 449 nm when OAM concentration is increased from 25.1 mmol to 50 mmol (Fig. 3C) and the comparison of spectra of different reaction aliquots at 2 hours (Fig. 3D). The decrease in growth rate with increasing alkylamine is consistent with the earlier observations in the synthesis of CdSe QDs42 as well as in the studies of their nucleation and growth rates.43 The kinetic results show that amine plays a different role than TOP in MSC synthesis. On one hand, a large excess of amines can coordinate with the cadmium precursor and slow down its conversion to MSCs. On the other hand, amine is required to trigger the nucleation of CdTe MSCs at relatively low temperatures.43 We note that the ∼430 nm species observed without amine could be the prenucleation complexes assembled from Cd(oleate)2 and TOPTe. As the reaction proceeded, the peak position remained constant at ∼430 nm but its intensity increased (Fig. S5A†). The ∼430 nm species is unlikely to be the polydisperse CdTe clusters since the peak position would shift with reaction time instead of remaining constant if different-sized CdTe clusters were formed.
The ratio between Cd and Te precursors also affects MSC formation. To investigate its effect, TOPTe was set to 1 mmol while Cd(oleate)2 was varied from 0.6 mmol to 4.8 mmol (Fig. 4). When half amount of Cd(oleate)2 was used (Cd:Te of 0.6:1), only 449 nm MSC was formed (Fig. 4A), similar to the case of 1.2:1 ratio (Fig. 4B). The two ratios showed a similar reaction rate (Fig. S7†), indicating the reaction rate was not limited by the Cd(oleate)2 conversion. Interestingly, no PL emission was observed for the 0.6:1 reaction (Fig. 4A), while faint green emission was present for the 1.2:1 reaction at 23 and 24 hours (Fig. 4B). It indicates the 0.6:1 reaction resulted in purer 449 nm MSCs without producing QDs impurity. Further increasing the ratio to 2.4:1 led to the formation of second magic-sized species and larger QDs, as shown in the decrease of the 449 nm absorbance as well as the appearance of a shoulder peak at ∼467 nm and a broad peak beyond 500 nm (Fig. 4C). Bright green emission centered at ∼530 nm was also observed for 23- and 24-hour aliquots due to the presence of QDs. More prominent differences were shown in the 4.8:1 ratio, where the 449 nm peak disappeared completely and the shoulder in the 2.4:1 sample became a distinct peak at 470 nm. The broad peak also redshifted from 510 nm to 525 nm, which was accompanied by stronger and sharper emission (Fig. 4D). Interestingly, the 4:8:1 reaction in toluene resembles the 1.2:1 reaction in ODE (Fig. 2). Both reactions lead to the formation of second magic size species in 24 hours. These experiments demonstrate that excess Cd(oleate)2 precursors can trigger the aggregation and ripening of 449 nm MSCs into larger species, which is probably due to the increased viscosity with more Cd(oleate)2.
