
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the 1T′/2H phases in WxMo1−xSe2 nanosheets†
Maria S.
Sokolikova
 a,
Gang
Cheng
a,
Mauro
Och
a,
Pawel
Palczynski
a,
Khalil
El Hajraoui
bc,
Quentin M.
Ramasse
bd and
Cecilia
Mattevi
*a
a,
Gang
Cheng
a,
Mauro
Och
a,
Pawel
Palczynski
a,
Khalil
El Hajraoui
bc,
Quentin M.
Ramasse
bd and
Cecilia
Mattevi
*a
aDepartment of Materials, Imperial College London, London SW7 2AZ, UK. E-mail: c.mattevi@imperial.ac.uk
bSuperSTEM Laboratory, SciTech Daresbury, Keckwick Lane, Daresbury WA4 4AD, UK
cYork NanoCentre & Department of Physics, University of York, York YO10 5DD, UK
dSchool of Physics and Astronomy & School of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
First published on 11th January 2023
Abstract
Controlling materials’ morphology, crystal phase and chemical composition at the atomic scale has become central in materials research. Wet chemistry approaches have great potential in directing the material crystallisation process to achieve tuneable chemical compositions as well as to target specific crystal phases. Herein, we report the compositional and crystal phase tuneability achieved in the quasi-binary WxMo1−xSe2 system with chemical and crystal phase mixing down to the atomic level. A series of WxMo1−xSe2 solid solutions in the form of nanoflowers with atomically thin petals were obtained via a direct colloidal reaction by systematically varying the ratios of transition metal precursors. We investigate the effect of selenium precursor on the morphology of the WxMo1−xSe2 material and show how using elemental selenium can enable the formation of larger and distinct nanoflowers. While the synthesised materials are compositionally homogeneous, they exhibit crystal phase heterogeneity with the co-existing domains of the 1T′ and 2H crystal phases, and with evidence of MoSe2 in the metastable 1T′ phase. We show at single atom level of resolution, that tungsten and molybdenum can be found in both the 1T′ and 2H lattices. The formation of heterophase 1T′/2H WxMo1−xSe2 electrocatalysts allowed for a considerable improvement in the activity for the acidic hydrogen evolution reaction (HER) compared to pristine, 1T′-dominated, WSe2. This work can pave the way towards engineered functional nanomaterials where properties, such as electronic and catalytic, have to be controlled at the atomic scale.
Introduction
Single atom catalysis as well as anisotropic transport at the atomic scale for neuromorphic devices to name a few, are amongst the new emerging materials science challenges where single atoms and atomic patterns can determine the overall materials properties. Layered transition metal dichalcogenides (TMDs), a class of functional 2D materials beyond graphene, exist in a variety of structural polymorphs with electronic properties ranging from semiconducting (2H)1,2 to metallic (1T)3 and semimetallic (1T′ and Td)2,4 for the same chemical composition. Studying the routes to achieve chemical composition and crystal phase engineering in these materials at the atomic scale can underpin the design of different properties which can be exploited in a broad range of nanoelectronic and electrochemical devices.The metastable polymorphs of TMDs are particularly interesting for application in the electrocatalytic hydrogen evolution reaction (HER)5,6 and electrochemical energy storage devices.7,8 In the case of the HER, the improved activity of the 1T′ phases of group VI TMDs is shown to originate from the activation of basal planes, which leads to the increased density of reaction sites.5,9 The activation mechanism is currently debated on, yet it is often linked to the intrinsic electron localisation around the transition metal zigzag chains10 and so to the inequivalence of the chalcogen sites that favours the proton adsorption on basal planes.11 For the applications in catalysis, increasing the density of active sites via phase engineering appears to be more promising than the alternative routes via the introduction of structural defects in the basal plane, such as pores12 or chalcogen vacancies,13 since the compromised structural integrity may have a negative effect on the durability of these catalyst materials. Additionally, in the heterophase TMD materials, it is found that the 1T′/2H boundaries outperform even pristine 1T′ materials, demonstrating faster HER kinetics and higher exchange currents. This is attributed to the effective accumulation of charges at the 1T′/2H homojunctions.14 Furthermore, the higher electrical conductivity of the semimetallic 1T′ phases compared to the semiconducting 2H counterpart aids reducing the resistive losses to achieve faster HER kinetics.9,15
Among numerous approaches to crystal phase engineering, tuning the chemical composition of TMD materials via heterovalent doping16 or alloying17–20 is actively explored as a route to induce the nucleation and stabilisation of the metastable phases in the host lattices. The heterovalent mixing often occurs in a limited range of compositions that are stable over segregation into the respective end-members.21–25 On the other hand, isovalent mixing in either cation or anion sublattice holds great potential for targeting wider ranges of chemical compositions.21,24 However, achieving the metastable crystal phases in such isovalent systems is challenging, and isovalent solid solutions‡ are generally reported in the thermodynamically stable phase.26–29 Heterophase ternary disulphides and diselenides reported in the literature are often produced by bottom-up vapour phase30,31 and liquid phase32 reactions from alkali metal or ammonium salts. In this case, the residual alkali metal or ammonium cations intercalated in the van der Waals gaps of TMD products help to stabilise the metastable 1T′ phase during the nucleation and growth stages.32,33 The limitation of such charge-mediated approaches is that the charge-stabilised metastable 1T′ phases may rapidly convert into the thermodynamically stable 2H phase once the charged species are removed.34,35
In the present work, we investigate how the direct formation of the metastable 1T′ crystal phase can be targeted in a quasi-binary MoSe2–WSe2 system via a bottom-up solution phase reaction. We confirm that WSe2 and MoSe2 are fully miscible and form a continuous series of WxMo1−xSe2 solid solutions in the entire range of compositions 0 ≤ x ≤ 1. Our data shows that, while being compositionally uniform, the WxMo1−xSe2 solid solutions display structural heterogeneity. Furthermore, we demonstrate via atomically resolved imaging that cation mixing effectively occurs in both these 1T′ and 2H crystal phase domains co-existing in the colloidal nanostructures. We observe a composition dependant 2H-to-1T′ transition in WxMo1−xSe2 with increasing x. Molybdenum can be stabilised in the host lattice of 1T′ WSe2 at concentrations of at least x = 0.5–1 with no evidence of segregation or cluster formation. We also provide an extended study of the repeatability of the proposed synthesis protocol and outline the material characteristics that need to be thoroughly investigated in order to gain unambiguous performance values when investigating the electrochemical, e.g. HER, activity of these complex WxMo1−xSe2 solid solutions. Our study suggests that the cation mixing and partial stabilisation of molybdenum in the 1T′ lattice of WSe2 helps to considerably improve the catalyst mass activity for the HER. This work highlights the possibility to control with atomistic precision the TMD solid solutions to enable tuneable properties, which are relevant in nanoelectronics and electrocatalysis.
Experimental section
Chemicals and stock solutions
Molybdenum hexacarbonyl (Mo(CO)6, ≥99.9% trace metals basis), tungsten hexacarbonyl (W(CO)6, 99.99% trace metals basis, excluding Mo), selenium (99.99% trace metals basis, 100 mesh), trioctylphosphine (TOP; 90%, technical grade), oleic acid (90%, technical grade), nitric acid (90%, ACS Reagent), and graphitic carbon foil (CF, 99.8%) were purchased from Sigma-Aldrich. Acetone (≥99%, technical grade), ethanol (96%, AnalaR NORMAPUR), and sulphuric acid (95%, AnalaR NORMAPUR) were purchased from VWR. Toray carbon paper (CP) was purchased from Alfa Aesar. Tungsten (Pure standard, 1000 μg mL−1 in H2O), molybdenum (Pure standard, 1000 μg mL−1 in H2O), and selenium (Pure standard, 1000 μg mL−1 in H2O, 2% HNO3) reference standards for ICP were purchased from PerkinElmer. All reagents were used without purification. Trioctylphosphine was stored in an Ar filled glovebox to prevent oxidation.1 M trioctylphosphine selenide (TOP:Se) stock solution was prepared by dissolving 5 mmol (395 mg) of Se powder in 5 mL of trioctylphosphine at room temperature under dry N2 atmosphere. 0.0125 M tungsten and molybdenum stock solutions were prepared by dissolving 0.125 mmol of the respective hexacarbonyl (either 33 mg of Mo(CO)6 or 44 mg of W(CO)6) in 10 mL of degassed oleic acid for 1 hour at 200 °C under vacuum. The ready stock solutions were stored in sealed glass vials in a drying desiccator.