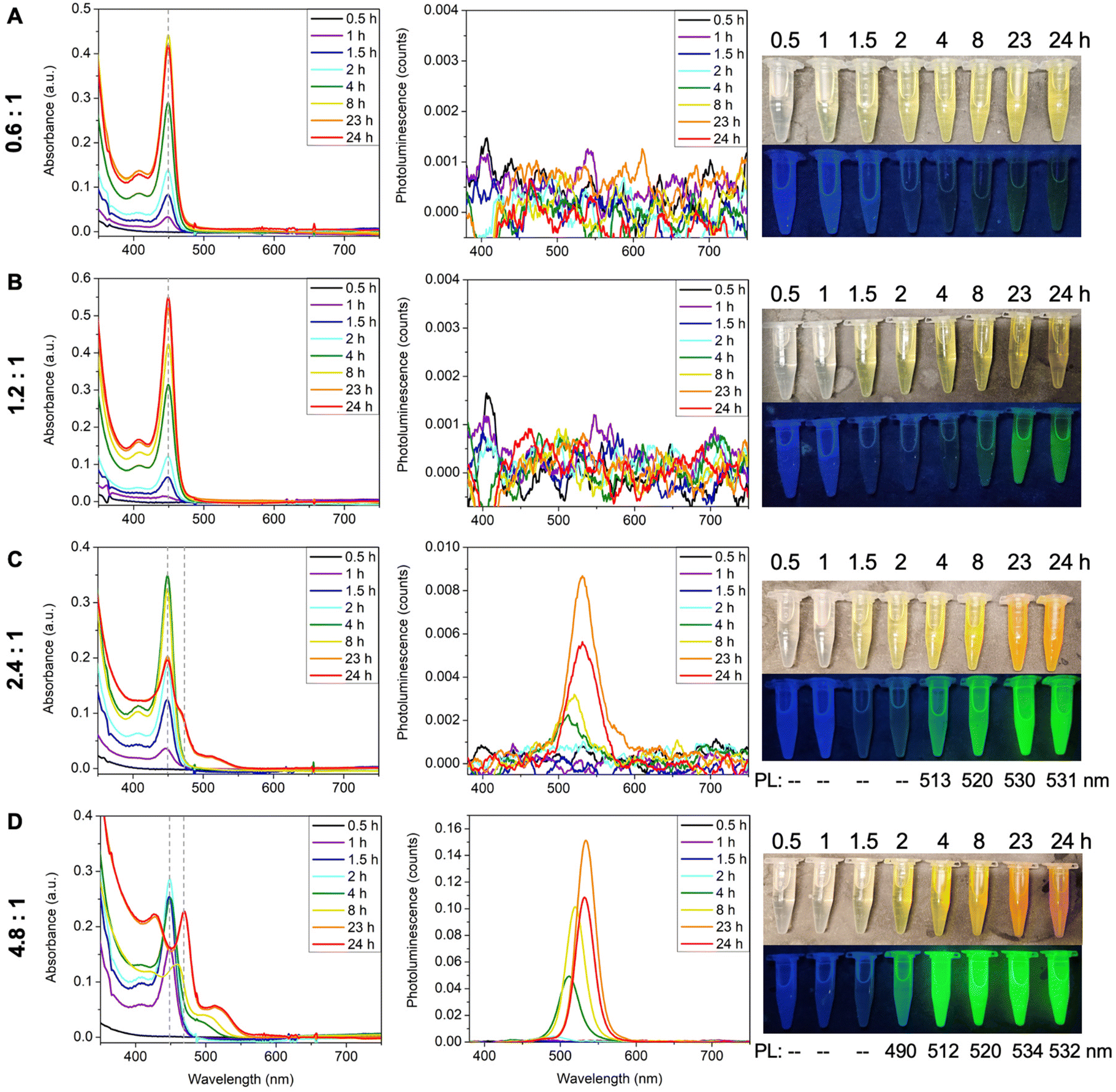 | ||
| Fig. 4 Effect of Cd:Te ratio on CdTe MSC formation. The amount of TOPTe is set to 1 mmol, while the Cd(oleate)2 is varied from (A) 0.6 mmol, (B) 1.2 mmol, (C) 2.4 mmol, and (D) 4.8 mmol. The reactions are monitored by absorption, emission, and photos of aliquots under ambient light and UV light. | ||
The effect of temperature is intuitive. When the reaction was performed at 50 °C, no CdTe clusters formed after one day, and the reaction solution remained colorless and non-emissive (Fig. 5A). Note that the faint blue emission of the aliquots is a result of background scattering of the UV light. Similarly, the peak at ∼400 nm region in the emission spectra is due to the scattering of excitation photons at 405 nm. When the reaction temperature was increased to 150 °C using ODE as the solvent, the 449 nm peak appeared in the first half hour (Fig. 5B, black line), and its absorbance increased to the maximum at 1.5 hours together with the onset of a broad peak centered at ∼530 nm. At 2 hours, the absorbance at 449 nm decreased, which completely disappeared at 4 hours. The broad peak continuously redshifted from ∼530 nm to ∼550 nm with increasing absorbance. Note that two small sharp peaks at constant positions of ∼480 nm and ∼550 nm coexisted with the broad QD peak, which implies the possible formation of the larger MSC species. The emission peak also redshifted from its initial appearance at 547 nm to its final position at 607 nm and the emission color evolved from green, yellow, orange, to red (Fig. 5B). Such continuous red shifts of absorption and emission peaks are characteristics of the previously studied CdTe QDs.44
 | ||
| Fig. 5 Effect of temperature on CdTe MSC formation. The reactions at (A) 50 °C and (B) 150 °C are monitored by absorption, emission, and photos of aliquots under ambient light and UV light, respectively. | ||
While CdTe MSCs formation depends on the interplay of different reaction parameters, our results suggest that oleylamine is a critical reagent to trigger MSC formation under current conditions. Oleylamine could play multiple roles in different stages of the reaction, including activating the Cd(oleate)2 precursor, facilitating the precursor conversion into CdTe monomers, triggering the nucleation at the lower temperature, guiding the assembly of monomers into well-defined clusters, and serving as surface ligands to stabilize the final MSCs.43 Other parameters, such as solvent, amount of TOP, Cd:Te ratio, and temperature affect the reaction rate, conversion yield, or aggregation pathways of MSCs, but they are less critical since these parameters do not alter the underlying chemistry for MSC formation. For example, increasing the amount of TOP simply increases the reaction rate and the yield of MSCs.