Materials preparation
All syntheses were carried out in a standard Schlenk line. Ternary compositions in the WxMo1−xSe2 system were achieved by taking the respective transition metal precursors, either in the form of hexacarbonyl powders or stock solutions described above, in the desired molar ratio. In a typical synthesis, 0.05 mmol of transition metal precursor was dissolved in oleic acid and the total volume of reaction mixture was kept at 8 mL; all details are provided in ESI Table S1.† The reaction mixture was first degassed under vacuum (0.5 mbar) at room temperature for one hour, and then heated up to the injection temperature (300 °C) under a constant flow of dry N2 gas. The selenium precursor was quickly injected into the reaction mixture, and the material was allowed to form over three hours. After the reaction time elapsed, the final product was separated from the unreacted precursors by centrifugation at 5000 rpm for 10 minutes, washed twice with a mixture of acetone and ethanol, and then redispersed in ethanol for storage. To prepare working electrodes for electrochemical testing, strips of carbon paper (5 cm2) were placed into the reaction flask prior to degassing the transition metal precursor in oleic acid. After the synthesis was completed, the WxMo1−xSe2 nanosheets anchored on carbon paper were repeatedly washed with ethanol and then gently heated (50 °C) on a hot plate to completely dry the electrodes.Characterisation
Transmission electron microscopy (TEM) images, high-resolution TEM (HR TEM) images, annular dark-field scanning transmission electron microscopy (ADF STEM) images, and STEM energy-dispersive X-ray spectroscopy (STEM EDS) elemental maps were acquired using a JEOL JEM-2100F field emission S/TEM equipped with an Oxford X-MaxN 80 mm2 SDD detector. DigitalMicrograph GMS3 software (Gatan) was used for HR TEM images processing and analysis. AZtecTEM software (Oxford Instruments) was used to visualise the elemental maps and to perform the quantitative analysis. Atomically resolved high-angle annular dark-field STEM (HAADF STEM) imaging was carried on a probe corrected Nion UltraSTEM microscope at 60 kV accelerating voltage with the beam convergence semi-angle of 30 mrad. The statistical analysis on the HAADF STEM images was carried using the Atomap36 and the TEMUL Toolkit37 Python packages. The HAADF STEM image simulations were done with the Dr Probe software.38 Scanning electron microscopy (SEM) images and SEM EDS elemental maps were recorded using a Zeiss LEO Gemini 1525 field emission SEM equipped with an Oxford x-act PentaFET precision EDS detector. INCA software (Oxford Instruments) was used to process the SEM EDS data. X-ray photoelectron spectroscopy (XPS) measurements were carried out using a Thermo Fisher K-Alpha+ spectrometer (Al Kα source). XPS spectra were analysed using the Avantage software (Thermo Scientific). Powder X-ray diffraction (XRD) data was collected using a Bruker D2 phaser diffractometer (Cu Kα source). Raman spectra were acquired using a Renishaw inVia Qontor confocal Raman microscope (laser excitation 532 nm, 1800 lines per mm grating). Inductively coupled plasma emission spectroscopy (ICP AES) was carried out on a Thermo Scientific iCAP 6000 series spectrometer. X-ray fluorescence (XRF) analysis was performed using a Malvern Panalytical Epsilon 4 spectrometer.Electrochemical testing
The electrochemical testing was performed in a standard three-electrode cell configuration with a commercial Ag/AgCl (3 M KCl) as a reference electrode (BASi MF-2052) and carbon foil as a counter electrode using a Gamry Interface 1000 potentiostat. All measurements were carried out in an acidic electrolyte (1 M H2SO4 in Milli-Q water, pH 0). The geometric active area of typical working electrodes ranged from 0.5 to 1 cm2. The applied potentials were referenced to the reversible hydrogen electrode (RHE) using the following equation: E(RHE) = E(Ag/AgCl) + 0.21. The polarisation curves were recorded in the range from 0 to −0.8 V vs. reference electrode at a scan rate of 5 mV s−1. The potentiostatic EIS measurements were carried out at the electrolysis operation potential with an amplitude of 20 mV, while sweeping the frequency in the range from 1 MHz to 10 mHz.Results and discussion
Colloidal synthesis and morphology of WxMo1−xSe2 nanoflowers
Quasi-binary WxMo1−xSe2 solid solutions with variable cation composition spanning the entire range 0 ≤ x ≤ 1 were synthesised via the direct reaction between transition metal precursors (W(CO)6, Mo(CO)6) and a selenium precursor (trioctylphosphine selenide complex, TOP:Se) in hot oleic acid (Fig. 1a), as described in detail in the Experimental section and in ESI Note 1.† The synthesis approach implemented in this study is a modified protocol we established for the colloidal 1T′ WSe2 nanoflowers.39 Crucially, in contrast to the protocols reported previously,29,40 the nucleation of cation-mixed solid solutions in the liquid phase was initiated by a swift injection of the chalcogen precursor into a hot solution of mixed transition metal precursor. This allowed us to drive the crystallisation reaction via the kinetically controlled route to attain the metastable 1T′ crystal phase. | ||
| Fig. 1 (a) A schematic illustration of the one-pot colloidal synthesis of binary WxMo1−xSe2 solid solutions. (b and c) Low-magnification TEM images of the WxMo1−xSe2 solid solutions x = 0.5 grown from elemental selenium and TOP:Se complex, respectively; scale bar 100 nm. (d) EDS W Mα, Mo Lα, Se Lα, and C Kα elemental maps for the WxMo1−xSe2 (x = 0.5) sample; scale bar 200 nm. (e) A diagram, demonstrating the correlation between the experimentally measured x′ and the nominal fraction of tungsten x in WxMo1−xSe2 samples. P stands for powder precursors (transition metal TM; selenium Se); SS denotes the precursor stock solution. Open symbols represent the mean values x′ with their respective standard deviations. The solid grey line corresponds to the x = x′ and is inserted as a visual guide. (f) A diagram, showing the variability of chemical compositions x′ immanent in TM P + Se SS synthesis. Open squares represent the statistics mean x′ and the respective standard errors for each nominal composition x. Synthesis of each composition x was repeated 4 times. | ||
In general, WxMo1−xSe2 grown in the solution phase acquire the morphology of nanoflowers that are composed of atomically thin individual nanosheets (petals) protruding from a dense core region, as shown in Fig. 1b, c and ESI, Fig. S3 and S4.† The Mo-rich compositions (x = 0.25) display a greater tendency to form continuous agglomerates of nanosheets, similar to those found in pristine MoSe2, while more isolated nanoflowers are seen in the W-rich compositions x = 1, 0.75 and 0.50 (ESI, Fig. S3a–c and S4d–f†). The mean diameters of WxMo1−xSe2 (x = 0.5) nanostructures vary in the range 150–200 nm, which is close to that of pristine WSe2 (ESI, Fig. S3a and b†). Transmission electron microscopy (TEM) imaging of the curled up petals of WxMo1−xSe2 nanoflowers reveals a gradual thinning to single layers towards the rims (ESI, Fig. S3d–f†). Elemental mapping at the nanometre scale confirms the uniform distribution of all three elements throughout the WxMo1−xSe2 nanoflowers, displaying no segregated domains (Fig. 1d and ESI, Fig. S4†).
We have investigated the effect of selenium precursor on the morphology of WxMo1−xSe2 nanoflowers. For all nominal compositions x, changing the selenium precursor to elemental selenium led to the formation of uniform, well-defined nanoflowers with rigid petals (Fig. 1b), appearing as straight high-contrast edges in scanning transmission electron microscopy (STEM) images (ESI, Fig. S4a–c†). In contrast, the materials synthesised from TOP:Se complex predominantly demonstrate the morphology of agglomerated nanoflowers and wrinkled nanosheets (Fig. 1b and ESI, Fig. S4d–f†). This suggests that a more reactive chalcogen source leads to the formation of more uniform nanostructures, in good agreement with previous works.39,41 Additionally, we attribute the perceived higher reactivity of elemental selenium to the in situ formation of highly active H2Se at elevated temperatures in hydrocarbon solvents,42,43 in contrast to less reactive and thermally robust tertiary phosphine selenide complexes (ESI Note 2†).