Previous mechanistic studies on CdE QDs synthesis (E = S, Se, or Te) shows that the reaction between Cd(II) carboxylate Cd(O2CR)2 and trialkylphosphine chalcogenide R′3P=E generates CdE QDs together with trialkylphosphine oxide R′3P=O and carboxylate acid anhydride O(COR)2 as the byproducts.38 This general reaction scheme should also be applicable to the CdTe MSC synthesis since the same types of precursors are employed in MSC reaction as in QD synthesis (eqn (1)).
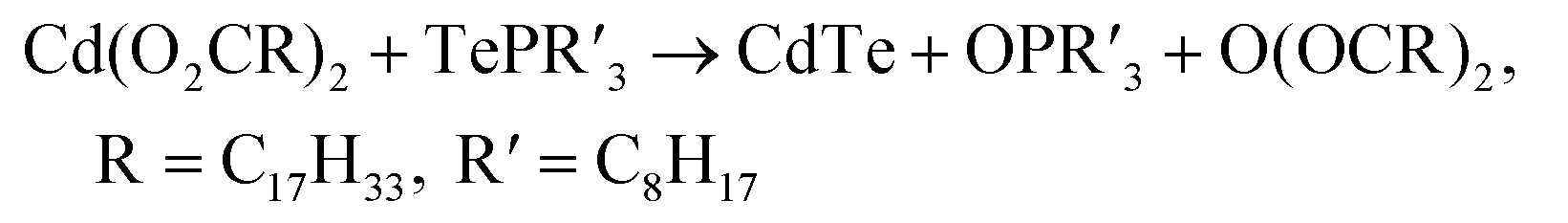 | (1) |
However, a high temperature (usually >200 °C) is required for the reaction between Cd(oleate)2 and TOPTe to proceed at a reasonable rate. This could be attributed to the high activation energy to form reaction intermediates (e.g., acyloxyphosphonium [RCOO-PR′3]+43) during the precursor conversion. The required high temperature for precursor conversion could result in the rapid nucleation and growth of generated CdTe monomers into large QDs instead of MSCs.
We hypothesize that a critical condition for MSC formation is the slow precursor conversion and monomer release rate at a relatively low reaction temperature. Such a condition could allow enough time for CdTe monomers to reorganize into well-defined cluster structures which can be stabilized at the reaction temperature. Oleyamine could facilitate the precursor conversion at a lower reaction temperature possibly through activating the Cd(oleate)2 precursors (e.g., by breaking the polymeric chain of [Cd(oleate)2]n into more reactive discrete Cd(oleate)2(NH2R)x unit) or stabilizing the reaction intermediates (e.g., formation of alkylamoniophosphonium [RHN-PR′3]+)43 thus lowering the activation energy of precursor conversion into CdTe unit at a lower temperature. Future NMR studies on the MSC formation process are needed to map out the reaction mechanism at the molecular level.