The chemical composition of the synthesised WxMo1−xSe2 materials can be effectively varied by tuning the molar ratio of transition metal precursors in the reaction mixture (ESI, Fig. S5†). However, the ability to precisely control the resulting chemical composition of the nanomaterials synthesised in liquid phase is challenging. The crystallisation pathways of nanomaterials are strongly affected by the synthesis conditions, such as local fluctuations of precursors concentrations, the finite rate of mass transport, and uneven heat distribution.44 These effects lead to the nucleation of diverse, locally favoured, structures. This eventually causes the inherent heterogeneity, including compositional variation, within the same batch. On the other hand, the synthesis protocol proposed in this work as well as in the previous reports on pristine colloidal TMDs use only small amounts of starting precursors (typically, less than 0.1 mmol),41,45,46 which can lead to the unavoidable variation of the W![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo molar ratio from synthesis to synthesis for the same nominal x. In order to assess the variability within the same batch, we analysed the chemical composition of WxMo1−xSe2 nanoflowers with the nominal values x = 1 (WSe2), 0.75, 0.50, 0.25, and 0 (MoSe2) by energy dispersive X-ray spectroscopy (EDS), acquiring the signal at various spots across each sample. The final chemical composition of each sample was determined by averaging the data from 3 to 10 spots, and the result is summarised in the ESI, Table S2.†Fig. 1e presents the correlation between the measured composition x′ and the nominal x for the WxMo1−xSe2 samples. Within each sample batch, the chemical composition varies only slightly (up to 7%), and the mean x′ of the WxMo1−xSe2 materials grown from mixed hexacarbonyls (TM P) is very close to the nominal value x, regardless of whether TOP:Se complex (Se SS) or elemental selenium (Se P) were used as selenium source. To assess the repeatability of the proposed synthesis protocol from TM P and Se SS, each growth was repeated 4 times, and the mean values x′ as well as their standard errors are presented in Fig. 1f. Using solubilised forms of transition metal precursors (TM SS, Experimental section) allowed for achieving better repeatability of the colloidal growth procedure (ESI, Fig. S6†). However, the measured compositions x′ were found to deviate from the nominal values x by up to 40% (Fig. 1e and ESI, Fig. S6†). It is likely that such a shift towards considerable molybdenum excess (x′ < x) is caused by a partial loss of W(CO)6 during the stock solution preparation due to its less effective solubilisation47 and higher volatility48,49 compared to Mo(CO)6. Because of this lack of control over the tungsten stock solutions concentrations, mixtures of tungsten and molybdenum hexacarbonyl powders were used as a mixed transition metal source (TM P) throughout this work, and the chemical composition x′ of each ensemble of binary WxMo1−xSe2 was established prior to electrocatalytic activity measurements. This detailed analysis of the actual sample composition suggests that in colloidal synthesis, each growth parameter can significantly affect the overall material composition, and that assessing the repeatability of experimental protocols is essential to understand whether there is any factor influencing the overall chemical composition of the synthesised materials.
:Mo molar ratio from synthesis to synthesis for the same nominal x. In order to assess the variability within the same batch, we analysed the chemical composition of WxMo1−xSe2 nanoflowers with the nominal values x = 1 (WSe2), 0.75, 0.50, 0.25, and 0 (MoSe2) by energy dispersive X-ray spectroscopy (EDS), acquiring the signal at various spots across each sample. The final chemical composition of each sample was determined by averaging the data from 3 to 10 spots, and the result is summarised in the ESI, Table S2.†Fig. 1e presents the correlation between the measured composition x′ and the nominal x for the WxMo1−xSe2 samples. Within each sample batch, the chemical composition varies only slightly (up to 7%), and the mean x′ of the WxMo1−xSe2 materials grown from mixed hexacarbonyls (TM P) is very close to the nominal value x, regardless of whether TOP:Se complex (Se SS) or elemental selenium (Se P) were used as selenium source. To assess the repeatability of the proposed synthesis protocol from TM P and Se SS, each growth was repeated 4 times, and the mean values x′ as well as their standard errors are presented in Fig. 1f. Using solubilised forms of transition metal precursors (TM SS, Experimental section) allowed for achieving better repeatability of the colloidal growth procedure (ESI, Fig. S6†). However, the measured compositions x′ were found to deviate from the nominal values x by up to 40% (Fig. 1e and ESI, Fig. S6†). It is likely that such a shift towards considerable molybdenum excess (x′ < x) is caused by a partial loss of W(CO)6 during the stock solution preparation due to its less effective solubilisation47 and higher volatility48,49 compared to Mo(CO)6. Because of this lack of control over the tungsten stock solutions concentrations, mixtures of tungsten and molybdenum hexacarbonyl powders were used as a mixed transition metal source (TM P) throughout this work, and the chemical composition x′ of each ensemble of binary WxMo1−xSe2 was established prior to electrocatalytic activity measurements. This detailed analysis of the actual sample composition suggests that in colloidal synthesis, each growth parameter can significantly affect the overall material composition, and that assessing the repeatability of experimental protocols is essential to understand whether there is any factor influencing the overall chemical composition of the synthesised materials.
Microstructure of WxMo1−xSe2 nanoflowers
Under the proposed synthesis conditions, the parent WSe2 and MoSe2 compounds crystalise in dissimilar crystal phases. Specifically, pristine WSe2 predominantly acquires the metastable monoclinic 1T′ phase, whereas pristine MoSe2 is found in the thermodynamically stable hexagonal 2H phase. The reason for the formation of the metastable TMD polymorphs during direct growth in solution phase is two-fold. First, the predicted energy difference between the metastable 1T′ and the thermodynamically stable 2H polymorphs in their monolayer limit is as small as 0.27 and 0.33 eV per MX2 for WSe2 and MoSe2, respectively.50 Second, the energy barrier for the 1T′-to-1H conversion exceeds 1 eV per MX2 for all group 6 TMDs.51 This suggests that the metastable 1T′ polymorphs can be attained under favourable synthesis conditions, and a few successful examples have been reported previously.39,46,52 Under our reaction conditions, MoSe2 may initially nucleate in the 1T′ phase. However, the reaction temperature (300 °C) considerably exceeds that of the 1T′-to-2H conversion (125–127 °C),30,53 and it is likely that any 1T′ phase, possibly present at the very early stages of the nucleation, fully converts into the 2H phase during the subsequent growth. The 1T′ phase of nanostructured WSe2 is more thermally robust, and only converts into the 2H phase at temperatures exceeding 390 °C.39The representative high-resolution TEM (HR TEM) images of the individual nanosheets comprising pristine WSe2 and MoSe2 nanoflowers are shown in ESI, Fig. S3g and i.† Colloidal WSe2 displays good crystallinity with the single crystal domains extended over tens of nm2. The contrasting stripes correspond to the one-dimensional zigzag chains of tungsten atoms along the x direction that are characteristic to the 1T′ phases.54–56 The experimental lattice constants determined from the HR TEM data are a = 5.77 Å and b = 3.32 Å and match well those of the 1T′ WSe2.57,58 Pristine MoSe2 demonstrates the mosaic-like nature of individual petals (ESI, Fig. S3i†). The crystalline domains show the hexagonal arrangement of uniformly spaced atomic columns with the characteristic d-spacing of 2.63 Å, corresponding to the (100) planes of MoSe2 (2.84 Å).
TEM imaging of WxMo1−xSe2 nanoflowers reveals the presence of segregated nm2-sized domains of the coexisting 1T′ and 2H crystal phases in the compositionally uniform solid solutions (ESI, Fig. S7a†). Due to the rich three-dimensional morphology of colloidal WxMo1−xSe2 nanoflowers, only the very rims of individual petals are accessible for atomic-scale imaging. While these single- to few-layered regions of the parent WSe2 and MoSe2 exhibit exclusively the 1T′ and 2H phases, respectively; the solid solutions display an inherently more polycrystalline nature with the numerous misaligned domains as shown in the overview HR TEM images in ESI, Fig. S3h and S7a.† The coexisting 1T′ and 2H crystal phase domains are repeatedly found in WxMo1−xSe2 with x = 0.75 and x = 0.50, while x = 0.25 mainly presents the 2H phase domains (ESI, Fig. S7b and c†). The lattice constants a = 5.8 Å and b = 3.23 Å of the 1T′ phase of WxMo1−xSe2 (x = 0.50) are close to these of pristine 1T′ WSe2 (a = 5.76 Å, b = 3.3 Å)39 and 1T′ MoSe2 (a = 5.98 Å, b = 3.25 Å).59 The 2H phase regions of the same WxMo1−xSe2 (x = 0.50) nanoflowers show the hexagonal lattice with the d-spacing of 2.82 Å, matching the (100) planes of both 2H WSe2 and 2H MoSe2 (ESI, Fig. S7a†).
The composition-dependent crystal phase heterogeneity was further examined using X-ray photoemission spectroscopy (XPS). Fig. 2a shows the high-resolution spectra of the W 4f and Mo 3d core level electrons of colloidal WxMo1−xSe2 solid solutions. The experimental spectra were calibrated using the C 1s level of adventitious carbon (284.8 eV) and were normalised to the same height to better highlight the evolution of spectral line shape with the nominal x increasing. Except for pristine MoSe2 (x = 0), the observed prominent W4+ and Mo4+ peaks cannot be fitted with a single doublet, supporting the coexistence of two disparate crystal phases in binary WxMo1−xSe2 solid solutions. Substitutional doping in TMDs typically manifests in uniform changes in the binding energy, reflecting the changes in chemical environment of the host atoms caused by doping,60,61 whereas the peak splitting has been previously reported in similar heterophase TMD systems.17,22,34,62 In pristine MoSe2 (x = 0), the Mo4+ 3d doublet is found at 228.8 and 232.0 eV and is consistent with the values reported for the semiconducting 2H MoSe2.41,53 On the other hand, two W4+ 4f doublets are present in the spectrum of pristine WSe2 (x = 1). The most prominent doublet at 31.9 and 34.0 eV is assigned to the semimetallic 1T′ crystal phase of WSe2, while the low intensity doublet found at 32.4 and 34.6 eV corresponds to the semiconducting 2H phase component.30,39 When forming a tungsten-rich WxMo1−xSe2 solid solution (x = 0.75), the W 4f lines remain largely unchanged and are resolved into two doublets with the energies close to those of the 1T′ and 2H phases of WSe2 seen in pristine WSe2 samples (Fig. 2a). The Mo 3d peaks, however, are significantly red shifted from the reference positions in pristine MoSe2, and the experimental spectrum is deconvoluted using two Mo4+ 3d doublets (Fig. 2a). The lower binding energy doublet at 228.3 and 231.4 eV, indicating the more metallic character of the Mo–Se bonding, matches well the 1T′ phase of MoSe2.53 The higher energy doublet at 228.9 and 232.1 eV originates from the 2H phase MoSe2. To our knowledge, this is the first time the metastable 1T′ phase of MoSe2 has been detected in the isovalently substituted material obtained by a direct solution-phase reaction. The previous reports concerned phase pure 1T′ MoSe2 single crystals synthesised from alkali cation containing intermediates53 and by salt-assisted conversion of the 2H crystals.31 In atomically thin nanosheets, similar stabilisation of molybdenum atoms in the 1T′ lattice has been observed in heterovalent RexMo1−xSe2.63 The splitting of the W 4f and the Mo 3d core levels into two sets of doublets is observed throughout the entire range of compositions x, and the peak positions remain nearly constant (Fig. 2a). As the fraction of tungsten x in WxMo1−xSe2 solid solutions decreases, the intensities of the 1T′ bands of both WSe2 and MoSe2 decrease, while the respective 2H bands become more dominant. This is summarised in the bar chart shown in Fig. 2b. The described splitting of the W 4f and the Mo 3d core levels suggests the presence of 2H MoSe2, 1T′ MoSe2, 2H WSe2 and 1T′ WSe2 in WxMo1−xSe2 solid solutions. The cation mixing occurs in both crystal phases, which is corroborated by the results of atomically resolved HAADF STEM imaging discussed later. This allows for tuning the chemical environment of both cations, effectively stabilising molybdenum in the lattice of 1T′ WSe2, as well as tungsten in the lattice of 2H MoSe2. Moreover, the dominating crystal phase of the heterophase WxMo1−xSe2 solid solutions appears to be dependent on their chemical composition x (Fig. 2b). This can enable tailoring of the electronic properties of WxMo1−xSe2, targeting the functional materials that are either more metallic (predominantly 1T′, x = 0.75) or alternatively more semiconducting (predominantly 2H, x = 0.25) in nature.
 | ||
| Fig. 2 (a) High-resolution XPS spectra of the W 4f and Mo 3d core level electrons of WxMo1−xSe2 nanoflowers. Open circles show the raw data, solid red lines are the fitting results. (b) A bar chart illustrating the progressive change from the 1T′-dominated to the 2H-dominated character of the WxMo1−xSe2 nanoflowers with the fraction of tungsten x decreasing. The fractions of the 1T′ and 2H crystal phase, as well as the fraction of oxidised material, were determined from the respective XPS components. | ||
The gradual change of the dominating crystal phase from the 2H to the 1T′ in the WxMo1−xSe2 solid solutions as the tungsten content x increases, is also corroborated by the results of powder X-ray diffraction (XRD) and Raman spectroscopy presented in ESI, Fig. S8.† Raman spectroscopy data confirms the coexistence of two distinct crystal phases in the quasi-binary WxMo1−xSe2 solid solutions. The most prominent vibration mode observed in the range 238.5–246.5 cm−1 for all intermediate compositions x suggests the presence of the 2H phase-based mixed-cation component (ESI, Fig. S8b†). This Raman signature progressively shifts from the characteristic A1g band of pristine 2H MoSe2 (236.6 cm−1) towards the unresolved A1g + E2g1 mode of 2H WSe2 (∼250 cm−1) as the x increases. Due to the similarity of MoSe2 and WSe2 crystal lattices, the Raman active modes in mixed-cation material are expected at intermediate frequencies compared to the parent phases. This is due to the random cation mixing in the transition metal sublattice, and similar shifts have been reported for micromechanically exfoliated 2H WxMo1−xSe2 monolayers27 and for colloidal 2H WxMo1−xSe2 nanostructures.29 The characteristic Raman modes of semimetallic 1T′ phases typically exhibit a very low intensity and are not easily resolved in the presence of the semiconducting 2H phase. The presence of mixed-cation 1T′ crystal phase can be inferred from two modes centred at ∼105 and 218 cm−1. Following the early reports on chemically exfoliated 1T′ MoS2 single layers,64 these modes are often labelled J1 and J3 and are reported to appear at ∼105 and 218 cm−1 in 1T′ WSe230,39 and in the range 108–114 and 198–228 cm−1 in 1T′ MoSe2,65,66 respectively. Since these J1 and J3 modes do not overlap with the strong vibration modes E1g and A1g of the 2H phase of either WSe2 or MoSe2, we selected them to track the presence of mixed-cation 1T′ component. Indeed, these peaks are the most prominent in W-rich WxMo1−xSe2 (x = 0.75 and 0.50) as shown in ESI, Fig. S8b.† This is in good agreement with the results of TEM and XPS studies that revealed the 1T′ phase being dominant in these compositions.