Conclusion
Using common reaction precursors, we systematically investigated different reaction parameters on the formation of CdTe MSCs as well as their PL properties. Reaction in toluene resulted in purer 449 nm MSCs, whereas ODE led to a mixture of 449 nm and 469 nm species as well as larger QDs. The lower viscosity of toluene compared to ODE can effectively prevent further aggregation of MSCs into larger clusters and QDs. Increasing the concentration of TOP simply increased the rate and yield of MSC formation without changing reaction pathways. OAM was further verified as a crucial reagent for CdTe MSCs formation, and a critical amount of OAM was required to convert the precursor complex into CdTe MSCs. Increasing the OAM above the critical amount slowed down the formation rate, possibly due to the stabilization of Cd complex by excess OAM. Purer 449 nm MSCs were obtained when an insufficient amount of Cd precursor was used, whereas a large amount of Cd precursor led to the aggregation of 449 nm MSCs into 469 nm magic-sized species and larger QDs, which is similar to the reaction performed in ODE. No clusters were formed at 50 °C due to the low precursor conversion rate. The formation rate of 449 nm MSC improved significantly at 150 °C, followed by their aggregation into large MSCs and QDs. While negligible PL was found in pure 449 nm MSCs, colorful emissions were observed when larger QDs were present in the reaction mixtures. The results enhance our understanding and are expected to facilitate further rational control of the synthesis and photoluminescent properties of CdTe MSCs.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was supported by the University of Florida start-up research funds.References
- C. R. Kagan, E. Lifshitz, E. H. Sargent and D. V. Talapin, Science, 2016, 353, 885–892 CrossRef CAS PubMed
.
- C. R. Kagan, L. C. Bassett, C. B. Murray and S. M. Thompson, Chem. Rev., 2021, 121, 3186–3233 CrossRef CAS PubMed
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2002, 124, 3343–3353 CrossRef CAS PubMed
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS
-
R. L. Johnston, Atomic and Molecular Clusters, CRC Press, 2002 Search PubMed
- G. S. H. Lee, D. C. Craig, I. Ma, M. L. Scudder, T. D. Bailey and I. G. Dance, J. Am. Chem. Soc., 1988, 110, 4863–4864 CrossRef CAS
- N. Herron, J. C. Calabrese, W. E. Farneth and Y. Wang, Science, 1993, 259, 1426–1428 CrossRef CAS
- T. Vossmeyer, G. Reck, B. Schulz, L. Katsikas and H. Weller, J. Am. Chem. Soc., 1995, 117, 12881–12882 CrossRef CAS
- V. N. Soloviev, A. Eichhöfer, D. Fenske and U. Banin, J. Am. Chem. Soc., 2001, 224, 285–289 CAS
- A. N. Beecher, X. Yang, J. H. Palmer, A. L. Lagrassa, P. Juhas, S. J. L. Billinge and J. S. Owen, J. Am. Chem. Soc., 2014, 136, 10645–10653 CrossRef CAS PubMed
- M. A. Green, E. D. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis, K. Bothe, D. Hinken, M. Rauer and X. Hao, Prog. Photovoltaics, 2022, 30, 687–701 Search PubMed
- J. M. Kurley, J.-A. Pan, Y. Wang, H. Zhang, J. C. Russell, G. F. Pach, B. To, J. M. Luther and D. V. Talapin, ACS Appl. Mater. Interfaces, 2021, 13, 44165–44173 CrossRef CAS
- D. Bera, L. Qian, T.-K. Tseng and P. H. Holloway, Materials, 2010, 3, 2260–2345 CrossRef CAS
- O. Chen, H. Wei, A. Maurice, M. Bawendi and P. Reiss, MRS Bull., 2013, 38, 696–702 CrossRef CAS
- A. Kasuya, R. Sivamohan, Y. A. Barnakov, I. M. Dmitruk, T. Nirasawa, V. R. Romanyuk, V. Kumar, S. V. Mamykin, K. Tohji, B. Jeyadevan, K. Shinoda, T. Kudo, O. Terasaki, Z. Liu, R. V. Belosludov, V. Sundararajan and Y. Kawazoe, Nat. Mater., 2004, 3, 99–102 CrossRef CAS PubMed