High resolution STEM imaging was employed to examine the cation distribution and the atomic structure of the synthesised WxMo1−xSe2 colloidal solid solutions in two representative x = 0.75 and x = 0.50 compositions. In order to provide atomic-scale insights into the microstructure of the two solid solutions, we focused on imaging the thinnest sample regions in the nanoflowers rims where monolayer domains can be observed (Fig. 3a and ESI, Fig. S10†). High angle annular dark field (HAADF) STEM images confirm the TEM observations whereby both the 1T′and 2H crystal phase domains are seen to coexist in the x = 0.75 and x = 0.50 solid solutions (Fig. 3a and ESI, Fig. S10†). This initial qualitative analysis was performed by a visual comparison of the experimental micrographs to the simulated HAADF signal intensity motifs of both MoSe2 and WSe2, in 1T′ and 2H crystal phases, as monolayers and bilayers (ESI, Fig. S9;† see Experimental section for details). 1T′/2H interfaces stabilised by vacancy lines are observed in the x = 0.75 solid solution and illustrate the stability of 1T′/2H heterophase monolayers (ESI, Fig. S10a and S11†). Within the intrinsically limited extent of atomic-scale STEM observations, nanoscale domains exhibiting the 1T′ crystal structure were observed more frequently in the x = 0.75 than x = 0.50 solid solutions, and predominantly in bilayers or multilayer domains (Fig. 3a), whereas the 2H domains were ubiquitously imaged in both cases, including as monolayers on the very edges of the nanoflower petals (ESI, Fig. S10a and c†). A more quantitative, statistical atom-by-atom examination of the observed HAADF intensities was carried out to provide further insights into the cation intermixing.67,68 Specifically, the 2H monolayer domains of both the x = 0.75 and x = 0.50 solid solutions were selected for the analysis (Fig. 3b and d), while stepped regions and areas exhibiting adventitious surface contamination were excluded. Three distinct types of atomic columns can unambiguously be recognised as 1Mo, 1W and 2Se, confirming cation intermixing at the atomic scale within single 2H domains of the x = 0.75 and x = 0.50 solid solutions. The assignment of the observed atomic columns to one of these types was performed based their relative intensity. While it would be difficult to provide overall statistics, reflecting the macroscopically measured compositions, due to the limited fields of view, the frequencies with which 1Mo and 1W columns are identified in images of the 2H domains in x = 0.75 and x = 0.50 samples are negatively correlated, that suggests a good agreement with the larger scale composition measurements (Fig. 3c and e). A similar intensity analysis on the 1T′ domains is complicated by the fact that multilayer or very restraint monolayer regions were observed in both the x = 0.75 and x = 0.50 WxMo1−xSe2 samples. However, the contrast variation along the metal chains might suggest that cation mixing effectively occurs in the 1T′ phase domains as well, as highlighted in the ESI, Fig. S10b and d.†
 | ||
| Fig. 3 (a) Atomically resolved HAADF STEM image of W0.75Mo0.25Se2 sample illustrating the co-existing 1T′ (purple box) and 2H (teal box) crystal phase domains (scale bar 5 nm). The respective fast Fourier transform (FFT) patterns of the highlighted areas are presented in the panels below. (b and d) Representative HAADF STEM images of the 2H phase monolayers in W0.75Mo0.25Se2 and W0.50Mo0.50Se2 samples (scale bar 1 nm). (c and e) Histograms showing the fractional HAADF intensities at the atomic positions in (b) and (d), respectively. | ||
Electrocatalytic HER activity of heterophase WxMo1−xSe2
Chemical composition engineering via substitutional doping and precise targeting of the metastable 1T′ phases in TMDs are being studied for application in electrocatalytic hydrogen generation.17,69,70 To assess the catalytic activity of quasi-binary WxMo1−xSe2 solid solutions, we prepared a set of working electrodes by growing the active material directly on carbon paper (CP). Thus, grown binary WxMo1−xSe2 forms dense arrays of atomically thin nanosheets, uniformly coating carbon fibres over a few cm2 areas of the substrate as illustrated in ESI, Fig. S12.† EDS elemental mapping shows the signal of W, Mo, and Se matching that of carbon, thus further confirming the uniform coverage of carbon fibres with active material (ESI, Fig. S12†).We found that inducing heterogeneous nucleation of binary WxMo1−xSe2 on CP did not lead to changes in their chemical composition. The composition x′ of WxMo1−xSe2 grown directly on CP is very close to that of the respective free-standing nanoflowers formed as a side-product in the solution phase (ESI, Fig. S13b and Table S3†). Raman spectroscopy confirms the presence of the 1T′ crystal phase in WxMo1−xSe2 formed on CP (ESI, Fig. S13a†). The fraction of the semimetallic 1T′ phase in WxMo1−xSe2 catalysts was estimated from the XPS data for each working electrode (ESI, Table S3†). To determine the mass loading of active material for each working electrode, binary WxMo1−xSe2 were dissolved off the carbon supports in concentrated nitric acid as described in ESI Note 3,† and the respective concentrations of tungsten, molybdenum and selenium measured using inductively coupled plasma atomic emission spectroscopy (ICP AES) were used to calculate the catalyst mass loading (in μg cm−2). This is summarised in the ESI, Table S4.†
The electrocatalytic activity of binary WxMo1−xSe2 solid solutions towards the HER was investigated in acidic electrolyte (1 M H2SO4, pH 0) as described in the Experimental section. The corresponding polarisation curves recorded in the range from 0 to −0.8 V vs. reference electrode are shown in Fig. 4a. The overpotential η required to reach the benchmarking catalytic current density of −10 mA cm−2 (η−10) depends on the chemical composition of our binary WxMo1−xSe2. Partial substitution of W with Mo results in the reaction overpotential η−10 lowering by 136 mV from 556 mV for x = 1 (pristine WSe2) to 420 mV for x = 0.50. Pristine semiconducting MoSe2 (x = 0) reaches the benchmarking current density at considerably lower overpotential (290 mV), which agrees well with the theoretical works predicting MoSe2 to be the most active catalyst for the HER among the other group 6 TMDs.71 It should be noted that the reaction overpotentials derived from geometric current densities strongly depend on the mass loading of active material per working electrode.72 Since the working electrodes were prepared by growing active material directly on the conductive supports, and so the catalyst mass loading was expected to vary, we present the mass normalised catalytic current densities to better highlight the effect of chemical composition x on the catalytic performance of binary WxMo1−xSe2 catalysts (Fig. 4d). Thus, normalised polarisation curves confirm the significant improvement of the catalytic activity of binary WxMo1−xSe2 compared to pristine WSe2 counterpart (Fig. 4d). In addition to the conventional metric η−10, we also report the mass activity of binary WxMo1−xSe2 as the overpotential η−20, at which the catalytic current density of −20 mA mg−1 is generated. The respective activity parameters are summarised in the ESI, Table S4.† Graphically, these catalytic performance metrics are correlated to the working electrodes characteristics in Fig. 4e. The Tafel plots derived from the polarisation curves are displayed in Fig. 4b. In the low overpotential region, the Tafel slopes b of binary WxMo1−xSe2 are close to that of pristine WSe2 (152 mV dec−1 for x = 1), whereas pristine MoSe2 (x = 0) exhibits faster reaction kinetics with the Tafel slope b of 137 mV dec−1. Additionally, the electrode kinetics under the HER operating conditions was investigated using electrochemical impedance spectroscopy (Fig. 4c). The charge transfer resistance in binary WxMo1−xSe2 catalysts (x = 0.75: 12 Ω; x = 0.50: 19 Ω; x = 0.25: 11 Ω) is noticeably lower than in pristine semimetallic 1T′ WSe2 (x = 1: 42 Ω) and semiconducting 2H MoSe2 (x = 0: 24 Ω). Lower charge transfer resistance is often linked to the higher electrical conductivity of the 1T/1T′ phases5,73 and has been previously reported for the doped and alloyed TMD catalysts with the enhanced catalytic activity.17,63,69
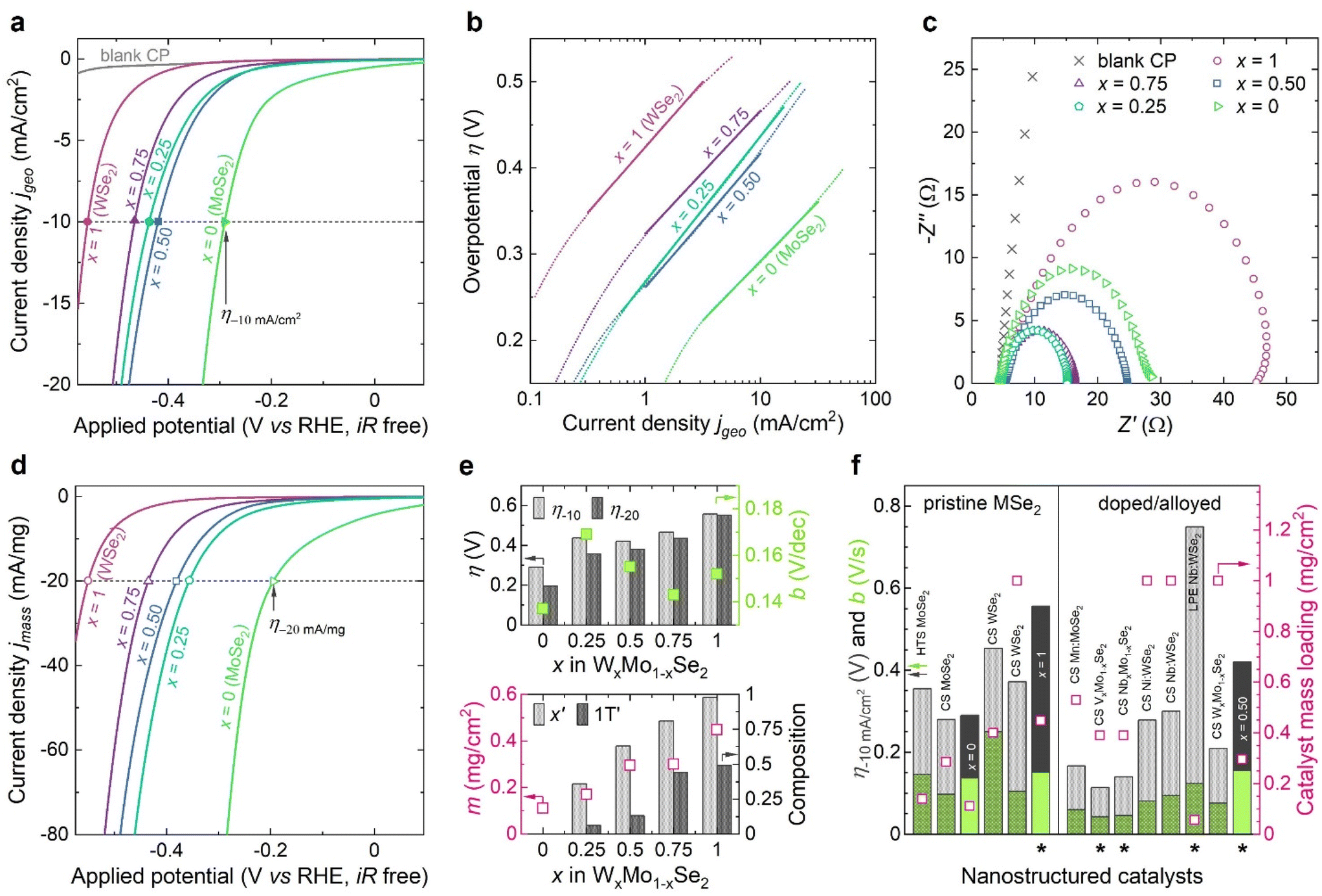 | ||
| Fig. 4 (a) Polarisation curves (scan rate 5 mV s−1) of the WxMo1−xSe2 nanosheets grown directly on CP for the HER in 1 M H2SO4 electrolyte. Filled scatters denote the overpotential η values required to achieve the benchmarking current density of −10 mA cm−2. (b) Tafel plots derived from the iR drop compensated polarisation curves in (a). (c) The impedance spectra of the WxMo1−xSe2 working electrodes. (d) Polarisation curves (scan rate 5 mV s−1) of the WxMo1−xSe2 working electrodes normalised by the active material mass loading. Open scatters denote the overpotential η values required to achieve the current density of −20 mA mg−1. (e) A comprehensive summary of the overall performance of binary WxMo1−xSe2 catalysts for the HER (top panel) linked to their chemical and crystal phase composition as well as mass loading (bottom panel). Top panel: overpotentials η (left y-axis, columns, light-grey −10 mA cm−2 and dark-grey −20 mA mg−1) and Tafel slopes b (right y-axis, filled squares from jgeo and open squares from jmass); x is the nominal fraction of tungsten in WxMo1−xSe2. Bottom panel: mass loading m (left y-axis, open squares, ICP AES); measured fraction of tungsten x′ (right y-axis, light-grey columns, EDS) and the fraction of the semimetallic 1T′ crystal phase (right y-axis, dark-grey columns, XPS); x is the nominal fraction of tungsten in WxMo1−xSe2. (f) Overpotentials η (left y-axis, grey columns) and Tafel slopes b (left y-axis, green columns) of the x = 0, 0.5, and 1 WxMo1−xSe2 samples compared to the nanostructured pristine and metal-doped MoSe2 and WSe2 catalysts reported in literature (ESI, Table S5†). The respective catalyst mass loadings are presented in open squares (right y-axis). HTS – hydrothermal synthesis, CS – colloidal synthesis, LPE – liquid phase exfoliation. Asterisks indicate the mixed 1T(1T′)/2H systems. | ||
Overall, we demonstrate that partial substitution with molybdenum at the tungsten sites of WSe2 and the formation of 1T′/2H heterophase WxMo1−xSe2 catalysts allow for improving the HER performance. The binary compositions reach the benchmarking catalytic current densities of −10 mA cm−2 and −20 mA mg−1 at significantly lower overpotentials compared to pristine WSe2 (Fig. 4e). While the reaction rate, as is evidenced by Tafel analysis, is nearly unchanged (Fig. 4e). Although the 2H MoSe2 demonstrates higher catalytic activity for the HER with the lowest reaction overpotential and Tafel slope, the experimental reports on the mixed-phase TMD catalysts showed that pristine MoSe2 was also the material most prone to oxidation among group 6 TMDs.74,75 Forming cation-mixed TMDs might help to mitigate this issue, while retaining high catalytic activity.
We compare the performance of our binary catalysts to that of both pristine and doped WSe2 and MoSe2 nanostructured materials reported in the literature (Fig. 4f and ESI, Table S5†). Our pristine WSe2 (x = 1) and MoSe2 (x = 0) are characterised by overpotentials η−10 and Tafel slopes b close to those of reported WSe2 and MoSe2 synthesised by colloidal and hydrothermal reactions. The x = 0.50, selected here as a representative binary solid solution, outperforms the exfoliated Nb-doped WSe2.76 Although the reported Ni- and Nb-doped colloidal WSe277 and 2H WxMo1−xSe240 express lower overpotentials, it should be taken into account that the active material mass loading in these studies is nearly 3 times higher than in our work, so the direct comparison must be done with caution.
Conclusions
In summary, we have synthesised WxMo1−xSe2 solid solutions with the optimized morphology of nanoflowers and chemical compositions tuneable in the entire range of x (0 ≤ x ≤ 1). The constituent chemical elements are distributed uniformly throughout individual nanoflowers while these cation-mixed WxMo1−xSe2 solid solutions present structural heterogeneity with the 1T′ and 2H crystal phases co-existing within the same nanostructure, and with evidence of MoSe2 in the metastable 1T′ phase. We demonstrate that the cation mixing effectively occurs in both crystal phases, and that the crystal phase composition of WxMo1−xSe2 solid solutions depends on their chemical composition x. Within the tungsten-rich compositions x = 0.75 and x = 0.50, molybdenum is effectively stabilised within the lattice of 1T′ WSe2, demonstrating an improved catalytic activity for the HER compared to the pristine 1T′ phase-dominated WSe2. This work can pave the way to engineer new solid solutions in a precise manner with atomistic control on lattice symmetry to tune properties such as electrical transport and catalysis at the nanoscale.Author contributions
M. S. S. and C. M. conceived the study, and M. S. S. designed the experiments. M. S. S. and G. C. prepared samples. M. S. S. performed SEM, TEM, EDS, XRD, ICP AES measurements and electrochemical testing, collated and analysed the data. G. C. performed XRF measurements and data analysis. G. C., M. O. and P. P. performed Raman measurements and data analysis. M. O. and P. P. performed XPS measurements and data analysis. K. E. H. and Q. M. R. performed HAADF STEM imaging and data analysis. C. M. supervised and guided the study. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the use of the characterization facilities within the Harvey Flower Suite for Electron Microscopy, the X-Ray Diffraction Suite, and the Advanced Photoelectron Spectroscopy Laboratory of the Department of Materials, Imperial College London. M. S. S. would like to acknowledge the funding through the President's PhD scholarship scheme at Imperial College London. C. G. acknowledges the China Scholarship Council for the PhD scholarship (grant #201808060492). C. M. would like to acknowledge the award of funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 819069) and the award of a Royal Society University Research Fellowship (UF160539) and the Research Fellow Enhancement Award 2017 (RGF\EA\180090) by the Royal Society UK.References
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed.
- D. Rhodes, D. A. Chenet, B. E. Janicek, C. Nyby, Y. Lin, W. Jin, D. Edelberg, E. Mannebach, N. Finney, A. Antony, T. Schiros, T. Klarr, A. Mazzoni, M. Chin, Y.-c. Chiu, W. Zheng, Q. R. Zhang, F. Ernst, J. I. Dadap, X. Tong, J. Ma, R. Lou, S. Wang, T. Qian, H. Ding, R. M. Osgood, D. W. Paley, A. M. Lindenberg, P. Y. Huang, A. N. Pasupathy, M. Dubey, J. Hone and L. Balicas, Nano Lett., 2017, 17, 1616–1622 CrossRef CAS PubMed.
- M. Kan, J. Y. Wang, X. W. Li, S. H. Zhang, Y. W. Li, Y. Kawazoe, Q. Sun and P. Jena, J. Phys. Chem. C, 2014, 118, 1515–1522 CrossRef CAS.