- M. J. Bowers, J. R. McBride and S. J. Rosenthal, J. Am. Chem. Soc., 2005, 127, 15378–15379 CrossRef CAS PubMed
- S. Kudera, M. Zanella, C. Giannini, A. Rizzo, Y. Li, G. Gigli, R. Cingolani, G. Ciccarella, W. Spahl, W. J. Parak and L. Manna, Adv. Mater., 2007, 19, 548–552 CrossRef CAS
- B. M. Cossairt and J. S. Owen, Chem. Mater., 2011, 23, 3114–3119 CrossRef CAS
- J. C. Newton, K. Ramasamy, M. Mandal, G. K. Joshi, A. Kumbhar and R. Sardar, J. Phys. Chem. C, 2012, 116, 4380–4389 CrossRef CAS
- S. M. Harrell, J. R. McBride and S. J. Rosenthal, Chem. Mater., 2013, 25, 1199–1210 CrossRef CAS
- A. S. Mule, S. Mazzotti, A. A. Rossinelli, M. Aellen, P. T. Prins, J. C. van der Bok, S. F. Solari, Y. M. Glauser, P. V. Kumar, A. Riedinger and D. J. Norris, J. Am. Chem. Soc., 2021, 143, 2037–2048 CrossRef CAS PubMed
- M. L. Steigerwald and L. E. Brus, Annu. Rev. Mater. Sci., 1989, 19, 471–495 CrossRef CAS
- I. A. Shuklov, I. S. Mikhel, A. V. Nevidimov, K. P. Birin, N. V. Dubrovina, A. A. Lizunova and V. F. Razumov, ChemistrySelect, 2020, 5, 11896–11900 CrossRef CAS
- A. L. Rogach, L. Katsikas, A. Kornowski, D. Su, A. Eychmüller and H. Weller, Ber. Bunsenges. Phys. Chem., 1997, 101, 1668–1670 CrossRef CAS
- S. F. Wuister, F. van Driel and A. Meijerink, Phys. Chem. Chem. Phys., 2003, 5, 1253–1258 RSC
- P. Dagtepe, V. Chikan, J. Jasinski and V. J. Leppert, J. Phys. Chem. C, 2007, 111, 14977–14983 CrossRef CAS
- P. Dagtepe and V. Chikan, J. Phys. Chem. A, 2008, 112, 9304–9311 CrossRef CAS PubMed
- A. D. Dukes, J. R. McBride and S. J. Rosenthal, Chem. Mater., 2010, 22, 6402–6408 CrossRef CAS
- M. Zanella, A. Z. Abbasi, A. K. Schaper and W. J. Parak, J. Phys. Chem. C, 2010, 114, 6205–6215 CrossRef CAS
- H. Xu, Y. Hou and H. Zhang, J. Nanopart. Res., 2017, 19, 189 CrossRef
- Y. Wang, Y. Zhou, Y. Zhang and W. E. Buhro, Inorg. Chem., 2015, 54, 1165–1177 CrossRef CAS PubMed
- Y. Zhou, R. Jiang, Y. Wang, H. W. Rohrs, N. P. Rath and W. E. Buhro, Inorg. Chem., 2019, 58, 1815–1825 CrossRef CAS PubMed
- M. Liu, K. Wang, L. Wang, S. Han, H. Fan, N. Rowell, J. A. Ripmeester, R. Renoud, F. Bian, J. Zeng and K. Yu, Nat. Commun., 2017, 8, 15467 CrossRef CAS PubMed
- C. Luan, J. Tang, N. Rowell, M. Zhang, W. Huang, H. Fan and K. Yu, J. Phys. Chem. Lett., 2019, 10, 4345–4353 CrossRef CAS PubMed
- Q. Shen, C. Luan, N. Rowell, M. Zhang, K. Wang, M. Willis, X. Chen and K. Yu, Inorg. Chem., 2021, 60, 4243–4251 CrossRef CAS
- W. W. Yu, Y. A. Wang and X. G. Peng, Chem. Mater., 2003, 4300–4308 CrossRef CAS
- Y. A. Yang, H. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., Int. Ed., 2005, 44, 6712–6715 CrossRef CAS PubMed
- H. Liu, J. S. Owen and A. P. Alivisatos, J. Am. Chem. Soc., 2007, 129, 305–312 CrossRef CAS
- J. S. Owen, E. M. Chan, H. Liu and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 18206–18213 CrossRef CAS
- J. S. Steckel, B. K. H. Yen, D. C. Oertel and M. G. Bawendi, J. Am.
Chem. Soc., 2006, 128, 13032–13033 CrossRef CAS
- C. M. Evans, M. E. Evans and T. D. Krauss, J. Am. Chem. Soc., 2010, 132, 10973–10975 CrossRef CAS PubMed
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS
- R. García-Rodríguez and H. Liu, J. Am. Chem. Soc., 2014, 136, 1968–1975 CrossRef
- D. V. Talapin, S. Haubold, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, J. Phys. Chem. B, 2001, 105, 2260–2263 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05808a |
| This journal is © The Royal Society of Chemistry 2023 |