- H. Xu, D. Han, Y. Bao, F. Cheng, Z. Ding, S. J. R. Tan and K. P. Loh, Nano Lett., 2018, 18, 5085–5090 CrossRef CAS PubMed.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- X. Wang, W. Ding, H. Li, H. Li, S. Zhu, X. Zhu, J. Dai, Z. Sheng, H. Wang, X. Zhu, Y. Sun and S. X. Dou, J. Mater. Chem. A, 2019, 7, 19152–19160 RSC.
- S. S. Chou, N. Sai, P. Lu, E. N. Coker, S. Liu, K. Artyushkova, T. S. Luk, B. Kaehr and C. J. Brinker, Nat. Commun., 2015, 6, 8311 CrossRef CAS PubMed.
- G. Lin, Q. Ju, X. Guo, W. Zhao, S. Adimi, J. Ye, Q. Bi, J. Wang, M. Yang and F. Huang, Adv. Mater., 2021, 33, 2007509 CrossRef CAS PubMed.
- X.-L. Fan, Y. Yang, P. Xiao and W.-M. Lau, J. Mater. Chem. A, 2014, 2, 20545–20551 RSC.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed.
- Y. Lee, N. Ling, D. Kim, M. Zhao, Y. A. Eshete, E. Kim, S. Cho and H. Yang, Adv. Funct. Mater., 2022, 32, 2105675 CrossRef CAS.
- G. Gao, Y. Jiao, F. Ma, Y. Jiao, E. Waclawik and A. Du, J. Phys. Chem. C, 2015, 119, 13124–13128 CrossRef CAS.
- Z. Luo, Y. Ouyang, H. Zhang, M. Xiao, J. Ge, Z. Jiang, J. Wang, D. Tang, X. Cao, C. Liu and W. Xing, Nat. Commun., 2018, 9, 2120 CrossRef PubMed.
- I. S. Kwon, I. H. Kwak, T. T. Debela, J. Y. Kim, S. J. Yoo, J.-G. Kim, J. Park and H. S. Kang, ACS Nano, 2021, 15, 14672–14682 CrossRef CAS PubMed.
- Q. Deng, X. Li, H. Si, J. Hong, S. Wang, Q. Feng, C. Hu, S. Wang, H. Zhang, K. Suenaga and H. Xu, Adv. Funct. Mater., 2020, 30, 2003264 CrossRef CAS.
- S.-Z. Yang, Y. Gong, P. Manchanda, Y.-Y. Zhang, G. Ye, S. Chen, L. Song, S. T. Pantelides, P. M. Ajayan, M. F. Chisholm and W. Zhou, Adv. Mater., 2018, 30, 1803477 CrossRef PubMed.
- J. A. Benda, Phys. Rev. B: Solid State, 1974, 10, 1409–1420 CrossRef CAS.
- Z. Hemmat, J. Cavin, A. Ahmadiparidari, A. Ruckel, S. Rastegar, S. N. Misal, L. Majidi, K. Kumar, S. Wang, J. Guo, R. Dawood, F. Lagunas, P. Parajuli, A. T. Ngo, L. A. Curtiss, S. B. Cho, J. Cabana, R. F. Klie, R. Mishra and A. Salehi-Khojin, Adv. Mater., 2020, 32, 1907041 CrossRef CAS PubMed.
- L. Zhang, T. Yang, X. He, W. Zhang, G. Vinai, C. S. Tang, X. Yin, P. Torelli, Y. P. Feng, P. K. J. Wong and A. T. S. Wee, ACS Nano, 2020, 14, 11140–11149 CrossRef CAS PubMed.
- A. Apte, A. Krishnamoorthy, J. A. Hachtel, S. Susarla, J. Yoon, L. M. Sassi, P. Bharadwaj, J. M. Tour, J. C. Idrobo, R. K. Kalia, A. Nakano, P. Vashishta, C. S. Tiwary and P. M. Ajayan, Nano Lett., 2019, 19, 6338–6345 CrossRef CAS PubMed.
- J.-H. Yang and B. I. Yakobson, Chem. Mater., 2018, 30, 1547–1555 CrossRef CAS.
- S. A. Dalmatova, A. D. Fedorenko, L. N. Mazalov, I. P. Asanov, A. Yu. Ledneva, M. S. Tarasenko, A. N. Enyashin, V. I. Zaikovskii and V. E. Fedorov, Nanoscale, 2018, 10(21), 10232–10240 RSC.
- Z. Wang, P. Liu, Y. Ito, S. Ning, Y. Tan, T. Fujita, A. Hirata and M. Chen, Sci. Rep., 2016, 6, 21536 CrossRef CAS PubMed.
- M. Zhang, J. Wu, Y. Zhu, D. O. Dumcenco, J. Hong, N. Mao, S. Deng, Y. Chen, Y. Yang, C. Jin, S. H. Chaki, Y.-S. Huang, J. Zhang and L. Xie, ACS Nano, 2014, 8, 7130–7137 CrossRef CAS PubMed.
- S. Tongay, D. S. Narang, J. Kang, W. Fan, C. Ko, A. v. Luce, K. X. Wang, J. Suh, K. D. Patel, V. M. Pathak, J. Li and J. Wu, Appl. Phys. Lett., 2014, 104, 012101 CrossRef.
- Y. Sun, K. Fujisawa, Z. Lin, Y. Lei, J. S. Mondschein, M. Terrones and R. E. Schaak, J. Am. Chem. Soc., 2017, 139, 11096–11105 CrossRef CAS PubMed.
- Z. Lai, Q. He, T. H. Tran, D. V. M. Repaka, D.-D. Zhou, Y. Sun, S. Xi, Y. Li, A. Chaturvedi, C. Tan, B. Chen, G.-H. Nam, B. Li, C. Ling, W. Zhai, Z. Shi, D. Hu, V. Sharma, Z. Hu, Y. Chen, Z. Zhang, Y. Yu, X. R. Wang, R. v. Ramanujan, Y. Ma, K. Hippalgaonkar and H. Zhang, Nat. Mater., 2021, 20(8), 1113–1120 CrossRef CAS PubMed.
- Z. Lai, Y. Yao, S. Li, L. Ma, Q. Zhang, Y. Ge, W. Zhai, B. Chi, B. Chen, L. Li, L. Wang, Z. Zheng, L. Gu, Y. Du and H. Zhang, Adv. Mater., 2022, 34, 2201194 CrossRef CAS PubMed.
- K. Yang, X. Wang, H. Li, B. Chen, X. Zhang, S. Li, N. Wang, H. Zhang, X. Huang and W. Huang, Nanoscale, 2017, 9, 5102–5109 RSC.
- Q. Liu, X. Li, Z. Xiao, Y. Zhou, H. Chen, A. Khalil, T. Xiang, J. Xu, W. Chu, X. Wu, J. Yang, C. Wang, Y. Xiong, C. Jin, P. M. Ajayan and L. Song, Adv. Mater., 2015, 27, 4837–4844 CrossRef CAS PubMed.
- S. J. R. Tan, I. Abdelwahab, Z. Ding, X. Zhao, T. Yang, G. Z. J. Loke, H. Lin, I. Verzhbitskiy, S. M. Poh, H. Xu, C. T. Nai, W. Zhou, G. Eda, B. Jia and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 2504–2511 CrossRef CAS PubMed.
- X. Zhu, D. Li, X. Liang and W. D. Lu, Nat. Mater., 2019, 18, 141–148 CrossRef CAS PubMed.
- M. Nord, P. E. Vullum, I. MacLaren, T. Tybell and R. Holmestad, Adv. Struct. Chem. Imaging, 2017, 3, 9 CrossRef PubMed.
- E. N. O'Connell, K. Moore, E. McFall, M. Hennessy, E. Moynihan, U. Bangert and M. Conroy, Microsc. Microanal., 2022, 28, 1444–1452 CrossRef PubMed.
- J. Barthel, Ultramicroscopy, 2018, 193, 1–11 CrossRef CAS PubMed.
- M. S. Sokolikova, P. C. Sherrell, P. Palczynski, V. L. Bemmer and C. Mattevi, Nat. Commun., 2019, 10, 712 CrossRef CAS PubMed.
- O. E. Meiron, V. Kuraganti, I. Hod, R. Bar-Ziv and M. Bar-Sadan, Nanoscale, 2017, 9, 13998–14005 RSC.
- D. Sun, S. Feng, M. Terrones and R. E. Schaak, Chem. Mater., 2015, 27, 3167–3175 CrossRef CAS.
- Z. Deng, L. Cao, F. Tang and B. Zou, J. Phys. Chem. B, 2005, 109, 16671–16675 CrossRef CAS PubMed.
- G. G. Yordanov, H. Yoshimura and C. D. Dushkin, Colloid Polym. Sci., 2008, 286, 813–817 CrossRef CAS.
- Q. Chen, Nano Lett., 2022, 22, 3–5 CrossRef CAS PubMed.
- W. Jung, S. Lee, D. Yoo, S. Jeong, P. Miró, A. Kuc, T. Heine and J. Cheon, J. Am. Chem. Soc., 2015, 137, 7266–7269 CrossRef CAS PubMed.
- Y. Sun, Y. Wang, D. Sun, B. R. Carvalho, C. G. Read, C. Lee, Z. Lin, K. Fujisawa, J. A. Robinson, V. H. Crespi, M. Terrones and R. E. Schaak, Angew. Chem., Int. Ed., 2016, 55, 2830–2834 CrossRef CAS PubMed.
- J. Li, G. Schreckenbach and T. Ziegler, J. Phys. Chem., 1994, 98, 4838–4841 CrossRef CAS.
- A. A. Blanchard, Chem. Rev., 1937, 21, 3–38 CrossRef CAS.
- M. J. S. Monte, A. R. R. P. Almeida and R. Notario, J. Therm. Anal. Calorim., 2018, 132, 1201–1211 CrossRef CAS.
- K.-A. N. Duerloo, Y. Li and E. J. Reed, Nat. Commun., 2014, 5, 1–9 CrossRef PubMed.
- X. Qian, J. Liu, L. Fu and J. Li, Science, 2014, 346, 1344–1347 CrossRef CAS PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- Y. Yu, G. Nam, Q. He, X. Wu, K. Zhang, Z. Yang, J. Chen, Q. Ma, M. Zhao, Z. Liu, F.-R. Ran, X. Wang, H. Li, X. Huang, B. Li, Q. Xiong, Q. Zhang, Z. Liu, L. Gu, Y. Du, W. Huang and H. Zhang, Nat. Chem., 2018, 10, 638–643 CrossRef CAS PubMed.
- S. W. Hla, V. Marinković, A. Prodan and I. Muševič, Surf. Sci., 1996, 352–354, 105–111 CrossRef CAS.
- J. Heising and M. G. Kanatzidis, J. Am. Chem. Soc., 1999, 121, 638–643 CrossRef CAS.
- X. R. Qin, D. Yang, R. F. Frindt and J. C. Irwin, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3490–3493 CrossRef PubMed.
- M. M. Ugeda, A. Pulkin, S. Tang, H. Ryu, Q. Wu, Y. Zhang, D. Wong, Z. Pedramrazi, A. Martín-Recio, Y. Chen, F. Wang, Z.-X. Shen, S.-K. Mo, O. v. Yazyev and M. F. Crommie, Nat. Commun., 2018, 9, 3401 CrossRef PubMed.
- W. Chen, X. Xie, J. Zong, T. Chen, D. Lin, F. Yu, S. Jin, L. Zhou, J. Zou, J. Sun, X. Xi and Y. Zhang, Sci. Rep., 2019, 9, 2685 CrossRef PubMed.
- F. Cheng, Z. Hu, H. Xu, Y. Shao, J. Su, Z. Chen, W. Ji and K. P. Loh, ACS Nano, 2019, 13, 2316–2323 CAS.
- W. Li, J. Huang, B. Han, C. Xie, X. Huang, K. Tian, Y. Zeng, Z. Zhao, P. Gao, Y. Zhang, T. Yang, Z. Zhang, S. Sun and Y. Hou, Adv. Sci., 2020, 7, 2001080 CrossRef CAS PubMed.
- J. Gao, Y. D. Kim, L. Liang, J. C. Idrobo, P. Chow, J. Tan, B. Li, L. Li, B. G. Sumpter, T.-M. Lu, V. Meunier, J. Hone and N. Koratkar, Adv. Mater., 2016, 28, 9735–9743 CrossRef CAS PubMed.
- V. Kochat, A. Apte, J. A. Hachtel, H. Kumazoe, A. Krishnamoorthy, S. Susarla, J. C. Idrobo, F. Shimojo, P. Vashishta, R. Kalia, A. Nakano, C. S. Tiwary and P. M. Ajayan, Adv. Mater., 2017, 29, 1703754 CrossRef PubMed.
- I. H. Kwak, I. S. Kwon, T. T. Debela, H. G. Abbas, Y. C. Park, J. Seo, J. Ahn, J. H. Lee, J. Park and H. S. Kang, ACS Nano, 2020, 14, 11995–12005 CrossRef CAS PubMed.
- S. Jiménez Sandoval, D. Yang, R. F. Frindt and J. C. Irwin, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3955–3962 CrossRef PubMed.
- I. S. Kwon, I. H. Kwak, T. T. Debela, H. G. Abbas, Y. C. Park, J. Ahn, J. Park and H. S. Kang, ACS Nano, 2020, 14, 6295–6304 CrossRef CAS PubMed.
- M. Naz, T. Hallam, N. C. Berner, N. McEvoy, R. Gatensby, J. B. McManus, Z. Akhter and G. S. Duesberg, ACS Appl. Mater. Interfaces, 2016, 8, 31442–31448 CrossRef CAS PubMed.
- O. L. Krivanek, M. F. Chisholm, V. Nicolosi, T. J. Pennycook, G. J. Corbin, N. Dellby, M. F. Murfitt, C. S. Own, Z. S. Szilagyi, M. P. Oxley, S. T. Pantelides and S. J. Pennycook, Nature, 2010, 464, 571–574 CrossRef CAS PubMed.
- L. P. Hansen, Q. M. Ramasse, C. Kisielowski, M. Brorson, E. Johnson, H. Topsøe and S. Helveg, Angew. Chem., Int. Ed., 2011, 50, 10153–10156 CrossRef CAS PubMed.
- I. S. Kwon, I. H. Kwak, J. Y. Kim, T. T. Debela, Y. C. Park, J. Park and H. S. Kang, ACS Nano, 2021, 15, 5467–5477 CrossRef CAS PubMed.
- I. H. Kwak, I. S. Kwon, G. M. Zewdie, T. T. Debela, S. J. Lee, J. Y. Kim, S. J. Yoo, J.-G. Kim, J. Park and H. S. Kang, ACS Nano, 2022, 16, 4278–4288 CrossRef CAS PubMed.
- C. Tsai, K. Chan, F. Abild-Pedersen and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 13156–13164 RSC.
- H. Vrubel, T. Moehl, M. Grätzel and X. Hu, Chem. Commun., 2013, 49, 8985 RSC.
- M. A. Lukowski, A. S. Daniel, C. R. English, F. Meng, A. Forticaux, R. J. Hamers and S. Jin, Energy Environ. Sci., 2014, 7, 2608–2613 RSC.
- A. Ambrosi, Z. Sofer and M. Pumera, Chem. Commun., 2015, 51, 8450–8453 RSC.
- A. Y. S. Eng, A. Ambrosi, Z. Sofer, P. Šimek and M. Pumera, ACS Nano, 2014, 8, 12185–12198 CrossRef CAS PubMed.
- X. Chia, N. A. A. Sutrisnoh, Z. Sofer, J. Luxa and M. Pumera, Chem. – Eur. J., 2018, 24, 3199–3208 CrossRef CAS PubMed.
- S. R. Kadam, A. N. Enyashin, L. Houben, R. Bar-Ziv and M. Bar-Sadan, J. Mater. Chem. A, 2020, 8, 1403–1416 RSC.
- D. F. Shriver, M. Weller, T. Overton, J. Rourke and F. Armstrong, Inorganic Chemistry, Oxford University Press, Oxford, 6th edn, 2014 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05631c |
| ‡ Throughout this manuscript, we prefer to use the conventional term ‘solid solution’ to highlight the substitutional nature of cation-mixed quasi-binary TMD, in which one cation (solute) occupies some of the lattice positions of the other cation (solution) in the host lattice; to differentiate from a broader term ‘alloys’, which can either be solid solutions or compounds with definite chemical composition and crystal structures.78 |
| This journal is © The Royal Society of Chemistry 2023 |